

Abstract
The mycobacterial D-arabinofuran is a common constituent of both cell wall mycolyl-arabinogalactan (AG) and the associated lipoarabinomannan (LAM), and thus accorded critical structural and immunological roles. Despite a well recognized importance, progress in understanding its full structural characteristics beyond the non-reducing terminal motifs has hitherto been limited by available analytical tools. An endogenous arabinanase activity recently isolated from Mycobacterium smegmatis was previously shown to be capable of releasing large oligoarabinosyl units from AG. Advanced tandem mass spectrometry utilizing both low and high energy collision induced dissociation now afforded a facile way to map and directly sequence the digestion products which were dominated by distinctive Ara18 and Ara19 structural units, together with Ara7 and lesser amount of Ara11 and Ara12. Significantly, evidence was obtained for the first time which validated the linkages and branching pattern of the previously inferred Ara22 structural motif of AG, on which the preferred cleavage sites of the novel arabinanase could be localized. The established linkage-specific MS/MS fragmentation characteristics further led to identification of a galactosamine substituent on the C2 position of a portion of the internal 3,5-branched Ara residue of the AG of M. tuberculosis, but not that of the non-pathogenic, fast growing M. smegmatis.
One of the most prominent macromolecular entities of all mycobacterial cell walls is the D-arabinofuran, a common constituent of both arabinogalactan (AG) and lipoarabinomannan (LAM) (1). In the chemical setting of AG, the central role of arabinan appears to be in maintaining the structural integrity of the cell wall proper by tethering the outer mycolic acid lipid barrier to the underlying peptidoglycan layer through the flexible glycosyl linkages of AG to form the mycolylarabinogalactan-peptidoglycan (mAGP) complex. Its most characteristic structural feature is a non-reducing terminal hexaarabinofuranosyl (Ara6) motif, Arafβ1→2Arafα1→5(Arafβ1→2Arafα1→3)Arafα1→5Arafα1→, where both the terminal β-sAraf and the penultimate 2-α-Araf serve as the anchoring points for the mycolic acids. In contrast, the non-reducing termini of the arabinan in LAM appear to be less strictly branched and a linear tetraarabinofuranosyl (Ara4) motif, Arafβ1→2Arafα1→5Arafα1→5Arafα1→, is known to coexist with the branched Ara6 termini. Through varying degree of mannose-capping at its non-reducing terminal β-Araf and attached at the reducing end to a lipomannan anchor, the arabinan in LAM plays instead pivotal immuno-modulatory functions in host-pathogen interactions and disease outcome (reviewed in (2, 3)).
Despite the well recognized importance of the mycobacterial arabinans and more than a decade of investigation following their first description (4, 5), structural details beyond the aforementioned non-reducing terminal motifs are still lacking. This severely hampers our delineation of the arabinosylation process. The homo-polymeric nature of the arabinan with intricate branching pattern and extreme size heterogeneity presents a major technical difficulty defying detailed structural analysis intended to reconstruct the intact polymer out of the chemically derived and characterized structural motifs. To date, principal findings from two complementary analytical approaches essentially conceptualize and confine our current understanding of the mycobacterial arabinan.
First, mass spectrometry (MS) analysis of the mild acid hydrolysates of the permethyl derivatives of arabinan from AG, has led to recognition of a speculative Ara22mer structural motif comprising two terminal Ara6 motifs (6). By virtue of the O-Me tag, it was possible to identify fragments from the non-reducing end formed by only a single arabinosyl cleavage. These included the expected single cleavages corresponding to Ara5, as well as Ara6, Ara7, Ara8 and Ara17-22 but not Ara9-16 (Fig. 1). In contrast, similar studies on LAM produced no evidence for the kind of branching and organization for AG-type arabinan. These seminal studies not only indicated some fundamental difference in the respective arabinan architecture but also gave rise to the current structural model in which the arabinan of AG was often depicted as comprising different alternative arrangement of three Ara22mers, to give a total of approximately 60–70 arabinosyl residues per galactan chain. Technically, this chemical strategy is powerful but degradative and tedious. Acid labile substituents could not be mapped and alternative arrangement superimposed on the Ara22mer framework cannot be delineated.
Fig. 1.
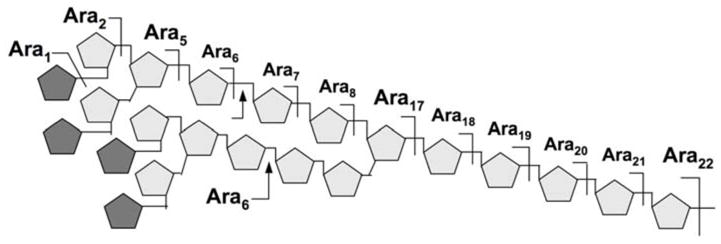
The Ara22mer structural model for AG. The proposed branching pattern was based primarily on the absence of Ara3,4 and Ara9-16 among the single chemical cleavage fragments detected by MS, as indicated above the 5-arabinan chain. Apart from the well established non-reducing terminal Ara6 motif, which can be released intact by the Cellulomonas arabinananse (indicated by arrows), the overall assembly of the arabinan motif remains speculative. The pentagons represent α-Araf residues, with darker shaded ones representing β-Araf.
A second powerful tool is provided in the form of a crude arabinanase preparation from a Cellulomonas species which was consistently shown to be capable of releasing the non-reducing end Ara6 and/or Ara4 (along with their mannose caps if present) from the arabinan while digesting the remaining into mostly a dimeric Arafα1→5Araf (Ara2) unit (7, 8). Thus a hallmark of digestion with this enzyme preparation on AG is the characteristic production of Ara6 and Ara2 whereas when applied to LAM, would additionally produced Ara4. The released arabinosyl oligomers could be rapidly profiled by high pH anion exchange chromatography on a Dionex LC system, as well as rapidly mapped and sequenced by MS and MS/MS analysis. Since its first application on mannose-capped LAM from wild type M. tuberculosis (7), the developed assay has since been effectively used to probe a variety of arabinan structures derived from different mycobacterial species, from drug resistant and other genetic mutants (9–12). A major limitation though is that it proves equally difficult to derive a full picture of the intact arabinan beyond the facile mapping of its terminal structural motifs.
Critical to gaining a better understanding at the structural level is the further development of analytical tools that would allow mapping and sequencing of larger arabinosyl motifs which retain all functionality including any additional non-arabinosyl substituents. A potential major breakthrough came when an endogenous M. smegmatis arabinanase activity that could release arabinan fragments of a size significantly larger than 10 oligoglucosyl units from AG was identified (13). The exact structures of the digestion products were then not reported although efforts have since been directed to purify and further characterize this novel arabinanase activity for both structural and functional studies. We report here how this enzyme has been effectively coupled to current generation of MALDI-MS instrumentation to provide new structural insight and direct evidence for the implicated Ara22 structural model of AG (Fig. 1) based on MALDI-MS mapping and MS/MS sequencing of the released intact oligoarabinosyl fragments. In the process, we defined the enzyme activity against AG and the precise location of a galactosamine substituent on the intact arabinan unit of AG from M. tuberculosis CSU20, which distinguishes it from that of M. smegmatis AG.
EXPERIMENTAL PROCEDURES
Chemical reagents
All chemical reagents were of the highest grade from Sigma/Aldrich unless otherwise specified. Milli-Q® water was used for all chemical reactions.
Preparation of soluble arabinogalactan
Mycobacterium smegmatis mc2155 was grown to mid-exponential phase and harvested using standard protocol (9). Wet cells (10 g) were delipidated with organic solvents and disrupted mechanically with the Soniprep 150 (Sanyo Gallenkamp PLC). The resulting homogeneous suspension was refluxed in 50% ethanol at 80ºC (3 × 2 h each). The combined supernatants were evaporated and digested with Proteinase K (Invitrogen; 1 mg/ml). After dialysis, cell wall core mAGP complex was obtained from the residual pellet after LAM, LM and PIMs extraction (9) and base-solubilized AG was prepared by a size exclusion chromatography using Sephacryl S-300 column (Amersham Biosciences) in water as previously described (4).
M. tuberculosis CSU20 was grown in glycerol alanine salts broth till late log phase and then harvested. Wet cells (10 g) were delipidated with chloroform/methanol/water (10:10:3) for 2 h at room temperature. For safety concern, growth and handling of M. tuberculosis till the delipidation step were carried out in a biosafety level 3 laboratory. The dried cellular residue was then suspended in breaking buffer containing protease inhibitor mixture (pepstatin A, phenylmethylsulfonyl fluoride, leupeptin, DNase and RNase in PBS) and disrupted mechanically using a French pressure cell. Triton X-114 was added to the cell lysate and LAM extraction was accomplished as described previously (12). The 27,000 g cell wall pellet was then further processed to generate base-solubilized AG (9), similar to the preparation of AG from M. smegmatis.
Purification of endogenous arabinanase
The endogenous arabinanase was obtained from M. smegmatis mc2155 as described previously (13) followed by partial purification. Briefly, M smegmatis mc2155 cell lysate from late mid log phase was centrifuged at 20,000 g for 1 h. The 60% (w/v) ammonium sulfate pellet was dissolved in 1.7 M ammonium sulfate in 10 mM phosphate buffer (pH 7.0, 2 mM MgCl2, 0.1 mM PMSF and 2 mM DTT) and applied to a 2.5 × 15 cm Phenyl Sepharose 6 hydrophobic interaction column (HI column, Amersham Biosciences). The column was eluted with a decreasing gradient of 1.7 M to 0 M ammonium sulfate in buffer spanning over 2 column volumes. Collected fractions corresponding to 0.4 M to 0 M ammonium sulfate were assayed for enzyme activity and those with highest activity were pooled and concentrated by Amicon Centriplus YM-10 (Millipore) at 4°C to a final protein concentration of 10 mg/ml.
Arabinanase activity assay and arabinan digestion
The arabinanase activity was assayed against 200 μg of AG as substrate, in 100 μl of 25 mM phosphate buffer (pH 7.0) at 37°C. Released arabinosyl oligomers were recovered from the reaction mixtures using an Amicon Microcon YM-30 (Millipore), centrifuged at 6500 g for 20 min, followed by washing the retentate three times with water. The respective carbohydrate contents in the flow through and retained fractions were quantified by phenol-sulfuric acid carbohydrate assay and the percentage of carbohydrate recovered in the flow through was taken as an indicator for the arabinanase activity. Typically only about 40–50% of the total carbohydrate in AG could be digested into the Microcon YM-30 flow through fractions after 4 h, using a substrate-to-enzyme ratio of 3:1 (μg carbohydrate : μg protein) in the total reaction buffer (100 μl). The retentate included the undigested galactan core to which the remnant of the arabinans were attached. Increasing further the enzyme protein concentration and/or prolonging the incubation time were found to afford only marginally better yield for the released products. All subsequent arabinanase digestions for structural analysis were therefore defaulted to these optimized conditions but increasing the total amount of AG substrate to 500 μg per digestion.
The arabinosyl oligomers recovered in the Microcon YM-30 flow through fraction were analyzed by MS directly or after further purification by eluting through a Superdex peptide 10/300 GL size exclusion HPLC column (Amersham Biosciences) with water, at a flow rate of 0.5 ml/min. The separation range of this column was estimated at around 200 to 5000 Da, as calibrated against maltooligosaccharides and hydrolyzed dextrans. The collected fractions were analyzed individually for enriched profiles of a particular range of size distribution, or pooled to obtain a ‘total’ profile of the released arabinosyl oligomers over the range of 5-30 arabinosyl residues. Alternatively, the arabinanase digestion mixtures were fractionated directly on a Bio-Gel P10 column (1 × 47.5 cm, Bio-Rad Lab) with 100 mM sodium acetate (pH 7.0). Fractions collected were assayed for carbohydrate content by phenol-sulfuric acid method and pooled accordingly for MALDI-MS analysis. The resulting profiles indicated that the major arabinosyl oligomers detected were similar to those centrifuged through the Microcon YM-30. Selected pooled fractions were further desalted and/or separated using a HyperSEP Hypercarb porous graphitic carbon (PGC) cartridge (ThermoHypersil-Keystone). Carbohydrate samples were stepwise eluted from the PGC cartridge with 25% aqueous acetonitrile (w/w) and 25% aqueous acetonitrile containing 0.1% trifluoroacetic acid (TFA), after initial wash with water.
Chemical derivatization for MS analysis
The arabinan fragments released by the arabinanase were either permethylated directly or pre-reduced and permethylated using the NaOH/dimethylsulfoxide slurry method as described (14). Permethylated samples as recovered by chloroform/water partitioning were analyzed directly or further loaded onto a reverse phase C18 Sep-Pak cartridge (Waters) for fractionation into stepwise acetonitrile- and methanol-eluted fractions. Selected samples were also peracetylated with acetic anhydride:pyridine (1:1, v/v) at 80°C for 2 h and analyzed directly after removal of the reagents under a stream of nitrogen, or after further chloroform/water partitioning.
MALDI-MS and MS/MS Analyses
MALDI-MS profiling and CID MS/MS sequencing were performed on either a Q-TOF Ultima MALDI (Micromass) or a 4700 Proteomics Analyzer (Applied Biosystems), both operated in reflectron positive ion mode. For acquisition on the Q/TOF instrument, the permethylated or peraectylated samples in acetonitrile were mixed 1:1 with α-cyano-4-hydroxycinnamic acid (CHCA) matrix (5 mg/ml in 50% acetonitrile/0.1% TFA) for spotting onto the target plate. MS survey and CID MS/MS data were manually acquired. Argon was used as the collision gas with a collision energy manually adjusted (between 100~200 V) to achieve optimum degree of fragmentation for the parent ions under investigation. For data acquisition on the TOF/TOF instrument (4700 Proteomics Analyzer), the permethyl or peracetyl derivatives in acetonitrile were mixed 1:1 with 2,5-dihydroxybenzoic acid (DHB) matrix (10 mg/ml in water) whereas underivatized samples in water were mixed 1:1 with 10 mg/ml DHB matrix in acetonitrile, for spotting onto the target plate. For high energy CID-MS/MS on the TOF/TOF, the potential difference between the source acceleration voltage and the collision cell was set at 2 or 3 kV to obtain optimum fragmentation. The indicated collision cell pressure was normally increased from 3 × 10−8 torr (no collision gas) to 5 × 10−7 torr, or up to a maximum of 2 × 10−6 torr (argon).
RESULTS
The arabinanase and arabinan of M. smegmatis AG
The arabinanase activity was initially tested against the AG substrates and digestion conditions were optimized as described in the Experimental Procedures section. To provide an overall digestion profile, the filtrate or the pooled HPLC fractions containing the released arabinosyl oligomers were analyzed directly by MALDI-MS, before and after chemical derivatization. The permethyl or peracetyl derivatives would typically afford a spectrum of better quality in comparison with that of a native sample. More importantly, by virtue of having an O-Me or O-Ac tag on each of the OH groups not involved in glycosidic linkage, fragment ions arising through in source prompt fragmentation can be readily distinguished from intact molecular ion species. Thus, each of the Aran oligomers detected in the spectra of the derivatives (Fig. 2) can be confidently ascribed to being present originally among the digestion products, and not derived from loss of Ara residues from larger oligomers during MS ionization.
Fig. 2.
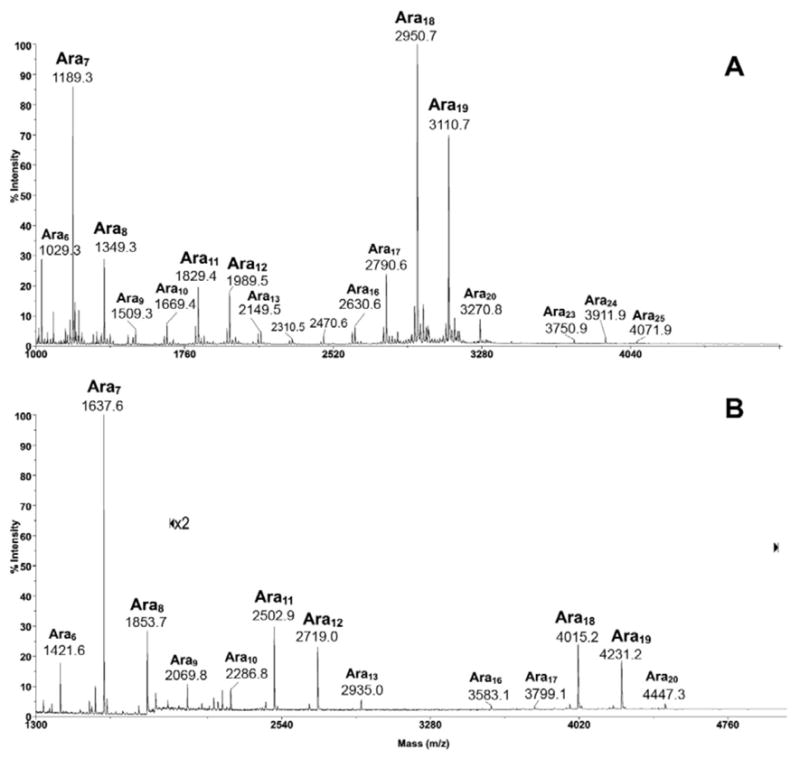
MALDI-MS profile of the permethyl (A) and peracetyl (B) derivatives of arabinosyl oligomers released from M. smegmatis AG by the endogenous M. smegmatis arabinanase. All molecular ions were detected as [M+Na]+.
As applied initially on solubilized AG from M. smegmatis, a typical arabinanase-digestion profile comprises three clusters of Aran signals centered at i) Ara7, ii) Ara11,12, and iii) Ara18,19 (Fig. 2). Although not absolutely quantitative, it is obvious from the signal intensity that Ara7 and Ara18,19 constitute the major digestion products. The permethyl derivatives, being smaller in size, allow a more ready detection of larger oligomers. The molecular ion signals afforded by Ara18,19 relative to those of the smaller oligomers are therefore significantly higher in intensity in the spectrum of the permethyl derivatives (Fig. 2A), in comparison with those afforded by the native (not shown) and peracetyl derivatives (Fig. 2B). Other than that, the rather similar MS profiles indicate that no significant chemical degradation was induced during the permethylation process.
To define the sequence and branching pattern of the major arabinosyl oligomers detected, each of the [M+Na]+ molecular ions afforded by the permethyl derivatives was selected for collision induced dissociation (CID) MALDI-MS/MS analysis. Typically for low energy CID MS/MS on Q/TOF, multiple glycosidic cleavages involving successive losses of arabinosyl residues from the parent ions can be observed. To avoid ambiguity in distinguishing a reducing terminal fragment ion from a non-reducing terminal ion derived from a homoploymer such as the arabinan, MS/MS were mostly performed on samples that were pre-reduced into oligoarabinosyl alditols before permethylation. The resulting molecular ions are therefore 16 u higher than the non-reduced counterparts.
For Ara7 (Fig. 3A), it is clear that loss of one and two Ara residues from the parent ions occurred readily through single glycosidic cleavage to give the Y ions at m/z 1031 and 871. Loss of 3 and 4 Ara residues could only be derived through double cleavages, giving rise to the Y ions at m/z 697 and 537, respectively, both of which carry a total of two free OH groups. Importantly, single cleavage loss of 5 Ara residues could again be detected at m/z 391, therefore indicative of a branch point. Similar pattern was afforded by Ara8 in which a distinctive non-reducing terminal branched Ara5 motif was likewise implicated (data not shown). The data is thus consistent with the Ara7 and Ara8 detected being derived from the well established non-reducing terminal motif of AG and that the endogenous M. smegmatis arabinanase appears to cleave at 2–3 arabinosyl residues from the branch point. This is to be contrasted with the Cellulomonas arabinanase, which preferentially releases Ara6. Another major difference is that instead of digesting the rest of the arabinan into Ara2 units, the M. smegmatis arabinanase can also release larger oligomers intact, as represented by Ara11,12 and Ara18,19 among the major products (Fig. 2).
Fig. 3.
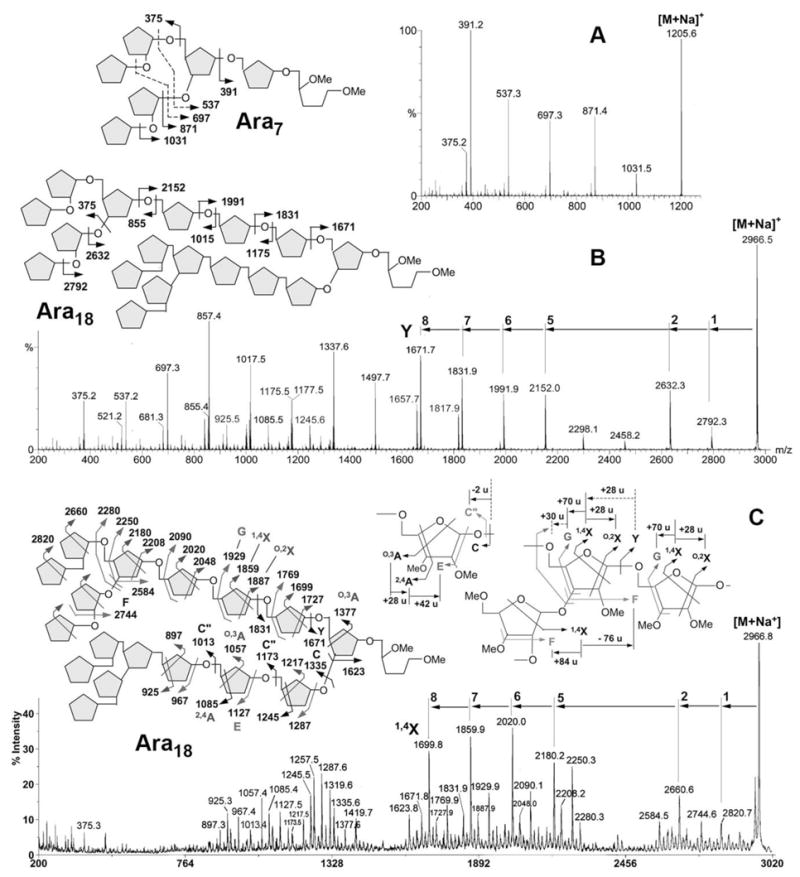
MALDI CID MS/MS analysis of permethylated oligoarabinosyl alditols representing the major arabinanase digestion products, Ara7 (A), and Ara18 (B, C). The major fragment ions afforded by the sodiated parent ions under low energy CID MS/MS (A, B) are mostly reducing terminal Y ions and non-reducing terminal C ions. The origins of single cleavage product ions are as indicated on the respective schematic drawings. Other cleavage ions are mostly derived from double cleavages, at 14 u lower than the expected single cleavage ions. For Ara7, the origins of the double cleavage ions are also indicated by dotted arrow. For Ara18, the 2,4A ions at m/z 925, 1085 and 1245 were afforded by both low (B) and high energy (C) CID-MS/MS. Assignment of the major fragment ions afforded by the latter is schematically illustrated on the Figure along with the adopted nomenclature and distinctive mass differences relating one characteristic series of ions to another.
The low energy CID MS/MS fragmentation pattern afforded by Ara18 (Fig. 3B) maintains the same characteristics with respect to loss of Ara1-5 residues from the parent ion. Namely, loss of 1, 2 and 5 residues but not 3 and 4 residues could occur through single cleavage. Beyond 5 residues, loss of 6, 7, and 8 Ara residues could similarly occur through single cleavage (m/z 1991, 1831 and 1671) but Y ions corresponding to loss of 9-16 Ara residues could only be derived through double cleavages (m/z 1497, 1337, 1177, 1017, 857, 697, 537, 377). In accordance with the original Ara22mer model (Fig. 1), the data thus implicates another branch point located proximal to the reducing end, from which two unique Ara8 units extend.
To seek further evidence for the deduced branching pattern, high energy CID MS/MS, which is known to favor cross ring cleavage ions and other satellite ions arising from concerted elimination of substituents around the ring (15–17), was additionally performed on a MALDI-TOF/TOF. Most of the fragment ions afforded are commonly attributed to single CID event and thus confined to the same glycosyl residue even when more than one bonds are ruptured. It is therefore anticipated that the mass spectrum will not contain fragment ions that arise by cleavages at more than one sites leading to the successive losses of glycosyl residues. A direct comparison against the low energy CID MS/MS spectra revealed that, instead of the usual Y ions, the corresponding cross-ring cleavage 1,4X ions at 28 u higher were clearly favored (Fig. 3C). As expected for Ara18, only the 1,4X ions resulting from loss of 1–2 and 5–8 Ara residues were detected whereas those ions that could only be derived via successive glycosidic cleavages at 2 or more distinct sites were not observed. More importantly for linkage assignment, detection of an O,2X ion at 28 u higher than the 1,4X is indicative of the loss of an Ara residue that is non-substituted at C2. In addition, concerted elimination of substituents at C2-C3 and C3-C4 gave rise to the satellite ions referred to as the F and G ions (16), respectively, at 84 u and 70 u higher than the 1,4X ion (see schematic drawing on Fig. 3C). Thus, a ready detection of the G and O,2X ions but not F ions for cleavages at each of the fifth to eighth Ara residues from non-reducing end (Fig. 3C) provides a good supporting evidence for a stretch of 5-linked Ara chain.
In contrast, only F and not G ion was produced for cleavage at the second Ara residue, as would be expected, since the non-reducing terminal Ara is known to be attached to C2 of this penultimate Ara residue. The next F ion in series at m/z 2584, which corresponds to loss of 2 Ara residues, places the terminal Ara2 unit to either C2- or C3- of a third Ara residue. In this case, it would be 3,5-disubstituted based on the presence of its G and O,2X ions at m/z 2250 and 2208, respectively. Taken together, careful delineation of the characteristic reducing end fragment ions described above allows an unambiguous linkage specific assignment of the non-reducing terminal Ara5 motif, which is extended at the reducing end by another three 5-linked Ara residues to give an Ara8 moiety. This assignment is further corroborated by the non-reducing end fragment ions, centered around m/z 900–1400.
Notably, the O,3A ions are specific to C5 substitution whereas the 2,4A ions and E ions further confirm that the C3 and C2 positions are not substituted, respectively. Thus the detection of these series of cleavage ions for the sixth to eighth Ara residues from the non-reducing end is in full accord with the conclusion made from assigning the reducing end fragment ions. At the ninth Ara residue, deduced to be the branched Ara, only O,3A (m/z 1377) and not 2,4A ion was found, which indicates that both C5 and C3 were substituted since a 2,4A ion would have been detected if only C5 and not C3 was substituted. Together with the F ion at m/z 1623, which corresponds to loss of 8 Ara residues from C3 (or C2), the detailed structure of the Ara18 can thus be definitively established, as drawn in Fig. 3C.
The consistent mass shifts observed in both low and high energy CID MS/MS analyses of Ara19 and Ara20 (data not shown) unequivocally demonstrate that these larger arabinosyl oligomers are based on the same Ara18 architecture but further extended by one and two Ara residues at the reducing end, respectively. The emerging picture therefore suggests that the endogenous M. smegmatis arabinanase prefers to cleave the arabinan at one to three Ara residues away from the reducing end of a branched 3,5-Ara site. Cleavage at either the site distal or proximal to the reducing end (sites I and II, Fig. 4A) would give Ara7 or Ara18,19, respectively, as the most abundant digestion products. Cleavages at both sites, on the other hand, would be expected to yield the less abundant but prominent Ara11, 12 products, as depicted schematically in Fig. 4A. This model predicts that the Ara11 and Ara12 products each comprises several possible isomeric structures arising from different permutation of the actual cleavage sites, which can be inferred from the MS/MS sequencing data afforded.
Fig. 4.
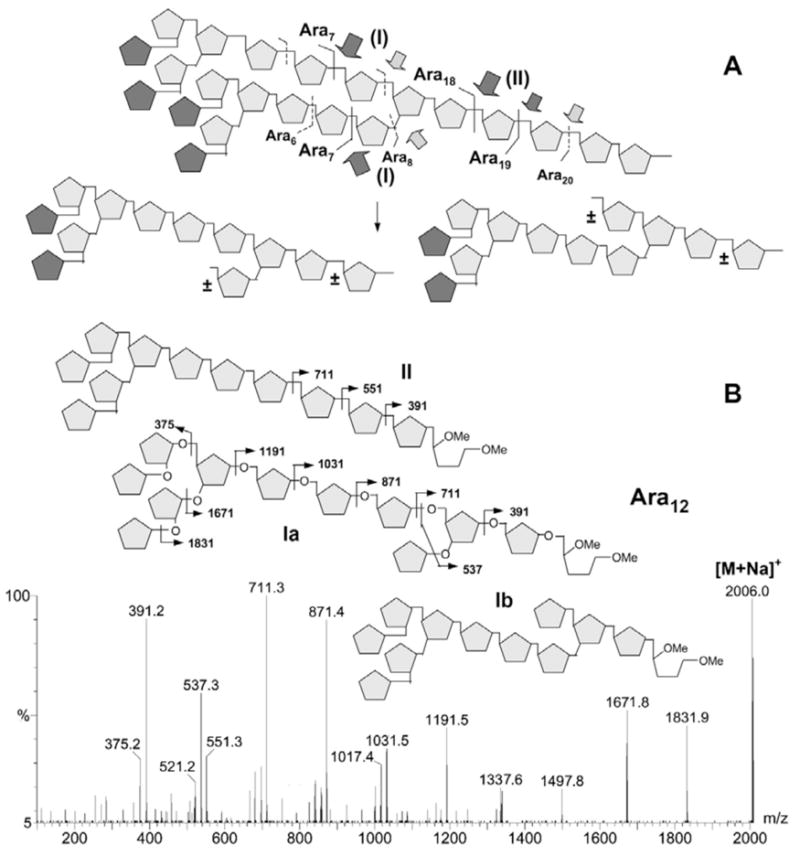
Deduced model for the preferred cleavage sites of the M. smegmatis arabinanase on the Ara22mer from AG (A) and low energy CID MS/MS analysis of one of the digestion products, Ara12 (B). In (A), the most preferred sites as deduced from the major digestion products detected (Fig. 2) are indicated by darker arrows. In addition to single-cleavages at either of the sites I and site II to give the major Ara6-8 and Ara18-20 products, respectively, the two isomeric products derived from cleavages at site II in combination with either of the sites I are shown. “±” denotes the Ara residues which may or may not be present depending on the exact cleavage sites. The lack of absolute specificity results in a heterogeneous mixture of isomeric Aran products, especially those corresponding to Ara11-12. The darker pentagon represents terminal β-Araf in the model drawn in (A) but cannot be distinguished from other pentoses by MS/MS analysis. In (B), the low energy CID MS/MS spectrum is consistent with the Ara12 carries a single Ara on either the C3- (structure Ia) or C5-position (structure Ib) of the internal branched 3,5-Ara residue. The additional presence of m/z 551 implies that the structural isomers resulting from removal of the entire Ara8 motif are also present (structure II, showing only one of the two possible isomers). The low energy CID MS/MS analysis alone cannot distinguish between the two alternatives of removing the Ara7,8 from either arm. However, for the more abundant structure I, additional high energy CID MS/MS analysis clearly identified the presence of both isomers (data not shown).
For low energy CID-MS/MS of Ara12 (Fig. 4B), the significant drop in intensity for the Y ion corresponding to loss of 9 Ara residues (m/z 551) relative to those derived from loss of 8 and 10 Ara residues (m/z 711 and 391, respectively) via single cleavage is indicative of a branch point located on the third Ara residue from the reducing end. As would be expected, removal of an Ara7 from either arm of a branched Ara19 by the arabinanase would leave a single Ara residue “stub” at the implicated branch point. Loss of 9 Ara residues from this Ara12 product (structures Ia and Ib in Fig. 4B) could thus occur only via double cleavages to give the Y ion at m/z 537. The presence of the single-cleavage Y ion at m/z 551 at lower intensity nevertheless implicates the existence of other isomeric Ara12 structures, one of which (structure II, Fig. 4B) could conceivably be derived from complete removal by the arabinanase of the entire Ara8 unit by cleaving right at the branch point of an Ara20. Analogous MS/MS analysis shows that the Ara11 isomers were likewise derived mostly from removal of either an Ara7 from Ara18 or Ara8 from Ara19 (data not shown). On the other hand, heterogeneity in the location of the branch point as counted from the non-reducing end, was not explicitly supported by either the low or high energy CID MS/MS data acquired. This is in accordance with the rather homogeneous Ara18,19 structure described earlier (Fig. 3C).
The arabinan of M. tuberculosis CSU20 AG
Having established the analytical methods and defined the arabinanase activities with respect to the digestion products given and their associated CID MS/MS fragmentation pattern, attention was turned to AG from M. tuberculosis CSU20. Unexpectedly, MALDI-MS mapping revealed that in addition to the regular Aran series of peaks similar to those afforded by AG from M. smegmatis, another previously not-observed, Aran-related signal series could be detected, which are best rationalized as Aran+X (Fig. 5). MALDI-MS analysis of the native sample (Fig. 5A) clearly defined the moiety X as an increment of 161 u, which was shifted to a difference of 287 u after peracetylation (Fig. 5B), and therefore consistent with it being a non-N-acetylated hexosamine (HexN) residue. Curiously, while the regular arabinosyl oligomers released by the arabinanase from the CSU20 AG were similarly clustered at Ara7, Ara11,12 and Ara18,19, the HexN containing Aran were mostly represented by Ara13,14 and Ara20, 21. These are respectively two additional Ara residues from the major non-HexN containing products with the conspicuous absence of one corresponding to Ara9 (Ara7+ 2 Ara residues). Both the analyses of native and peracetyl derivatives indicate that the HexN carrying Aran species constitute a significant proportion of the digestion products but are apparently of less abundance than their non-HexN containing counterparts.
Fig. 5.
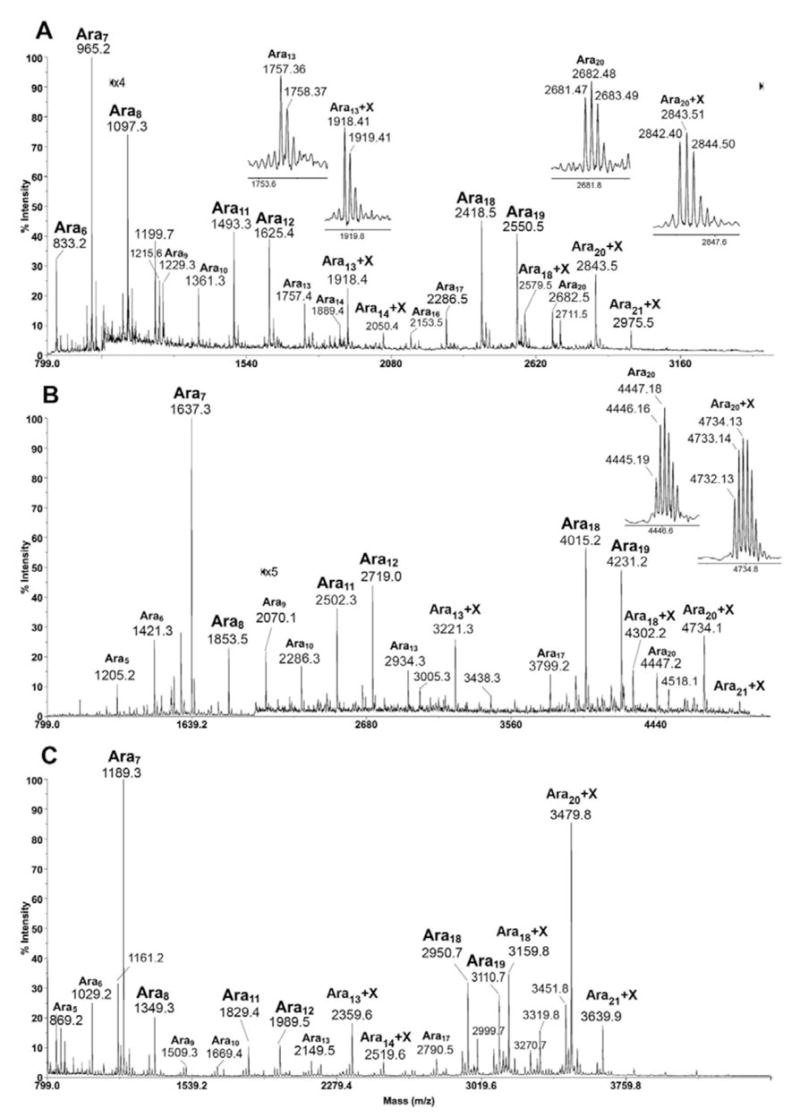
MALDI-MS profile of the native (A), peracetyl (B) and permethyl (C) derivatives of arabinosyl oligomers released from M. tuberculosis CSU20 AG by arabinanase. The HexN carrying peaks are tentatively assigned as Aran+X ion series here, which include Ara12-14+X and Ara17-21+X. Direct permethylation apparently enhanced the sensitivity of detection of the Aran+X series, leading to detection of species larger than Ara21+X (C). The isotopic clusters of X containing Aran were selectively magnified to show clearly the mass increment from the original Aran counterpart.
As noted previously (18), permethylation of the peracetyl derivatives is expected to convert the free amine into –NMeAc. This was indeed observed with the mass increment now shifted to 245 u, corresponding to the expected residual mass for permethylated HexNAc (data not shown). Further linkage analysis of the resulting products confirmed the identity of a terminal GalNAc and not GlcNAc (data not shown). On the other hand, direct permethylation of glycans containing free amine will lead to ready incorporation of 3 methyl groups on the amine, converting it into positive charge carrying quaternary ammonium. Consequently, a mass increment of 209 u would be observed instead, which corresponds to an HexN(Me3)+ substituent concomitant with losing the sodium cation adduct. Introducing a permanent positive charge into the molecules apparently also enhanced their being detected by MALDI-MS and hence an increase in signal intensity. It is clear from the resulting spectrum (Fig. 5C) that the major HexN carrying Aran products correspond to Ara18,20,21 and Ara13,14.
In contrast to the high energy CID-MS/MS spectra afforded by the permethyl derivatives of Aran, the cluster of signals that would correspond to the non-reducing terminal fragment ions of the HexN(Me3)+ carrying Ara18 are clearly missing (Fig. 6A). It can be further established from careful examination that the nominal monoisotopic masses of all fragment ions afforded are of even m/z values and thus consistent with them carrying a single nitrogen atom. This observation provides the first indication that the HexN substituent is located proximal to the reducing end. Being derivatized by permethylation into trimethylated quaternary ammonium, the charge localization on HexN(Me3)+ could conceivably favor the formation of ion series carrying the substituent.
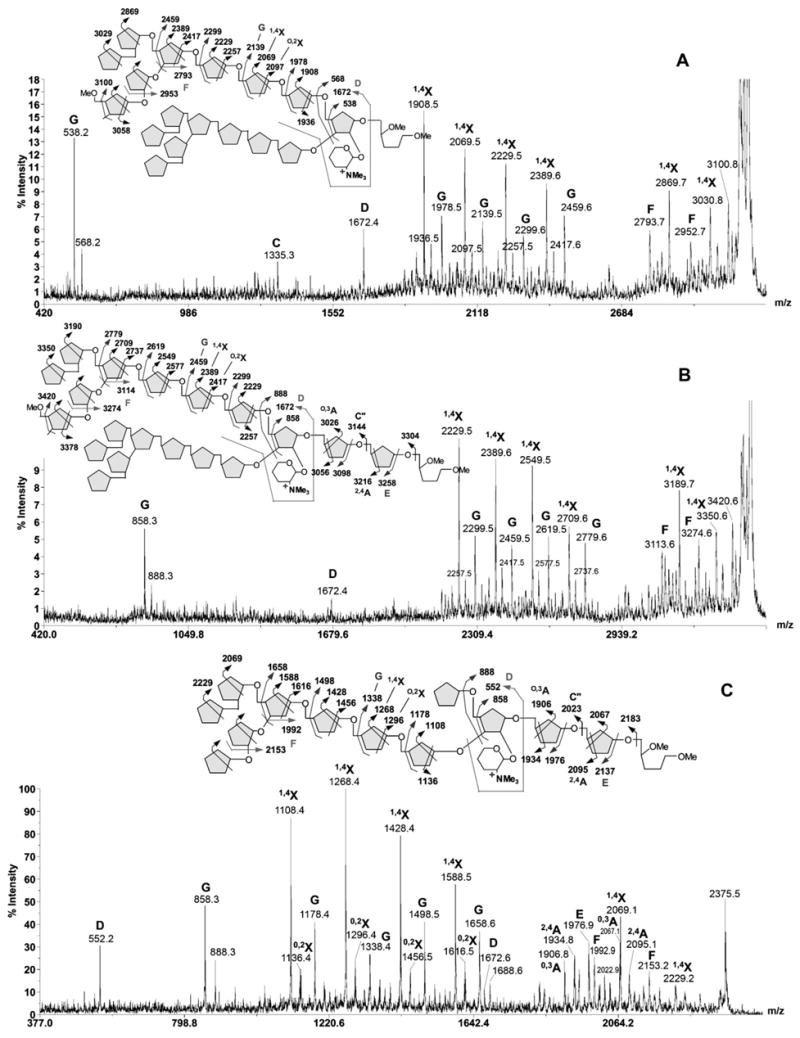
Fig. 6.
High energy CID MS/MS analysis of permethylated Ara18+GalN (A), Ara20 +GalN (B) and Ara13+GalN (C), as performed on MALDI-TOF/TOF. The sample was pre-reduced to give oligoarabinosyl alditols before permethylation for MALDI-MS/MS analysis. Major fragment ions observed are assigned as labeled and schematically illustrated. For simplicity, only one of the major isomeric structures is shown in (C).
Thus, for Ara18+HexN, loss of one and two Ara residues gave the first cluster of signals proximal to the parent ion. The dominant 1,4X ion signals at m/z 3030 and 2869 are both accompanied by signals at 76 u lower, corresponding to the F ions described above. After a gap of 2 Ara residues, the 1,4X ion series resume at m/z 2388, 2228, 2068 and 1908, corresponding to loss of 5 to 8 Ara residues, respectively. Each is accompanied by the O,2X ion (+28 u) and G ion (+70 u), a pattern similar to that produced by non-HexN substituted Ara series and thus indicate that the HexN-substituted Ara18 may share the same structural architecture as the non-substituted Ara18. Importantly, loss of both Ara8 arms gave rise to a prominent G ion at m/z 538, which localizes the HexN substitution to the reducing end Ara2 unit. A final supporting evidence comes from the ion at m/z 1672, which can be assigned as the double cleavage ion as depicted in the schematic drawing. In analogy to what was consistently observed in high energy CID MS/MS analysis of N-glycans, this ion can be referred to as the D ion (19, 20) resulting from glycosidic cleavage at a branched glycosyl residue concomitant with elimination of its C3-substituent. Although similar phenomenon has not been examined in furanose, it is nevertheless consistent with elimination of the Ara8 moiety from the 3-position of a 3,5-branched Ara residue located next to the reducing end Ara, as in the already established Ara18 structure. Such interpretation would also unequivocally localize the HexN substituent to the remaining C2-position of the branched Ara residue.
The Ara20+HexN component (Fig. 6B) afforded a fragmentation pattern very similar to that of Ara18+HexN. Notably, similar loss of 1, 2, 5–8 Ara residues gave rise to the corresponding 1,4X ion series, together with the O,2X ions, F ions and G ions. The G ion deriving from the inner branched 3,5-Ara residue was shifted from m/z 538 to 858, corresponding exactly to an increment of 2 Ara residues whereas the D ion at m/z 1672 remains unchanged. The fragmentation pattern afforded therefore strongly indicated that the extra 2 Ara residues are located at the reducing end, most likely representing further 5-linked Ara extension from the Ara18+HexN structure, as drawn. Supporting evidence were found in other minor peaks clustering around the first two 1,4X ions, which may be assigned as the HexN-containing, non-reducing terminal C”, O,3A and 2,4A ions, deriving from cleavages at the second and third Ara residues from the reducing end. Since Ara20+HexN represents the most abundant HexN-containing, arabinanase digested product, it may be deduced that the presence of HexN substituent on the branched Ara residue probably disfavor arabinanase digestion at this site and shifted the preferred cleavage site to two Ara residues further away, making the Ara20+HexN as the major digestion product. Accordingly, the favored product resulting from additional removal of the terminal Ara7 by the arabinanase is Ara13+HexN rather than Ara11+HexN.
Earlier high energy CID-MS/MS analyses have shown that the major Ara11,12 products represent a further trimming away of the non-reducing terminal Ara7 motif from either arm of the Ara18,19 structures, leaving either the 3- or 5-position of the 3,5-branched Ara with a single Ara residue although other minor isomers are evidently present. It could be inferred that the Ara13+HexN is likewise based on such structural architecture but with additional 2 Ara residues at the reducing end, similar to the case of Ara20+HexN. Indeed, the strong signal at m/z 858 (Fig. 6C) provides the first indication that the HexN-containing reducing terminal structure, after losing both extensions on the 3- and 5- position of the branched Ara, is the same as that of Ara20+HexN. This is further supported by the expected series of 1,4X, O,2X, F, and G ions. Importantly, additional HexN-containing, non-reducing terminal C”, O,3A, 2,4A and E ions, deriving from cleavages at the second and third Ara residues from the reducing end can also be easily identified. Finally, the D ions are detected at m/z 552 and 1672, supporting the presence of both isomers, namely with either arm extended by ether a single Ara or a complete Ara8 unit.
The identification and subsequent sequence determination of Ara13+HexN and Ara20+HexN by MS/MS indicate that the HexN substituent is located proximal to the reducing end, at the inner branched Ara residue. This is consistent also with the absence of ions corresponding to Ara7-9+HexN since the Ara7 was deduced to be derived from non-reducing end. Following the same rationale, further digestion of Ara20+HexN with the Cellulomonas arabinanase should remove the non-reducing terminal Ara6 and leave behind an Ara2 unit on both arms, which may or may not be further digested away. Indeed, it was found that the major product after further arabinanase digestion was Ara6+HexN, which afforded the molecular ion signal at m/z 1238 after permethylation (Fig. 7A). High energy CID MS/MS (Fig. 7B) produced a predicable fragmentation pattern similar to those described above. Since the product was not pre-reduced, the characteristic G ion at m/z 858 was shifted to 16 u lower at m/z 842. This is accompanied by a full array of fragment ions carrying the HexN(Me3)+ moiety, as assigned on Fig 7B. Importantly, the D ion at m/z 392 identifies a non-extended 5-arm although the presence of the other isomer cannot be ruled out. Together, the data is consistent with trimming away of the Ara8 extension on the 5-arm but retaining an Ara2 unit on the 3-arm. This model is compatible with the digestion of other Aran+HexN product since as small as Ara4 +HexN and at least up to Ara9+HexN were detected (Fig. 7A). However, these lower abundant products are likely to represent a mixture of isomeric products instead of discrete structures and further MS/MS analysis was not attempted.
Fig. 7.
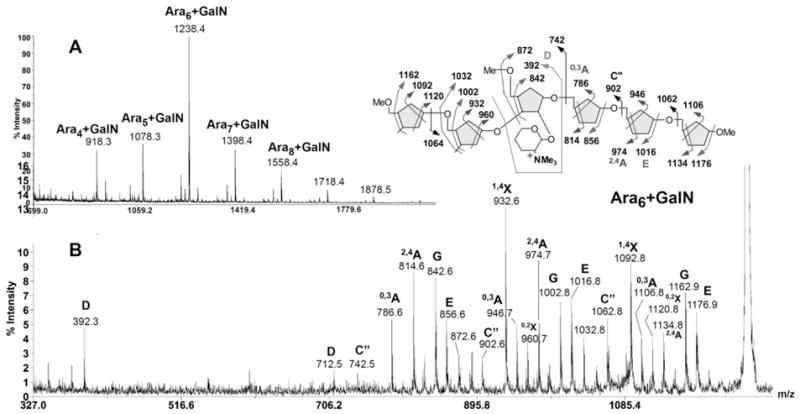
MALDI-MS mapping of the Cellulomonas arabinanase digested Aran+GalN series (A) and high energy CID MS/MS analysis of the major product, Ara6+GalN (B). After additional Cellulomonas arabinanase digestion, the permethyl derivatives of GalN-containing products were isolated from those non-GalN containing Aran series by reverse phase C18 solid phase extraction cartridge. The trimmed Aran+GalN products were eluted in the later methanol fraction after acetonitrile wash.
DISCUSSION
Recent advances in mass spectrometry have invigorated the field of complex glycan mapping and sequencing. While significant attention has been focused on protein glycosylation particularly of mammalian type, much less has been devoted to advancing the analysis of cell wall associated polymeric glycans. Sequencing of the latter by MS techniques has always been daunted by its large size and often the same mass values of their constituents. Homopolymeric structures such as the glucan, galactan, mannan and arabinan do not carry particularly favorable sites of cleavage and the nature of problem often boils down to delineating its linkage and branching details. NMR techniques aside, their analyses by MS were invariably performed on chemically derivatized and/or partially degraded products. In the context of mycobacterial arabinan, our previous MS analysis of the characteristic fragments derived by either partial acid hydrolysis (4–6, 21–23) or arabinanase digestion (7, 8, 10–12), is representative of the few viable approaches commonly adopted. Our current work essentially built on these earlier successes but benefited from two significant advances.
The first stemmed from the availability of the endogenous arabinanase isolated from M. smegmatis (13), which was shown unequivocally here to be capable of releasing oligoarabinosides larger than the previous Cellulmonas arabinanase did. Based on the relatively well defined structural model of AG, we could deduce the specificity of this M. smegmatis arabinanase which appears to go after sites at one or more Ara residues from a branch point. In AG, there will be two such distinct sites, denoted here as the one that is distal (I) or proximal (II) to the reducing end (Fig. 4A). The Cellulomonas arabinanase prefers site I and produces predominantly the non-reducing terminal Ara6 (8). It is unclear if it also cleaves at site II since an internal Ara6 not capped with β2-Ara has not been identified among the digestion products. The same crude enzyme preparation also contains other arabinanase activities that will cleave most of the internal arabinan into Ara2. It is possible that the released Ara6, which is either capped by Man or simply the β2-Ara, would be resistant to this further digestion and thus survive as distinctive digestion products. In contrast, the M. smegmatis arabinanase prefers site II and seems to be devoid of activities that would further degrade the arabinan to Ara2. However, co-existing but incomplete activity against site I leads to recovery of both Ara7 and Ara18,19, along with Ara11,12 resulting from enzymatic cleavages at both sites.
The ability of the M. smegmatis arabinanase to release intact Ara18,19 allowed, for the first time, structural analysis on this large molecule without the uncertainty of losing other labile substituents through partial acid hydrolysis as employed previously. The mere fact that such a product was detected at all is a testament to previous work, which correctly deduced the branching pattern through a more laborious, indirect and sample consuming method (6). An equally important development though is the timely advent of MS instruments that enable high sensitivity and high mass range MS/MS that is applicable to such molecules. We have previously used MALDI-Q/TOF to perform MALDI-MS/MS on the Cellulomonas arabinanase digested products, namely the Ara6 (24), and we have now extended this to Ara18-20. As shown here, although this itself is powerful, it lacks in capability to give more definitive information with respect to the linkage.
A high energy CID MS/MS as performed on a MALDI-TOF/TOF fills the technical gap, although much more complicated in spectral assignment. We have, in fact, demonstrated through this work the first systematic assignment of the numerous cleavage ions afforded by a furanose-based glycan. We showed that what have been established as the fragmentation characteristics of high energy CID MS/MS on permethyl derivatives (15, 25) are equally applicable here albeit with refinement. In addition to the nomenclature proposed by Domon and Costello (26), which is widely used in all glycan MS/MS studies, we adopted additionally both the D ion nomenclature for the concerted double cleavage with specific elimination of 3-substituent (19) and the nomenclature proposed by Spina et al for the satellite ions involving concerted elimination of adjacent substituents around the ring (16). A point to note though is that while G ion arises by elimination of the exocyclic C3 and C4 substituents of a pyranose, the corresponding exocyclic C4 substituent of a pentofuranose is in fact C5 and its substituent (Fig. 3C). This is actually more similar to elimination of the exocyclic C4 and C5 substituents of a pyranose, which was noted before and additionally named H ion (20). Further, the commonly observed 1,5X ion for ring cleavage on a pyranose is now referred to as the 1,4X ion for the furanose based arabinan.
Although many ions not previously encountered in the analysis of pyranose based N-glycans (20) were additionally detected and could be rationalized, we have chosen here to focus mainly on those that are most abundant, reproducible, and sequence informative. For the furanoses, the trios of reducing terminal 1,4X, O,2X and G ions qualify as such and could be well corroborated by the non-reducing terminal O,3A, 2,4A and E ions, in all applicable cases. These were validated here against the reasonably well accepted Ara22 model, as well as other smaller Ara4-6mers, both synthetic and naturally derived (unpublished data), to make a convincing case of assignment. Fortuitously for the GalN-containing Aran, the localization of a fixed positive charge on the GalN proximal to the reducing end has favored the formation of these reducing terminal ions and these alone are sufficient to critically establish the novel structural motif, described in full for the first time. It also represents one true unique application of MALDI TOF/TOF where the low energy CID afforded by Q/TOF was found to give unsatisfactory fragmentation. It further illustrates how MS/MS could be most useful to define the location of additional substituent(s) in a comparative mapping study.
The GalN substituent is the most conspicuous distinguishing factor for an otherwise similar architecture for the arabinan of M. smegamtis AG versus that of M. tuberculosis. In agreement with an earlier work (18), our data unambiguously showed that it is a terminal substituent on the branched Ara residue. Previous analysis on the galactosaminyl arabinosides isolated through acid hydrolysis has implicated a ±Ara-5(GalN-2)Ara-5Ara motif (18), which is consistent with current model assuming that the 3-linked branched might be readily lost through acid hydrolysis whereas the glycosidic bond of the galactosaminylated Ara was somewhat stabilized by the positive charge carried on the free amine, as suggested previously. The use of a less destructive arabinanase digestion coupled with the established MS/MS sequencing method enabled us now to precisely localize it onto the Ara18-20 framework.
Unlike AG which yielded mostly Ara7 and Ara18,19 when digested with the endogenous arabinanase, the M. smegmatis LAM gave a full range of arabinosyl oligomers from Ara6 to Ara30 and beyond, without a clearly dominating product (27). This appears to be consistent with a long held view that there is probably no single unique branching pattern for the arabinans of LAM. The analytical methods established here are necessary to further substantiate this model especially in conjunction with detailed analyses of the various truncated LAM variants synthesized by the genetically manipulated mutants defective in LAM biosynthesis. Our initial data has already generated a working model in which the M. smegmatis LAM was depicted as having the linear stretches extending out from the basic Ara18-20 framework similar to that of AG (27). The full picture and structural details with respect to the arabinan of LAM will, however, only be revealed when each of the oligoarabinosyl units released by the endogenous arabinanase could be adequately resolved and then sequenced.
Abbreviations
- Araf
arabinofuranose
- AG
arabinogalactan
- CID
Collision Induced Dissociation
- mAGP
mycolylarabinogalactan peptidoglycan complex
- LAM
lipoarabinomannan
- LM
lipomannan
- MS
mass spectrometry
- MALDI
matrix-assisted laser-desorption ionization
- PIMs
phosphatidylinositol mannosides
- Q/TOF
Quadrupole/Time-Of-Flight
- TFA
trifluoroacetic acid
Footnotes
This work was supported by a Taiwan NSC grant 94-2311-B-001-071 to K.K.; NIH Grant AI-37139 to D.C.; and AI-33706 to M.M. Mass spectrometry analyses were performed at the National Core Facilities for Proteomics located at the Institute of Biological Chemistry, Academia Sinica, supported by a Taiwan NSC grant (94-3112-B-001-009-Y) and the Academia Sinica.
References
- 1.Brennan PJ. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2003;83:91–7. doi: 10.1016/s1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 2.Chatterjee D, Khoo KH. Mycobacterial lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology. 1998;8:113–20. doi: 10.1093/glycob/8.2.113. [DOI] [PubMed] [Google Scholar]
- 3.Briken V, Porcelli SA, Besra GS, Kremer L. Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol Microbiol. 2004;53:391–403. doi: 10.1111/j.1365-2958.2004.04183.x. [DOI] [PubMed] [Google Scholar]
- 4.Daffe M, Brennan PJ, McNeil M. Predominant structural features of the cell wall arabinogalactan of Mycobacterium tuberculosis as revealed through characterization of oligoglycosyl alditol fragments by gas chromatography/mass spectrometry and by 1H and 13C NMR analyses. J Biol Chem. 1990;265:6734–43. [PubMed] [Google Scholar]
- 5.Chatterjee D, Bozic CM, McNeil M, Brennan PJ. Structural features of the arabinan component of the lipoarabinomannan of Mycobacterium tuberculosis. J Biol Chem. 1991;266:9652–60. [PubMed] [Google Scholar]
- 6.Besra GS, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. A new interpretation of the structure of the mycolyl-arabinogalactan complex of Mycobacterium tuberculosis as revealed through characterization of oligoglycosylalditol fragments by fast-atom bombardment mass spectrometry and 1H nuclear magnetic resonance spectroscopy. Biochemistry. 1995;34:4257–66. doi: 10.1021/bi00013a015. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee D, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. Structural definition of the non-reducing termini of mannose-capped LAM from Mycobacterium tuberculosis through selective enzymatic degradation and fast atom bombardment-mass spectrometry. Glycobiology. 1993;3:497–506. doi: 10.1093/glycob/3.5.497. [DOI] [PubMed] [Google Scholar]
- 8.McNeil MR, Robuck KG, Harter M, Brennan PJ. Enzymatic evidence for the presence of a critical terminal hexa-arabinoside in the cell walls of Mycobacterium tuberculosis. Glycobiology. 1994;4:165–73. doi: 10.1093/glycob/4.2.165. [DOI] [PubMed] [Google Scholar]
- 9.Escuyer VE, Lety MA, Torrelles JB, Khoo KH, Tang JB, Rithner CD, Frehel C, McNeil MR, Brennan PJ, Chatterjee D. The role of the embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J Biol Chem. 2001;276:48854–62. doi: 10.1074/jbc.M102272200. [DOI] [PubMed] [Google Scholar]
- 10.Khoo KH, Douglas E, Azadi P, Inamine JM, Besra GS, Mikusova K, Brennan PJ, Chatterjee D. Truncated structural variants of lipoarabinomannan in ethambutol drug-resistant strains of Mycobacterium smegmatis. Inhibition of arabinan biosynthesis by ethambutol. J Biol Chem. 1996;271:28682–90. doi: 10.1074/jbc.271.45.28682. [DOI] [PubMed] [Google Scholar]
- 11.Khoo KH, Tang JB, Chatterjee D. Variation in mannose-capped terminal arabinan motifs of lipoarabinomannans from clinical isolates of Mycobacterium tuberculosis and Mycobacterium avium complex. J Biol Chem. 2001;276:3863–71. doi: 10.1074/jbc.M004010200. [DOI] [PubMed] [Google Scholar]
- 12.Torrelles JB, Khoo KH, Sieling PA, Modlin RL, Zhang N, Marques AM, Treumann A, Rithner CD, Brennan PJ, Chatterjee D. Truncated structural variants of lipoarabinomannan in Mycobacterium leprae and an ethambutol-resistant strain of Mycobacterium tuberculosis. J Biol Chem. 2004;279:41227–39. doi: 10.1074/jbc.M405180200. [DOI] [PubMed] [Google Scholar]
- 13.Xin Y, Huang Y, McNeil MR. The presence of an endogenous endo-D-arabinase in Mycobacterium smegmatis and characterization of its oligoarabinoside product. Biochim Biophys Acta. 1999;1473:267–71. doi: 10.1016/s0304-4165(99)00204-4. [DOI] [PubMed] [Google Scholar]
- 14.Dell A, Reason AJ, Khoo KH, Panico M, McDowell RA, Morris HR. Mass spectrometry of carbohydrate-containing biopolymers. Methods Enzymol. 1994;230:108–32. doi: 10.1016/0076-6879(94)30010-0. [DOI] [PubMed] [Google Scholar]
- 15.Stephens E, Maslen SL, Green LG, Williams DH. Fragmentation characteristics of neutral N-linked glycans using a MALDI-TOF/TOF tandem mass spectrometer. Anal Chem. 2004;76:2343–54. doi: 10.1021/ac030333p. [DOI] [PubMed] [Google Scholar]
- 16.Spina E, Sturiale L, Romeo D, Impallomeni G, Garozzo D, Waidelich D, Glueckmann M. New fragmentation mechanisms in matrix-assisted laser desorption/ionization time-of-flight/time-of-flight tandem mass spectrometry of carbohydrates. Rapid Commun Mass Spectrom. 2004;18:392–8. doi: 10.1002/rcm.1350. [DOI] [PubMed] [Google Scholar]
- 17.Mechref Y, Novotny MV, Krishnan C. Structural characterization of oligosaccharides using MALDI-TOF/TOF tandem mass spectrometry. Anal Chem. 2003;75:4895–903. doi: 10.1021/ac0341968. [DOI] [PubMed] [Google Scholar]
- 18.Draper P, Khoo KH, Chatterjee D, Dell A, Morris HR. Galactosamine in walls of slow-growing mycobacteria. Biochem J. 1997;327(Pt 2):519–25. doi: 10.1042/bj3270519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey DJ. Structural determination of N-linked glycans by matrix-assisted laser desorption/ionization and electrospray ionization mass spectrometry. Proteomics. 2005;5:1774–86. doi: 10.1002/pmic.200401248. [DOI] [PubMed] [Google Scholar]
- 20.Yu SY, Wu SW, Khoo KH. Distinctive characteristics of MALDI-Q/TOF and TOF/TOF tandem mass spectrometry for sequencing of permethylated complex type N-glycans. Glycoconj J. 2006;23:355–69. doi: 10.1007/s10719-006-8492-3. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee D, Lowell K, Rivoire B, McNeil MR, Brennan PJ. Lipoarabinomannan of Mycobacterium tuberculosis. Capping with mannosyl residues in some strains. J Biol Chem. 1992;267:6234–9. [PubMed] [Google Scholar]
- 22.Prinzis S, Chatterjee D, Brennan PJ. Structure and antigenicity of lipoarabinomannan from Mycobacterium bovis BCG. J Gen Microbiol. 1993;139:2649–58. doi: 10.1099/00221287-139-11-2649. [DOI] [PubMed] [Google Scholar]
- 23.Khoo KH, Dell A, Morris HR, Brennan PJ, Chatterjee D. Inositol phosphate capping of the nonreducing termini of lipoarabinomannan from rapidly growing strains of Mycobacterium. J Biol Chem. 1995;270:12380–9. doi: 10.1074/jbc.270.21.12380. [DOI] [PubMed] [Google Scholar]
- 24.Zhang N, Torrelles JB, McNeil MR, Escuyer VE, Khoo KH, Brennan PJ, Chatterjee D. The Emb proteins of mycobacteria direct arabinosylation of lipoarabinomannan and arabinogalactan via an N-terminal recognition region and a C-terminal synthetic region. Mol Microbiol. 2003;50:69–76. doi: 10.1046/j.1365-2958.2003.03681.x. [DOI] [PubMed] [Google Scholar]
- 25.Morelle W, Slomianny MC, Diemer H, Schaeffer C, Dorsselaer AV, Michalski JC. Fragmentation characteristics of permethylated oligosaccharides using a matrix-assisted laser desorption/ionization two-stage time-of-flight (TOF/TOF) tandem mass spectrometer. Rapid Commun Mass Spectrom. 2004;18:2637–2649. doi: 10.1002/rcm.1668. [DOI] [PubMed] [Google Scholar]
- 26.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5:397–409. [Google Scholar]
- 27.Shi L, Berg S, Lee A, Spencer JS, Zhang J, Vissa V, McNeil MR, Khoo KH, Chatterjee D. The carboxy terminus of EmbC from Mycobacterium smegmatis mediates chain length extension of the arabinan in lipoarabinomannan. J Biol Chem. 2006;281:19512–26. doi: 10.1074/jbc.M513846200. [DOI] [PubMed] [Google Scholar]