

Abstract
IFN-γ is a pro-inflammatory cytokine which also acts as a potent immunomodulatory agent. In this study, a replication-deficient recombinant avian (fowlpox) virus was engineered to express the murine IFN-γ gene (rF-IFN-γ) with the rationale of delivering concentrated levels of the cytokine to a local tissue microenvironment. Subcutaneous (s.c.) rF-IFN-γ administration resulted in IFN-γ production that (1) was restricted to the tissue microenvironment of the injection site and (2) was biologically active as evidenced by a significant increase of class I MHC expression levels in s.c. growing tumors following rF-IFN-γ administration. Infection of a highly tumorigenic murine cell line, MC38, with rF-IFN-γ functioned as an effective tumor cell-based vaccine by protecting mice from the formation of primary tumors and from subsequent tumor challenge. The cell-based vaccine was completely ineffective if mice were vaccinated with MC38 cells either pretreated with recombinant IFN-γ or infected with the wild-type fowlpox virus. Analysis of the regional lymph nodes draining the site of injection of the rF-IFN-γ based tumor cell vaccine revealed the presence of tumor specific cell lysis (CTL) as well as significant amount of lysis directed at NK-sensitive YAC-1 cells. Flow cytometric analyses coupled with functional assays confirmed the sustained presence of NK1.1+ cells within those draining lymph nodes for up to 5 days following rF-IFN-γ injection. Mice treated with NK cell-depleting antibodies prior to the injection of the rF-IFN-γ-infected MC38 tumor cells were not protected from primary tumor growth; analysis of the lymph nodes draining the injection site in NK-depleted mice, revealed an accompanying loss of the tumor specific CTL activity. The findings provide evidence that NK cells, known for their contributions to host innate immunity, also provide immunoregulatory signals required for the development of an adaptive immune response which, in turn, protected vaccinated mice against tumor growth.
INTRODUCTION
Recombinant interferon-gamma protein (rIFN-γ) is a potent pro-inflammatory cytokine that exerts a variety of anti-proliferative and immunomodulatory actions which have implicated it as a potential anti-cancer agent. Among its immunomodulatory effects are (a) the upregulation of major histocompatibility complex (MHC), Fas molecules and tumor associated antigens on tumor cells (1, 2) potentially making tumors better targets for the immune system, (b) the inhibition of the production of immunosuppressive factors such as TGF-β(3) and PGE2 (4) and (c) potent anti-angiogenic effects which inhibit tumor metastasis (5, 6). Despite those multiple actions that could negatively impact tumor growth, the effectiveness of rIFN-γ as a single anticancer agent has been disappointing. Clinical studies of patients diagnosed with late-stage cancers have revealed some antitumor responses (7, 8), but only at doses of rIFN-γ that are associated with severe and treatment-limiting systemic toxicities.
Unlike the type I interferons, IFN-γ is produced by a restricted group of cells that include T cells activated during adaptive immunity as well as NK and NK T cells that provide IFN-γ for the innate phase of the host immune response. Those observations indicate that IFN-γ actions are primarily local and, indeed, IFN-γ has been shown to enhance TH1 polarization by: (1) facilitating IL-12 production by DCs primed by microbial products (9) and (2) synergizing with T cell receptor (TCR) signals to maximally induce T-bet which controls IL-12 receptor expression in naive T cells and acts as a master regulator of TH1 differentiation (10). Several investigators have proposed that a more rational approach might be to deliver high concentrations of IFN-γ to the local tissue environment to exploit its antigen upregulating, angiostatic and immunomodulatory actions while avoiding the toxicities associated with its systemic administration. Indeed, research groups have engineered various recombinant viruses (11-15) and DNA-based technology (16) as delivery platforms to target IFN-γ to predetermined tissues. Those approaches have been shown to be safe, well-tolerated and capable of inducing anti-tumor immunity in experimental animals (14, 16). For example, the intratumoral administration of recombinant retro- or adenoviruses expressing the IFN-γ gene revealed an excellent safety profile coupled with an enhanced tumor immunogenicity which correlated with disease stabilization in patients diagnosed with advanced melanoma (17, 18). Other studies have shown that genetically modifying transformed cells to produce a type I interferon, particularly IFN-α, can also reduce their tumorigenicity (19, 20).
Fowlpox virus, a member of the Avipoxvirus genus, with a double-stranded DNA genome of about 300 kb, has been developed as a live virus vector capable of delivering vaccines and immune adjuvants safely in mammalian hosts (11, 13, 21, 22). Although the replication of fowlpox-based recombinant viruses is blocked in mammalian cells, inserted genes under transcriptional control of early promoters are expressed with high fidelity. Recent preclinical and clinical studies have shown the ability to administer recombinant fowlpox viruses multiple times without inducing neutralizing host immunity (21, 23). A recombinant fowlpox virus expressing murine IFN-γ (rF-IFN-γ) is described in this study. Local IFN-γ production following a single rF-IFN-γ injection selectively expanded the lymphoid cell populations, particularly the NK1.1+ cells, in the regional draining lymph nodes. Those NK1.1+ cells exhibited unique YAC-1 lytic characteristics and provided crucial regulatory signals for priming of an antigen-specific T cell response that protected vaccinated mice from the establishment of a primary tumor. Finally, in vivo antibody depletion studies provided additional argument that the presence of NK cells provided critical immunoregulatory signals which supported the generation of an antitumor adaptive host immune response.
MATERIALS AND METHODS
Animals, cell lines and reagents
Six- to eight-week-old, female C57BL/6 (B6) mice (H-2b) were obtained from NCI-Frederick, Frederick, MD. Mice were housed and maintained in microisolator cages under pathogen-free conditions.
The murine MC38 and B16/F10 (H-2b), colon adenocarcinoma and melanoma cell lines, respectively were maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing high glucose and 10% heat-inactivated fetal bovine serum (FBS). MC32A tumor cells, a subline of the MC38 cells transfected to express human carcinoembryonic antigen (CEA) (24), were grown in the same supplemented media. EL4, a murine T-cell lymphoma cell line (H-2b) and YAC-1 (mouse, A/Sn, lymphoma) tumor cell line which is sensitive to NK mediated lysis, were maintained in RPMI 1640 supplemented with 10% heat-inactivated FBS and complete T cell media (21), respectively.
Lyophilized rIFN-γ was obtained from PBL Laboratories, Inc. (Piscataway, NJ) (carrier-free, specific activity = 1.02 ×107 units/mg protein) and stored at -80EC until use. Prior to injection, rIFN-γ was reconstituted in saline containing 1% mouse serum. Reconstituted murine IFN-γ was also stored at -80EC, and its biological activity was checked every 3-6 months for its ability to upregulate MHC antigen expression on a variety of murine tumor cell lines.
Construction of rF-IFN-γ
The recombinant fowlpox virus expressing the murine IFN-γ gene was constructed by inserting the foreign gene sequences into the BamH1 J region of the genome of the POXVAC-TC (Schering Corporation, Kenilworth, NJ, USA) strain of fowlpox virus as described (25). Expression of murine IFN-γ in the recombinant fowlpox construct is under the control of the p40 promoter (25). IFN-γ production was confirmed by the in vitro infection of MC38 tumor cells as previously described (21). MC38 cells were harvested and washed twice with Opti-MEM media (Life Technologies Inc., Gaithersburg, MD). Two- 4×106 cells were added to 15 ml polypropylene conical tubes and pelleted by centrifugation. Cell pellets were suspended in 300 μl of Opti-MEM media to which the indicated MOI (in 10 μl) of rF-IFN-γ was added. Cells were incubated for 1 hr at 37°C, washed with complete medium twice and suspended in 10 ml of complete medium in T-25 flasks. At different time points, media supernatant was removed, stored at − 20°C and IFN-γ levels were measured using a murine IFN-γ ELISA (Endogen, Inc., Cambridge, MA). Both uninfected MC38 cells and MC38 cells infected with wild-type fowlpox (FP-WT) served as controls. Serum IFN-γ levels were measured using the same ELISA.
Flow Cytometry
Flow cytometry was used to determine the level of expression of the MHC molecules on the tumor cell surface. Briefly, cells that had been either infected with rF-IFN-γ or FP-WT or incubated with rIFN-γ (1-10 ng/ml, 48 hr) were harvested and washed twice with cold Ca2+-Mg2+-free DPBS. Cells were resuspended in Ca2+-Mg2+-free DPBS at a concentration of 0.5–1.0 × 106 cells/ml. In order to block the Fc receptors, 1 μg of the unlabeled CD16/CD32 Fc γ III/II receptor antibody (clone 2.4G2, rat IgG2b) was added to each sample. Cells were then incubated with 2 μg of FITC-labeled antibodies against H-2Kb (clone AF6-88.5, mouse IgG2a, κ), H-2Db (clone 28-14-8, mouse IgG2b, κ) and I-Ab (clone AF6-120.1, rat (Lou) IgG2a, κ) molecules or appropriate control antibodies (clone R19-15, mouse IgG2a, κ and clone 25-35, mouse IgG2b, κ)(PharMingen, Inc., San Diego, CA) for 1 h at 4°C. Cells (10,000-20,000) were analyzed using a Becton Dickinson FACScan or an LSR II. To determine changes of MHC expression of tumors treated with rF-IFN-γ in vivo, mice received an s.c. injection on their lower flank consisting of 3 × 105 of either MC38 or B16/F10 cells. When the tumors had achieved a volume of 75-100 mm3, an intratumoral injection of 1×107 pfu/50 μl rF-IFNk-γ or FP-WT or vehicle (HBSS) was administered. Twenty-four hours later, tumors were excised, cut into smaller pieces and incubated in 5 ml of complete medium containing 15 mg of Collagenase D (Roche, Nutley, NJ) for 1 hour. Every 20 minutes cells were mechanically separated by vigorous pipetting and at the end of the incubation period, cells were washed with cold Ca2+-Mg2+-free DPBS, centrifuged and counted. Flow cytometry was performed as described above.
Regional lymph nodes, consisting of the subiliac, para-aortic, and sacral nodes, were surgically isolated from groups of B6 mice following a single s.c. injection of rF-IFN-γ, FP-WT, rIFN-γ or vehicle. Lymph nodes from each treatment group were combined, dissected into small pieces and incubated in 3.0 ml of complete medium containing collagenase D, as previously described. Cells were pelleted by centrifugation and washed twice with cold Ca2+-Mg2+-free DPBS. Cell aliquots from each group were resuspended and analyzed using the following antibodies: APC-Cy7-anti-mouse CD3e (clone 145-2C11), PE-Cy7-anti-mouse CD19 (clone 1D3), PE-anti-mouse NKG2D (clone CX5, rat IgG1, κ) (www.ebioscience.com), APC-anti-mouse NK1.1 (NKR-P1B and NKR-P1C) (PK136) (mouse IgG2a). Control antibodies included APC-Cy7 hamster IgG1 (anti-TNP), PE-Cy7 mouse IgG2a, PE-rat IgG1 (clone R3-34) and APC-mouse IgG2a (clone X13). In order to block the Fc receptors, 1 μg of the unlabeled CD16/CD32 Fc γ III/II receptor antibody (clone 2.4G2, rat IgG2b) was added to each sample. Cells (200,000-500,000) were analyzed using Becton-Dickinson LSR II.
NK-mediated lytic assay
Lysis of the NK sensitive YAC-1 cells was measured using either the 4-h Cr-51 or overnight In-111 release assays. YAC-1 cells (2-4 × 106) were labeled with 250 μCi of Na2[51Cr]O4 (PerkinElmer, Shelton, CT) or with 3.7 MBq of sodium [111In] (Amersham, Arlington Heights, IL, USA) in serum-free RPMI 1640 for 30 min at 37°C. After each labeling, the cells were washed and plated in 96-well round bottom plates (Falcon) at a concentration of 5×103 cells/well in RPMI complete medium with 10% FBS. Effector cells, consisting of unfractioned or NK-enriched fractions from LNs or spleens, were added at varying effector to target (E:T) ratios. Depending on whether the YAC-1 cells were labeled with Cr-51 or In-111, the plates were incubated for 4-hr or overnight at 37°C after which the plates were centrifuged (200 × g) for 1 minute, supernatants harvested and the radioactivity counted using a Beckman 5500 gamma counter (Beckman Scientific Instruments, Irvine, CA). Specific cellular cytotoxicity was determined by the following formula: percentage of specific lysis = (experimental cpm - spontaneous cpm) / (maximum cpm – spontaneous cpm) × 100. Maximum cpm was obtained by adding Triton X-100 (0.25%) (Sigma, St. Louis, MO) to the target cells in the absence of effector cells. Spontaneous cpm was the radioactivity from wells with target cells alone. EL-4 cells were used as negative control target cells. All assays were done in triplicate.
In selected experiments, NK cells were purified from either lymph nodes or spleens using an NK cell negative selection isolation kit (Dynal Biotech ASA, Oslo, Norway) that included rat IgG depleting antibodies to CD4, CD5, CD8α, CD19, Ly-6G and Ter-119. Flow cytometric analyses revealed that >85% of the cells purified from either lymph nodes or spleens expressed NK1.1. In other experiments, different agents were analyzed for their ability to block LN-cell mediated YAC-1 lysis during the overnight lytic assay. Lymph node effector cells were preincubated for 1-2 hr in the presence of 100 nM concanamycin A (CMA) (Sigma, St. Louis, MO), 50 μg of the following antibodies: hamster anti-mouse CD178 (Fas Ligand, CD95 Ligand) (clone MFL3, hamster IgG1, κ) (PharMingen, Inc.), hamster anti-mouse CD120b (TNF receptor Type II, p75)(clone TR75-32, hamster IgG1, λ1), hamster anti-mouse CD120a (TNF receptor Type I/p55)(clone MABTNFR1-B1, mouse IgG2a, κ). The CMA-treated effectors were washed prior to, whereas, the neutralizing antibodies were present during the assay.
In vitro T cell stimulation and CTL assay
The MC38 tumor-specific CTL line was generated by isolating the regional lymph nodes from mice administered rF-IFN-γ-infected MC38 cells. Lymph node cells were counted and added to T-25 flasks in 20 ml complete media at a ratio of 50:1 lymphocytes to irradiated (20,000 rads) MC38 tumor cells. Seven days later the cells were counted using trypan blue and 2-5×105 cells were restimulated with 1×105 irradiated MC38 cells and 5×105 fresh, irradiated splenocytes used as feeder cells in 2.0 ml/well of a 24-well plate. After an additional seven days of in vitro stimulation, T cells were harvested, MC38 and EL4 cells were labeled with Cr-51 and 4-hr cytolytic assays were carried out at predetermined E:T ratios.
Tumor Studies
Tumor studies included the s.c. administration of MC38, B16/F10 or MC32A tumor cells harvested from in vitro propagated cell lines. In other experiments, MC38 tumor cells were infected ex vivo at the indicated MOI with either rF-IFN-γ or FP-WT as previously outlined. Following infection, the cells were washed with complete medium twice, resuspended in 10 ml of complete medium and placed in T-25 flasks and grown in vitro for 2-3 days. Another group of mice received MC38 cells that were grown in vitro for 48 hrs in complete medium supplemented with 10 ng/ml of rIFN-γ. In all cases, mice received a single injection of 3-5 × 105 cells in 0.1 ml administered s.c. into the lower flank and checked 2x/week for tumor growth.
In Vivo Depletion of Immune Cells
The following were used for the in vivo immune cell depletion studies: GK1.5, an anti-mouse L3T4 (rat IgG2b) (CD4); 2.43, an anti-mouse Lyt 2.2 (rat IgG2b) (CD8) MAb; anti-mouse NK1.1 (PK136, mouse IgG2a), rabbit anti-mouse/rat asialo GM1 polyclonal antibody (Cedarlane Laboratories, Lmtd. Hornby, Ontario, Canada); and Ly49D&H (gift from Drs. John Ortaldo and Robin Winkler-Pickett). GK1.5, 2.43 and PK136 were administered i.p. alone or in combination at a dose of 100 μg/day for 5 consecutive days. The anti-asialo GM1 polyclonal antibody was reconstituted according to the manufacturer’s instructions and mice received a single i.v. injection of 50 μl of the reconstituted antibody. Ly49D&H was diluted to 1.0 mg/ml in PBS and 0.2 ml (200 μg) was given as a single i.p. injection. Flow cytometric analyses of splenocytes revealed depletion of >98% of the CD4 and CD8 T cells following injection of the appropriate depleting antibodies. Short-term (4-hr) Cr-51 release cytotoxicity assay using YAC-1 cells as targets was used to verify NK cell depletion following the injection of PK136, anti-asialo-GM1 and Ly49D&H. In all cases, the percentage of YAC-1 killing by splenocytes (200:1 E:T) was <2.0% in those mice treated with the NK depleting antibodies. NK-mediated YAC-1 lysis of splenocytes isolated from normal mice ranged between 8-12% (200:1 E:T ratio). T and NK cell depletion was maintained with weekly injections.
Statistical Methods
Survival estimates among treatment groups were generated using Kaplan-Meier survival analyses. Differences in survivor functions between treatment groups were assessed using the log-rank test. Statistical significance of YAC-1 lysis and the number of NK1.1+, NK1.1+/NKG2D+ cells was based on Student’s two-tailed t test. All p values are two-sided and not adjusted for the multiplicity of evaluation performed on the data. Analyses were conducted using STATA software (version 1.0, Stata Corp., College Station, TX) and statistical significance was set at a P value of <0.05.
RESULTS
Production of biologically active IFN-γ by rF-IFN-γ-infected cells
Murine colon carcinoma (MC38) cells infected with rF-IFN-γ (1.0 MOI) produced approximately 150 ng IFN- γ/106 cells/day. No detectable IFN-γ levels were found in the supernatants from either uninfected or FP-WT-infected MC38 cells. Upregulation of MHC class I expression on the surface of tumor cells was used as readout to establish that biologically active IFN-γ was produced following rF-IFN-γ infection (Table 1). Infection of either MC38 or B16 melanoma cells significantly increased MHC Class I molecule (H-2Kb and H-2Db) expression levels. A slight increase was observed following FP-WT infection. Supernatants from rF-IFN-γ-infected MC38 and B16/F10 cells also upregulated H-2b antigens on both tumor cell lines, providing additional evidence of the production of biologically active IFN-γ (Table 1). Moreover, the extent of those increases were comparable to those measured following the addition of 10 ng/ml of rIFN-γ protein (Table 1).
Table 1.
MHC upregulation on MC38 and B16/F10 tumor cells following rF-IFN-γ infection.1
| MHC Cell Surface Expression
|
|||||
|---|---|---|---|---|---|
| H-2Kb | H-2Db | I-Ab | |||
| MC38 | none | N/A | 61.8 [ 68.9] 2 | 30.2 [54.4] | neg |
| FP-WT | 6.25 MOI | 70.1 [ 69.3] | 41.5 [45.8] | neg | |
| rF-IFN-γ | 6.25 MOI | 93.1 [177.6] 3 | 77.5 [45.8] 3 | neg | |
| 2x dilution supernatant | 95.5 [165.6] 3 | 92.2 [69.2] 3 | neg | ||
| rIFN-γ | 10 ng/ml | 98.5 [89.8] 3 | 96.2 [47.6] 3 | neg | |
|
| |||||
| B16/F10 | None | N/A | 9.1 [14.1] | 3.4 [12.2] | neg |
| FP-WT | 6.25 MOI | 13.4 [18.8] | 9.7 [17.7] | neg | |
| rF-IFN-γ | 6.25 MOI | 98.8 [92.2] 3 | 97.5 [52.6] 3 | neg | |
| 2x dilution supernatant | 87.8 [67.6] 3 | 81.5 [44.0] 3 | neg | ||
| rIFN-γ | 10 ng/ml | 98.5 [89.8] 3 | 96.2 [47.6] 3 | neg | |
MC38 and B16/F10 tumor cells were either infected with rF-IFN-γ or FP-WT at the indicated MOI or treated in vitro with supernatants from rF-IFN-γ-infected MC38 or by the exogenous addition of rIFN-γ as outlined in the Materials and Methods.
Values represent the percentage of cells expressing the indicated MHC class I antigen measured by FACS. Data in parentheses are the mean channel fluorescence intensity. Data are from a single experiment that was repeated with similar results. neg, negative.
P<0.05 (vs. untreated cells).
To test whether rF-IFN-γ can upregulate MHC class I expression in vivo, mice bearing s.c. MC38 tumors (volume of 75-100 mm3; 8-12 day post-injection) received a single intratumoral injection of rF-IFN-γ or FP-WT or HBSS. Class I MHC expression levels were increased approximately 2-fold, as evidenced by MFI increases, on only those tumors isolated from mice given a single intratumoral injection of rF-IFN-γ (data not shown). The B16/F10 s.c. tumors in B6 mice provided a more convincing assessment of the ability of rF-IFN-γ to enhance MHC expression. Constitutive H-2b expression was low on the B16/F10 tumors cells with <10% of the tumor cells expressing H-2Kb and -Db (Fig. 1, panels A and B, respectively). Intratumoral inoculation with either HBSS or FP-WT did not change H-2b expression levels, whereas at 24 hrs post-rF-IFN-γ injection, the percentage of H-2Kb as well as H-2Db expressing B16/F10 tumor cells were increased to >90% with accompanying increases in MFI providing in vivo evidence for the production of biologically active IFN-γ.
FIG. 1.
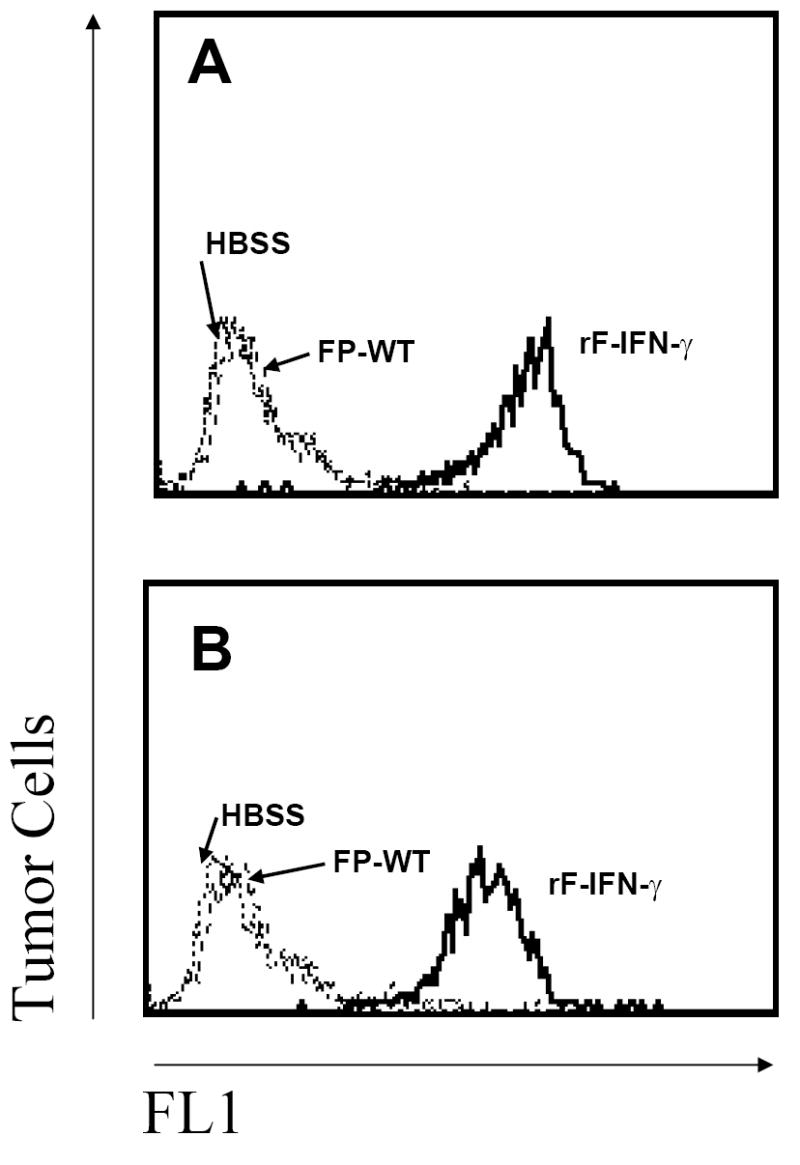
Enhanced H-2b expression levels on B16/F10 tumors following intratumoral rF-IFN-γ injection. Mice received 3×105 B16/F10 cells which resulted in 75-100 mm3 s.c. tumors 8-12 days later at which time they were divided into three treatment groups (3/group). Each mouse received a single intratumoral injection (50 μl) of HBSS (dotted lines) or 108 pfu of either rF-IFN-γ (solid lines) or FP-WT (dashed lines). Tumors were excised 24 hrs later and (A) H-2Kb and (B) H-2Db expression levels analyzed by flow cytometry (see Materials and Methods). Data are from a single experiment that was repeated with similar results.
Serum IFN-γ and changes in the regional lymph nodes after injection of FP-WT, rF-IFN-γ or rIFN-γ
Recombinant fowlpox viruses are, for the most part, non-lytic and replication incompetent with their infectivity range limited to the needle tract (26). This restricts the production of any inserted gene product, in this case, IFN-γ, to the local tissue microenvironment. To confirm this, mice received a single s.c. injection of 108 pfu rF-IFN-γ or 1, 5 or 10 μg rIFN-γ. Serum IFN-γ levels were measured at 1-48 hr post-injection. No detectable serum IFN-γ levels were found in mice that received rF-IFN-γ. In contrast, mice that received a single s.c. injection of 1, 5 or 10 μg rIFN-γ had peak serum IFN-γ levels of 7, 83 and 121 ng/ml, respectively, at 1-h post-injection.
Because IFN-γ production from rF-IFN-γ was restricted to the injection site, any immune-related actions resulting would be examined in the regional lymph nodes (LNs) draining that site. LNs isolated from untreated B6 mice contain 2-3 million cells/node. A single s.c. injection of either rIFN-γ or rF-IFN-γ significantly increased lymph node cellularity (Fig. 2A). In the case of rIFN-γ treatment, the increase in the total number of cells/node was transient, peaking at 24 hrs and approaching baseline levels by 72 hr. Total number of cells/node draining the site of rF-IFN-γ administration was increased 2.5-fold at 72 hrs and that enrichment was sustained for 5-6 days (Fig. 2A). Changes in the number of immune cell subsets, i.e., T, B and NK cells, within the LNs of mice administered FP-WP, rF-IFN-γ and rIFN-γ mirrored the changes in total cells/node. For example, at 24 hr post-injection of FP-WT, rF-IFN-γ or rIFN-γ, the regional LNs contained significantly higher numbers of CD3+ (Fig, 2B), CD19+ (Fig. 2C) and/or NK1.1+ (Fig.2D) cells, but at 72 hrs, only the LN from mice treated with rF-IFN-γ had sustained elevated numbers of each of the immune cell subsets. Interestingly, the number of NK1.1+ cells remaining in the regional LN 72 hr (Fig. 2D) after rF-IFN-γ was approximately 10-fold higher than any other treatment.
FIG. 2.

Increased LN cellularity and changes in CD3+, CD19+ and NK1.1+ cells following injection of rF-IFN-γ, FP-WT or rIFN-γ. (A) Mice (3-4/group) received single s.c. injections of either 5 μg rIFN-γ (circles) or 108 pfu FP-WT (triangles) or rF-IFN-γ (squares). Regional LNs were isolated 1-7 days later and the total number of cells/node determined using a hemocytometer. (B-D) Total number of CD3+ T cells (B), CD19+ B cells (C) and NK1.1+ cells (D) within the regional LN at 24 (open bars) and 72 hr (filled bars) of mice given a single s.c. injection of the indicated immune adjuvant. Data are the mean ± SEM from a representative experiment which was repeated with similar results. * P<0.05 (vs. untreated mice).
Flow cytometry identified NK1.1+ and NK1.1+/NKG2D+ cells in LNs 72 hrs post-injection of FP-WT, rF-IFN-γ or rIFN-γ - treated mice (Fig.3). Within the LNs of untreated mice the percentage of cells expressing NK1.1 was <1% (Fig. 3B) with 14.7% of the NK1.1+ cells co-expressing NKG2D (Fig. 3F), an activation/cytotoxicity receptor on murine NK cells (27). Administration of FP-WT or rIFN-γ induced no significant changes in either the total number of NK1.1+ or NK1.1+/NKG2D+ cells. In contrast, rF-IFN-γ injection induced a significant (P<0.05 vs untreated mice) enrichment of NK1.1+ and NK1.1+/NKG2D+ cells in the regional LNs. The percent NK1.1+ LN cells increased from 0.8% in untreated mice to 3.3% (Fig. 3C) in mice administered rF-IFN-γ Furthermore, the percentage of those NK1.1+ cells which also expressed NKG2D rose from 14.7% in untreated mice to 42.6% in mice injected with rF-IFN-γ (Fig. 3G).
FIG. 3.

Changes in NK1.1+ and NK1.1+/NKG2D+ cells in the regional LN following a single injection of FP-WT, rF-IFN-γ or rIFN-γ. LNs were harvested from B6 mice (3-6/group) three (3) days after a single injection of HBSS, FP-WT rF-IFN-γ or rIFN-γ as described above. Panel A establishes the staining pattern for the isotype control and the region of interest for further analyses. Panels B-E illustrates the percentage of total LN cells that expressed NK1.1+ and panels F-I represent the percentage of the NK1.1+ cells co-expressing NKG2D. Data are the mean ± SEM from a representative experiment which was repeated with similar results.
Identification of NK-mediated YAC-1 lysis in regional LNs draining the rF-IFN-γ injection site
While NK cells are usually thought to be restricted to the spleen and circulation during steady-state conditions, the flow cytometric data suggest that local IFN-γ production, particularly following rF-IFN-γ administration, results in a significant enrichment of activated NK1.1 cells within the regional LNs. LN were isolated from mice 72 hr post-injection of FP-WT, rF-IFN-γ or rIFN-γ and analyzed for their ability to lyse an NK sensitive target, YAC-1 cells, in both short-term (4 h) and overnight assays (Fig. 4). No appreciable YAC-1 lysis was detected in the LNs of mice injected with either FP-WT or rIFN-γ in either the short-term (Fig. 4A) or overnight (Fig. 4B) assays. In the short-term 4 h cytolytic assay, there was a measurable level of YAC-1 killing by the LN from rF-IFN-γ treated mice (fig. 4A). However, the most dramatic amount of YAC-1 killing (>30% @ 200:1 E:T) was seen in an overnight assay using unfractionated LN cells from mice administered rF-IFN-γ (Fig. 4B).
FIG. 4.
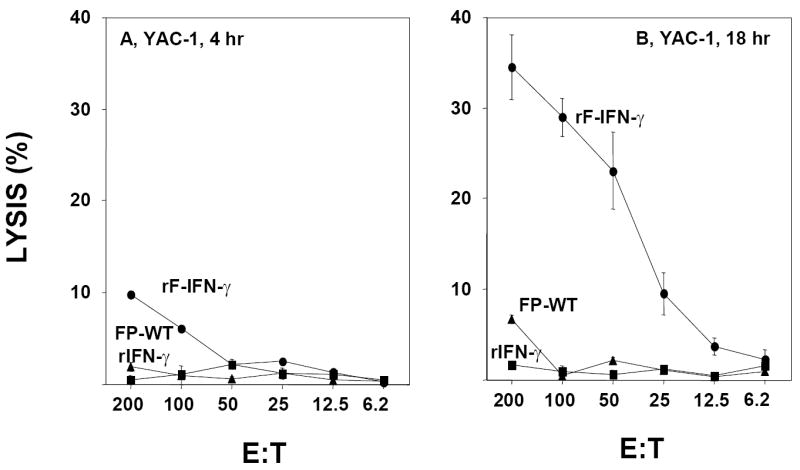
YAC-1 cytolysis within the regional LNs of mice following a single injection of 108 pfu FP-WT (triangles), 108 pfu rF-IFN-γ (circles) or 5 μg rIFN-γ (squares). Regional LNs were isolated 72 hrs post-injection and tested for YAC-1 lysis in a 4-hr (A) and overnight (B) assays. Data are presented as the mean ± SEM from triplicate wells at each E:T ratio and are from a representative experiment performed 4-5 times with similar results.
Subsequent studies determined (1) the temporal aspects of YAC-1 lysis, (2) whether YAC-1 lysis was, indeed, localized to the regional LN draining the rF-IFN-γ injection site, (3) possible molecular events which may be responsible for YAC-1 lysis and (4) the effects of NK cell depletion on YAC-1 killing in the overnight assay. Mice received a single s.c. injection of rF-IFN-γ, FP-WT, rIFN-γ or HBSS and LN-mediated YAC-1 lysis was measured over 7 days (Fig. 5A). At 24 hrs post-injection of either rF-IFN-γ or rIFN-γ protein, the regional LNs of those mice contained enriched levels of YAC-1 lysis measured in the overnight assay (Fig. 5A). The difference was that the enrichment present in the regional LNs draining the site of mice injected with rIFN-γ was transient, peaking at 24 hrs post-injection and returning to baseline levels by 72 hrs. Delivery of IFN-γ as rF-IFN-γ resulted in sustained levels of YAC-1 killing in the draining LNs over 5-6 days with peak lysis occurring 72 hrs post-injection. The temporal aspect of YAC-1 lysis in the LN from mice administered rF-IFN-γ was very reminiscent of the changes seen in total cells and the number of NK1.1+ cells/node in the same treatment group (Fig. 2A and D). Enhanced YAC-1 lysis was present only in the regional LNs that drained the rF-IFN-γ injection site. No YAC-1 lysis was observed in LNs distal (contralateral) to the rF-IFN-γ injection site (Fig. 5B). LN cells from mice injected with rF-IFN-γ were isolated and co-incubated in the presence of an anti-FasL antibody, CMA or anti-TNF-α antibodies with significant inhibition of YAC-1 lysis (P<0.05 vs. no treatment) associated with the addition of the anti-TNF-α antibodies (Fig. 5C). Mice were administered three different NK depleting antibodies – anti-asialo-GM1, anti-NK1.1 and Ly49D&H - prior to rF-IFN-γ administration. Since both anti-asialo-GM1 (28) and anti-NK1.1 (29) also deplete rare T cell populations, the Ly49D&H which selectively depletes activated NK cells (27, 30) was added. The ability of the LN cells to lyse YAC-1 targets was completely lost in those mice receiving any of the three NK-depleting antibodies (Fig. 5D). Likewise, splenocytes isolated from those same mice also lost their ability to recognize and lyse YAC-1 cells in a conventional 4hr-Cr-51-release assay (data not shown). Therefore, both phenotypic and functional analyses provide a strong argument that the YAC-1 lysis identified in the regional LNs draining the rF-IFN-γ injection site was, indeed, NK cell-mediated.
FIG. 5.
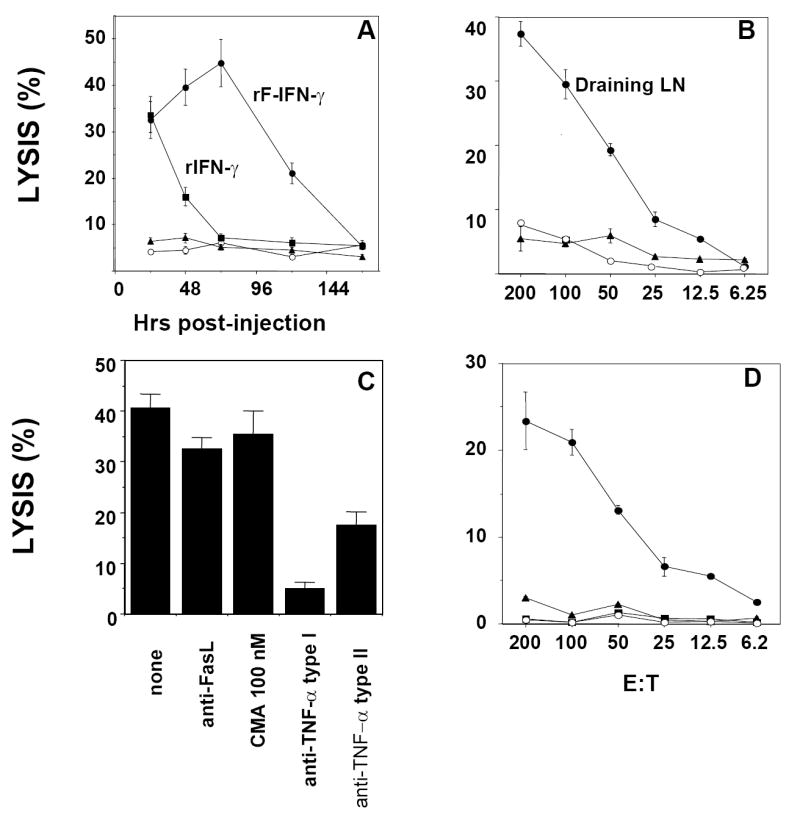
Description of the overnight YAC-1 targeted lytic activity in the regional LNs. (A) Mice (3-4/group) received single s.c. injections of either 5 μg rIFN-γ (squares) or 108 pfu FP-WT (triangles) or rF-IFN-γ (closed circles). Untreated mice (n=6) received a single s.c. injection of 100 μl HBSS (open circles). Regional LNs were isolated after 24-168 hrs and YAC-1 lysis (E:T, 200:1) measured. (B) Mice (3-4/group) received a single injection (s.c.) of rF-IFN-γ (108 pfu) and control mice (n=6, open circles) received HBSS. LNs draining (circles) or distal (contralateral, triangles) to the s.c. injection site were isolated 72 hrs post-injection and YAC-1 lysis (E:T, 200:1) measured. (C) Mice (n=12) that received a single s.c injection of 108 pfu rF-IFN-γ were sacrificed 72 h later, LNs removed, combined and an overnight YAC-1 lytic assay (E:T 200:1) was performed. Treatment with the different potential inhibitors was described in the Materials and Methods. (D) Mice (3-5/group) received the NK cell-depleting antibodies, anti-asialo-GM (triangles), anti-NK1.1 (squares) or Ly49D&H (open circles) prior to receiving a single s.c. injection of 108 pfu rF-IFN-γ. LN cells were isolated, combined and YAC-1 lysis measured in an overnight assay. Untreated mice are represented by the closed circles. All data are presented as the mean ± SEM from triplicate wells from a representative experiment that was repeated with similar results.
IFN-γ produced by rF-IFN-γ-infected tumor cells prevents in vivo tumor development
Next, studies were designed to examine whether IFN-γ administration in the presence of antigen might influence host adaptive immunity. MC38 tumor cells were treated ex vivo with 6.25 MOI of either FP-WT or rF-IFN-γ or 10 ng/ml rIFN-γ prior to their s.c. injection into syngeneic B6 mice. Both rF-IFN-γ and rIFN-γ enhanced class I MHC expression levels on the MC38 tumor cells (Table 1). Despite higher H-2b expression levels, only those mice receiving rF-IFN-γ infected MC38 tumor cells were protected from primary tumor growth (Fig. 6A, P<0.05). Six of 10 mice within that treatment group remained tumor-free 8 weeks after tumor cell inoculation. Moreover, when those tumor-free mice were challenged with uninfected MC38, approximately 30% successfully rejected tumor growth with no such protection against B16/F10 tumor growth (Fig. 6B). The findings argue that a single vaccination, consisting of rF-IFN-γ-infected MC38 tumor cells, generated sufficient antitumor immunity to protect those mice from subsequent tumor challenge. Both the protection from tumor growth as well as the generation of tumor-specific host immunity could not be explained simply by the upregulation of MHC molecules (31) on the tumor cell surface following rIFN-γ treatment. Sustained in vivo IFN-γ production in the microenvironment encompassing the injection site of the rF-IFN-γ-infected MC38 tumor cells seemed to be essential in preventing tumor growth.
FIG. 6.
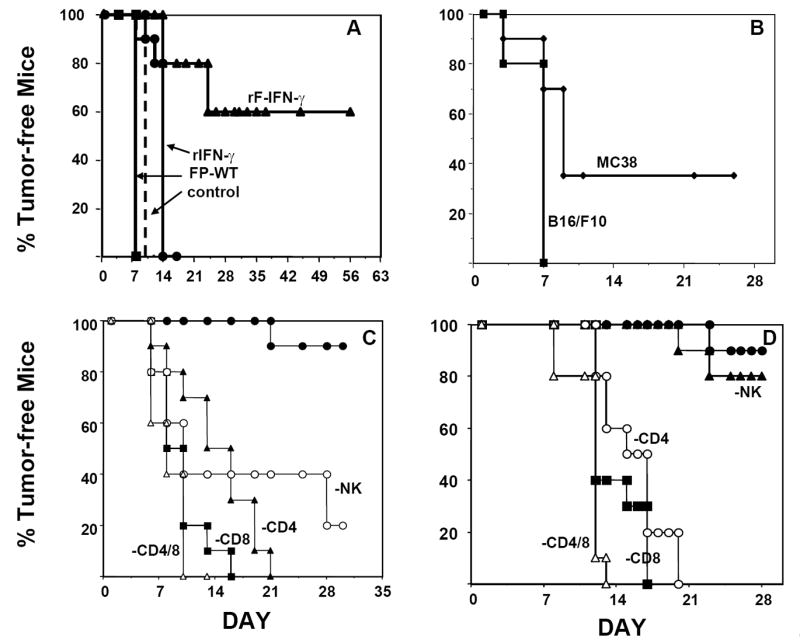
rF-IFN-γ infection protects mice from tumor development: effects of immune cell depletion in naïve and vaccinated mice. (A) MC38 tumor cells were infected with 6.25 MOI of either FP-WT (squares) or rF-IFN-γ (triangles) or treated in vitro with 10 ng/ml rIFN-γ (circles) (see Materials and Methods). Control mice were injected with uninfected MC38 cells (dashed line). All mice (n=20/group) received a tumor burden of 3×105 cells and were monitored daily for tumor appearance (minimum volume approx. 20-30 mm3). Results are presented as the percentage of tumor-free mice. (B) Tumor-free mice from the mice vaccinated with rF-IFN-γ-infected MC38 cells were challenged s.c. on the opposite side with 3×105 of either MC38 (diamonds) or B16/F10 (squares) cells and tumor appearance checked daily. (C) Prior to the injection of rF-IFN-γ infected MC38 tumor cells, mice (10/group) were administered the following immune cell-depleting antibodies: GK1.5 (anti-CD4, triangles), 2.43 (anti-CD8, squares), anti-asialo-GM1 (anti-NK, open circles), GK1.5/2.43 (open triangles) (see Materials and Methods). Control mice (solid circles) received a single injection of rF-IFN-γ-infected MC38 tumor cells alone. The percentage of tumor-free mice was determined as explained in panel A. (D) Mice (10/group) received two s.c. vaccinations of rF-IFN-γ (10 MOI)-infected MC32A (CEA-expressing) tumor cells (5×105/injection). Prior to challenge with 3×105 parental MC32A tumor cells, mice were administered GK1.5 (open circles), 2.43 (anti-CD8, squares), anti-asialo-GM1 (closed triangles), GK1.5/2.43 (open triangles) or an isotype control antibody (closed circles). Mice were monitored daily for tumor appearance (minimum volume approx. 20-30 mm3) and the results are presented as described in A. Data in all panels are the combined results from two separate experiments.
Immune cell depletion studies attempted to identify those cell population(s) which conferred protection against primary tumor growth in mice administered rF-IFN-γ-infected MC38 tumor cells. Mice were administered immune cell depleting antibodies prior to their inoculation with rF-IFN-γ-infected MC38 cells (Fig. 6C). Protection from primary tumor growth was lost with CD4 or CD8 or their combined depletion. Interestingly, protection from the growth of rF-IFN-γ-infected MC38 tumors was also partially lost in mice depleted of their NK1.1+ cells with the PK136 antibody. Those observations suggested that protection from the growth of primary MC38 tumors infected with rF-IFN-γ required the presence of both innate and adaptive immune cells.
Role(s) of the different immune cell subsets in the protection of vaccinated mice were subsequently addressed. Mice received two vaccinations consisting of rF-IFN-γ-infected MC32A tumor cells that had been engineered to express the human CEA antigen (80-95% CEA+). CEA-expressing tumor cells were chosen in order to provide a stronger rejection antigen absent in the parental MC38 tumor cells. Indeed, 90% of the vaccinated mice remained tumor-free following challenge with parental MC32A tumor cells (Fig. 6D). In mice depleted of CD4, CD8 or both T cell subsets following the second vaccination, protection from subsequent tumor rechallenge with the MC32A tumor cells was lost. In contrast, NK cell depletion in vaccinated mice was not accompanied with a loss of protection against MC32A tumor challenge (Fig. 6D), indicating that resident effector T cell memory responses capable of protecting the host from tumor rechallenge does not require the presence of NK cells.
Tumor-specific CTL activity in the regional LNs: relationship with NK cells
Local delivery of IFN-γ via a recombinant fowlpox virus, rF-IFN-γ, enriches the regional LNs with immune cells (Fig. 2 and 3), particularly, NK cells and when administered with antigen, i.e., rF-IFN-γ-infected MC38 tumor cells, can induce a protective T cell response (Fig. 6A-C). It was hypothesized that the LNs draining the site of injection of MC38 cells infected with either FP-WT or rF-IFN-γ or treated in vitro with rIFN-γ might contain cells capable of antigen-specific tumor cell lysis.
Mice received a single s.c. injection of MC38 cells that were previously infected with FP-WT or rF-IFN-γ or treated ex vivo with rIFN-γ Seventy-two hours after injection, regional lymph nodes were isolated from mice of each treatment group and analyzed for lysis MC38 and EL4 cells in short-term and overnight assays (Fig. 7). Within the LNs draining the site of injection of the rF-IFN-γ-infected MC38 cells was found MC38-specific cell-mediated lysis using either a 4-hr (panel A) or overnight assays (panel C). In both cases, MC38-specific lysis was weak (8-12%), but statistically significant (P<0.05 vs. lysis from mice injected with FP-WT or rIFN-γ-treated MC38 cells), and could be selectively enhanced by in vitro stimulation with irradiated MC38 cells (Fig. 7A, insert). No measurable lysis was observed with EL4 cells as targets (Fig. 7B and D).
FIG. 7.

Tumor cell specific cytolytic activity in the regional LNs of mice following injection of rF-IFN-γ-infected MC38 cells. MC38 cells were either infected with 10 MOI of FP-WT (triangles) or rF-IFN-γ (circles) or treated in vitro with 10ng rIFN-γ/ml (squares) prior to injection s.c. into mice (3×105 cells/injection). Seventy-two hrs after injection, regional LNs were isolated and tested for lysis against MC38 (A and C) and EL4 (panels B and D) in 4-hr (A and B) and overnight (panels C and D) assays. Panel A, insert: LN cells from mice injected with rF-IFN-γ-infected MC38 cells were stimulated in vitro in the presence of irradiated MC38 tumor cells (see Materials and Methods) prior to 4-hr cytolytic assays with either MC38 (circles) or EL4 (triangles) cells as targets. Data are presented as the mean ± SEM from triplicate wells at each E:T ratio and are from a representative experiment performed 2-3 times with similar results.
The presence of a large number of NK cells together with tumor specific CTLs within the regional LNs draining the site of injection of rF-IFN-γ-treated MC38 tumor cells prompted the examination as to whether any functional relationships existed between those cells. To address this question, mice were administered anti-asialo-GM, anti-NK1.1 or Ly49D&H (Fig. 8, panels B-D) prior to the injection of rF-IFN-γ-infected MC38 cells. The low, but measurable tumor-specific lysis in the regional LNs of mice injected with rF-IFN-γ-infected MC38 tumors can be specifically enriched by in vitro stimulation with re-exposure to tumor antigen (i.e., irradiated MC38 tumor cells)(Fig. 8A). Depletion of NK cells by the administration of any of the three antibodies completely abrogated the ability of those mice develop a primary tumor-specific T cell response (Fig. 8, panels B-D). Those findings implicate NK cells as a crucial component in the development of a primary T cell response to MC38 tumors, in the draining lymph nodes of mice receiving rF-IFN-γ-infected MC38 tumors.
FIG. 8.

In vivo NK cell depletion inhibits the development of an antigen-specific CTL response. Mice (3-5/group) received the NK cell-depleting antibodies, anti-asialo-GM1 (panel B), anti-NK1.1 (panel C) or Ly49D&H (panel D) as described in the Materials and Methods. Untreated mice which received an isotype control antibody are shown in panel A. All mice then received a single s.c. injection of rF-IFN-γ-infected (10 MOI) MC38 tumor cells. Three (3) days later the draining LNs were removed, combined and stimulated in vitro in the presence of irradiated MC38 cells (see Materials and Methods). Cells were harvested and 4-h lytic assays were carried out with MC38 (circles) and EL4 (triangles) cells as targets. All data are presented as the mean ± SEM from triplicate wells from a representative experiment that was repeated with similar results.
DISCUSSION
The present study underscores the advantages of targeted delivery of IFN-γ to a prescribed tissue microenvironment. Administration of IFN-γ as either a recombinant protein, rIFN-γ, or as a recombinant avipox virus, rF-IFN-γ, induces profound expansions of immune cells within the local, regional LNs. Several distinct advantages are gained, however, by delivering IFN-γ as rF-IFN-γ rather than injecting the rIFN-γ protein. First, the expansion of immune cells, particularly NK1.1+ cells, within the regional LNs was sustained for 5-6 days in comparison to a transient (approx. 24 hrs) expansion after rIFN-γ injection. Second, the enrichment within the regional nodes was achieved without any measurable circulating IFN-γ levels, thus negating any side effects associated with high doses and/or multiple injections of the rIFN-γ protein. And, finally, the combined administration of rF-IFN-γ, not rIFN-γ, with antigen (i.e., highly tumorigenic MC38 tumor cells) induced sufficient tumor-specific adaptive T cell immunity to protect mice against primary tumor growth and subsequent tumor challenge.
In the past, evaluations of rIFN-γ as a single antitumor agent have been predicated on the ability to deliver sufficient amounts of the cytokine to the tumor site. Upregulation of MHC, Fas and tumor associated antigens on tumor cells, inhibition of immunosuppressive factors and potent anti-angiogenic effects have all been document with systemic IFN-γ administration and have provided the argument for the immunomodulatory and, perhaps, antitumor actions of this cytokine. However, clinical studies revealed severe treatment-limiting toxicities that have blunted the use of rIFN-γ in cancer treatment. Yet, IFN-γ remains a potent proinflammatory cytokine as do questions on how best to exploit its actions. Our rationale was to target IFN-γ production to a tissue microenvironment via rF-IFN-γ injection, thus localizing its proinflammatory actions that would induce a controlled migration of host immune cells to the injection site. Under those conditions, with the addition of antigen, in the form of rF-IFN-γ-infected MC38 tumor cells, tumor specific CTL activity was generated in the antigen-stimulated LNs. Furthermore, T cell depletion studies established their requirement to protect mice from the growth of rF-IFN-γ-infected MC38 tumor cells. Thus, the findings of this study might provide the rationale to reexamine IFN-γ as a classic immune adjuvant when delivered with antigen to a selected tissue microenvironment.
The ability of rF-IFN-γ to act as an immune adjuvant was dependent on: (1) the sustainability of IFN-γ production at the injected site, consistent with using a fowlpox-based vector as a delivery vehicle and (2) the cellular enrichment (Fig.2), particularly NK1.1+ cells (Fig. 5A) within the regional lymph nodes of mice receiving rF-IFN-γ. Several intriguing findings focused our attention on the NK1.1+ cell population in the regional lymph nodes of mice injected with rF-IFN-γ. In vivo NK1.1 depletion studies suggested that they play an important role(s) in (1) the protection against primary tumor growth (Fig. 6C) and (2) the generation of a tumor-specific host immune response (Fig. 8). In contrast, in a tumor challenge experiment of vaccinated [rF-IFN-γ-infected MC32A (CEA-expressing) cells] mice, the ability to reject tumor was not lost with NK1.1+ cell depletion (Fig. 6D). Those findings indicate a role(s) for NK1.1+ cells in T cell priming but not for the recall of memory T cells.
The results argue that injection of rF-IFN-γ-infected MC38 initiates signals for the extravasation of NK1.1+ cells from the circulation to the site of inflammation. Traditionally, NK cells function as peripheral effector cells that recognize and kill stressed, transformed and virally infected cells, their principle role in innate immunity (32, 33). Whether NK1.1+ cells recognize and kill rF-IFN-γ-infected MC38 tumor cells, contributing to the protection of mice against primary tumor growth is arguable. Infection of MC38 tumor cells with rF-IFN-γ enhances MHC class I expression (Table 1, Fig. 1) and might be expected to inhibit NK cell cytotoxicity through engagement of NK cell receptors containing a tyrosine-based inhibitory motif (34). Yet, when mice were depleted of NK cells, protection from the growth of rF-IFN-γ-infected MC38 tumor cells was lost (Fig. 6C). One explanation is that the NK1.1+ cells within the antigen-stimulated LNs are not tumoricidal, but their presence supports the development of tumor-specific adaptive immunity. Indeed, priming of naïve T cells to tumor-specific antigen occurs within secondary lymph sites and requires the presence of mature DCs. Within sites of chronic inflammation (i.e., allergen-induced atopic eczema/dermatitis syndrome), NK cells are in close contact with resident DCs (35) which results in DC maturation and local IL-12 production (36) driving a TH1 cellular response. In the present study, flow cytometric analyses revealed that not only were the antigen-stimulated lymph nodes draining the rF-IFN-γ injection site enriched for NK1.1+ cells, but a significant percentage co-expressed the NKG2D-activating receptor, the primary cytotoxicity receptor for murine NK cells (Fig. 3). Usually, following NKG2D receptor-ligand interaction cytolysis by the NK, NKT and T cells is perforin-mediated (37, 38), however, no perforin-mediated lysis was detected. Unfractionated LN cells from mice injected with rF-IFN-γ lysed YAC-1 cells in a TNF-α-dependent manner (Fig. 5C). A previous study (39) reported NK-dependent DC maturation/migration through a TNF-dependent mechanism. Absence of that pathway in NK cell-depleted mice would be expected to interrupt DC maturation and severely impair the cross-talk between innate and adaptive immune responses. Whether those findings signal a difference between “helper” (40) and “effector” NK cells or indicate a particular NK cell subset will be addressed in future studies.
More recent data have shown that NK cells also provide other “helper” function(s) to the adaptive immune system. Their presence in secondary lymphoid sites, particularly, antigen-stimulated LNs, provides a local IFN-γ source that assists in T-cell priming (41-44). One could speculate that the local IFN-γ production, by virtue of rF-IFN-γ injection, might substitute some needed “helper” functions in the NK cell-depleted mice, such as IL-12 or IL-18 induction in the microenvironment, and support some measurable T cell priming. However, depletion of NK cells prior to the administration of the cell-based vaccine (i.e., rF-IFN-γ-infected-MC38 tumor cells) led to a complete loss in the ability to generate a primary antigen-specific T cell response (Fig. 8). Those findings suggest that other NK-mediated events are required for T-cell priming which is consistent with recent evidence of an IL-18-dependent soluble factor released by activated NK which promotes DC maturation (45). Thus, NK cells provide crucial signals which can (1) impact DC editing and (2) provide a local source of IFN-γ and the absence of those signals due to NK cell depletion can severely impact T-cell priming (46, 47). Indeed, patients with deficits in NK cell function have impaired induction of virus-specific immune response and a heightened susceptibility to recurrent viral infection (48). Therefore, it seems that NK cells are important early innate cells involved at multiple steps leading to the priming of adaptive T cell responses. An understanding of the cellular signals delivered to the innate immune system and how they shape the events upstream of the DC-T helper cell and/or NK-cytotoxic T cell interactions could contribute to purposefully driving a particular adaptive immune response and facilitate the design of novel immunization schema.
Acknowledgments
The authors would like to thank Drs. John Ortaldo and Robin Winkler-Pickett for their helpful discussion and Dr. Ortaldo for his critical reading of the manuscript. The authors would also like to thank Debra Weingarten for her excellent editorial assistance. Finally, the authors acknowledge the excellent technical assistance of Garland Davis and Eileen Thompson which was an important contribution for the completion of these studies. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- rIFN-γ
recombinant interferon-gamma
- MHC
major histocompatibility complex
- rF-IFN-γ
recombinant fowlpox virus expressing murine IFN-γ
- FP-WT
fowlpox wild-type
- DMEM
Dulbecco’s Modified Eagle Medium
- LNs
lymph nodes
- s.c.
subcutaneous
- MFI
mean fluorescence intensity
- MOI
multiplicity of infection
- DPBS
Dulbecco’s phosphate buffered saline
- HBSS
Hanks’ Balanced Salt Solution
- pfu
plaque-forming units
References
- 1.Kantor J, Tran R, Greiner JW, Pestka S, Shively J, Schlom J. Modulation of carcinoembryonic antigen mRNA levels in human colon carcinoma cells by recombinant γ interferon. Cancer Res. 1989;49:2651–2655. [PubMed] [Google Scholar]
- 2.Schmitz J, Reali E, Hodge JW, Patel AC, Davis G, Schlom J, Greiner JW. Identification of an interferon-γ-inducible carcinoembryonic antigen (CEA) CD8+ T-cell epitope, which mediates tumor killing in CEA transgenic mice. Cancer Res. 202;62:5058–5064. [PubMed] [Google Scholar]
- 3.Myers L, Croft M, Kwon BS, Mittler RS, Vella AT. Peptide-specific CD8 T regulatory cells use IFN-γ to elaborate TGF-β-based suppression. J Immunol. 2002;174:7625–7632. doi: 10.4049/jimmunol.174.12.7625. [DOI] [PubMed] [Google Scholar]
- 4.Walker W, Rotondo D. Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-γ synthesis. Immunol. 2002;111:298–305. doi: 10.1111/j.1365-2567.2004.01810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, Kleinman HK, Reaman GH, Tosato G. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182:155–162. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickerson EB, Akhtar N, Steinberg H, Wang Z-Y, Lindstrom MJ, Padilla ML, Auerbach R, Helfand SC. Enhancement of the antiangiogenic activity of interleukin-12 by peptide targeted delivery of the cytokine to alphavbeta3 integrin. Mol Cancer Res. 2004;2:663–673. [PubMed] [Google Scholar]
- 7.Foon KA, Sherwin SA, Abrams PG, Stevenson HC, Holmes P, Maluish AE, Oldham RK, Herberman RB. A phase I trial of recombinant gamma interferon in patients with cancer. Cancer Immunol Immunother. 1985;20:193–197. doi: 10.1007/BF00205575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quesada JR, Kurzrock R, Sherwin SA, Gutterman JU. Phase II studies of recombinant interferon g in metastatic renal cell carcinoma. J Biol Response Mod. 1987;6:20–27. [PubMed] [Google Scholar]
- 9.Snijders A, Kalinski P, Hilkens CM, Kapsenberg ML. High-level IL-12 production by human dendritic cells requires two signals. Int Immunol. 1998;10:1593–1598. doi: 10.1093/intimm/10.11.1593. [DOI] [PubMed] [Google Scholar]
- 10.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 11.Leong KH, Ramsay AJ, Boyle DB, Ramshaw IA. Selective induction of immune responses by cytokines coexpressed in recombinant fowlpox virus. J Virol. 1994;68:8125–8130. doi: 10.1128/jvi.68.12.8125-8130.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puisieux I, Odin L, Poujol D, Moingeon P, Tartaglia J, Cox W, Favrot M. Canarypox virus-mediated interleukin 12 gene transfer into murine mammary adenocarcinoma induces tumors suppression and long-term antitumoral immunity. Human Gene Ther. 1998;9:2481–2491. doi: 10.1089/hum.1998.9.17-2481. [DOI] [PubMed] [Google Scholar]
- 13.Rautenschlein S, Sharma JM, Winslow BJ, McMillen J, Junker D, Cochran M. Embryo vaccination of turkeys against Newcastle disease infection with recombinant fowlpox virus constructs containing interferons as adjuvants. Vaccine. 2000;18:426–433. doi: 10.1016/s0264-410x(99)00254-6. [DOI] [PubMed] [Google Scholar]
- 14.Gattacceca F, Pilatte T, Billard C, Monnet I, Moritz S, Le Carrou J, Eloit M, Jaurand M-C. Ad-IFN gamma induces antiproliferative and antitumoral responses in malignant mesothelioma. Clin Cancer Res. 2002;10:3298–3304. [PubMed] [Google Scholar]
- 15.Legrand FA, Verardi PH, Chan KS, Peng Y, Jones LA, Yilma TD. Vaccinia viruses with a serpin gene deletion and expressing IFN-gamma induce potent immune responses without detectable replication in vivo. PNAS (USA) 2005;102:2940–2945. doi: 10.1073/pnas.0409846102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curnis F, Gasparri A, Sacchi A, Cattaneo A, Magni F, Corti A. Targeted delivery of IFN-γ to tumor vessels uncouples antitumor from counteregulatory mechanisms. Cancer Res. 2002;65:2906–2913. doi: 10.1158/0008-5472.CAN-04-4282. [DOI] [PubMed] [Google Scholar]
- 17.Fujii S, Huang S, Fong TC, Ando D, Burrows F, Jolly DJ, Nemunaitis J, Honn DS. Induction of melanoma-associated antigen systemic immunity upon intratumoral delivery of interferon-gamma retroviral vector in melanoma patients. Cancer Gene Ther. 2000;7:1220–1230. doi: 10.1038/sj.cgt.7700224. [DOI] [PubMed] [Google Scholar]
- 18.Khorana AA, Rosenblatt JD, Sahasrabudhe DM, Evans T, Ladrigan M, Marquis D, Rosell K, Whiteside T, Phillippe S, Acres B, Slos P, Squiban P, Ross M, Kendra K. A phase I trial of immunotherapy with intratumoral adenovirus-interferon-gamma (TG1041) in patients with malignant melanoma. Cancer Gene Ther. 2003;10:251–259. doi: 10.1038/sj.cgt.7700568. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar S, Flores I, De Rosa C, Ozzello L, Ron Y, Pertka S. Injection of irradiated B16 melanoma genetically modified to secrete IFN-a causes regression of an established tumor. Int J Oncology. 1995;7:17–24. doi: 10.3892/ijo.7.1.17. [DOI] [PubMed] [Google Scholar]
- 20.Kaido T, Bandu MT, Maury C, Ferrantini M, Belardelli F, Gresser I. IFN-alpha1 gene transfection completely abolishes the tumorigenicity of murine B16 melanoma cells in in allogeneic DBA/2 mice and decreases their tumorigenicity in syngeneic C57BL/6 mice. Int J Cancer. 1995;60:221–229. doi: 10.1002/ijc.2910600216. [DOI] [PubMed] [Google Scholar]
- 21.Kass E, Panicali DL, Mazzara G, Schlom J, Greiner JW. Granulocyte/macrophage-colony stimulating factor produced by recombinant avian poxviruses enriches the regional lymph nodes with antigen-presenting cells and acts as an immunoadjuvant. Cancer Res. 2001;61:206–214. [PubMed] [Google Scholar]
- 22.Reali E, Canter D, Zeytin H, Schlom J, Greiner JW. Comparative studies of avipox-GM-CSF versus recombinant GM-CSF protein as immune adjuvants with different vaccines platforms. Vaccine. 2005;23:2909–2921. doi: 10.1016/j.vaccine.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 23.Marshall JL, Gulley JL, Arlen PM, Beetham PK, Ysang KY, Slack R, Hodge JW, Doren S, Grosenbach DW, Hwang J, Fox E, Odogwu L, Park S, Panicali D, Schlom J. Phase I study of sequential vaccinations with fowlpox-CEA (6D)-TRICOM alone and sequentially with vaccinia-CEA (6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–731. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 24.Robbins PF, Kantor J, Salgaller M, Horan Hand P, Fernstein PD, Schlom J. Transduction and expression of the human carcinoembryonic antigen (CEA) gene in a murine colon carcinoma cell line. Cancer Res. 1991;51:3757–3762. [PubMed] [Google Scholar]
- 25.Jenkins S, Gritz L, Fedor C, O’Neill EM, Cohen LK, Panicali DL. Formation of lentivirus particles by mammalian cells infected with recombinant fowlpox virus. AIDS Res Hum Retroviruses. 1991;7:991–998. doi: 10.1089/aid.1991.7.991. [DOI] [PubMed] [Google Scholar]
- 26.Jourdier T-M, Moste C, Bonnet M-C, Delisle F, Tafani JP, Devauchelle P, Tartaglia J, Moingeon P. Local immunotherapy of spontaneous feline fibrosarcoma using recombinant poxviruses expressing interleukin 2 (IL2) Gene Ther. 2003;10:2126–2132. doi: 10.1038/sj.gt.3302124. [DOI] [PubMed] [Google Scholar]
- 27.George TC, Mason LH, Ortaldo JR, Kumar V, Bennett M. Positive recognition of MHC class I molecules by the Ly49D receptor of murine NK cells. J Immunol. 1999;162:2035–2043. [PubMed] [Google Scholar]
- 28.Suttles J, Schwarting GA, Stout RD. Flow cytometric analysis reveals the presence of asialo GM1 on the surface membrane of alloimmune cytotoxic T lymphocytes. J Immunol. 1986;136:1586–1591. [PubMed] [Google Scholar]
- 29.Karlhofer FM, Yokoyama WM. Stimulation of murine natural killer (NK) cells by a monoclonal antibody for the NK1.1 antigen. J Immunol. 1991;146:3662–3673. [PubMed] [Google Scholar]
- 30.Lee S-H, Zafer A, de Repentigny Y, Lothay R, Trembley ML, Gros P, Duplay P, Webb JR, Vidal SM. Transgenic expression of the activating natural killer receptor Ly49H confers resistance to Cytomegalovirus in genetically susceptible mice. J Exp Med. 2003;197:515–526. doi: 10.1084/jem.20021713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber JS, Rosenberg SA. Effects of murine class I major histocompatibility complex expression on antitumor activity of tumor-infiltrating lymphocytes. J Natl Cancer Inst. 1990;82:755–761. doi: 10.1093/jnci/82.9.755. [DOI] [PubMed] [Google Scholar]
- 32.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–201. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 34.Mason LH, Gosselin P, Anderson SK, Fosler WE, Ortaldo JR, McVicar DW. Differential tyrosine phosphorylation of inhibitory versus activating Ly49 receptors and their recruitment of SHP-1 phosphatase. J Immunol. 1997;159:4187–4196. [PubMed] [Google Scholar]
- 35.Buentke EL, Heffler C, Wilson JL, Wallin RP, Lofman C, Chambers BJ, Ljunggren HG, Scheynius A. Natural killer and dendritic cell contact in lesional atopic dermatitis skin-Malassezia-influenced cell interactions. J Invest Dermatol. 202;119:850–857. doi: 10.1046/j.1523-1747.2002.00132.x. [DOI] [PubMed] [Google Scholar]
- 36.Borg C, Jalil A, Laderach D, Laderach D, Maruyama K, Charrier S, Ryffel B, Cambi A, Figdor C, Vainchenker W, Galy A, Caignard A, Zitvogel L. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization. Blood. 2004;104:3267–3275. doi: 10.1182/blood-2004-01-0380. [DOI] [PubMed] [Google Scholar]
- 37.Hayakawa Y, Kelley JM, Westwood JA, Darcy PK, Diefenbach A, Rauler D, Smyth MJ. Tumor rejection mediated by NKG2D receptor-ligand interaction is dependent upon perforin. J Immunol. 2002;169:5377–381. doi: 10.4049/jimmunol.169.10.5377. [DOI] [PubMed] [Google Scholar]
- 38.Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor formation. J Exp Med. 2005;202:583–588. doi: 10.1084/jem.20050994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood. 2005;106:2252–2258. doi: 10.1182/blood-2005-03-1154. [DOI] [PubMed] [Google Scholar]
- 40.Cooper MA, Fehniger TA, Fuchs A, Colonna M, Caligiuri MA. NK cell and DC interactions. TRENDS in Immunol. 2004;25:47–52. doi: 10.1016/j.it.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 41.Moretta L, Ferlazzo G, Mingari MC, Melioli G, Moretta A. Human natural killer cell function and their interactions with dendritic cells. Vaccine. 2003;21(S2):38–42. doi: 10.1016/s0264-410x(03)00197-x. [DOI] [PubMed] [Google Scholar]
- 42.Mailliard RB, Son Y-I, Redlinger R, Coates PT, Giermasz A, Morel PA, Storkus WJ, Kalinski P. Dendritic cells mediate NK cell help for Th1 and CTL responses: Two-signal requirement for the induction of NK cell helper function. J Immunol. 2001;171:2366–2373. doi: 10.4049/jimmunol.171.5.2366. [DOI] [PubMed] [Google Scholar]
- 43.Stewart TJ, Smyth MJ, Fernando GJP, Frazer IH, Leggatt GR. Inhibition of early tumor growth requires Ja18-positive (Natural Killer T) Cells. Cancer Res. 2003;63:3058–3060. [PubMed] [Google Scholar]
- 44.Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nature Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 45.Semino C, Angelini G, Poggi A, Rubartelli A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factors HMGB1. Blood. 2005;106:609–616. doi: 10.1182/blood-2004-10-3906. [DOI] [PubMed] [Google Scholar]
- 46.Kalinski P, Giermasz A, Nakamura Y, Basse P, Storkus WJ, Kirkwood JM, Mailliard RB. Helper role of NK cells during the induction of anticancer responses by dendritic cells. Mol Immunol. 2005;42:535–539. doi: 10.1016/j.molimm.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 47.Andoniou CE, van Dommelen SLH, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposito M. Interaction between conventional dendritic cells and natural killer cell is integral to the activation of effective immunity. Nature Immunol. 2005;6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- 48.Biron CA. Activation and function of natural killer cell responses during viral infections. Curr Opin Immunol. 1997;9:24–34. doi: 10.1016/s0952-7915(97)80155-0. [DOI] [PubMed] [Google Scholar]