

Abstract
Alamethicin is a well-studied channel-forming peptide that has a prototypical amphipathic helix structure. It permeabilizes both microbial and mammalian cell membranes, causing loss of membrane polarization, and leakage of endogenous contents. Antimicrobial peptide-lipid systems have been studied quite extensively and have led to significant advancements in membrane biophysics. These studies have been performed on lipid bilayers that are generally charged or zwitterionic and restricted to a thickness range of 3 – 5 nm. Bilayers of amphiphilic diblock copolymers are a relatively new class of membranes that can have significantly different material properties compared with those of lipid membranes. In particular, they can be made un-charged, non-zwitterionic, and much thicker than their lipid counterparts. In an effort to extend studies of membrane-protein interactions to these synthetic membranes, we have characterized the interactions of alamethicin and several other membrane-active peptides with diblock copolymer bilayers. We find that although alamethicin is too small to span the bilayer, the peptide interacts with, and ruptures thick polymer membranes.
Introduction
It is now clear that a self-assembled bilayer of amphiphiles can be much thicker than the 3-5 nm that is typical of lipid bilayers. Indeed, diblock copolymers of the appropriate amphiphilic proportions assemble in water into vesicles – polymersomes – and can be made with hydrophobic cores up to 20 nm thick1. Copolymer membranes can also be made uncharged, non-zwitterionic, mechanically stronger, and chemically more inert than lipid bilayers. For many biotechnology applications, polymersomes appear to offer a number of advantages1. Analogies with biological membranes have guided much of the work on polymersomes and initial work with detergents2 has demonstrated a scaled resistance to disruption. A theoretical analysis of the energetics of inclusions in polymeric membranes suggests that it should be possible to incorporate membrane proteins into polymersomes even when there is a substantial mismatch between the hydrophobic thickness of the protein and bilayer3. Indeed, at least two membrane proteins (porins) have been successfully incorporated in a copolymer membrane4. However interactions of diblock copolymers with well-characterized model protein and peptide systems have not yet been explored. It is not known, in particular, to what extent general features of protein-lipid bilayer interactions carry over to thick, neutral, non-zwitterionic block copolymer membranes.
We describe in this paper studies of the action of the membrane-active peptide alamethicin on polymersomes. Alamethicin is a well-studied antimicrobial peptide of 21 amino acids from the fungus, Trichoderma viride. It adopts a helical structure in both monomeric and helix-bend-helix dimeric forms, that self-assemble in membranes into voltage-dependent5 ion conducting helix bundles. Alamethicin is also an antimicrobial agent6 that acts both on microbes and mammalian cells by permeabilizing the cell membrane causing leakage of endogenous contents and membrane depolarization7. The interaction of alamethicin with lipid bilayers has served as an important system for the study of basic properties of membrane-protein interactions, ion channels, and antimicrobial peptides and has continued to be studied quite extensively5,8-14. Past work has led to an elegant model for the action of alamethicin and other antimicrobial peptides10,15,16 in which bilayer thinning leads to the formation of peptide-stabilized pores.
The polymer vesicles used in our experiments are composed of two different amphiphilic diblock copolymers, OE7 and OB18, which form membranes with 8 nm and 15 nm thick cores, respectively (Fig. 1)17,18. When dry films of the copolymers are hydrated in water, polymersomes form spontaneously19. Both copolymer membranes are significantly thicker than the ∼4 nm thickness of a typical phosphatidyl choline (PC) lipid bilayer20.
Figure 1. Relative size of peptide and membranes.
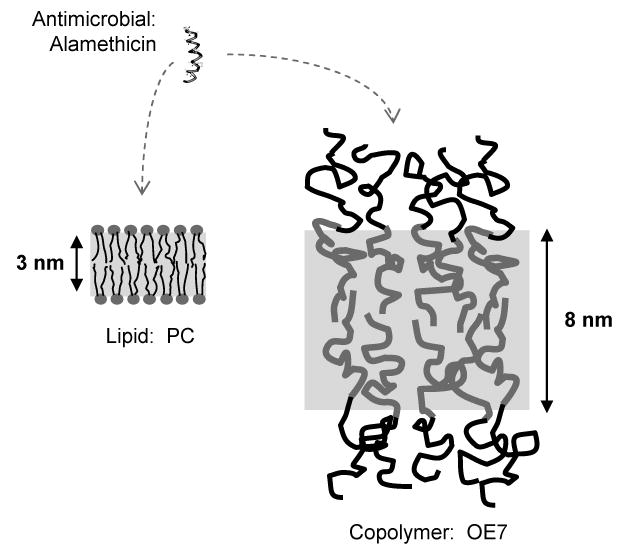
Alamethicin forms an amphiphilic helix that is only a few angstroms shorter than the ∼3 nm typical hydrophobic core thickness of lipid membranes. The hydrophobic core thickness of the OE7 (PEO-PEE) diblock copolymer membrane is around 8 nm as determined by cryo-TEM and the corresponding thickness for OB18 (PEO-PBD) is ∼14 nm (not shown)18.
We analyzed alamethicin-polymersome interactions with a host of complementary techniques. Peptide binding was monitored by a spectral shift of the fluorophore LAURDAN and by circular dichroism (CD), vesicle permeabilization or lysis was monitored by calcein leakage, and changes to vesicle and bilayer morphology were monitored by optical microscopy, dynamic light scattering, and neutron scattering. Given the importance of electrostatic interactions (for both charged and zwitterionic lipids) in the alamethicin-lipid system and the ability of alamethicin to span the lipid bilayer hydrophobic core, it is not clear a priori whether alamethicin would interact with polymer vesicles, which are neutral, non-zwitterionic, and have substantially thicker bilayers (Fig. 1). However, we found that alamethicin binds to both the thinner (OE7) and thicker (OB18) membranes and induces lysis of vesicles at high peptide concentrations. We also found evidence of bilayer thinning prior to lysis, which is reminiscent of the behavior observed for lipid bilayers. The disruptive effects of alamethicin on neutral polymer vesicles indicate unanticipated interactions that suggest, for lipid membranes, more generic amphipathic insertion mechanisms.
Materials and Methods
Chemicals
The diblock copolymers OE7, (polyethylene oxide)40-(polyethylethylene)37 (number-averaged molecular weight Mn = 3,700 g/mol), and OB18, (polyethylene oxide)80-(polybutadiene)125 (Mn = 10,400), were synthesized by Hillmyer and Bates18,21. LAURDAN was purchased from Molecular Probes (Eugene, Oregon), and a stock solution of 20 mg/ml was made in chloroform, from which aliquots of 0.2 mg/ml (in chloroform) were prepared for use in actual experiments. Egg PC, calcein, alamethicin, melittin, polymyxin and mastoparan were from Sigma (St. Louis, MO). The alamethicin from Sigma (product # A4665) consists of the neutral alamethicin F50 peptide with trace amounts of the negatively charged alamethicin F30 22. Alamethicin and mellitin were dissolved in 20 % methanol to 1 mg/ml, except for the neutron scattering experiments where 10 mg/ml stock solutions of alamethicin were made in 100% methanol. Deuterated water (Sigma) was used to prepare phosphate buffered saline (PBS) solutions for neutron scattering experiments. Polymyxin and mastoparan were dissolved in distilled de-ionized water to make 1mg/ml stock solutions.
Vesicle preparation
Vesicles for all of the experiments were made by film rehydration19. Briefly, 1 ml of a 1 mg/ml solution of polymer or lipid dissolved in chloroform was uniformly coated on the inside wall of a glass vial. For the LAURDAN spectral shift experiment, 0.1 mol % LAURDAN was mixed with lipid or polymer in chloroform. The chloroform was evaporated in a stream of nitrogen and the film allowed to dry in vacuum for 6-8 hours. For LAURDAN and light scattering experiments, vesicles were swelled by adding 1 ml PBS to the film and placing the vials in a 60° C oven for 8 hours, which led to budding of vesicles off the glass wall. After 5 cycles of freeze-thawing in dry ice and water at 37°C, the vesicles were made monodisperse by passing the suspension repeatedly through a vesicle extruder with a pore size (diameter) of 100 nm or 200 nm (Avestin Inc., Ottawa, Ontario, Canada). Average size and polydispersity were measured by dynamic light scattering. For neutron scattering experiments, vesicles were swelled in deuterated PBS. For the calcein leakage assay23, 50 mM calcein was made in Tris buffer and the pH was adjusted to 7.4 with NaOH. Vesicles were swelled by adding this solution to polymer or lipid films and following the above protocols. Non-encapsulated calcein was removed by dialysis prior to vesicle extrusion. Since there was no significant change in volume after dialysis, we have assumed the final polymer concentration was equal to the starting concentration. For microscopy, the polymer or lipid film was rehydrated in a ∼150 mOsm sucrose/H2O solution, freeze-thawed 5 times in dry ice and water at 37°C, and viewed under phase contrast after dilution into PBS. In all cases for solutions of vesicle, molar concentration refers to the molar concentration of polymer molecules.
Fluorescence spectroscopy of LAURDAN
LAURDAN-incorporated OE7 vesicles were added to 1.5 ml of PBS in a cuvette equipped with a small magnetic stir bar. The final concentration of polymersomes was either 2.5 or 5 μM, and peptide was added in steps to obtain various concentration ratios of polymer or lipid to peptide. LAURDAN fluorescence was monitored with a Photon Technology International (Lawrenceville, NJ) spectrofluorometer.
Calcein leakage assay
Calcein loaded vesicles were suspended in 1.5 ml of buffer (10 mM Tris/HCl,. 3 mM EDTA, NaCl, pH 7.4). The amount of NaCl was chosen to match the osmolarity of the calcein solution used to prepare the vesicles and was in the range of 150 - 200 mM. The final polymer concentration was between 5 and 10 μM. Peptides were added in fixed concentration steps. The sample was stirred with a magnetic stir bar for a short time immediately after adding peptides to ensure uniform mixing. Permeabilization or rupture was monitored through the increase in calcein fluorescence. 0.6 to 1 % of Triton X100 detergent was used to cause complete lysis of vesicles. Normalized fluorescence (FN) was defined by FN = (F – FB)*100/(FD – FB), where FB is the baseline fluorescence before adding peptide and FD is the fluorescence obtained after detergent lysis.
Circular dichroism
For circular dichroism (CD) measurements, a suspension of 200 nm diameter OE7 vesicles (42 μM OE7), was added to a 300 μl quartz cuvette with 1 mm optical path length. Alamethicin was added to the cuvette in steps, and ellipticity (for wavelengths between 200 and 260 nm, at 1 nm interval) was measured on an AVIV 62DS spectropolarimeter. Background signal from the buffer was subtracted before processing of the data. The helical content of the peptide was monitored from the ellipticity of the solution at 222 nm. For each peptide concentration, the ellipticity of the sample containing vesicles and alamethicin (θ222) was subtracted from the corresponding ellipticity for a suspension of pure alamethicin (θ222,Alam).
Dynamic light scattering
Dynamic light scattering (DLS) was performed on a Protein Solutions, DynaPro MS/X instrument (Proterion Corp, Piscataway, NJ). Samples were placed in a quartz cuvette with an effective sample volume of 50 μl. The concentration of vesicles used was typically 3 to 5 μM in PBS. Peptides were added to the cuvette and rapidly mixed by drawing the solution in and out of the pipette several times. The samples were placed in the DLS instrument immediately after mixing. The DynaPro Dynamics software included with the instrument was used to extract the distribution of vesicle sizes (hydrodynamic radii) from the light scattering signal. This software uses a proprietary algorithm to determine the distribution of particle sizes that is similar to the algorithm used in the Dynals software24.
Microscopy and micromanipulation
Video microscopy was performed on a Nikon TE-300 inverted microscope equipped with phase contrast. Image collection through a 40x or 60x objective lens was accomplished with a CCD video camera mounted on the front port of the microscope. A custom manometer with pressure transducers (Validyne, Northridge, CA) was used for controlling and maintaining pressure to the micropipette as in 25, 19. Micropipettes, with tip diameters of 5 or 10 microns, were prepared as in 25, 19. Fractional membrane thinning was determined from the increase in membrane projection inside the micropipette by: % thinning = r·Δl/(2R2 + r·li), where Δl is the change in membrane projection inside the micropipette, li is the initial aspirated length, r is the pipette radius, and R is the vesicle radius. This formula is based on the assumption that the hydrophobic core of the membrane is incompressible and that the volume change of the vesicle is negligible 26.
Neutron scattering
Neutron scattering measurements from polymersomes were performed at the time-of-flight small angle diffractometer (TOF-SAD) at IPNS, Argonne national laboratory, Argonne, Il. The time-resolved detector used in the TOF-SANS instrument enables simultaneous measurement of scattering in a large Q (wavenumber) range from a single pulse of polychromatic neutrons, thus reducing the time the sample has to be kept in the neutron beam. Scattering density contrast between the polymer bilayer and the surrounding medium was obtained by preparing the vesicles in deuterated PBS. Experiments were performed at 25 °C in circular quartz cells of 2 mm path length. The concentration of vesicles was 2 mg/ml and the samples were placed in the neutron beam for at least 6 hours to collect reliable scattering statistics.
Results
Blue-shifted LAURDAN Emission
LAURDAN is an amphiphilic fluorophore that has been used to assess interfacial perturbations and phase changes in both liposomes27 and polymersomes19. When LAURDAN incorporates into membranes, the molecule's lauric acid chain pulls the chromophore into the hydrophobic core of the bilayer just beneath the lipid/water (polymer/water) interface. Fluorescence excitation leads to a localized charge separation that couples dissipatively to surrounding dipoles. As a result, the LAURDAN emission spectrum is sensitive to the strength of this coupling. For example, changes that result in decreased water accessibility, e.g. as a result of adding organic solvents, will result in a blue-shift of the emission spectrum19.
Addition of alamethicin to LAURDAN labeled PC vesicles caused a blue-shift in the LAURDAN emission spectrum (Fig. 2A). Likewise, when alamethicin was added to LAURDAN-containing polymer vesicles, a blue-shift was also seen (Fig. 2A,B). This clearly indicates that alamethicin is able to penetrate the polymersome's hydrophilic brush and partition into the bilayer interface. Since the alamethicin was dissolved in methanol, control experiments were performed in which identical amounts of methanol (up to 0.01 volume %) were added without alamethicin. No spectral shift was observed in this case. For PC vesicles, the blue-shift was detectable above a threshold molar ratio of alamethicin to PC of approximately 1:20 (Fig. 2A). The effect saturated at higher concentrations. In the absence of peptide, the LAURDAN emission spectrum from OE7 vesicles was red-shifted relative to the spectrum from PC vesicles (maxima at 480 nm and 470 nm for OE7 and PC, respectively). This suggests that LAURDAN is exposed to a more polar environment in the OE7 membrane. No blue shift was seen below 1:5 molar ratio of alamethicin to OE7.
Figure 2. LAURDAN spectral shift.

(A) Addition of alamethicin to PC, OE7 and OB18 vesicles causes LAURDAN emission maxima to blue shift. The data for PC is shown in the inset. A threshold concentration below which alamethicin did not result in a detectable blue shift was observed for PC and OE7. (B) Emission spectra of LAURDAN incorporated in OE7 vesicles with different concentrations of alamethicin. Note that the curves intersect at a common point. The arrows denote directions of increasing molar ratio of alamethicin to OE7. (C) Two-population fits for blue-shifted LAURDAN spectra. The fits were determined with the formula Fi = αF0 + (1-α)F∞, where α is the fitting parameter, F0 is the spectrum in the absence of alamethicin, and F∞ is the spectrum at a saturating concentration of alamethicin, which we took to be 2:1 alamethicin to OE7. The values for α determined from the fits are shown in the table on the right. Representative fits are shown in the graph on the left. (D) LAURDAN blue-shift for three other membrane-active peptides added to OE7 vesicles. Melittin and mastoparan are two antimicrobial peptides that are also known to permeabilize lipid vesicles. They both show weak blue-shifts, indicating at least some interaction with the OE7 bilayer. Polymyxin, an anti-bacterial agent that binds to lipopolysaccharides found in gram-negative bacterial cell walls, does not affect the spectral properties of LAURDAN in the bilayer.
All of the LAURDAN-OE7 emission spectra intersect at the same point as the alamethicin concentration is varied (Fig. 2B). This behavior was not observed in PC-LAURDAN spectra nor in OB18-LAURDAN spectra. A simple explanation for this behavior is that at intermediate levels of alamethicin the samples consist of two populations: one has an unshifted LAURDAN spectrum and the other has a blue-shifted spectrum. As alamethicin is varied the relative abundance of the two populations varies from 100% unshifted population (e.g. no alamethicin added) to 100% blue-shifted population (e.g. 2.5 μM alamethicin). To test this we fitted each spectrum to a linear combination of the unshifted (no alamethicin) and blue-shifted (5 μM alamethicin) spectra (Fig. 2C). All of the spectra gave good fits to within experimental error (representative fits are shown in Fig. 2C) indicating that there are indeed two distinct populations. This could reflect two different populations of LAURDAN within each vesicle, e.g. due to two different peptide-copolymer phases in the membrane, or alternatively it may reflect two different LAURDAN-containing polymer structures, e.g. coexisting polymersomes and micelles.
LAURDAN incorporates into PC and OE7 bilayers at a fairly low concentration of 0.05 mol %. At similar LAURDAN concentrations in OB18 membranes the fluorescence was very low and noisy. Spectral shift experiments in OB18 were therefore performed with 1 mol % LAURDAN. LAURDAN has an emission peak at 470 nm in OB18, which is closer to the emission peak of LAURDAN in PC than that of LAURDAN in OE7. This may indicate that for OB18 the LAURDAN molecules are trapped deep within the polymer brush and are well-shielded from water. Alamethicin causes a strong blue-shift in OB18 membranes that saturates at approximately 1:1 ratio of peptide to polymer (Fig. 2A).
In addition to alamethicin, three other antimicrobials were added to LAURDAN containing OE7 vesicles (Table 1). Polymyxin did not alter the spectral properties of LAURDAN in any way. Mastoparan and mellitin, two peptides that are known to form toroidal pores in lipid membranes, showed relatively weak blue-shifts (Fig. 2D).
Table 1.
| PC | OE7 | OB18 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Peptide | LAURDAN Emission Peak | Calcein Leakage | DLS | LAURDAN Emission Peak | Calcein Leakage | DLS | LAURDAN Emission Peak | Calcein Leakage | DLS |
| Alamethicin | Blue-Shifts | Leaks | Ruptures | Blue-Shifts | Leaks | Ruptures | Blue-Shifts | No Leak | No Change |
| Melittin | Blue-Shifts | Leaks | Ruptures | Blue-Shifts | No Leak | No Change | No Change | No Leak | No Change |
| Mastoparan | Blue-Shifts | Leaks | No Change | Blue-Shifts | No Leak | No Change | No Change | -- | No Change |
| Polymyxin | -- | No Leak | No Change | No Change | No Leak | No Change | No Change | -- | No Change |
Calcein Leakage
Calcein leakage is a well-established assay for studying vesicle permeabilization23. Vesicles loaded with a high concentration (50 mM) of the fluorescent compound calcein show substantial self-quenching. Permeabilization or rupture leads to a drop in calcein concentration and an increase in fluorescence. For our experiments, fluorescence emission was normalized by the fluorescence after detergent lysis. Addition of a high concentration of alamethicin to calcein-loaded PC vesicles showed rapid and complete leakage (Fig. 3A), which is consistent with previous studies28. Surprisingly, we found that alamethicin was also able to permeabilize OE7 vesicles (Fig. 3B). Addition of alamethicin to a molar ratio of 1:2 (alamethicin:OE7) caused a partial increase in fluorescence (Fig. 3B). The kinetics varied from sample to sample of OE7 vesicles (data not shown). However, over a period of half an hour, the extent of leakage was comparable (within 20%) across samples. The order of addition did not matter, i.e. addition of OE7 vesicles to an alamethicin solution gave similar results, ruling out the possibility that the incomplete lysis is due to a transient high concentration of peptide upon addition of peptide to the polymersome solution. Alamethicin did not cause any leakage from OB18 vesicles, even at concentrations of 1:1 (Fig. 3C).
Figure 3. Representative graphs of alamethicin-induced calcein leakage.

Fluorescence was normalized by the fluorescence level after detergent lysis (0.6 % Triton X 100). The arrows indicate the instant at which the peptide or detergent was added. The concentration of peptide is represented as a molar ratio (alamethicin to polymer or lipid). (A) Alamethicin induces 100% leakage from 200 nm PC vesicles. (B) Alamethicin induces partial calcein leakage from 200 nm OE7 vesicles. (C) Alamethicin does not induce calcein leakage from 200 nm OB18 vesicles.
Leakage from OE7 vesicles was not observed below a threshold molar ratio of ∼ 1:4 (alamethicin:OE7) (Fig. 4). In addition, unlike the case for PC vesicles, complete leakage from OE7 vesicles occurred only at extremely high alamethicin concentrations (5:1 or more). When alamethicin was added at intermediate concentrations in steps of 2 μM to a solution of calcein-loaded OE7 vesicles (15 μM OE7) the extent of leakage varied with each addition of peptide. Sometimes adding alamethicin caused no further increase in fluorescence while the fluorescence level would increase with the next dose of peptide. These observations are consistent with sub-populations of vesicles with varying degrees of susceptibility to alamethicin.
Figure 4. Calcein leakage from OE7 vesicles as a function of alamethicin concentration.

Below a threshold molar ratio of about 0.25, alamethicin does not cause any leakage from OE7. Complete leakage is not observed except at very high concentrations. The amount of leakage varies between different samples of OE7 polymersomes, with a maximum variation of about 20%.
We also looked at the effects of mellitin, mastoparan, and polymyxin on calcein-loaded OE7 polymersomes. None of these peptides showed any evidence of calcein leakage for molar ratios up to 1:1 (data not shown).
Circular Dichroism
The calcein leakage results indicate that alamethicin permeabilizes OE7 vesicles. However this permeabilization occurs only at relatively high peptide concentrations. This may indicate that alamethicin has a low affinity for the polymer bilayer. Alternatively, it may indicate that a very large amount of bound alamethicin is required for polymersome permeabilization. The former view is supported by the LAURDAN experiments, which showed a significant blue-shift for OE7 polymersomes only at a high alamethicin concentration. Partitioning of alamethicin into lipid bilayers is associated with an increase in the helical structure of the peptide, which can be detected by a corresponding increase in ellipticity (e.g. 29). We therefore carried out circular dichroism measurements on suspensions of OE7 polymersomes with various levels of alamethicin (Fig 5). There was a sharp increase in ellipticity at a molar concentration ratio of ∼1:3 (alamethicin:OE7). If we assume that, as in the case of lipid bilayers, binding of alamethicin to OE7 bilayers is associated with an increase in helical content of the peptide, then this result suggests that the peptide has very weak affinity for the bilayer at low peptide concentrations. Interestingly, the sharp rise in ellipticity at a threshold concentration suggests binding to the copolymer membrane is a strongly cooperative process. There is also a second increase in ellipticity at very high peptide concentration (∼1.5:1 alamethicin:OE7). This could indicate a change in phase of the copolymer-alamethicin mixture to a non-bilayer form that is able to bind more peptide.
Figure 5. Circular dichroism measurements of alamethicin in the presence of OE7 polymersomes.

The ellipticity at 222 nm (θ222) was determined for various molar ratios of alamethicin to OE7 and subtracted from the corresponding ellipticity for a solution of alamethicin alone (θ222,Alam). For all measurements, the OE7 concentration was 42 μM.
Microscopy and micromanipulation
To directly visualize the effects of alamethicin on vesicles, large (∼15 μm), phase dark liposomes and polymersomes were prepared in which the lumen was loaded with a sucrose solution that matched the osmolarity of the phosphate buffer on the outside (∼ 290 mOsm). The refractive index difference between the vesicle interior and exterior provided contrast for phase contrast microscopy. When alamethicin at 1:2 concentration ratio was added to PC and OE7 vesicles while they were being imaged under the microscope, vesicles were observed to lyse. To study individual vesicles in more detail, micro-manipulation techniques were used30,31. Alamethicin at 0.05 mg/ml was loaded into a micropipette and moved into a chamber containing lipid or polymer vesicles. The pipette was then positioned close to a vesicle and peptide was allowed to diffuse out. For OE7 vesicles, membrane disintegration was observed at one spot on the membrane, followed by an abrupt and dramatic collapse of the whole vesicle (Fig. 6A). Vesicle shape fluctuations occurred just before collapse. To study OE7 lysis more closely, an apparatus with two micropipettes was used. An OE7 vesicle was held aspirated by a micropipette while peptide was allowed to diffuse from a second pipette that was positioned next to the vesicle (Fig. 6B). We found that as alamethicin was released, the amount of OE7 membrane that was pulled into the pipette increased with time until the vesicle ultimately ruptured. At the same time, however, the radius of the vesicle did not change (Fig. 6B). This apparent increase in membrane area may be due to incorporation of alamethicin into the bilayer, with concomitant membrane thinning. (Similar observations for PC vesicles in the presence of other peptides, have been made32.) Bilayer thinning was calculated from the increase in projection inside the micropipette (see methods). We found that the percentage thinning at the time of rupture was a decreasing function of the size of the vesicles (Fig. 7). Since the alamethicin was allowed to diffuse freely from the second micropipette in all experiments, this observation could be due to the high local ratio of alamethicin to polymer in smaller vesicles. The range of thinning calculated is from 2 % to 14 % of the original thickness. These polymersomes have been reported to rupture at pipette-imposed area strains of ∼20%19.
Figure 6. Micropipette delivery of alamethicin.
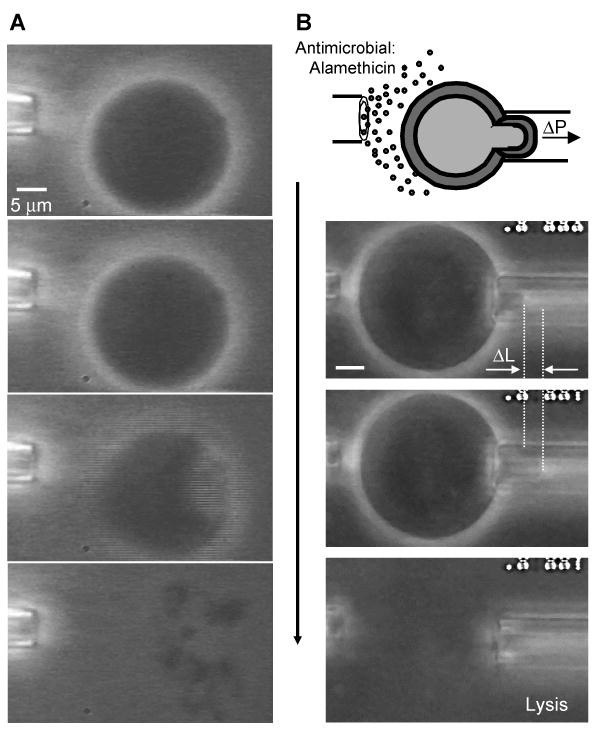
(A) Alamethicin was allowed to diffuse freely from a micropipette placed near an OE7 vesicle. Phase contrast was achieved by loading the lumen with a sucrose solution that was equi-osmotic to the surrounding PBS buffer. The pipette contained alamethicin at 0.05 mg/ml. No phase loss was observed before disintegration of the vesicle. Instead, a dramatic collapse occurred, triggered by the loss of membrane integrity at one spot on the vesicle. (B) An OE7 vesicle was held in a micropipette and alamethicin was allowed to diffuse from a second pipette. The amount of membrane aspirated into the pipette increased with time until vesicle rupture while the radius of the vesicle remained unchanged. (The scale is the same as in A.)
Figure 7. Membrane thinning due to micropipette delivery of alamethicin.

(A) Increase in the membrane projection inside the micropipette as a function of time. Zero time denotes the frame before any noticeable change was observed in the length of the membrane projection. The curve terminates at the point of rupture. (B) The increase in membrane surface area due to the increase in membrane inside the micropipette can be used to estimate the extent of bilayer thinning if we assume the hydrophobic core of the bilayer is incompressible. The thinning just prior to rupture is plotted as the percentage of initial membrane thickness vs. vesicle diameter.
For PC vesicles exposed to 0.05 mg/ml alamethicin, loss of phase contrast was gradual and preceded loss of membrane integrity (Fig. 8A). Smaller vesicles trapped inside larger PC vesicles were expelled, indicating the presence of large pores. As noted above, phase loss and lysis were simultaneous for OE7 vesicles. When alamethicin at these same concentrations was delivered locally to OB18 vesicles by a micropipette no change in morphology or phase contrast was observed. However when OB18 vesicles were transported into a bath of 0.8 mg/ml alamethicin, phase loss and membrane curling were observed (Fig. 8B). Alamethicin appears to rupture OB18 vesicles only at extremely high concentrations of peptide.
Figure 8.

(A) Alamethicin was allowed to diffuse locally near a PC vesicle. Loss of phase contrast was observed before vesicle disintegration. Smaller vesicles trapped inside the larger vesicle were pushed out prior to complete lysis of the large vesicle. (B) Phase loss followed by lysis of an OB18 vesicle placed in a bath of 0.8 mg/ml alamethicin. OB18 is inert to alamethicin except at very high concentrations of peptide.
Dynamic Light Scattering
To examine the effect of alamethicin on the morphology of the smaller vesicles used in the LAURDAN and calcein experiments, we used dynamic light scattering. Data was analyzed assuming scattering from spherical objects, which were characterized by their hydrodynamic radii. It is possible that after addition of peptide, part of the light scattering was in fact from non-spherical bilayer or micellar structures. Nevertheless, representation of the scattering data as a distribution of hydrodynamic radii provides a convenient measure of changes in vesicle morphology. Representative size distribution histograms for 100 nm vesicles in the presence of alamethicin are shown in Fig. 9. For PC vesicles, a change in the distribution was only observed for an alamethicin to PC molar ratio above 1:20 (data not shown). Populations of larger and smaller sizes were evident, suggesting rupture and re-aggregation. For OE7, there appeared to be a threshold molar ratio of roughly 1:4 (alamethicin to polymer) below which no significant change in the size distribution was observed. Adding alamethicin to OB18 vesicles did not cause any significant change in the vesicle size distribution even at 1:1 molar ratios. Mellitin, mastoparan, and polymyxin did not show any effect on light scattering from OE7 vesicles (data not shown).
Figure 9. Dynamic light scattering from 200 nm vesicles.

The size distribution is plotted as mass fraction (assuming scattering from spherical particles) vs. radius. (A) The size distribution shifts to higher values after addition of alamethicin to PC vesicles. This indicates possible rupture and re-aggregation. (B) The size distribution broadens on adding alamethicin to OE7 vesicles. (C) There is very little change in the size distribution after addition of alamethicin to OB18 vesicles.
Neutron Scattering
To examine the effect of alamethicin at small length scales (of order the bilayer thickness), neutron scattering was used (e.g.33). Addition of alamethicin caused a significant shift in the scattering profile for molar ratios above 1:4 (Fig. 10A). With increasing alamethicin, the location of the scattering intensity minimum in the I vs. Q plot (Qmin) shifted towards larger wave numbers. The corresponding length scale 2π/Qmin shifted by ∼30 Å over the range of alamethicin concentrations studied (Fig. 10B). If we interpret this length scale as corresponding to an effective bilayer thickness, then this would give a thickness of OE7 vesicles in the absence of alamethicin of 11.6 nm. Since this includes the hydrated corona, it is not surprising that it is thicker than the 8 nm core thickness. At molar ratios of 1:3 and 1:2 the effective bilayer thickness is 8.9 nm and 7.8 nm, respectively. Note however that, at least for the samples containing alamethicin, other copolymer morphologies such as micelles may be present. In this case the length scale 2π/Qmin may not have a simple interpretation in terms of a bilayer thickness.
Figure 10.

(A) Scattering intensity vs. wave number (Q) from neutron scattering experiments. The first minimum shifts towards larger Q (smaller length scale) with increasing alamethicin. (B) Length scale associated with the first minimum in the scattering intensity (2π/Qmin), as a function of the molar ratio of peptide to polymer.
Discussion
The results here are the first to demonstrate interactions between a membrane-active peptide and thick, neutral, non-zwitterionic copolymer bilayers. We find that alamethicin, a well-characterized pore-forming antimicrobial peptide, partitions into amphiphilic copolymer membranes and causes vesicle rupture. Using a variety of different experimental techniques – LAURDAN blue-shift, calcein leakage, circular dichroism, dynamic light scattering, and neutron scattering – we consistently found a threshold concentration of ∼1:4 – 1:3 molar ratio of alamethicin to OE7 copolymer below which this peptide does not affect these vesicles. The circular dichroism and LAURDAN data suggest that below this threshold the peptide does not partition into the copolymer bilayer. Partial leakage from calcein-encapsulated vesicles indicates permeabilization and/or rupture, while the change in vesicle size distribution observed by DLS strongly supports the rupture hypothesis. The LAURDAN, calcein, and DLS results for OE7 as well as PC and OB18 vesicles are summarized in Table 1. Large OE7 vesicles visualized under the microscope in phase contrast were also seen to rupture upon adding alamethicin. Loss of phase contrast prior to rupture, which would indicate permeabilization, was never observed.
A reasonable hypothesis that accounts for many of the results is that above a critical concentration, alamethicin irreversibly binds to and ruptures a subpopulation of OE7 vesicles, and the number of ruptured vesicles increases with increasing concentration of alamethicin. LAURDAN blue-shift experiments further support this mechanism. A continuous blue-shift in the LAURDAN-OE7 fluorescence emission spectrum was observed as the alamethicin concentration was increased above a 1:4 molar ratio. This blue-shift can be decomposed into a linear combination of two spectra: one being the initial spectrum in the absence of any alamethicin, and the other the saturated spectrum at very high concentrations of alamethicin (5:1). This observation suggests there are two populations of LAURDAN at intermediate concentrations of alamethicin: ruptured and unruptured vesicles perhaps.
Further insights into the mechanism of interaction and rupture at large and small length scales are found in the micropipette aspiration and neutron scattering experiments, respectively. In the micropipette experiments, the radius of the aspirated vesicle remained constant while the membrane projection inside the micropipette increased until the vesicle finally ruptured. We interpret this as being due to an increase in the vesicle surface area as a result of alamethicin partitioning into the hydrophobic-hydrophilic interface of the copolymer membrane. Since the hydrophobic core is incompressible, the area increase is accompanied by a thinning of the bilayer. Neutron scattering from 200 nm OE7 vesicles gave results that were consistent with an increase in membrane thinning with increasing alamethicin. However, in light of the above results, the neutron scattering data at the higher alamethcin concentrations most likely represent scattering from a mixture of intact and ruptured vesicles. Further information regarding the structure of the ruptured vesicles will be required to provide a more realistic analysis of the structure factor.
The above picture of alamethicin interaction with OE7 polymersomes is reminiscent of a model for the interaction of alamethicin and other antimicrobial peptides with lipid bilayers15. In this model, alamethicin adsorbs to the membrane surface and intercalates between the lipid head groups. This results in an effective increase in membrane tension and bilayer thinning. As the concentration of alamethicin is increased, a threshold is reached beyond which it is energetically favorable for peptide to form membrane-spanning pores in a barrel stave fashion. This leads to bilayer permeabilization and, at high alamethicin concentrations, vesicle lysis. In contrast with the case of lipid bilayers, we did not find any evidence that alamethicin forms stable pores in polymersomes. Within the context of the above model, this may be because alamethicin cannot span the 8 nm thick hydrophobic core of the OE7 bilayer. In addition, our CD results indicate that alamethicin does not gradually partition into the polymer bilayer but instead shows a strongly cooperative transition at a threshold concentration.
We also found that alamethicin interacts with OB18 vesicles, which have a core that is roughly twice that of OE7. A very strong LAURDAN blue-shift was observed when alamethicin was added to LAURDAN-containing OB18 vesicles although, since we had to use a much higher concentration of LAURDAN (20-fold higher), it is difficult to compare these results with the PC and OE7 data. At molar concentrations of alamethicin to OB18 as high as 1:1 there was no evidence of lysis in calcein leakage experiments and no evidence of a change in vesicle morphology in DLS experiments. In microscopy experiments, phase loss and lysis of OB18 vesicles was observed only at extremely high concentrations of alamethicin. It thus appears that OB18 is largely inert to alamethicin, presumably as a result of the larger thickness of the OB18 bilayer.
Three other antimicrobial peptides, mellitin, polymyxin, and mastoparan, were tested for interaction with OE7 membranes. Melittin34, from bee venom, forms pores in lipid vesicles. Polymyxin35, a powerful anti-bacterial agent, kills bacteria by preferentially binding to lipopolysaccharides found in the cell walls of gram-negative bacteria. Mastoparan36, from wasp venom, is a human nerve cell de-granulizer and is also known to form pores and cause lipid flip-flop in liposomes. Mastoparan and melittin were found to interact with OE7 vesicles in LAURDAN blue-shift experiments, but they did not lead to vesicle permeabilization or rupture. Polymyxin did not appear to have any effect on polymersomes. These results are summarized in Table 1. At present we do not have an explanation for the differences between these peptides and alamethicin in their reactivity towards OE7 bilayers. However the fact that the alamethicin that we have used is predominantly neutral, with a minority component that is negatively charged, whereas the above peptides are positively charged, may be important.
The results described here are an important step towards developing peptides and proteins that can stably function in polymer bilayers. The fact that alamethicin is able to penetrate the neutral hydrophilic polymer brush and partition into the thick hydrophobic cores of polymersomes suggests that synthetic peptides could be designed that further mimic the behavior of alamethicin in lipid bilayers. In particular, derivatives of alamethicin that form longer amphipathic helices might form stable barrel stave pores that span the polymer bilayer. On the other hand, our observation that some peptide-polymersome systems are stable and do not lead to vesicle lysis suggests that, in contrast with lipid vesicles, polymersomes could be used to target, encapsulate, and/or deliver high concentrations of antimicrobial peptides to specific microbial cell surfaces or specific tissues. Finally, given the powerful computational tools that are available for the analysis of copolymer assemblies3,37,38, the alamethicin-polymersome system provides a new tool for testing models of membrane-protein interactions.
Acknowledgments
Funding was provided by The Nanotechnology Institute of Pennsylvania (M.G, D.E.D), NSF-MRSEC (D.E.D), NIH R01 HL67286 (P.A.J) and NIH R21 (D.E.D). We thank Harry Bermudez for assistance with the micropipette aspiration experiments and Bhodana Discher for assistance with circular dichroism measurements.
References
- 1.Discher BM, Won YY, Ege DS, Lee JC, Bates FS, Discher DE, Hammer DA. Science. 1999;284:1143. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- 2.Pata V, Ahmed F, Discher DE, Dan N. Langmuir. 2004;10:3888. doi: 10.1021/la035734e. [DOI] [PubMed] [Google Scholar]
- 3.Pata V, Dan N. Biophys J. 2003;85:2111. doi: 10.1016/s0006-3495(03)74639-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meier W, Nardin C, Winterhalter M. Angew Chem Int Ed Engl. 2000;39:4599. [PubMed] [Google Scholar]
- 5.Duclohier H, Wroblewski H. J Membr Biol. 2001;184:1. doi: 10.1007/s00232-001-0077-2. [DOI] [PubMed] [Google Scholar]
- 6.Kamysz W, Okroj M, Lukasiak J. Acta Biochim Pol. 2003;50:461. [PubMed] [Google Scholar]
- 7.Ritov VB, Tverdislova IL, Avakyan T, Menshikova EV, Leikin Yu N, Bratkovskaya LB, Shimon RG. Gen Physiol Biophys. 1992;11:49. [PubMed] [Google Scholar]
- 8.Spaar A, Munster C, Salditt T. Biophys J. 2004;87:396. doi: 10.1529/biophysj.104.040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kessel A, Tieleman DP, Ben-Tal N. Eur Biophys J. 2004;33:16. doi: 10.1007/s00249-003-0345-4. [DOI] [PubMed] [Google Scholar]
- 10.Chen FY, Lee MT, Huang HW. Biophys J. 2003;84:3751. doi: 10.1016/S0006-3495(03)75103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tieleman DP, Hess B, Sansom MS. Biophys J. 2002;83:2393. doi: 10.1016/s0006-3495(02)75253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bak M, Bywater RP, Hohwy M, Thomsen JK, Adelhorst K, Jakobsen HJ, Sorensen OW, Nielsen NC. Biophys J. 2001;81:1684. doi: 10.1016/S0006-3495(01)75822-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang HW. Novartis Found Symp. 1999;225:188. [PubMed] [Google Scholar]
- 14.Dan N, Safran SA. Biophys J. 1998;75:1410. doi: 10.1016/S0006-3495(98)74059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang HW, Chen FY, Lee MT. Phys Rev Lett. 2004;92:198304. doi: 10.1103/PhysRevLett.92.198304. [DOI] [PubMed] [Google Scholar]
- 16.Lee MT, Chen FY, Huang HW. Biochemistry. 2004;43:3590. doi: 10.1021/bi036153r. [DOI] [PubMed] [Google Scholar]
- 17.Discher DE, Eisenberg A. Science. 2002;297:967. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 18.Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE. Macromolecules. 2002;35:8203. [Google Scholar]
- 19.Lee JC, Bermudez H, Discher BM, Sheehan MA, Won YY, Bates FS, Discher DE. Biotechnol Bioeng. 2001;73:135. doi: 10.1002/bit.1045. [DOI] [PubMed] [Google Scholar]
- 20.Lipowsky R, Sackmann E, editors. Structure and Dynamics of Membranes - From Cells to Vesicles, vol 1 of the Handbook of Biological Physics. Elsevier Science; Amsterdam: 1995. pp Kindly note pages 19 and 229 of this reference for thicknesses of phospholipid membranes. [Google Scholar]
- 21.Hillmyer MA, Bates FS. Macromolecules. 1996;29:6994. [Google Scholar]
- 22.Kirschbaum J, Krause C, Winzheimer RK, Bruckner H. J Pept Sci. 2003;9:799. doi: 10.1002/psc.535. [DOI] [PubMed] [Google Scholar]
- 23.Kendall DA, MacDonald RC. J Biol Chem. 1982;257:13892. [PubMed] [Google Scholar]
- 24.Golden, A.
- 25.Aranda-Espinoza H, Bermudez H, Bates FS, Discher DE. Phys Rev Lett. 2001;87:208301. doi: 10.1103/PhysRevLett.87.208301. [DOI] [PubMed] [Google Scholar]
- 26.Kwok R, Evans E. Biophys J. 1981;35:637. doi: 10.1016/S0006-3495(81)84817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagatolli LA, Gratton E. Biophys J. 2000;78:290. doi: 10.1016/S0006-3495(00)76592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuzaki K, Shioyama T, Okamura E, Umemura J, Takenaka T, Takaishi Y, Fujita T, Miyajima K. Biochim Biophys Acta. 1991;1070:419. doi: 10.1016/0005-2736(91)90082-j. [DOI] [PubMed] [Google Scholar]
- 29.Kikukawa T, Araiso T. Arch Biochem Biophys. 2002;405:214. doi: 10.1016/s0003-9861(02)00396-x. [DOI] [PubMed] [Google Scholar]
- 30.Evans E, Heinrich V, Ludwig F, Rawicz W. Biophys J. 2003;85:2342. doi: 10.1016/s0006-3495(03)74658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi MJ, Kang SH, Kim S, Chang JS, Kim SS, Cho H, Lee KH. Peptides. 2004;25:675. doi: 10.1016/j.peptides.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Longo ML, Waring AJ, Hammer DA. Biophys J. 1997;73:1430. doi: 10.1016/S0006-3495(97)78175-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balgavy P, Dubnickova M, Kucerka N, Kiselev MA, Yaradaikin SP, Uhrikova D. Biochim Biophys Acta. 2001;1512:40. doi: 10.1016/s0005-2736(01)00298-x. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. Biophys J. 2001;81:1475. doi: 10.1016/S0006-3495(01)75802-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiese A, Munstermann M, Gutsmann T, Lindner B, Kawahara K, Zahringer U, Seydel U. J Membr Biol. 1998;162:127. doi: 10.1007/s002329900350. [DOI] [PubMed] [Google Scholar]
- 36.Arbuzova A, Schwarz G. Biochim Biophys Acta. 1999;1420:139. doi: 10.1016/s0005-2736(99)00098-x. [DOI] [PubMed] [Google Scholar]
- 37.Muller M, Katsov K, Schick M. Biophys J. 2003;85:1611. doi: 10.1016/S0006-3495(03)74592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller M, Katsov K, Schick M. J Chem Phys. 2002;116:2342. [Google Scholar]