

Abstract
A novel series of ligands with substitutions at the 5-position on phenyl ring A and at the 4′-position on phenyl ring B of 2′-(2-((dimethylamino)methyl)-4′-(2-fluoro- alkoxy)phenylthio)benzenamine (4′-2-fluoroethoxy derivatives, 28–31 and 4′-3-fluoro propoxy derivatives, 40–42) were prepared and tested as serotonin transporter (SERT) imaging agents. The new ligands displayed high binding affinities to SERT (Ki ranging from 0.07 to 1.5 nM). The corresponding 18F labeled compounds, which can be prepared readily, showed excellent brain uptake and retention after iv injection in rats. The hypothalamus region showed high uptake values between 0.74 to 2.2 % dose/g at 120 min post iv injection. Significantly, the hypothalamus to cerebellum ratios (target to non-target ratios) at 120 min were 7.8 and 7.7 for [18F]28 and [18F]40, respectively. The selective uptake and retention in the hypothalamus, which has a high concentration of SERT binding sites, demonstrated that [18F]28 and [18F]40 are promising PET (positron emission computed tomography) imaging agents for mapping SERT binding sites in the brain.
Introduction
The association between serotonin and depression has been well documented. Patients with major depression show distinct alterations in serotonergic neuronal function.1–3 Selective serotonin reuptake inhibitors (SSRIs), targeting serotonin transporter (SERT) binding sites in the brain, are currently being prescribed to treat millions of patients with depression.2,4 There is an urgent need to find a simple method to measure the drug occupancy (or the lack thereof) of the target sites in the brains of non-responders. This could be accomplished by developing PET (positron emission computed tomography) and SPECT (single photon emission computed tomography) imaging agents to target SERT in the brain. These imaging agents would be able to study the interaction of psychoactive drugs with specific binding sites and therefore monitor their effectiveness in occupying the targeted binding sites in the living human brain. SERT may also play an important role as a surrogate marker for changes in serotonin transmission in Parkinson’s disease, drug abuse and other mental illnesses. A simple method of imaging SERT would also further our understanding of these conditions.
The ability to image SERT binding sites could significantly improve the management of patients on SSRIs. First of all, PET imaging techniques could provide a direct measurement of drug occupancy at the target site. This would enable physicians to reduce side-effects by titrating SSRI dosage to reach > 60–70% occupancy.5–11 A minimum necessary dose could be individually estimated; personalized medicine can be achieved. Secondly, it has been observed that there is a lag time of 2 to 6 weeks before the anti-depressant onset of action of SSRIs and that at least 30% of patients are non-responders. It is believed that SSRIs directly inhibit serotonin reuptake in the synapse thus leading to an increase in serotonin neurotransmission.12 Yet, the lag time and the percentage of non-responders indicate that there is no single mechanism that is responsible for depression. There are many alternative explanations, and new treatment targets have been suggested to overcome the lack of drug response.3,4,13–15 By estimating the drug occupancy of SERT binding sites by the antidepressant used for treatment, in vivo PET or SPECT imaging studies could assist in the development of more effective approaches to the treatment of depression.
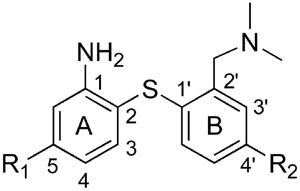
There are several types of core structures which have been employed in developing SERT imaging agents, i.e. nitroquipazines,16–19 (+)-McN5652,12,20,21 tropanes22–26 and biphenylthiols.27–36 Due to a higher serotonin transporter binding selectivity and ease of synthesis, in the past few years a large number of biphenylthiol derivatives have been prepared and tested as potential SERT imaging agents.27–33 Several analogs of biphenylthiol derivatives, such as [123I]ADAM (2) have shown excellent binding affinity and selectivity and have been successfully used for SPECT imaging in humans.37 (see Chart 1) Comparable PET imaging agents for SERT, based on the same biphenylthiol core, have also been reported (see Table 1). Wilson’s group developed [11C]DASB(1)27 which has now become a standard tracer for PET imaging of SERT in humans.8,10,38–40 Yet, the practical challenges of using a 11C tracer (T1/2 = 20 min) on a routine basis have limited its application to major medical centers which possess an on-site cyclotron and a radiochemistry team. In contrast, 18F (T1/2 = 109 min) labeled tracers can be prepared by regional radiopharmacies and distributed within any major metropolitan area. Thus, 18F labeled SERT imaging agents could potentially serve many more patients. In the past few years, several 18F derivatives of biphenylthiols have been reported including AFM(4),41 ACF(5)36 and 5-FADAM(3).42,43 (Chart 1). The most promising candidate is 5-FADAM (3). However, due to radiochemical limitations, which require a nucleophilic substitution of [18F]fluoride on a less activated aromatic ring, the radiochemical yield of [18F]3 is relatively low (< 5%).44 In order to improve the yield and study the structure-activity relationship of this series of biphenylthiol derivatives, we evaluated 4′-(2-fluoroethoxy)- and 4′-(3-fluoropropoxy)- derivatives with 4′-substitutions on phenyl ring B. These compounds provide an alternative approach for introduction of [18F]fluoride via a simple nucleophilic reaction readily achievable in high yields and in a short time period. The reaction could be performed in an automated radiolabeling synthesizer which is commonly used in centralized radiopharmacies for making [18F]FDG. In this paper, we report the preparation and characterization of the first examples of 18F labeled 4′-substituted (ring B substituted) biphenylthiol derivatives targeting SERT for PET imaging.
Chart 1.

Chemical structures and binding affinities of serotonin transporter ligands (1–5): aWilson et al.,10,27, bChoi et al.,35,37,47, cShiue et al.,42, dHuang et al., 33, eOya et al.,36. It is important to note that there are at least two different nomenclatures for the same molecule 3. Depending on the method for counting the positions on ring A, 5-FADAM (3) could also be named as 4-FADAM42,43,46,54.
Table 1.
Binding affinities of new biphenylthiols for monoamine transporters*
 | |||||
|---|---|---|---|---|---|
|
Ki (nM)
|
|||||
| Compound | R1 | R2 | SERT | NET | DAT |
| 28 | H | O(CH2)2F | 0.25 ± 0.02 | 7.5 ± 0.7 | 340 ± 64 |
| 29 | F | O(CH2)2F | 0.10 ± 0.01 | 37 ± 5 | >1000 |
| 30 | Cl | O(CH2)2F | 0.03 ± 0.001 | 97 ± 3 | 847 ± 73 |
| 31 | Br | O(CH2)2F | 0.05 ± 0.01 | 114 ± 8 | >1000 |
| 40 | H | O(CH2)3F | 1.4 ± 0.2 | 12 ± 2 | 299 ± 9 |
| 41 | F | O(CH2)3F | 0.95 ± 0.13 | 95 ± 9 | >1000 |
| 42 | Br | O(CH2)3F | 0.15 ± 0.02 | >1000 | >1000 |
| 47 | H | O(CH2)2OH | 1.1 ± 0.04 | 11 ± 1 | 351 ± 49 |
| 48 | F | O(CH2)2OH | 1.2 ± 0.1 | 56 ± 5 | >1000 |
| 49 | H | O(CH2)3OH | 1.3 ± 0.3 | 7 ± 0.2 | 443 ± 68 |
| 50 | Br | O(CH2)3OH | 0.21 ± 0.02 | 279 ± 25 | >1000 |
| 43 | H | OCH3 | 5 ± 0.4 | 14 ± 1 | 576 ± 31 |
| 44 | H | OH | 17 ± 2 | 33 ± 3 | 659 ± 38 |
In vitro binding assays (n = 3) were employed to determine inhibition constants (Ki ± SEM) with membrane preparations of three different groups of LLC-PK1 cells, each expressing one specific monoamine transporter, either SERT, DAT or NET.47
Chemistry
A wealth of information on the synthesis of biphenylthiols (1–5) has been reported in the past few years.27,28,30–33,45,46 The new series of biphenylthiols 28–42 were successfully prepared using schemes 1–3. The synthesis of fluorinated biphenylthiols 28–31 is illustrated in scheme 1. Commercially available 2-amino-5-methoxy benzoic acid 6 was readily converted to the corresponding thiol 7 via diazotization followed by replacement of the diazonium group by a sulfur atom. The unstable thiol 7 (due to a rapid oxidation of the thiol group) was immediately coupled with various nitrobenzenes 8–11 to give benzoic acids 12–15 in moderate to good yields (ranging from 55 to 70 % yield). The carboxylic acids were converted to the amides 16–19 by first treating with thionyl chloride to produce the acid chlorides and the subsequent reaction of acid chlorides with N,N-dimethylamine. The methoxy group of amides 16–19 were readily demethylated with BBr3 to produce the corresponding phenolic compounds 20–23. The free OH-groups of phenolic compounds 20–23 were alkylated with 1-bromo-2- fluoroethane to provide 2-fluorethoxy compounds 24–27. The simultaneous reduction of nitro and amide groups yielded the desired non-radioactive compounds 28–31.
Scheme 1.

Preparation of 2-fluoroethoxy derivativesa
aReagents and conditions: (a) 1) NaNO2, 2) KS2COEt, 3) HCl; (b) Na/EtOH, (c) 1) SOCl2, 2) (CH3)2NH, CHCl3; (d)1.0 M BBr3 in DCM, MW, 100 °C or − 78°C - rt, 8h; (e) 1-bromo-2-fluoroethane, DMF, K2CO3, MW; (f) BH3-THF, THF, 4–5 h.
Scheme 3.

Preparation of 4′-methoxy, 4′-hydroxy, 4′-(2-hydroxyethoxy)- and 4′-(3-hydroxypropoxy)- derivativesa
aReagents and conditions: (a) BH3-THF.THF, 4–5 h; (b) 2-bromo-1-ethanol, K2CO3, DMF, MW, 150°C, 15 min.
Similarly, three nonradioactive 3-fluorpropoxy compounds 40–42 were synthesized from intermediate phenolic compounds 20–23 (scheme 2). Alkylation of the free 4′-OH group with 3-bromopropanol yielded compounds 32–34. The amide 34 was directly converted to the fluoride 39 by treatment with DAST. Alternatively, the amides 32 and 33 were first transformed into their corresponding mesylates 35 and 36 by reacting with methanesulfonyl chloride. These mesylates were further converted to the fluorides 37, 38 by microwave irradiation with TBAF. The simultaneous reduction of amide and nitro groups furnished the desired 3-fluoropropoxy compounds 40–42.
Scheme 2.

Preparation of 3-fluoropropoxy derivativesa
aReagents and conditions: (a) 3-bromo-1-propanol, K2CO3, DMF, MW, 15 min; (b) MsCl, TEA, DCM, 1 h; (c) TBAF, THF, MW, 20 min; (d) BH3-THF.THF, 4–5 h; (e) DAST, DCM, −78 °C - rt, 1 h.
Compounds 43 and 44 were obtained by reduction of both amide and nitro groups of intermediate compounds 16 and 20 (scheme 3). Similarly compounds 47 and 48 were synthesized in two steps from 20 and 21 by alkylation with 2-bromoethanol followed by reduction. Similarly, reduction of 32 and 34 yielded the desired compounds 49 and 50. The purity of all nonradioactive compounds used for the bio-assay was confirmed from HPLC analysis using two different systems. All bio-assay involved compounds reported in this paper showed greater than 95% purity in both systems (see supporting information).
We have also prepared the O-mesylate derivatives for 18F radiolabeling. Methanesulfonate precursors 57–60 were prepared as shown in scheme 4. The phenolic compounds 20–23 were alkylated with 2-bromoethanol to give compounds 45–46 and 51–52. Selective reduction of amide group by BH3-THF followed by an introduction of methanesulfonyl group provided the desired precursors 57–60. The synthesis of three more methanesulfonate precursors 64–66 in two similar steps is shown in scheme 4.
Scheme 4.

Preparation of O-mesylated precursors and 18F radiolabelinga
a Reagents and conditions: (a) 2-bromoethanol, K2CO3, DMF, MW; (b) BH3-THF, THF, 1.5 h; (c) methanesulfonyl chloride, TEA, DCM, 1 – 2 h; (d) K[18F]F, K222, DMSO; (e) SnCl2.
A similar radiolabeling approach, as reported previously, was used for 2-fluoroethoxy and 3-fluoro-propoxy derivatives (Scheme 4). The radiolabeling was accomplished by starting with mesylate precursors (57–60 or 64–66) reacted with [18F]fluoride in the presence of Kryptofix 222 and potassium carbonate in DMSO at 100°C for 5–10 min. The resulting 18F labeled nitro intermediates were directly reduced with tin chloride in hydrochloric acid/EtOH to provide desired labeled compound [18F]28–31 or [18F]40–42 (Scheme 4). The crude product was purified by HPLC. The procedure took 60–120 min and the specific activity at the end of synthesis was 280–1050 mCi/μmol (n = 3) (Radiochemical purity > 99%, radiochemical yield = 10–35 % decay corrected). The purified product showed an HPLC profile consistent with the “cold” carrier (a typical example of HPLC profile is shown in the supporting information).
Using the O-mesylates as the starting material, the 18F labeling was successfully achieved for [18F]28–31 and [18F]40–42 in reasonable radiochemical yield and high radiochemical purity. The labeling reactions have not been optimized further; however, it is likely that the two-step one-pot reaction can be adapted for automation in a higher level (in multi-Curries of 18F) of preparation.
Biological studies
In vitro binding assays were carried out using membrane homogenates prepared from three different LLC-PK1 cell lines, each over-expressing one of the monoamine transporters. As expected, the binding affinities of the 5-halogen and 4′-2-fluoroethoxy or 4′-3-fluoropropoxy substituted derivatives to SERT were excellent (Ki = 0.03 to 0.95 nM) with a rank of potency of Br ≥ Cl > F (Table 1). Interestingly, an opposite trend in NET affinity was seen. The binding affinities toward NET decreased with the size of the halogen atom (F > Cl > Br). The analogs which were substituted with heavier halogens (Cl or Br), i.e. 30, 31 and 42, showed the weakest NET affinities (Ki = 97, 114, and > 1,000 nM, respectively). The 5-H substituted analogs, 28 and 40 as compared to the 5-halogen substituted analogs, displayed slightly lower SERT binding affinities and showed some NET binding (Ki = 0.25 and 1.4 nM for SERT and 7.5 and 12 nM for NET, respectively). This, however, is not believed to be a significant impediment towards their potential as SERT selective radiotracers since it is known that the concentration of the NET binding sites (Bmax for NET) is at least four fold less than that of SERT in the brain48,49. Thus, the new 5-H substituted analogs, 28 and 40, will likely be sufficiently selective for imaging SERT binding sites and will be further evaluated. It is also apparent that the addition of a carbon atom to form the 4′-3-fluoropropoxy substituted derivatives results in lower SERT and NET binding affinities as compared to the corresponding 4′-2-fluoroethoxy derivatives. Binding affinities of these derivatives toward DAT as compared to SERT were much lower (Ki >300 nM). The results were consistent with the binding profiles reported for other 5-methyl or 5-halogen substituted biphenylthiol derivatives reported previously (ring A substituted derivatives).27,31–33,35,36,47 To our knowledge, this is the first time such a structure-activity relationship has been observed for the biphenylthiol series of compounds. It also suggests that relative binding affinities between SERT and NET are highly dependent on the substitution groups on ring A and ring B. Further exploration of this structure-activity relationship could enhance the selectivity between SERT and NET.
To test whether these potential imaging agents could bind to SERT binding sites in the brain, we measured brain uptake and regional brain distribution of the [18F] labeled compounds in rats after an iv injection. The whole body biodistribution data, an example of which can be seen in the supplemental information, was relatively similar to previously reported labeled biphenylthiols and the in vivo de-fluorination for these agents was relatively low (based on the relatively low bone uptake). The brain uptake between 30 min and 120 min provides information on the relative brain washout. Biodistribution of all the [18F] labeled compounds, 28, 29, 30, 31, 40, 41 and 42, in normal rats showed that they readily penetrated the intact blood brain barrier with initial brain uptakes between 1.12 and 2.23 % dose/organ at 30 min post iv injection (Table 2). We were surprised at the high brain uptake and persistent retention of [18F]29, 30, 31, 41 and 42 at 120 min after injection (Table 2). It is unlikely that this relatively prolonged retention is caused by any single factor, but rather it is most likely caused by a combination of factors such as lipophilicity, affinity, molecular weight, metabolism, and/or presence of a selective efflux mechanism for the tracer. Ring A 5-halogen (F, Cl and Br) substituted derivatives all displayed high brain uptake but slow brain washout. On the contrary, ring A 5-H substituted compounds, [18F]28 and 40, displayed a significant washout from the brain (1.85 vs 0.63 % dose/organ for 28 and 1.23 vs 0.59 % dose/organ for 41 at 30 vs 120 min, respectively). Such high brain uptake and washout has not been observed in any other 18F labeled biphenylthiol derivatives reported in the literature.28,30,33,36,42 Clearly, the 4′-2-fluoroethoxy- or 4′-3-fluoropropoxy- substitution on phenyl ring B has led to different in vivo brain kinetics.
Table 2.
Biodistribution of [18F] labeled SERT tracers were evaluated in rats after an iv injection. Brain uptake (% dose/organ), hypothalamus uptake (% dose/g), and hypothalamus to cerebellum ratio are shown.
| Brain uptake (% dose/organ)
|
|||||
|---|---|---|---|---|---|
| Compound | 30 min | 120 min | |||
| 28 | 1.85 ± 0.15 | 0.63 ± 0.15 | |||
| 29 | 2.23 ± 0.22 | 2.57 ± 0.18 | |||
| 30 | 1.72 ± 0.35 | 1.46 ± 0.16 | |||
| 31 | 1.88 ± 0.18 | 2.16 ± 0.14 | |||
| 40 | 1.23 ± 0.18 | 0.59 ± 0.10 | |||
| 41 | 1.43 ± 0.06 | 1.32 ± 0.01 | |||
| 42 | 1.12 ± 0.06 | 1.15 ± 0.10 | |||
|
| |||||
| Hypothalamus uptake (% dose/g)
|
Hypothalamus to cerebellum ratio
|
||||
| Compound | 30 min | 120 min | Compound | 30 min | 120 min |
|
|
|
||||
| 28 | 1.62 ± 0.10 | 0.74 ± 0.18 | 28 | 3.79 ± 0.30 | 7.83 ± 2.96 |
| 29 | 1.76 ± 0.16 | 2.22 ± 0.28 | 29 | 2.78 ± 0.32 | 4.34 ± 0.60 |
| 30 | 1.31 ± 0.28 | 1.19 ± 0.11 | 30 | 2.36 ± 0.68 | 3.77 ± 0.40 |
| 31 | 1.19 ± 0.10 | 1.78 ± 0.24 | 31 | 2.04 ± 0.07 | 3.70 ± 0.58 |
| 40 | 1.22 ± 0.16 | 0.75 ± 0.18 | 40 | 3.50 ± 0.67 | 7.67 ± 2.60 |
| 41 | 1.05 ± 0.12 | 1.09 ± 0.09 | 41 | 2.66 ± 0.56 | 4.36 ± 0.45 |
| 42 | 0.85 ± 0.08 | 1.01 ± 0.21 | 42 | 1.93 ± 0.82 | 4.17 ± 0.48 |
|
|
|
||||
Regional brain distribution of [18F]28, 29, 30, 31, 40, 41 and 42 showed that the hypothalamus, an area containing a high density of SERT binding sites, had the highest uptake (Table 2). The hypothalamus to cerebellum ratios displayed a significant increase from 30 to 120 min, and there was a more pronounced change for the ring A 5-H substituted compounds, [18F]28 and 40. [18F]28 displayed a hypothalamus to cerebellum ratio of 3.79 vs 7.83 at 30 vs 120 min, respectively while [18F]40 showed a ratio of 3.5 vs 7.67 (Table 2). Although [18F]28 and 40 had lower hypothalamus uptake compared to [18F]29, 30, 31, 41 and 42, the hypothalamus to cerebellum ratios of [18F]28 and 40 were higher due to faster washout from the cerebellum compared to washout from the hypothalamus. Although [18F]28 and 40 possess slightly lower in vitro binding affinities than the other compounds in their respective series, it is not clear why [18F]28 or 40 is able to dissociate faster from the non-target region. It is known that in vitro experiments cannot always predict the behavioral kinetics and success of a tracer. Nevertheless, the favorable in vitro and in vivo kinetic properties of [18F]28 and 40 make them more desirable compared to other derivatives in the series as potential SERT imaging agents. The absolute values of hypothalamus to cerebellum ratios for [18F]28 and 40 were comparable to those of [11C]127 and [18F]342 (hypothalamus uptake at 60 min was 0.62 and 0.73 % dose/g and hypothalamus to cerebellum ratio at 60 min was 8.89 and 5.98, respectively). The absolute hypothalamus uptake values in the brain were also comparable to those of the above two tracers.27,42
In order to demonstrate that the uptake in the hypothalamus region of the brain for this series of 18F labeled derivatives was indeed due to SERT specific binding, we performed a blocking study using several of the [18F] labeled compounds including the most promising ligands, [18F]28 and [18F]40 (data not shown). Rats were pretreated with either 2 mg/kg (+)McN5652, GBR-12909, or nisoxetine, iv 5 min prior to the tracer administration. At two hours after the tracer injection, uptake in each brain region was compared between saline-pretreated (control) and drug-pretreated rats. Only (+)McN5652, a selective SERT ligand, was able to significantly block the uptake in the hypothalamus region, an area containing a high density of SERT binding sites. As expected, the other drugs, nisoxetine, a NET ligand, and GBR-12909, a DAT ligand, showed no significant inhibition of selective uptake in the hypothalamus region (data not shown). The results will be published in the future as part of full characterization of these promising SERT imaging agents.
Discussion
The biphenylthiol core has several unique advantages for developing new SERT imaging agents. First of all, the biphenylthiol core is relatively easy to prepare. It is also a relatively simple molecule containing no optical center. Finally, biphenylthiol derivatives show excellent binding affinities to SERT (in the nM or sub-nM range), while their affinities to the two other monoamine transporters, DAT and NET, are generally lower. Up to this point, most of the reported structure-activity studies of biphenylthiols have focused on the 4- or 5- position of the 2-aminophenyl ring (ring A) (Chart 1). The effects of adding substitution groups on phenyl ring B has not been fully explored. We developed the 4′-2-fluoroethoxy or 4′-3-fluoropropoxy substituted derivatives in order to provide a simple site for 18F labeling. We have demonstrated that the substitution groups described in this paper successfully preserved SERT binding affinity and the respective derivatives showed selective SERT binding with favorable in vivo kinetics in the hypothalamus of the rat brain. We were surprised to find that biodistribution was greatly improved, resulting in better SERT localization (higher target to non-target, ie, hypothalamus/cerebellum ratios) as well as the opportunity to fine-tune the retention time in the brain. The changes in the in vivo pharmacokinetics were novel and unexpected. This is the first time that these types of 4′-2-fluoroethoxy or 4′-3-fluoropropoxy substituted biphenylthiol derivatives have been prepared as PET imaging agents targeting SERT binding sites.
It is important to balance between the brain uptake and washout out rates for SERT binding sites in the brain and selectivity between SERT and NET binding sites. It appears that the 5-H ring A substituted compounds, [18F]28 and 40, showed the best kinetic properties. Even though these two 5-H ring A substituted compounds, [18F]28 and 40, displayed reasonable binding affinities to SERT, they displayed a lower selectivity between SERT and NET (Ki = 0.25 and 1.4 nM for SERT and 7.5 and 12 nM for NET, respectively). We are currently carrying out additional studies of the in vivo binding selectivity by using PET imaging studies in rats. Initial data suggests that the binding in the brain is reversible and the hypothalamus uptake can be selectively displaced by a challenge dose of IDAM during imaging studies of SERT binding sites in the rat brain (data not shown).
Recently, we have reported two other novel ligands with a 4′-substitution (iodo or methoxy group) on the phenyl ring B of biphenylthiol: 5-chloro-2-(2′-((dimethylamino) methyl)-4′-iodophenylthio)benzenamine and 2-(2′-((dimethylamino) methyl)-4′-methoxyphenylthio)-5-iodobenzenamine as potential serotonin transporter (SERT) imaging agents.45 These new iodinated ligands also displayed extremely high binding affinities to SERT (Ki = 0.22 ± 0.09 and 0.11 ± 0.04 nM, respectively), with very low binding affinities to DAT and NET. They showed good brain uptakes and prolonged retention after iv injection in rats. Significantly, they also showed excellent uptake and prolonged retention in the hypothalamus where the SERT concentration is the highest. The hypothalamus/cerebellum ratios for the latter 125I labeled agents were 3.97, 5.57 and 5.06 at 1, 2 and 4 hr, respectively. Adding the 4′-iodo- group to phenyl ring B appeared to reduce the rate of clearance from the brain, and the kinetics favored uptake and retention in the hypothalamus. These observations are very consistent with what we have observed for the 4′-2-fluoroethoxy and 4′-3-fluoropropoxy derivatives reported in this paper. More recently, an abstract was presented describing the synthesis and testing of [11C]5-hydroxymethyl and 4′-bromo derivatives of the same core structure as SERT imaging agent.50 The results also confirm our observation that substitution at the 4′-position on the B ring in general changes the in vivo kinetics of SERT binding in the brain.
In conclusion, seven novel biphenylthiol derivatives, [18F] 28, 29, 30, 31, 40, 41 and 42, were prepared and tested as serotonin transporter imaging agents for PET. These new ligands have a 4′-2-fluoroethoxy or 4′-3-fluoropropoxy substitution group on phenyl ring B which maintains the binding affinity and selectivity. These substitutions also dramatically changed the kinetics of in vivo biodistribution and improved the hypothalamus uptake and retention, a desirable property for a PET imaging agent. These novel ligands, especially the 5-H ring A substituted compounds, [18F]28 and 40, showed excellent kinetic properties. Considering this, we are confident that these ligands and their analogs with similar substitutions would be good potential candidates for studying serotonin transporters with in vivo PET imaging.
Experimental Section
I. Chemistry
General Procedure (A) for the Preparation of Thiodiarylbenzoic acid
To a solution of 2-amino-5-methoxybenzoic acid (6) (2.5 g, 15 mmol) in 1.6 mL of 50% sodium hydroxide, water (22 mL) and NaNO2 (1.03 g, 15 mmol) were slowly added. The resulting mixture was poured into a mixture of concentrated HCl (5 mL) and 7 g of ice with external cooling with salt/ice. The mixture was stirred at 0°C for 1 h and neutralized by an addition of potassium acetate (~1–2 g). The mixture was slowly added with vigorous stirring to a solution of O-ethylxanthic acid, potassium salt (7 g, 43.7 mmol) in 25 mL water at 80 °C. The temperature was maintained at 80 °C during the addition and stirring was continued until the evolution of N2 gas had subsided. After cooled down to room temperature by external cooling by an ice bath, the mixture was acidified by an addition of concentrated HCl and extracted by dichloromethane (20 mL × 2) under argon atmosphere. The organic layer was dried with sodium sulfate under argon and the solvent was removed in vacuo. The crude product, 2-mercapto-5-methoxybenzoic acid (7), was quickly dissolved in 20 ml of hot ethanol and added to a mixture of nitrobenene (8–11) (16.5 mmol) and sodium ethoxide (0.76 g sodium in 35 mL ethanol). The resulting solution was heated to reflux for 2 hrs. After cooling, the solvent was removed in vacuo and water was added to the residue. The solution was acidified by an addition of concentrated HCl and extracted with ethyl acetate. The combined organic layers were washed with water and dried with sodium sulfate. The solvent was removed in vacuo. The residue was purified by silica gel column (ethylacetate/methanol 10%) to yellow a solid.
5-Methoxy-2-(2-nitrophenylthio)-benzoic acid (12) was prepared from 8 as a yellow solid in 77% yield according to general procedure A. 1H NMR (CD3OD) δ 8.16 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.45 (d, J = 2.9 Hz, 1H), 7.43-7.37 (m, 1H), 7.32-7.24 (m. 1H), 7.17 (dd, J = 8.5 Hz, 2.9 Hz, 1H), 6.90 (dd, J = 8.0 Hz, 1.4 Hz, 1H), 3.90 (s, 3H). HRMS calcd for C14H11NO5SNa [M++ Na] 328.0256, obsd 328.0253.
2-(4-Fluoro-2-nitrophenylthio)-5-methoxybenzoic acid (13). Crude material was used for the amidation reaction.
2-(4-Chloro-2-nitrophenylthio)-5-methoxybenzoic acid (14) was prepared from 10 as a yellow solid in 57% yield according to general procedure A. 1H NMR (CD3OD) δ 8.17 (d, J = 1.9 Hz, 1H), 7.57-7.50 (m, 2H), 7.31 (d, J = 2.0 Hz, 1H), 7.11 (dd, J = 5.9 Hz, 2.7 Hz, 1H), 6.79 (d, J = 8.7 Hz, 1H), 3.90 (s, 3H).
2-(4-Bromo-2-nitrophenylthio)-5-methoxybenzoic acid 15 was prepared from 11 as a yellow solid in 55% yield according to general procedure A. 1H NMR (CDCl3) δ 13.4 (s, 1H), 8.39 (s, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 2 Hz, 1H), 7.26 (dd, J = 8.6 Hz, J = 2 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 3.91 (s, 3H).
General Procedure (B) for the Preparation of Amides
A solution of benzoic acid (12–15) (5.2 mmol) and thionyl chloride (1.0 mL, 14 mmol) in chloroform (50 mL) was heated to reflux for 2 h. The solvent and excess thionyl chloride were removed in vacuo. The residue was dissolved in chloroform (20 mL) and added to the 2M solution of dimethylamine in tetrahydrofuran (10 mL) at 0 °C. The mixture was stirred at room temperature for 3 hrs. The solvent was removed and the residue was dissolved in ethyl acetate and washed with water and brine. The organic layer was passed through a short pad of silica gel eluting with ethyl acetate to give the products as yellow oils.
5-Methoxy-2-(2-nitrophenylthio)-N,N-dimethylbenzamide (16) was prepared from 12 as a yellow oil in 71% yield according to general procedure B. 1H NMR (CDCl3) δ 8.18 (dd, J = 8.2 Hz, 1.5 Hz, 1H), 7.5 (d, J = 8.5 Hz, 1H), 7.41-7.32 (m, 1H), 7.23-7.14 (m, 1H), 7.02-6.93 (m, 3H), 3.88 (s, 3H), 3.02 (s, 3H), 2.86 (s, 3H). HRMS calcd for C16H16N2NaO4S [M++ Na] 355.0728, obsd 355.0738.
2-(4-Fluoro-2-nitrophenylthio)-5-methoxy-N,N-dimethylbenzamide (17) was prepared from 13 as a yellow oil in 70% yield according to general procedure B. 1H NMR (CDCl3) δ 7.90 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.17-7.08 (m, 1H), 7.07-6.90 (m, 3H), 3.88 (s, 3H), 3.03 (s, 3H), 2.86 (s, 3H). HRMS calcd for C16H15FN2NaO4S [M++ Na] 373.0634, obsd 373.0625.
2-(4-Chloro-2-nitrophenylthio)-5-methoxy-N,N-dimethylbenzamide (18) was prepared from 14 as a yellow oil in 72% yield according to general procedure B. 1H NMR (CDCl3) δ 8.17 (d, J = 2.1 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.32 (m, 1H), 7.03-6.86 (m, 3H), 3.88 (s, 3H), 3.03 (s, 3H), 2.86 (s, 3H). HRMS calcd for C16H15ClN2NaO4S [M++ Na] 389.0339, obsd 389.0359.
2-(4-Bromo-2-nitrophenylthio)-5-methoxy-N,N-dimethylbenzamide (19) was prepared from 15 as a yellow oil in 68% yield according to general procedure B. 1H NMR (CDCl3) δ 8.31 (d, J = 2.2 Hz, 1H), 7.39–7.50 (m, 2H), 6.92–7.02 (m, 2H), 6.80 (d, J = 8.8 Hz, 1H), 3.87 (s, 3H), 3.02 (s, 3H), 2.85 (s, 3H).
General Procedure (C) for the Deprotection of Methoxy Group
A solution of (16–19) (0.78 mmol) in dichloromethane (3.0 mL) was added with the 1.0 M solution of BBr3 in dichloromethane (1.1 mL) The mixture was irradiated with microwaves at 100°C for 20 min and washed with water (2 mL). The organic layer was dried (Na2SO4) and purified by silica gel column eluting with either ethyl acetate or methanol/dichloromethane (1/19) to yield yellow oil. Alternatively, a solution of amide was added with 1.0 M solution of BBr3 in dichloromethane at − 78 ºC and the temperature was raised slowly to room temperature and stirred for overnight. The workup described earlier gave the product as yellow oil.
5-Hydroxy-2-(2-nitrophenylthio)-N,N-dimethylbanzamide (20) was prepared from 16 as a yellow oil in 85% yield according to general procedure C (eluting with ethyl acetate). 1H NMR (CDCl3) δ 9.01 (s. 1H), 8.19 (dd, J = 8.2 Hz, 1.5 Hz, 1H), 7.48-7.36 (m, 2H), 7.22-7.18 (m, 1H), 6.96-6.91 (m, 2H), 6.85 (dd, J = 8.4 Hz, 2.7 Hz, 1H), 3.06 (s, 3H), 2.90 (s, 3H). HRMS calcd for C15H15N2O4S [M++ H] 319.0753, obsd 319.0761.
2-(4-Fluoro-2-nitrophenylthio)-5-hydroxy-N,N-dimethylbanzamide (21) was prepared from 17 as a yellow oil in 83% yield according to general procedure C (eluting with methanol/dichloromethane mixture). 1H NMR (CD3OD) δ 7.97 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.35-7.26 (m, 1H), 7.06-6.97 (m, 2H), 6.84 (d, J = 2.6 Hz, 1H), 3.00 (s, 3H), 2.85 (s, 3H). HRMS calcd for C15H14FN2O4S [M++ H] 337.0658, obsd 337.0663.
2-(4-Chloro-2-nitrophenylthio)-5-hydroxy-N,N-dimethylbanzamide (22) was prepared from 18 as a yellow oil in 80% yield according to general procedure C (eluting with methanol/dichloromethane mixture). 1H NMR (CD3OD) δ 8.21 (d, J = 2.3 Hz, 1H), 7.53-7.45 (m, 2H), 7.03-6.94 (m, 2H), 6.85 (d, J = 2.6 Hz, 1H), 3.00 (s, 3H), 2.86 (s, 3H).
2-(4-Bromo-2-nitrophenylthio)-5-hydroxy-N,N-dimethylbenzamide (23) was prepared from 19 as a yellow oil in 93% yield according to general procedure C (eluting with methanol/dichloromethane mixture). 1H NMR (CDCl3/CD3OD 10%) δ 8.23 (d, J = 2.2 Hz, 1H), 7.38 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 6.83 (dd, J = 8.4 Hz, 2.6 Hz 1H), 6.70-6.75 (m, 2H), 2.91 (s, 3H), 2.77 (s, 3H).
General Procedure (D) for the Alkylation with 1-bromo-2-fluoroethane
A solution of (20–23) (0.126 mmol) and 1-bromo-2-fluoroethane (25 mg, 0.197 mmol) in DMF (2 mL) was added with potassium carbonate (400 mg, 2.9 mmol). The mixture was irradiated with microwaves at 150°C for 15 min and added to water (10 mL) and extracted with ethyl acetate (5 mL × 2). The combined organic layers were dried (Na2SO4) and purified by silica gel column (ethyl acetate or methanol/dichloromethane 1/19) to yield yellow oil.
5-(2-Fluoroethoxy)-2-(2-nitrophenylthio)-N,N-dimethylbenzamide (24) was prepared from 20 as a yellow oil in 95% yield according to general procedure D (eluting with ethyl acetate). 1H NMR (CDCl3) δ 8.18 (dd, J = 8.2 Hz, 1.5 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.40-7.32 (m, 1H), 7.20 (m, 1H), 7.04-6.93 (m, 3H), 4.77 (dt, J = 47.4 Hz, 4.0 Hz, 2H), 4.28 (dt, J = 27.5 Hz, 4.0 Hz, 2H), 3.03 (s, 3H), 2.86 (s, 3H). HRMS calcd for C17H18 FN2O4S [M++ H] 365.0971, obsd 365.0982.
5-(2-Fluoroethoxy)-2-(4-fluoro-2-nitrophenylthio)-N,N-dimethylbenzamide (25) was prepared from 21 as a yellow oil in 91% yield according to general procedure D (eluting with ethyl acetate). 1H NMR (CDCl3) δ 7.90 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.18-6.84 (m, 4H), 4.79 (dt, J = 47.3 Hz, 4.0 Hz, 2H), 4.28 (dt, J = 27.6 Hz, 4.0 Hz, 2H), 3.03 (s, 3H), 2.86 (s, 3H). HRMS calcd for C17H17F2N2O4S [M++ H] 383.0877, obsd 383.0858.
2-(4-Chloro-2-nitrophenylthio)-5-(2-fluoroethoxy)-N,N-dimethylbenzamide (26) was prepared from 22 as a yellow oil in 96% yield according to general procedure D (eluting with methanol/dichloromethane mixture). 1H NMR (CDCl3) δ 8.18 (d, J = 2.3 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.32 (dd, J = 8.8 Hz, 2.3 Hz, 1H), 7.04 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.96 (d, J = 2.7 Hz, 1H), 6.88 (d, J = 8.8 Hz, 1H), 4.79 (dt, J = 47.5 Hz, 4.0 Hz, 2H), 4.29 (dt, J = 27.6 Hz, 4.0 Hz, 2H), 3.02 (s, 3H), 2.86 (s, 3H). HRMS calcd for C17H17ClFN2O4S [M++ H] 399.0582, obsd 399.0564.
2-(4-Bromo-2-nitrophenylthio)-5-(2-fluoroethoxy)-N,N-dimethylbenzamide (27) was prepared from 23 as a yellow oil in 89% yield according to general procedure D (eluting with methanol/dichloromethane mixture). 1H NMR (CDCl3) δ 8.32 (d, J = 2.1 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.44 (dd, J = 8.6 Hz, 2.1 Hz, 1H), 7.03 (dd, J = 8.4 Hz, 2.6 Hz, 1H), 6.95 (d, J = 2.6 Hz, 1H), 6.80 (d, J = 8.6Hz, 1H), 4.79 (dt J = 47.4 Hz, 4.05 Hz, 2H), 4.30 (dt J = 27.6 Hz, 4.0 Hz, 2H), 3.02 (s, 3H), 2.85 (s, 3H).
General Procedure (E) for Simultaneously Reduction of Nitro and Amide Groups
To a solution of (24–27), (37–39) and other starting materials having both nitro and amide groups (0.1 mmol) in THF at 0°C, 1.0 M BH3-THF (10–15 equivalent) was added. The mixture was heated to reflux for 4.5 – 5 h. After cooled down, 0.5 mL concentrated HCl was cautiously added and the solvent was removed in vacuo. Water (5 mL) was added to the residue and the mixture was heated to reflux for 30 min. To the mixture 1N NaOH was added to adjust the pH to basic (pH 10–11) and it was extracted with ethyl acetate (5 mL x 2). The organic layer was dried (Na2SO4) and purified by silica gel column (2% methanol in dichloromethane to 5% methanol in dichloromethane).
2-(2-((Dimethylamino)methyl)-4-(2-fluoroethoxy)phenylthio)benzenamine (28) was prepared from 24 as a colorless oil in 63% yield according to general procedure E. 1H NMR (CDCl3) δ 7.38 (dd, J = 7.7 Hz, 1.5 Hz, 1H), 7.19-7.11 (m, 1H), 6.96-6.92 (m, 2H), 6.72-6.66 (m, 3H), 4.71 (dt, J = 47.4 Hz, 4.0 Hz, 2H), 4.51 (brs, 2H), 4.17 (dt, J = 27.8 Hz, 4.0 Hz, 2H), 3.57 (s, 2H), 2.30 (s, 6H). HRMS calcd for C17H22FN2OS [M++ H] 321.1437, obsd 321.1442; HPLC >98%.
2-(2-((Dimethylamino)methyl)-4-(2-fluoroethoxy)phenylthio)-5-fluoro-benzen amine (29) was prepared from 25 as a colorless oil in 62% yield according to general procedure E. 1H NMR (CDCl3) δ 7.45-7.38 (m, 1H), 6.91 (d, J = 8.6 Hz, 1H), 6.86 (d, J = 2.6 Hz, 1H), 6.69 (dd, J = 8.6 Hz, 2.8 Hz, 1H), 6.45-6.36 (m, 2H), 4.85 (brs, 2H), 4.71 (dt, J = 47.2 Hz, 4.1 Hz, 2H), 4.16 (dt, J = 27.7 Hz, 4.0 Hz, 2H), 3.53 (s, 2H), 2.30 (s, 6H). HRMS calcd for C17H21F2N2OS [M++ H] 339.1343, obsd 339.1342; HPLC >98%.
5-Chloro-2-(2-((Dimethylamino)methyl)-4-(2-fluoroethoxy)phenylthio)benze namine (30) was prepared from 26 as a colorless oil in 52% yield according to general procedure E. 1H NMR (CDCl3) δ 7.33 (d, J = 8.8 Hz, 1H), 6.95 (d, J = 8.8 Hz, 1H), 6.87(d, J = 2.8 Hz), 6.73-6.62 (m, 3H), 4.72 (dt, J = 47.3 Hz, 4.1 Hz, 2H), 4.16 (dt, J = 27.8 Hz, 4.0 Hz, 2H), 3.52 (s, 2H), 2.29 (s, 6H). HRMS calcd for C17H21ClFN2OS [M++ H] 355.1047, obsd 355.1054; HPLC >96%.
5-Bromo-2-(2-((Dimethylamino)methyl)-4-(2-fluoroethoxy)phenylthio)benzen amine (31) was prepared from 27 as a colorless oil in 51% yield according to general procedure E. 1H NMR (CDCl3) δ 7.23 (d, J = 8.0 Hz, 1H), 6.65–6.97 (m, 5H), 4.74 (br s, 2H), 4.73 (dt J = 47.2 Hz, 4.0 Hz, 2H), 4.14 (dt J = 27.8 Hz, 4.0 Hz, 2H), 3.53 (s, 2H), 2.33 (s, 6H); HRMS calcd for C17H20BrFN2OS [M+] 398.0464, obsd 398.0457; HPLC >98%.
2-(2-((Dimethylamino)methyl)-4-(3-fluoropropoxy)phenylthio)benzenamine (40) was prepared from 37 as a colorless oil in 54% yield according to general procedure E. 1H NMR (CDCl3) δ 7.38 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 7.19-7.10 (m, 1H), 6.94 (d, J = 8.6 Hz, 1H), 6.88 (d, J = 2.7 Hz, 1H), 6.72 (m, 1H), 6.69-6.63 (m, 2H), 4.62 (dt, J = 47.0 Hz, 5.8 Hz, 2H), 4.50 (brs, 2H), 4.05 (t, J = 6.1 Hz, 2H), 3.54 (s, 2H), 2.30 (s, 6H), 2.13 (dt, J = 26.0 Hz, 5.9 Hz, 2H). HRMS calcd for C18H24FN2OS [M++ H] 335.1593, obsd 335.1590; HPLC >99%.
2-(2-((Dimethylamino)methyl)-4-(3-fluoropropoxy)phenylthio)-5-fluorobenze namine (41) was prepared from 38 as a colorless oil in 57% yield according to general procedure E. 1H NMR (CDCl3) δ 7.45-7.37 (m, 1H), 6.91 (d, J = 8.6 Hz, 1H), 6.82 (d, J = 2.7 Hz, 1H), 6.67 (dd, J = 8.6 Hz, 2.7 Hz, 1H), 6.45-6.35 (m, 2H), 4.83 (brs, 2H), 4.62 (dt, J = 47.0 Hz, 5.8 Hz, 2H), 4.04 (t, J = 6.1 Hz, 2H), 3.53 (s, 2H), 2.30 (s, 6H), 2.13 (dt, J = 26.0 Hz, 6.0 Hz, 2H). HRMS calcd for C18H23F2N2OS [M++ H] 353.1499, obsd 353.1494; HPLC >98%.
5-Bromo-2-(2-((dimethylamino)methyl)-4-(3-fluoropropoxy)phenylthio)benze namine (42) was prepared from 39 as a colorless oil in 39% yield according to general procedure E. 1H NMR (CDCl3/CD3OD) δ 7.34 (d, J = 2.2 Hz, 1H), 7.08 (d, J = 8.6 Hz, 1H), 6.90-6.79 (m, 3H) 6.72 (dd, J = 8.6, 1.4 Hz, 1H), 4.55 (dt, J = 44 Hz, 5.8 Hz 2H), 4.35 (s, 2H), 4,09 (t, J = 6.1 Hz, 2H), 2.79 (s, 6H), 2.19-2.00 (m, 2H). HRMS calcd for C18H23BrFN2OS [M++ H] 413.0698, obsd 413.0705; HPLC >95%.
2-(2-((Dimethylamino)methyl)-4-(methoxy)phenylthio)benzenamine (43) was prepared from 16 as a colorless oil in 61% yield according to general procedure E. 1H NMR (CDCl3) δ 7.38 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 7.19-7.10 (m, 1H), 6.95 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 2.7 Hz, 1H), 6.72-6.64 (m, 3H), 4.53 (brs, 2H), 3.76 (s, 3H), 3.55 (s, 2H), 2.30 (s, 6H). HRMS calcd for C16H21N2OS [M++ H] 289.1375, obsd 289.1376; HPLC >98%.
4-(2-Aminophenylthio)-3-dimethylaminomethylphenol (44) was prepared from 20 as a colorless oil in 57% yield according to general procedure E. 1H NMR (CDCl3) δ 7.32 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.18-7.10 (m, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.81 (d, J = 2.7 Hz, 1H), 6.71 (d, J = 7.6 Hz, 2H), 6.58 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 4.29 (brs, 2H), 3.58 (s, 2H), 2.32 (s, 6H). HRMS calcd for C15H19N2OS [M++ H] 275.1218, obsd 275.1211; HPLC >97%.
2-(4-(2-Aminophenylthio)-3-dimethylaminomethylphenoxy)ethanol (47) was prepared from 45 as a colorless oil in 50% yield according to general procedure E. 1H NMR (CDCl3) δ 7.39 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.19-7.11 (m, 1H), 6.96-6.90 (m, 2H), 6.73-6.65 (m, 3H), 4.51 (brs, 2H), 4.06-4.01 (m, 2H), 3.94-3.90 (m, 2H), 3.54 (s, 2H), 2.30 (s, 6H). HRMS calcd for C17H23N2O2S [M++ H] 319.1480, obsd 319.1479; HPLC >97%.
2-(4-(2-Amino-4-fluoro-phenylthio)-3-dimethylaminomethylphenoxy)ethanol (48) was prepared from 46 as a colorless oil in 47% yield according to general procedure E. 1H NMR (CDCl3) δ 7.45-7.37 (m, 1H), 6.94-6.87 (m, 2H), 6.69 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 6.44-6.36 (m, 2H), 4.81 (brs, 2H), 4.04 (m, 2H), 3.93 (m, 2H), 3.54 (s, 2H), 2.30 (s, 6H). HRMS calcd for C17H22F2N2O2S [M++ H] 337.1386, obsd 337.1401; HPLC >95%.
3-(4-(2-Aminophenylthio)-3-dimethylaminomethylphenoxy)-1-propanol (49) was prepared from 32 as a colorless oil in 49% yield according to general procedure E. 1H NMR (CDCl3) δ 7.37 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.19-7.11 (m, 1H), 6.96-6.89 (m, 2H), 6.72-6.60 (m, 3H), 4.52 (brs, 2H), 4.08 (t, J = 5.9 Hz, 2H), 3.84 (t, J = 5.9 Hz, 2H), 3.56 (s, 2H), 2.30 (s, 6H), 2.01 (quintet, J = 5.9 Hz, 2H). HPLC >96%.
3-(4-(2-Amino-4-bromophenylthio)-3-((dimethylamino)methyl)phenoxy)prop an-1-ol (50) was prepared from 34 as a colorless oil in 70% yield according to general procedure E. 1H NMR (CDCl3/CD3OD) δ 7.23 (m, 1H), 6.95 (d, J = 8.6 Hz, 1H), 6.87-6.66 (m, 4H) 4.8 (br s, 2H), 4.08 (t, J = 6.0 Hz 2H), 3.84 (t, J = 6.0 Hz, 2H), 3.54 (s, 2H), 2.30 (s, 6H), 2.07-1.95 (m, 2H).
General Procedure (F) for Alkylation with Bromoethanol and Bromopropanol
A solution of (20–23) (0.239 mmol) and 2-bromoethananol or 3-bromopropanol (0.32 mmol) in DMF (10 mL) was added with potassiun carbonate (80 mg). The mixture was heated at 100°C over night and added to water (50 mL) and extracted with ethyl acetate (20 mL × 2). The organic layer was dried (Na2SO4) and purified by silica gel column (ethyl acetate) to yield yellow oil. Alternatively, the above mixture was irradiated with microwaves at 150°C for 15 min followed by the work-up described earlier gave the desired product.
5-(3-Hydroxypropoxy)-2-(2-nitrophenylthio)-N,N-dimethylbenzamide (32) was prepared from 20 as a yellow oil in 87% yield according to general procedure F. 1H NMR (CDCl3) δ 8.18 (dd, J = 8.2 Hz, 1.5 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.40-7.32 (m, 1H), 7.23-7.15 (m, 1H), 7.03-6.93 (m, 3H), 4.19 (t, J = 6.0 Hz, 2H), 3.88 (m, 2H), 3.02 (s, 3H), 2.86 (s, 3H), 2.08 (m, 2H). HRMS calcd for C18H21N2O5S [M++ H] 377.1171, obsd 377.1156.
2-(4-Fluoro-2-nitrophenylthio)-5-(3-hydroxypropoxy)-N,N-dimethylbenzami de (33) was prepared from 21 as a yellow oil in 85% yield according to general procedure F. 1H NMR (CDCl3) δ 7.90 (dd, J = 8.4 Hz, 2.7 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.13-7.08 (m, 1H), 7.03-6.90 (m, 3H), 4.18 (t, J = 6.0 Hz, 2H), 3.87 (m, 2H), 3.02 (s, 3H), 2.86 (s, 3H), 2.07 (m, 2H). HRMS calcd for C18H20FN2O5S [M++ H] 395.1077, obsd 395.1051.
2-(4-Bromo-2-nitrophenylthio)-5-(3-hydroxypropoxy)-NN, -dimethylbenzami de (34) was prepared from 23 as a yellow oil in 70% yield according to general procedure F. 1H NMR (CDCl3) δ 8.31 (d, J = 2.2 Hz, 1H), 7.49-7.42 (m, 2H), 7.01-6.93 (m, 2H), 6.80 (d, J = 8.4 Hz, 1H), 4.21 (t, J = 5.6 Hz 2H), 3.87 (dd, J = 5,8, 5.6 Hz, 2H), 3.01 (s, 3H), 2.85 (s, 3H).
5-(2-Hydroxyethoxy)-2-(2-nitrophenylthio)-N,N-dimethylbenzamide (45) was prepared from 20 as a yellow oil in 94% yield according to general procedure F. 1H NMR (CDCl3) δ 8.18 (dd, J = 8.2 Hz, 1.4 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.40-7.32 (m, 1H), 7.19-7.15 (m, 1H), 7.01 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.96-6.92 (m, 2H), 4.17-4.10 (m, 2H), 4.03-3.93 (m, 2H), 3.02 (s, 3H), 2.86 (s, 3H). HRMS calcd for C15H19N2O5S [M++ H] 363.1015, obsd 363.1045.
2-(4-Fluoro-2-nitrophenylthio)-5-(2-hydroxyethoxy)-N,N-dimethylbenzamide (46) was prepared from 21 as a yellow oil in 93% yield according to general procedure F. 1H NMR (CDCl3) δ 7.90 (dd, J = 8.2 Hz, 2.6 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.18-7.08 (m, 1H), 7.05-6.90 (m, 3H), 4.15 (m, 2H), 4.01 (m, 2H), 3.03 (s, 3H), 2.88 (s, 3H). HRMS calcd for C17H18FN2O5S [M++ H] 381.0920, obsd 381.0912.
2-(4-Chloro-2-nitrophenylthio)-5-(2-hydroxyethoxy)-N,N-dimethylbenzamide (51) was prepared from 22 as a yellow oil in 89% yield according to general procedure F. 1H NMR (CDCl3) δ 8.17 (d, J = 2.1 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.31 (dd, J = 8.5 Hz, 2.2 Hz, 1H), 7.03 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.88 (d, J = 8.8 Hz, 1H), 4.17-4.13 (m, 2H), 4.01-3.99 (m, 2H), 3.02 (s, 3H), 2.86 (s, 3H).
2-(4-Bromo-2-nitrophenylthio)-5-(2-hydroxyethoxy)-N,N-dimethylbenzamide (52) was prepared from 23 as a yellow oil in 76% yield according to general procedure F. 1H NMR (CDCl3) δ 8.31 (d, J = 2.2 Hz, 1H), 7.49 (d, J = 8.7 Hz, 1H), 7.65 (dd, J = 8.7 Hz, 2.2 Hz, 1H), 7.02 (dd, J = 8.7 Hz, 2.8 Hz, 1H), 6.95 (d, J = 2.8 Hz, 1H), 8.81 (d, J = 8.7 Hz, 1H), 4.15 (m, 2H), 3.99 (m, 2H), 3.02 (s, 3H), 2.85 (s, 3H).
General Procedure (G) for Selective Reduction of Amide Group
To a solution of amides (45-52 and 32-34) (0.18 mmol) in THF (10 mL) at 0°C, 1.0 M BH3-THF (5 eqiuivalent) was added. The mixture was heated to reflux for 1.5-2 hrs. To the mixture 0.5 mL concentrated HCl was cautiously added and the solvent was removed in vacuo. Water (5 mL) was added to the residue and the mixture was heated to reflux 30 min. To the mixture 1N NaOH was added to adjust the pH to basic (pH 10) and extracted with ethyl acetate (5 ml × 2). The organic layer was dried (Na2SO4) and purified by silica gel column (2% methanol in dichloromethane to 5% methanol in dichloromethane) to provide the product as yellow oil.
2-(3-(Dimethylamino)methyl)-(2-nitrophenylthio)phenoxy)ethanol (53) was prepared from 45 as a pale yellow oil in 70% yield according to general procedure G. 1H NMR (CDCl3) δ 8.25 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.34-7.30 (m, 2H), 7.22- 7.14 (m, 1H), 6.90 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.66 (dd, J = 8.0 Hz, 1.4 Hz, 1H), 4.18 (t, J = 4.0 Hz, 2H), 4.00 (t, J = 4.0 Hz, 2H), 3.50 (s, 2H), 2.19 (s, 6H). HRMS calcd for C17H21N2O4S [M++ H] 349.1222, obsd 349.1215.
2-(3-((Dimethylamino)methyl)-(4-fluoro-2-nitrophenylthio)phenoxy)ethanol (54) was prepared from 46 as a pale yellow oil in 69% yield according to general procedure G. 1H NMR (CDCl3) δ 7.97 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 2.7 Hz, 1H), 7.11-7.02 (m, 1H), 6.91 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 6.64 (m, 1H), 4.18 (t, J = 4.0 Hz, 2H), 4.00 (t, J = 4.0 Hz, 2H), 3.48 (s, 2H), 2.18 (s, 6H). HRMS calcd for C17H20FN2O4S [M++ H] 367.1128, obsd 367.1121.
2-(4-Chloro-2-nitrophenylthio)-3-((dimethylamino)methyl)phenoxy)ethanol (55) was prepared from 51 as a pale yellow oil in 66% yield according to general procedure G. 1H NMR (CDCl3) δ 8.24 (d, J = 2.3 Hz, 1H), 7.47 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 2.3 Hz, 1H), 7.23 (d, J = 2.3 Hz, 1H), 6.91 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.59 (d, J = 8.8 Hz, 1H), 4.17 (m, 2H), 4.00 (m, 2H), 3.47 (s, 3H), 2.18 (s, 3H). HRMS calcd for C17H20ClN2O4S [M++ H] 383.0832, obsd 383.0817.
2-(4-Bromo-2-nitrophenylthio)-3-((dimethylamino)methyl)phenoxy)ethanol (56) was prepared from 52 as a pale yellow oil in 71% yield according to general procedure G. 1H NMR (CDCl3) δ 8.38 (d, J = 2.2 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.38 (dd, J = 8.6 Hz, 2.2 Hz, 1H), 7.29 (d, J = 2.8 Hz, 1H), 6.91 (dd, J = 8.8 Hz, 2.8 Hz, 1H), 6.52 (d, J = 8.8 Hz, 1H), 4.17 (t, J = 4.3 Hz, 2H), 4.00 (t, J = 4.3 Hz, 2H), 3.47 (s, 2H), 2.23 (s, 6H).
3-(3-(Dimethylamino)methyl)-(2-nitrophenylthio)phenoxy)propanol (61) was prepared from 32 as a pale yellow oil in 61% yield according to general procedure G. 1H NMR (CDCl3) δ 8.25 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.34-7.29 (m, 2H), 7.22-7.11 (m, 1H), 6.89 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.66 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 4.21 (t, J = 6.0 Hz, 2H), 3.89 (t, J = 5.9 Hz, 2H), 3.49 (s, 2H), 2.19 (s, 6H), 2.08 (quintet, J = 6.0 Hz, 2H). HRMS calcd for C18H23N2O4S [M++ H] 363.1379, obsd 363.1406.
3-(3-(Dimethylamino)methyl)-(4-flouro-2-nitrophenylthio)phenoxy)propanol (62) was prepared from 33 as a pale yellow oil in 63% yield according to general procedure G. 1H NMR (CDCl3) δ 7.97 (dd, J = 8.4 Hz, 2.7 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.27 (m, 1H), 7.12-7.02 (m, 1H), 6.89 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.65 (dd, J = 9.0 Hz, 5.2 Hz, 1H), 4.21 (t, J = 6.0 Hz, 2H), 3.90 (t, J = 5.9 Hz, 2H), 3.47 (s, 2H), 2.18 (s, 6H), 2.08 (quintet, J = 5.9 Hz, 2H). HRMS calcd for C18H22FN2O4S [M++ H] 381.1284, obsd 381.1262.
3-(4-(4-Bromo-2-nitrophenylthio)-3-((dimethylamino)methyl)phenoxy)propa n-1-ol (63) was prepared from 34 as a pale yellow oil in 73% yield according to general procedure G. 1H NMR (CDCl3/CD3OD) δ 8.37 (d, J = 2.2 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.38 (dd, J = 8.5 Hz, 2.2 Hz, 2H), 7.28 (d, J = 3.0 Hz, 1H), 6.89 (dd, J = 8.8, 3.0 Hz, 1H), 6.52 (d, J = 8.8 Hz, 1H), 4.20 (t, J = 6.0 Hz 2H), 3.86 (t, J = 6.0 Hz, 2H), 3.47 (s, 2H), 2.18 (s, 6H), 2.14-2.05 (m, 2H).
General Procedure (H) for Mesylation
To the solution of (32–33, 53–56 and 61–63) (0.11 mmol) in dichloromethane (5 mL) and triethylamine (45 mg, 0.44 mmol), methanesulfonyl chloride (30 mg, 0.26 mmol) was added. The solution was stirred at room temperature for 2h and washed with water (2 mL) and brine (2 mL). The organic layer was dried (Na2SO4) and solvent was removed in vacuo. Purified by silica gel column (dichloromethane/methanol 19/1) to give product as yellow oils.
3-(3-(Dimethylcarbamoyl)-4-(2-nitrophenylthio)phenoxy)propyl methanesulfonate (35) was prepared from 32 as a yellow oil in 88% yield according to general procedure H. 1H NMR (CDCl3) δ 8.18 (dd, J = 8.2 Hz, 1.4 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.41-7.32 (m, 1H), 7.23-7.15 (m, 1H), 7.02-6.93 (m, 3H), 4.46 (t, J = 6.0 Hz, 2H). 4.16 (t, J = 5.8 Hz, 2H), 3.04 (s, 3H), 3.02 (s, 3H), 2.86 (s, 3H), 2.28 (quintet, J = 5.9 Hz, 2H).
3-(3-(Dimethylcarbamoyl)-4-(4-fluoro-2-nitrophenylthio)phenoxy)propyl methanesulfonate (36) was prepared from 33 as a yellow oil in 85% yield according to general procedure H. 1H NMR (CDCl3) δ 7.90 (dd, J = 8.4 Hz, 2.7 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.18-7.09 (m, 1H), 7.01-6.90 (s, 3H), 4.47 (t, J = 6.0 Hz, 2H), 4.16 (t, J = 5.8 Hz, 2H), 3.03 (s, 3H), 3.02 (s, 3H), 2.86 (s, 3H), 2.27 (quintet, J = 6.0 Hz, 2H).
2-(3-((Dimethylamino)methyl)-(2-nitrophenylthio)phenoxy)ethyl methanesulfonate (57) was prepared from 53 as a yellow oil in 94% yield according to general procedure H. 1H NMR (CDCl3) δ 8.25 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.35-7.27 (m, 2H), 7.22-7.17 (m, 1H), 6.89 (dd, J = 8.5 Hz, 2.9 Hz, 1H), 6.65 (dd, J = 8.0 Hz, 1.3 Hz, 1H), 4.63-4.59 (m, 2H), 4.36-4.31 (m, 2H), 3.50 (s, 2H), 3.12 (s, 3H), 2.19 (s, 6H).
3-((Dimethylamino)methyl-4-(4-fluoro-2-nitrophenylthio)phenoxy)ethyl methanesulfonate (58) was prepared from 54 as a yellow oil in 93% yield according to general procedure H. 1H NMR (CDCl3) δ 7.97 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 2.7 Hz, 1H), 7.13-7.03 (m, 1H), 6.89 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 6.61 (dd, J = 9.0 Hz, 5.2 Hz, 1H), 4.60 (t, J = 4.0 Hz, 2H), 4.33 (t, J = 4.0 Hz, 2H), 3.47 (s, 2H), 3.12 (s, 3H), 2.17 (s, 6H). HRMS calcd for C18H22FN2O6S2 [M++ H] 445.0903, obsd 445.0920.
2-(4-(4-Chloro-2-nitrophenylthio)-3-((dimethylamino)methyl)phenoxy)ethyl methanesulfonate (59) was prepared from 55 as a yellow oil in 93% yield according to general procedure H. 1H NMR (CDCl3) δ 8.24 (d, J = 2.2 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 2.3 Hz, 1H), 7.23 (d, J = 2.3 Hz, 1H), 6.89 (dd, J = 8.5 Hz, 2.6 Hz, 1H), 6.58 (d, J = 8.7 Hz, 1H), 4.61 (m, 2H), 4.33 (m, 2H), 3.47 (s, 2H), 3.12 (s, 3H), 2.18 (s, 6H). HRMS calcd for C18H22ClN2O6S2 [M++ H] 461.0608, obsd 461.0641.
2-(4-(4-Bromo-2-nitrophenylthio)-3-((dimethylamino)methyl)phenoxy))ethyl methanesulfonate (60) was prepared from 56 as a yellow oil in 70% yield according to general procedure H. 1H NMR (CDCl3) δ 8.38 (d, J = 2.2 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.38 (dd, J = 8.5 Hz, 2.2 Hz, 1H), 7.29 (d, J = 2.8 Hz, 1H), 6.89 (dd, J = 8.6 Hz, 2.8 Hz, 1H), 6.51 (d, J = 8.6 Hz, 1H), 4.60 (t, J = 4.5 Hz, 2H), 4.35 (t, J = 4.5 Hz, 2H), 3.48 (s, 2H), 3.11 (s, 3H), 2.18 (s, 6H).
2-(3-Dimethylamino)methyl)-(2-nitrophenylthio)phenoxy)propylmethanesulf onate (64) was prepared from 61 as a yellow oil in 90% yield according to general procedure H. 1H NMR (CDCl3) δ 8.25 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.35-7.14 (m, 3H), 6.88 (dd, J = 8.5 Hz, 2.8 Hz, 1H), 6.66 (dd, J = 8.0 Hz, 1.1 Hz, 1H), 4.48 (t, J = 6.1 Hz, 2H), 4.18 (t, J = 5.8 Hz, 2H), 3.49 (s, 2H), 3.03 (s, 3H), 2.27 (quintet, J = 6.0 Hz, 2H), 2.19 (s, 6H).
2-(4-(3-(Dimethylamino)-4-Fluoro-2-nitrophenylthio)phenoxy)propyl methanesulfonate (65) was prepared from 62 as a yellow oil in 87% yield according to general procedure H. 1H NMR (CDCl3) δ 7.97 (dd, J = 8.4 Hz, 2.7 Hz, 1H), 7.47 (d, J = 8.5 Hz, 1H), 7.27 (m, 1H), 7.12-7.03 (m, 1H), 6.88 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 6.69-6.62 (m, 1H), 4.48 (t, J = 6.0 Hz, 2H), 4.18 (t, J = 5.8 Hz, 2H), 3.47 (s, 2H), 3.03 (s, 3H), 2.27 (quintet, J = 5.9 Hz, 2H), 2.18 (s, 6H). HRMS calcd for C19H24FN2O6S2 [M++H] 459.1080, obsd 459.1066.
3-(4-(4-Bromo-2-nitrophenylthio)-3-((dimethylamino)methyl)phenoxy)propyl methanesulfonate (66) was prepared from 63 as a yellow oil in 85% yield according to general procedure H. 1H NMR (CDCl3/CD3OD) δ 8.37 (d, J = 2.2 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.39 (dd, J = 8.8 Hz, 2.2 Hz, 2H), 7.28 (br s, 1H), 6.88 (dd, J = 8.8, 2.8 Hz, 1H), 6.52 (d, J = 8.8 Hz, 1H), 4.47 (t, J = 6.0 Hz 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.56 (s, 2H), 3.00 (s, 3H), 2.32-2.18 (m, 2H), 2.10 (s, 6H).
General Procedure (I) for Fluorination Reaction
A mixture of mesylates (0.14 mmol), 1.0 M TBAF in THF (5 eqiuv., 0.7 mL) and THF (1.3 mL) were irradiated under microwave at 150 °C for 15 min. The solvents were evaporated in vacuo and diluted with 20 mL of dichloromethane, washed with 2N HCl. The organic layer was dried (Na2SO4) and solvent was removed. The crude mixture was purified by silica gel chromatography (1/30 methanol/dichloromethane) to give products as yellow oil.
5-(3-Fluoropropoxy)-2-(2-nitrophenylthio)-N,N-dimethylbenzamide (37) was prepared from 35 as a pale yellow oil in 71% yield according to general procedure I. 1HNMR (CDCl3) δ 8.18 (dd, J = 8.2 Hz, 1.4 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.40-7.32 (m, 1H), 7.25-7.10 (m, 1H), 7.02-6.93 (m, 3H), 4.66 (dt, J = 47.0 Hz, 5.7 Hz, 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.02 (s, 3H), 2.86 (s, 3H), 2.21 (dt, J = 26.2 Hz, 5.9 Hz, 2H). HRMS calcd for C18H20FN2O4S2 [M++H] 379.1128, obsd 379.1112.
2-(4-Fluoro-2-nitrophenylthio)-5-(3-fluoropropoxy)-N,N-dimthylbenzamide (38) was prepared from 36 as a pale yellow oil in 67% yield according to general procedure I. 1H NMR (CDCl3) δ 7.90 (dd, J = 8.3 Hz, 2.6 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.18-7.08 (m, 1H), 7.02-6.90 (m, 3H), 4.66 (dt, J = 47.0 Hz, 5.7 Hz, 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.03 (s, 3H), 2.86 (s, 3H), 2.21 (dt, J = 26.2 Hz, 5.9 Hz, 2H). HRMS calcd for C18H19F2N2O4S2 [M++H] 397.1034, obsd 397.1036.
2-(4-Bromo-2-nitrophenylthio)-5-(3-fluoropropoxy)-NN, -dimethylbenzamide (39). To a solution of 34 (15 mg, 0.033 mmol) in dichloromethane (1 mL), DAST(10 mg) was added at −78 °C. The mixture was slowly warmed up to rt (1 h). The mixture was directly placed on preparatory TLC plate (silica gel) for purification. (dichloromethane/ethyl acetate 1/1). Colorless oil (12 mg, 80%); 1H NMR (CDCl3): δ 8.31 (d, J = 2.2 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H), 7.05-6.89 (m, 2H), 6.80 (d, J = 8.6 Hz, 1H), 4.65 (dt, J = 47.0 Hz, 5.7 Hz, 2H), 4,16 (t, J = 6.0 Hz, 2H), 3.10 (s, 3H), 2.85 (s, 3H), 2.33-2.03 (m, 2H).
II. Radiochemistry
General Procedure for F-18 radiolabeling ([18F]28–31, 40–42)
[18F]Fluoride was produced by a cyclotron using 18O(p,n)18F reaction and passed through a Sep-Pak Light QMA cartridge as an aqueous solution in [18O]-enriched water. The cartridge was dried by airflow, and the 18F activity was eluted with 2 mL of Kryptofix 222 (K222)/K2CO3 solution (13.2 mg of K222 and 3.0 mg of K2CO3 in CH3CN/H2O 1.12/0.18). The solvent was removed at 100 °C under an argon stream. The residue was azeotropically dried with 1 mL of anhydrous CH3CN twice at 100 °C under an argon stream. A solution of mesylate precursor (57–60, 64–66) (2 to 3 mg) in DMSO (0.3 mL) was added to the reaction vessel containing the dried 18F activities. The mixture was heated at 100°C for 3 min. Water (5 mL) was added and the mixture was passed through preconditioned Waters OASIS® HLB (3 cc) cartridge. The cartridge was washed with water (10 mL) and labeled compound was eluted with ethanol (2 mL). To the ethanol solution, 0.5 ml 2N HCl and SnCl2 (50 mg) was added and the mixture was heated at 80 °C for 4 to 6 min. Water (5 mL) was added and the mixture was passed through preconditioned Waters OASISreg; HLB (3 cc) cartridge. The cartridge was washed with water (20 mL) and labeled compound was eluted with CH3CN (2 mL). Eluted compound was purified by HPLC. [Phenonemex Gemini C-18 semi-prep column (10 × 250 mm, 5 micron), CH3CN/ammonium formate buffer (50 mM) 6/4, flow rate 4 mL/min. tR = 6 to 15 min]. To determine radiochemical purity (RCP) and specific activity, analytical HPLC was used [Phenomenex Gemini C18 analytical column (4.6 × 250 mm, 5 micron), CH3CN/ammonium formate buffer (10 mM) 8/2; Flow rate 1 mL/min; tR = 4.0 to 6.5 min. Specific activity was estimated by comparing UV peak intensity of purified F-18 labeled compound with reference non-radioactive compound of known concentration.
III. Pharmacology
In vitro binding assays
Membrane homogenates, containing one of the three monoamine transporters (referred to as LLC-DAT, LLC-NET, and LLC-SERT, which were expressed in a common parental cell line LLC-PK1) were prepared and used for the binding assays.51 Competitive binding assays were performed in a final volume of 0.5 mL. Aliquots of membrane suspensions (100 μL, corresponding to 30–80 μg protein) were mixed with 50 mM Tris-HCl, pH 7.4, 120 mM NaCl, 5 mM KCl and 0.1% bovine serum albumin, radioligand (either 0.06 nM [125I]N-(3′-iodopropen-2′-yl)-2 beta-carbomethoxy-3 beta-(4-chlorophenyl) tropane (DAT ligand)52, 0.09 nM [125I] 5-iodo-2-[[2-2-[(dimethyl amino)methyl]phenyl]thio]benzyl alcohol (SERT ligand)53 or 0.06 nM [125I] iodo-nisoxetine (NET ligand)49, and 8–10 concentrations (10−10 to 10−5M) of competing drugs. Nonspecific binding was defined with 2.5 μM (+)McN5652 for SERT, 1.9 μM GBR-12909 for DAT, and 3.2 μM nisoxetine for NET assays. Incubation was carried out for 1 hr at room temperature, and the bound ligand was collected on glass fiber filters presoaked with 1% polyethylenimine (Sigma, St. Louis, MO) and counted in a gamma counter (Packard 5000, Downers Grove, IL). Results of competition experiments were subjected to nonlinear regression analysis using MS excel.
Biodistribution in rats
Three rats per group were used for each biodistribution study. While under isoflurane anesthesia, 0.2 ml of sterile water with 1 mg/mL BSA and 10–100 μCi of radioactive tracer was injected into the femoral vein. The rats were sacrificed at the time indicated cervical dislocation while under anesthesia. Organs of interest were removed and weighed and the radioactivity was counted. The percent dose per organ was calculated by comparing the tissue counts to counts of 1% of the initial dose (aliquots of the injected material diluted 100 times) measured at the same time. Samples from different brain regions [cortex (CX), striatum (ST), hippocampus (HP), cerebellum (CB) and hypothalamus (HY)] were dissected, weighed and counted. The percentage dose/g of each sample was calculated by comparing sample counts with the counts of the diluted initial dose as described above. The ratio of specific binding in each region was obtained by dividing percentage dose/g of each region by that of the cerebellum [region/CB]. The cerebellum (CB) was used as a background region because it contains relatively few serotonin transporters. The standard deviation for the target to non-target ratios was calculated by the following equation: (Mean1/Mean2) × [(SD1/Mean1)2 + (SD2/Mean2)2]1/2.
In vivo competitive binding in the regional uptake of [18F]42 was investigated by pretreating animals respectively with competing drugs (2 mg/kg, each, iv at 5 min prior to injection of a tracer dose of [18F]42). The competing drugs included GBR-12909, nisoxetine and (+)McN5652. Regional brain distributions were determined at 120 min after [18F]42 injection as described above. The reduction of regional specific binding in the drug-pretreated rats was compared to that of control animals that had been pretreated with saline (data not shown).
Supplementary Material
The 1H spectra of some bio-assay involved compounds, HPLC purity analysis data for all bio-assay involved compounds in two different HPLC systems, radiochemistry data, full sets of biodistribution data in normal mice for [18F]28 and [18F]42. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by a grant awarded from the National Institutes of Health (R01-MH068782, H.F.K.). The authors thank Dr. Carita C. Huang for her editorial assistance.
Footnotes
Abbreviations: Single photon emission computed tomography, SPECT; Positron emission tomography, PET; 2-Fluoro-2-deoxy-D-glucose, FDG; Serotonin transporter, SERT; Dopamine transporter, DAT; Norepinephrine transporter, NET; (+) trans-1,2,3,5,6,10-beta-hexahydro-6-[4-(methylthio)phenyl[pyrrolo- [2,1-a]isoquinoline, (+)-McN5652; ;3-amino-4-(2-dimethylaminomethylphenylsulfanyl) benzonitrile, DASB; 5-iodo-2′-(2-((dimethylamino)methyl)phenylthio)benzenamine, ADAM; N,N-dimethyl- 2-(2-amino-4-[18F]fluorophenylthio)benzylamine, 5-FADAM; 2-[(2-amino-4-fluoro methylphenyl)thio]-N,N-dimethylbenzenemethanamine, AFM; 2-[(2-amino-4-chloro- 5-fluorophenyl)thio]-N,N-dimethyl-benzenmethanamine, ACF
References
- 1.Owens MJ. Selectivity of antidepressants: from the monoamine hypothesis of depression to the SSRI revolution and beyond. J Clin Psychiatry. 2004;4:5–10. [PubMed] [Google Scholar]
- 2.Baghai TC, Moller HJ, Rupprecht R. Recent progress in pharmacological and non-pharmacological treatment options of major depression. Curr Pharm Des. 2006;12:503–15. doi: 10.2174/138161206775474422. [DOI] [PubMed] [Google Scholar]
- 3.Holtzheimer PE, 3rd, Nemeroff CB. Advances in the treatment of depression. NeuroRx. 2006;3:42–56. doi: 10.1016/j.nurx.2005.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemeroff CB. The burden of severe depression: A review of diagnostic challenges and treatment alternatives. J Psychiatr Res. 2006 doi: 10.1016/j.jpsychires.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K, Houle S. Occupancy of serotonin transporters by paroxetine and citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry. 2001;158:1843–1849. doi: 10.1176/appi.ajp.158.11.1843. [DOI] [PubMed] [Google Scholar]
- 6.Ginovart N, Wilson AA, Meyer JH, Hussey D, Houle S. [11C]-DASB a tool for in vivo measurement of SSRI-induced occupancy of the serotonin transporter: PET characterization and evaluation in cats. Synapse. 2003;47:123–133. doi: 10.1002/syn.10155. [DOI] [PubMed] [Google Scholar]
- 7.Suhara T, Takano A, Sudo Y, Ichimiya T, Inoue M, Yasuno F, Ikoma Y, Okubo Y. High levels of serotonin transporter occupancy with low-dose clomipramine in comparative occupancy study with fluvoxamine using positron emission tomography. Arch Gen Psychiatry. 2003;60:386–391. doi: 10.1001/archpsyc.60.4.386. [DOI] [PubMed] [Google Scholar]
- 8.Kim JS, Ichise M, Sangare J, Innis RB. PET Imaging of Serotonin Transporters with [11C]DASB: Test-Retest Reproducibility Using a Multilinear Reference Tissue Parametric Imaging Method. J Nucl Med. 2006;47:208–214. [PubMed] [Google Scholar]
- 9.Frankle WG, Narendran R, Huang Y, Hwang DR, Lombardo I, Cangiano C, Gil R, Laruelle M, Abi-Dargham A. Serotonin transporter availability in patients with schizophrenia: a positron emission tomography imaging study with [11C]DASB. Biol Psychiatry. 2005;57:1510–6. doi: 10.1016/j.biopsych.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 10.Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB, Arango V, Mann JJ. Acute occupancy of brain serotonin transporter by sertraline as measured by [11C]DASB and positron emission tomography. Biol Psychiatry. 2006;59:821–8. doi: 10.1016/j.biopsych.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Guilloteau D, Chalon S. PET and SPECT exploration of central monoaminergic transporters for the development of new drugs and treatments in brain disorders. Curr Pharm Des. 2005;11:3237–45. doi: 10.2174/138161205774424744. [DOI] [PubMed] [Google Scholar]
- 12.Parsey RV, Hastings RS, Oquendo MA, Huang YY, Simpson N, Arcement J, Huang Y, Ogden RT, Van Heertum RL, Arango V, Mann JJ. Lower serotonin transporter binding potential in the human brain during major depressive episodes. Am J Psychiatry. 2006;163:52–8. doi: 10.1176/appi.ajp.163.1.52. [DOI] [PubMed] [Google Scholar]
- 13.Binder EB, Holsboer F. Pharmacogenomics and antidepressant drugs. Ann Med. 2006;38:82–94. doi: 10.1080/07853890600551045. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida K, Takahashi H, Higuchi H, Kamata M, Ito K, Sato K, Naito S, Shimizu T, Itoh K, Inoue K, Suzuki T, Nemeroff CB. Prediction of antidepressant response to milnacipran by norepinephrine transporter gene polymorphisms. Am J Psychiatry. 2004;161:1575–80. doi: 10.1176/appi.ajp.161.9.1575. [DOI] [PubMed] [Google Scholar]
- 15.Nemeroff CB, Mayberg HS, Krahl SE, McNamara J, Frazer A, Henry TR, George MS, Charney DS, Brannan SK. VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology. 2006;31:1345–55. doi: 10.1038/sj.npp.1301082. [DOI] [PubMed] [Google Scholar]
- 16.Mathis CA, Biegon A, Taylor SE, Enas JD, Hanrahan SM. [I-125]5-Iodo-6-nitro-2-piperazinylquinoline: a potent and selective ligand for the serotonin uptake complex. Eur J Pharmacol. 1992;210:103–104. doi: 10.1016/0014-2999(92)90658-q. [DOI] [PubMed] [Google Scholar]
- 17.Mathis CA, Taylor SE, Biegon A, Enas JD. [125I]5-Iodo-6-nitroquipazine: a potent and selective ligand for the 5-hydroxytryptamine uptake complex I in vitro studies. Brain Res. 1993;619:229–235. doi: 10.1016/0006-8993(93)91616-z. [DOI] [PubMed] [Google Scholar]
- 18.Jagust WJ, Eberling JL, Biegon A, Taylor SE, VanBrocklin HF, Jordan S, Hanrahan SM, Roberts JA, Brennan KM, Mathis CA. Iodine-123-5-iodo-6-nitroquipazine: SPECT radiotracer to image the serotonin transporter. J Nucl Med. 1996;37:1207–1214. [PubMed] [Google Scholar]
- 19.Karramkam M, Dolle F, Valette H, Besret L, Bramoulle Y, Hinnen F, Vaufrey F, Franklin C, Bourg S, Coulon C. Synthesis of a fluorine-18-labelled derivative of 6-nitroquipazine, as a radioligand for the In vivo serotonin transporter imaging with PET. Bioorg Med Chem. 2002;10:2611–2623. doi: 10.1016/s0968-0896(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 20.Scheffel U, Szabo Z, Mathews WB, Finley PA, Dannals RF, Ravert HT, Szabo K, Yuan J, Ricaurte GA. In vivo detection of short- and long-term MDMA neurotoxicity--a positron emission tomography study in the living baboon brain. Synapse. 1998;29:183–92. doi: 10.1002/(SICI)1098-2396(199806)29:2<183::AID-SYN9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 21.Parsey RV, Kegeles LS, Hwang DR, Simpson N, Abi-Dargham A, Mawlawi O, Slifstein M, Van Heertum RL, Mann JJ, Laruelle M. In vivo quantification of brain serotonin transporters in humans using [11C]McN 5652. J Nucl Med. 2000;41:1465–77. [PubMed] [Google Scholar]
- 22.Scheffel U, Dannals RF, Cline EJ, Ricaurte GA, Carroll FI, Abraham P, Lewin AH, Kuhar MJ. [123/125I]RTI-55 and an in vivo label for the serotonin transporter. Synapse. 1992;11:134–139. doi: 10.1002/syn.890110206. [DOI] [PubMed] [Google Scholar]
- 23.Laruelle MA, Giddings SS, Zea-Ponce Y, Charney DS, Neumeyer JL, Baldwin RM, Innis RB. Methyl 3beta-(4-[125I]iodophenyl)tropane-2beta-carboxylate in vitro binding to dopamine and serotonin transporters under “physiological” conditions. J Neurochem. 1994;62:978–986. doi: 10.1046/j.1471-4159.1994.62030978.x. [DOI] [PubMed] [Google Scholar]
- 24.Bergstrom KA, Halldin C, Hall H, Lundkvist C, Ginovart N, Swahn CG, Farde L. In vitro and in vivo characterisation of nor-beta-CIT: a potential radioligand for visualisation of the serotonin transporter in the brain. Eur J Nucl Med. 1997;24:596–601. doi: 10.1007/BF00841395. [DOI] [PubMed] [Google Scholar]
- 25.Stehouwer JS, Plisson C, Jarkas N, Zeng F, Voll RJ, Williams L, Martarello L, Votaw JR, Tamagnan G, Goodman MM. Synthesis, radiosynthesis, and biological evaluation of carbon-11 and fluorine-18 (N-fluoroalkyl) labeled 2beta-carbomethoxy-3beta-(4′-(3-furyl)phenyl)tropanes and -nortropanes: candidate radioligands for in vivo imaging of the serotonin transporter with positron emission tomography. J Med Chem. 2005;48:7080–3. doi: 10.1021/jm0504095. [DOI] [PubMed] [Google Scholar]
- 26.Plisson C, Jarkas N, McConathy J, Voll RJ, Votaw J, Williams L, Howell LL, Kilts CD, Goodman MM. Evaluation of carbon-11-labeled 2beta-carbomethoxy-3beta-[4′-((Z)-2-iodoethenyl)phenyl]nortropane as a potential radioligand for imaging the serotonin transporter by PET. J Med Chem. 2006;49:942–6. doi: 10.1021/jm050799v. [DOI] [PubMed] [Google Scholar]
- 27.Wilson AA, Ginovart N, Schmidt M, Meyer JH, Threlkeld PG, Houle S. Novel radiotracers for imaging the serotonin transporter by positron emission tomography: synthesis, radiosynthesis, and in vitro and ex vivo evaluation of 11C-labeled 2-(phenylthio)araalkylamines. J Med Chem. 2000;43:3103–10. doi: 10.1021/jm000079i. [DOI] [PubMed] [Google Scholar]
- 28.Vercouillie J, Mavel S, Galineau L, Ragusa T, Innis R, Kassiou M, Chalon S, Dolle F, Besnard JC, Guilloteau D, Emond P. Synthesis and in vitro evaluation of novel derivatives of diphenylsulfide as serotonin transporter ligands. Bioorg Med Chem Lett. 2006;16:1297–300. doi: 10.1016/j.bmcl.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Zhen J, Reith ME, Dutta AK. Design, synthesis, and preliminary SAR study of 3- and 6-side-chain-extended tetrahydro-pyran analogues of cis- and trans-(6-benzhydryl-tetrahydropyran-3-yl)-benzylamine. Bioorg Med Chem. 2006 doi: 10.1016/j.bmc.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 30.Zessin J, Deuther-Conrad W, Kretzschmar M, Wust F, Pawelke B, Brust P, Steinbach J, Bergmann R. [11C]SMe-ADAM, an imaging agent for the brain serotonin transporter: synthesis pharmacological characterization and microPET studies in rats. Nucl Med Biol. 2006;33:53–63. doi: 10.1016/j.nucmedbio.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Jarkas N, Votaw JR, Voll RJ, Williams L, Camp VM, Owens MJ, Purselle DC, Bremner JD, Kilts CD, Nemeroff CB, Goodman MM. Carbon-11 HOMADAM: a novel PET radiotracer for imaging serotonin transporters. Nucl Med Biol. 2005;32:211–24. doi: 10.1016/j.nucmedbio.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Jarkas N, McConathy J, Votaw JR, Voll RJ, Malveaux E, Camp VM, Williams L, Goodman RR, Kilts CD, Goodman MM. Synthesis and characterization of EADAM: a selective radioligand for mapping the brain serotonin transporters by positron emission tomography. Nucl Med Biol. 2005;32:75–86. doi: 10.1016/j.nucmedbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 33.Huang Y, Bae SA, Zhu Z, Guo N, Roth BL, Laruelle M. Fluorinated diaryl sulfides as serotonin transporter ligands: synthesis, structure-activity relationship study, and in vivo evaluation of fluorine-18-labeled compounds as PET imaging agents. J Med Chem. 2005;48:2559–70. doi: 10.1021/jm0400808. [DOI] [PubMed] [Google Scholar]
- 34.Oya S, Kung MP, Acton PD, Mu M, Hou C, Kung H. A new single-photon emission computed tomography imaging agent for serotonin transporters: [123I] IDAM, 5-Iodo-2-((2-((dimethylamino)methyl)phenyl)thio)benzyl alcohol. J Med Chem. 1999;42:333–335. doi: 10.1021/jm9806751. [DOI] [PubMed] [Google Scholar]
- 35.Oya S, Choi SR, Hou C, Mu M, Kung MP, Acton PD, Siciliano M, Kung HF. 2-((2-((Dimethylamino)methyl)phenyl)thio)-5-iodophenylamine (ADAM): An Improved Serotonin Transporter Ligandx. J Med Biol. 2000;27:249–254. doi: 10.1016/s0969-8051(00)00084-6. [DOI] [PubMed] [Google Scholar]
- 36.Oya S, Choi SR, Coenen H, Kung HF. New PET Imaging Agent for the Serotonin Transporter: [18F]ACF (2-[(2-Amino-4-chloro-5-fluorophenyl)thio]-N, N-dimethyl-benzenmethanamine) J Med Chem. 2002;45:4716–4723. doi: 10.1021/jm020167y. [DOI] [PubMed] [Google Scholar]
- 37.Newberg AB, Amsterdam JD, Wintering N, Ploessl K, Swanson RL, Shults J, Alavi A. 123I-ADAM binding to serotonin transporters in patients with major depression and healthy controls: a preliminary study. J Nucl Med. 2005;46:973–7. [PubMed] [Google Scholar]
- 38.Cannon DM, Ichise M, Fromm SJ, Nugent AC, Rollis D, Gandhi SK, Klaver JM, Charney DS, Manji HK, Drevets WC. Serotonin transporter binding in bipolar disorder assessed using [11C]DASB and positron emission tomography. Biol Psychiatry. 2006;60:207–17. doi: 10.1016/j.biopsych.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Frankle WG, Slifstein M, Gunn RN, Huang Y, Hwang DR, Darr EA, Narendran R, Abi-Dargham A, Laruelle M. Estimation of serotonin transporter parameters with 11C-DASB in healthy humans: reproducibility and comparison of methods. J Nucl Med. 2006;47:815–26. [PubMed] [Google Scholar]
- 40.Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N, Goulding V, Kennedy J, Wilson AA. Brain serotonin transporter binding potential measured with carbon 11-labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry. 2004;61:1271–9. doi: 10.1001/archpsyc.61.12.1271. [DOI] [PubMed] [Google Scholar]
- 41.Huang Y, Narendran R, Bischoff F, Guo N, Zhu Z, Bae SA, Lesage AS, Laruelle M. A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis- 4-methoxycyclohexyl)methanone. J Med Chem. 2005;48:5096–9. doi: 10.1021/jm050263+. [DOI] [PubMed] [Google Scholar]
- 42.Shiue GG, Choi SR, Fang P, Hou C, Acton PD, Cardi C, Saffer JR, Greenberg JH, Karp JS, Kung HF, Shiue CY. N, N-dimethyl-2-(2-amino-4-(18)F-fluorophenylthio)-benzylamine (4-(18)F-ADAM): an improved PET radioligand for serotonin transporters. J Nucl Med. 2003;44:1890–7. [PubMed] [Google Scholar]
- 43.Huang Y, Narendran R, Bae SA, Erritzoe D, Guo N, Zhu Z, Hwang DR, Laruelle M. A PET imaging agent with fast kinetics: synthesis and in vivo evaluation of the serotonin transporter ligand [11C]2-[2-dimethylaminomethylphenylthio)]-5-fluorophenylamine ([11C]AFA) Nucl Med Biol. 2004;31:727–38. doi: 10.1016/j.nucmedbio.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Shiue GG, Fang P, Shiue CY. Synthesis of N, N-dimethyl-2-(2-amino-4-[18F]fluorophenylthio)benzylamine as a serotonin transporter imaging agent. Appl Radiat Isot. 2003;58:183–191. doi: 10.1016/s0969-8043(02)00271-3. [DOI] [PubMed] [Google Scholar]
- 45.Oya S, Choi SR, Kung MP, Kung HF. 5-Chloro-2-(2′-((dimethylamino)methyl)-4′-iodophenylthio)benzenamine: a new serotonin transporter ligand. Nucl Med Biol. 2007;34:129–39. doi: 10.1016/j.nucmedbio.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kung HF, Newman S, Choi SR, Oya S, Hou C, Zhuang ZP, Acton PD, Plossl K, Winkler J, Kung MP. 2-(2-(dimethylaminomethyl)phenoxy)-5-iodophenylamine: an improved serotonin transporter imaging agent. J Med Chem. 2004;47:5258–64. doi: 10.1021/jm049917p. [DOI] [PubMed] [Google Scholar]
- 47.Choi SR, Hou C, Oya S, Mu M, Kung MP, Siciliano M, Acton PD, Kung HF. Selective in vitro and in vivo binding of [(125)I]ADAM to serotonin transporters in rat brain. Synapse. 2000;38:403–12. doi: 10.1002/1098-2396(20001215)38:4<403::AID-SYN5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 48.Kung MP, Kung HF. Mass effect of injected dose in small rodent imaging by SPECT and PET. Nucl Med Biol. 2005;32:673–8. doi: 10.1016/j.nucmedbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Kung MP, Choi SR, Hou C, Zhuang ZP, Foulon C, Kung HF. Selective binding of 2-[125I]iodo-nisoxetine to norepinephrine transporters in the brain. Nucl Med Biol. 2004;31:533–41. doi: 10.1016/j.nucmedbio.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Jarkas N, Voll RJ, Williams L, Votaw JR, Goodman MM. 17th Interantional Synposium on Radiopharmaceutical Sciences. Elsevier; Aachen, Germany: 2007. Synthesis and evluation of a serotonin transporter (SERT) PET imaging agent: 11C-BHOMADAM. Abs # p208. [Google Scholar]
- 51.Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1994;269:7124–7130. [PubMed] [Google Scholar]
- 52.Kung MP, Essman WD, Frederick D, Meegalla S, Goodman M, Mu M, Lucki I, Kung HF. IPT: a novel iodinated ligand for the CNS dopamine transporter. Synapse. 1995;20:316–324. doi: 10.1002/syn.890200405. [DOI] [PubMed] [Google Scholar]
- 53.Kung MP, Hou C, Oya S, Mu M, Acton PD, Kung HF. Characterization of [123I]IDAM as a novel single-photon emission tomography tracer for serotonin transporters. Eur J Nucl Med. 1999;26:844–53. doi: 10.1007/s002590050458. [DOI] [PubMed] [Google Scholar]
- 54.Fang P, Shiue GG, Shimazu T, Greenberg JH, Shiue CY. Synthesis and evaluation of N, N-dimethyl-2-(2-amino-5-[18F]fluorophenylthio)benzylamine (5-[18F]-ADAM) as a serotonin transporter imaging agent. Appl Radiat Isot. 2004;61:1247–54. doi: 10.1016/j.apradiso.2004.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 1H spectra of some bio-assay involved compounds, HPLC purity analysis data for all bio-assay involved compounds in two different HPLC systems, radiochemistry data, full sets of biodistribution data in normal mice for [18F]28 and [18F]42. This material is available free of charge via the Internet at http://pubs.acs.org.