

Abstract
Selenoenzymes have a central role in maintaining cellular redox potential. These enzymes have selenenylsulfide bonds in their active sites that catalyze the reduction of peroxides, sulfoxides and disulfides. The selenol/disufide exchange reaction is common to all of these enzymes and the active site redox potential reflects the ratio between the forward and reverse rates of this reaction. The preparation of enzymes containing selenocysteine (Sec) is experimentally challenging. As a result, little is known about the kinetic role of selenols in enzyme active sites, and the redox potential of a selenenylsulfide or diselenide bond in a protein has not been experimentally determined. In order to fully evaluate the effects of Sec on oxidoreductase redox potential and kinetics, glutaredoxin 3 (Grx3) and all three Sec variants of its conserved 11CXX14C active site were chemically synthesized. Grx3, Grx3(C11U) and Grx3(C14U) exhibited redox potentials of −194, −260 and −275 mV, respectively. The position of redox equilibrium between Grx3(C11U-C14U) (−309 mV) and thioredoxin (Trx) (−270 mV) suggests a possible role for diselenide bonds in biological systems. Kinetic analysis is consistent with the hypothesis that the lower redox potentials of the Sec variants result primarily from the greater nucleophilicity of the active site selenium rather than its role as either a leaving group or a ‘central atom’ in the exchange reaction. The 102 to 104-fold increase in the rate of Trx reduction by the seleno-Grx3 analogs demonstrates that Oxidoreductases containing either selenenylsulfide or diselenide bonds can have physiologically compatible redox potentials and enhanced reduction kinetics in comparison with their sulfide counterparts.
Introduction
The unique role of selenium in biochemical systems was demonstrated by the discovery that selenocysteine (Sec, U) is an essential part of the active site of the antioxidation enzyme glutathione peroxidase (GPx).1,2 The number of newly discovered selenoproteins grows steadily, triggering increased research interest in the biological functions of these enzymes.3,4 Sec is viewed as the 21st amino acid in the natural repertoire,3,4 and has been found in various prokaryotic and eukaryotic proteins. The pKa of the selenol group (5.7 in free Sec) is 3 units lower than that of the thiol group (8.5 in free Cys) and both are expected to be ~1 pKa, unit higher within a polypeptide chain.5 In addition, the selenolate anion is a stronger nucleophile than the analogous thiolate, and has been suggested to be a better leaving group.6 Many Sec residues in enzymes form transient selenenylsulfide bonds with proximal cysteine residues as part of their catalytic mechanisms. Although the redox potential of a selenenylsulfide bond in proteins has not been experimentally determined, its value was predicted to be within the range between the redox potentials of the disulfide and diselenide bonds.3 Additionally, diselenide bonds have not been rigorously characterized in proteins, and it has been suggested that thioredoxin, the most reducing oxidoreductase in E. coli7 would be unable to reduce the diselenide bond, leading to the assumption that diselenide bonds are nonexistent in natural proteins.8,9 However, the Sec-rich protein from zebrafish, zSe1Pa, was found to contain 17 Sec residues,10 suggesting that zSe1Pa may contain a number of diselenide bonds. Sec residues, are often found in the active site of selenoenzymes, and have been found to be essential for their activity. Mutating the Sec residue to Cys in selenoenzymes usually results in decreased catalytic activity of up to 1000-fold.6,11,12 Similarly, a Cys to Sec mutation in methionine-R-sulfoxide reductase (MsrB2 and MsrB3) resulted in 100-fold increase in the catalytic activity of the enzyme,8 demonstrating the advantages of Sec in enzyme engineering.
The selenoproteins Sep15 and Se1M have homology to thioredoxin, defining a new class of selenoenzymes.13,14 The thiol/disulfide oxidoreductases of the thioredoxin superfamily, include the thioredoxins (Trx), glutaredoxins (Grx), protein disulfide isomerase (PDI), and bacterial protein-folding factor (DsbA),15 all sharing a similar molecular architecture, known as the Trx fold (α/β fold).16,17 They also share the same active site motif -Cys-Xaa-Xaa-Cys- (CXXC), with the pKa, of the N-terminal Cys thiol being lower than that of free Cys,18 ranging from 3.4 in DsbA19,20 to 6.3 in Trx.21,22 The selenoproteins Sep15 and Se1M have CXU and CXXU sequences in their active site.13,14,23 Although replacement of selenium by sulfur in the active site is expected to substantially modify the redox potential, no accurate potentials of these proteins have been determined. Furthermore, the effect of such replacement on the catalytic activity of oxidoreductases has not been studied.
The broad range of redox potentials, from −125 mV in DsbA to −270 mV in Trx,7 reflects the variability of the protein environment around the conserved active site motif, CXXC, and its geometry. Indeed, site-directed mutagenesis experiments have shown that changing the two X amino acids between the two Cys residues in this motif can significantly alter the redox potential.24,25,26,27,28 The observed redox potential of proteins in the Trx superfamily reflects the relative stability of the oxidized (disulfide) and reduced (dithiol) forms of the protein. On the basis of the protein sequence, it is difficult to predict the relative stability of these two forms.29 For example, both Grx130 and Grx3,31 which act as hydrogen donors to ribonucleotide reductase (RR), share the same active site sequence (CPYC) and have 33% sequence identity.32,33 Yet, their redox potentials vary by 35 mV.7
The incorporation of Sec into proteins represents a non-trivial challenge. In Nature, this process is highly regulated, with the UGA termination codon being used to specify the insertion of Sec.34,35 However, it is difficult to prepare selenoproteins using this approach with the traditional recombinant expression systems.36 An alternative strategy employs Sec instead of Cys in the growth medium of a cysteine-auxotrophic environment.37 This technique was used to prepare a Trx(Cys32Sec-Cys35Sec) mutant with only partial replacement (80%) of all Cys residues in the protein by Sec.37 An alternative approach involves the chemical modification of wild-type proteins, as was achieved, by Hilvert et al in the preparation of selenosubtilisin.38 Unfortunately, the chemical modification approach is limited to very few cases where a specific active-site residue is sufficiently prone to selective chemical modifications.
Chemical synthesis is an attractive strategy for incorporating Sec into polypeptides, as demonstrated by the solid-phase peptide synthesis (SPPS) of Sec variants of IL-8 by Clark-Lewis and coworkers.39 This approach was also used to study Sec fragments of Grx1(10–17) and analogs of α-conotoxin ImI.40,41,42 The SPPS approach is significantly enhanced by the native chemical ligation (NCL) strategy, which allows the assembly of unprotected peptides to produce large proteins.43,44 The NCL methodology has proven useful in the efficient preparation of many proteins,44,45 and was recently applied to the semisynthesis of selenoproteins using Sec instead of Cys at the ligation site.46,47,48,49,50 However, incorporation of Sec at positions other than the ligation site has not been demonstrated.
Here we report on the first synthesis and characterization of Sec substitutions into an oxidoreductase, yielding enzymes that contain selenenylsulfide and diselenide bonds. We have chosen Grx3 as a model oxidoreductase due to its intermediate redox potential within the Trx superfamily.7 An efficient chemical synthesis of Grx3 was developed using conformationally assisted native chemical ligation51 followed by selective oxidation of the active site residues and alkylation of the cysteine at the ligation site. The native Grx3 and three active site Sec analogs were then synthesized to determine the effects of selenenylsulfide and diselenide bonds on the redox potential and equilibration kinetics of oxidoreductase enzymes.
Results
Synthesis of Grx3 analogs
To probe the role of Sec in oxidoreductase enzymes, we chemically synthesized four proteins: “wild-type” Grx3 (sGrx3), Grx3(C11U), Grx3(C14U), and Grx3(C11U-C14U). The Grx3 analogs were assembled from two synthetic peptide fragments by native chemical ligation (NCL, Figure 1).43 To facilitate ligation, Ala38, a solvent exposed residue in the middle of the sequence, was substituted with Cys. Machine-assisted Boc-SPPS was used to assemble the Sec containing thioester peptides, since it has been shown that incorporation of Boc-Sec(4-MeBzl)-OH has low levels of epimerization and the 4-MeBzl side chain protecting group can be removed cleanly by HF.42 Thus, an efficient procedure for the synthesis of Boc-Sec(4-MeBzl)-OH was developed, analogous to that reported by van der Donk for Fmoc-Sec(4-MeOBzl)-OH (Scheme 1).48 Notable changes to the method include the synthesis of the (4-MeBzl)-diselenide, using elemental selenium under bubbling CO(g),52 and the mild deprotection of the methyl ester using Me3SnOH.53 This strategy furnished 10 g of the desired product in 45% overall yield. Four thioester (-COSR) peptides: Grx3(1-37)-COSR, Grx3(1-37)(C11U)-COSR, Grx3(1-37)(C14U)-COSR, Grx3(1-37)(C11U-C14U)-COSR, and a single N-terminal Cys peptide, Grx3(38-82)(A38C)-OH were synthesized and purified to yield 50–100 mg of each peptide.
Figure 1.
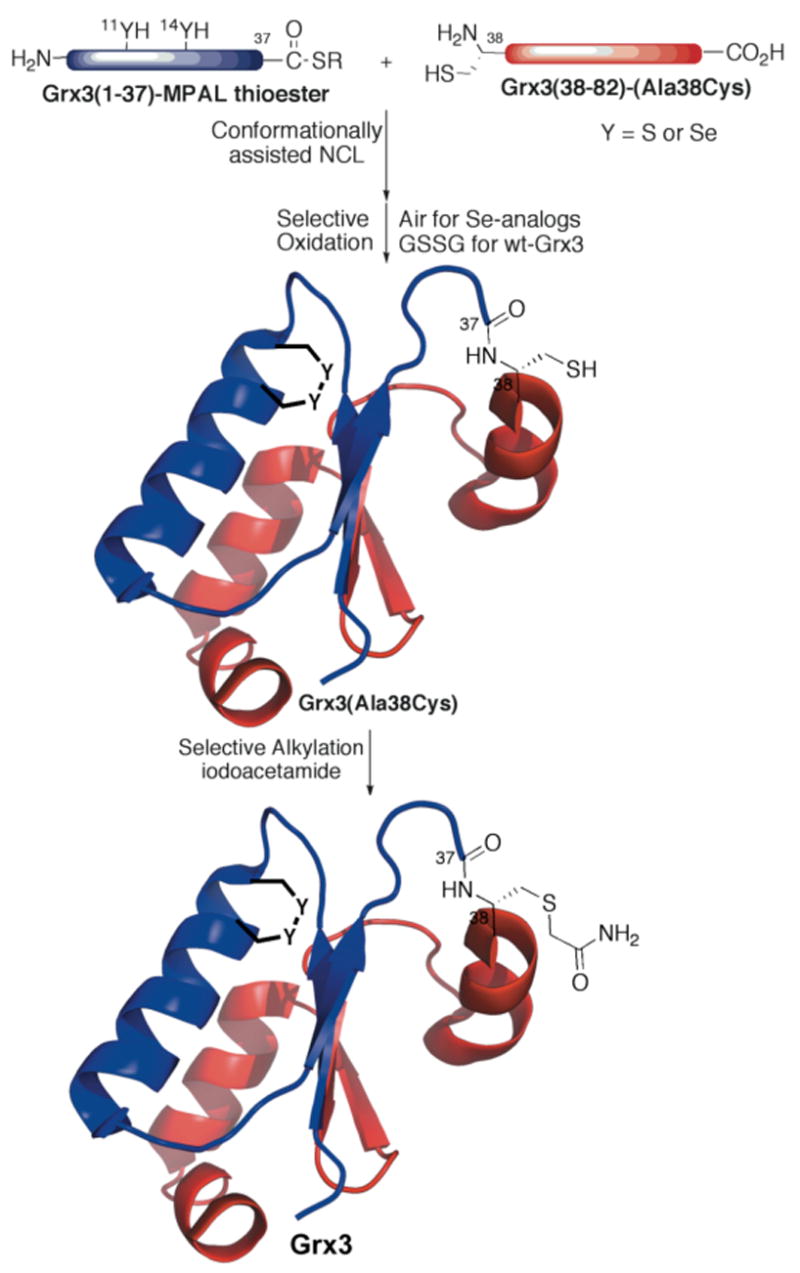
Conformationally assisted NCL for chemical synthesis of Grx3 combined with selective oxidation and alkylation. For NCL the thioester-peptide Grx3(1-37)-MPAL (containing either Cys (Y=S) or Sec (Y=Se), at positions 11 and/or 14), mixed with the Cys-peptide Grx3(C38-82). After purification, the ligated product was oxidized to form the disulfide bond (or selenenylsulfide or diselenide bond) to protect them from the alkylation with iodoacetamide. (MPAL = β-mercaptoproparionyl-Leu).
Scheme 1.

The synthetic route used to prepare Boc-(4-MeBzl)Sec-OH, 7.
Conformationally assisted ligation of Grx3 analogs
Preparation of all Grx3 analogs via NCL was carried out under folding conditions.51 The progress of the reaction was monitored by analytical HPLC, indicating that the reaction was complete within 4 h, compared with 16 h under denaturating conditions (6 M GnHCl), affording the desired proteins in high yields (see Figure S1 in the Supporting Information). A typical reaction mixture included 10 mg of the thioester-peptide analog (~1.2 equiv) and 10 mg Cys-peptide in 800 μL phosphate buffer (200 mM, pH 7.8, ~3 mM peptide) with 12 μL (1.5% v/v) thiophenol.
Selective thermodynamic or kinetic active site oxidation and alkylation with iodoacetamide
The active site of Grx3(C11U-C14U-A38C) was found to be oxidized in the presence of thiophenol while the ligation site Cys38 remained reduced.54 In contrast, the mono-Sec analogs, Grx3(C11U-A38C) and Grx3(C14U-A38C), were partially reduced in the presence of thiophenol, and the active site of Grx3(A38C) was obtained predominantly in its reduced form. Following purification, the proteins were dissolved at 0.1 mg/mL in 200 mM phosphate buffer (pH 7.8) and stirred open to air for 30 min. This procedure resulted in exclusive oxidation of the active site selenenylsulfide bonds. Since the ligated Grx3(A38C), like the wt-Grx3, is an oxidizing enzyme,7,55 this procedure failed, yielding a disulfide dimer at Cys38 rather than the desired active site disulfide monomer. This problem was solved by a kinetically controlled oxidation of the active site using the Grx3 substrate, oxidized glutathione (GSSG). The addition of 1 equiv of GSSG resulted in rapid conversion to the active site disulfide product. All active site oxidized proteins were alkylated at Cys38 without purification using excess iodoacetamide (~1000-fold) and were purified by HPLC (see Figure S2 in the Supporting Information).
Equilibration kinetics and redox potential determination
The redox potential of oxidoreductases is typically determined using end-point analysis at equilibrium with a known redox pair, such as recombinant E. coli Trx (E0=−270 mV, Scheme 2).7 However, the high reactivity of reduced seleno-Grx3 analogs to trace oxidants made this approach impossible. Instead, the equilibration of equimolar Grx3 analogs (oxidized) and Trx (reduced) was easily followed as a function of time. First, the folded proteins were prepared by dissolving 1 mg of each Grx3 analog and 1 mg of Trx in separate tubes using 100 μL argon-degassed potassium phosphate buffer. For each individual time point equimolar oxidized Grx3 analog and reduced Trx were mixed at room temperature and stopped by acid-quenching. The amount of reduced and oxidized Trx was determined using HPLC (Figure 2). The data was fit (Figure 3) to the second-order rate equation (see SI), which contains a term for a linear background oxidation These data are summarized in Table 1. The developed rate equation (see SI) enabled calculation of the second-order rate constants of the forward reaction, k1 and of the reverse reaction, k−1, (Table 1 and Scheme 2) and the equilibrium constant, Keq (Eq. 1). Using the Nernst equation, (Eq. 2), the redox potential differences between Trx (−270 mV) and each Grx3 analog were also determined and summarized in Table 1.
Scheme 2.

Direct protein-protein redox equilibria between Trx and Grx3 analogs.
Figure 2.
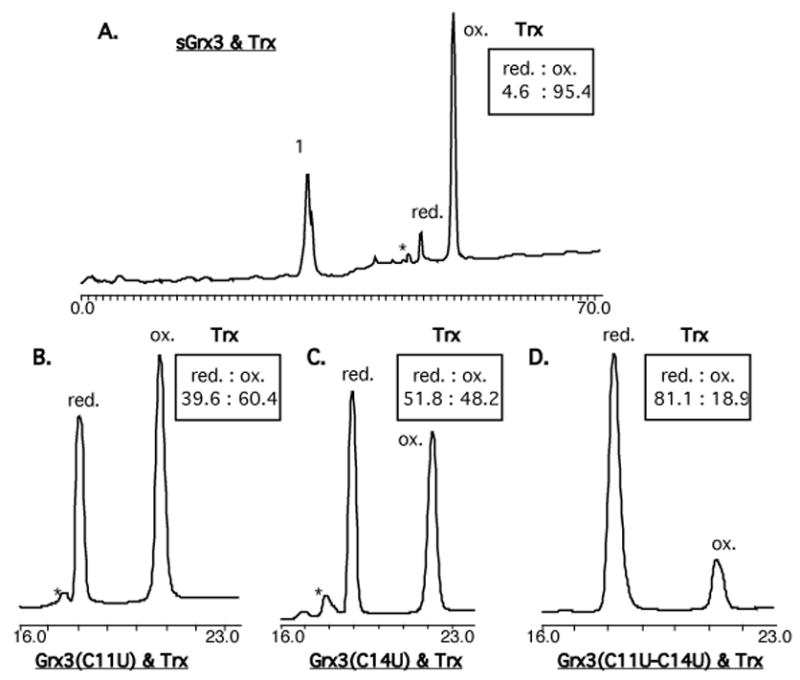
HPLC chromatograms of species present after reaching equilibrium between equimolar amounts of reduced Trx and oxidized Grx3 analogs: A. sGrxS, B. Grx3(C11U), C. Grx3(C14U), D. Grx3(C11U-C14U). Peak 1 contains the inseparable oxidized and reduced forms of sGrxS. An asterisk indicates the position of an oxygenated (M+16) methionine residue.7
Figure 3.
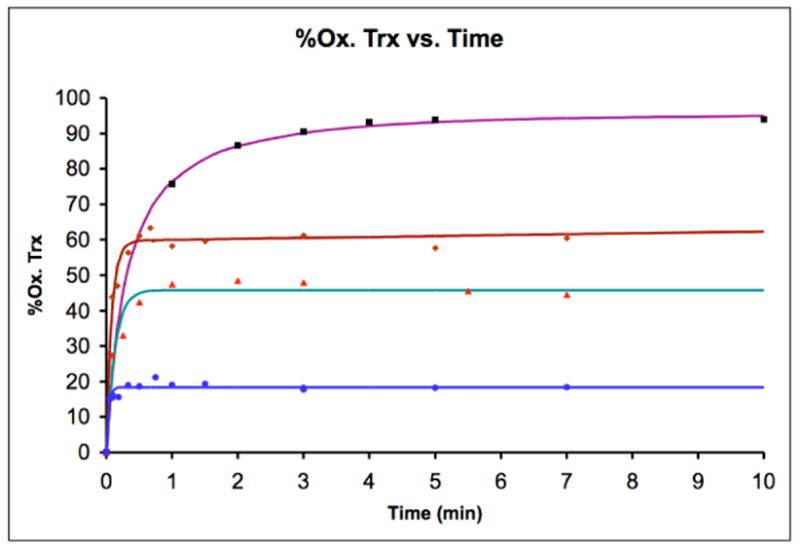
The percentage of oxidized Trx formed as a function of time in the redox equilibrium of reduced Trx and oxidized Grx3 analogs: sGrxS (■); Grx3(C11U) (
 ); Grx3(C14U) (
); Grx3(C14U) (
 ); Grx3(C11U-C14U) (
); Grx3(C11U-C14U) (
 ).
).
Table 1.
Redox potential and kinetic parameters obtained from direct protein-protein equilibria between reduced Trx and oxidized Grx3 analogs. The second-order rate constants (k1 and k−1) as well as the equilibrium constant, Keq were calculated by fitting the kinetic data. Redox potential of Grx3 analogs was calculated from Keq.
| Entry | Protein | E0(mV) | Keq. | k1(M−1s−1) | k−1(M−1s−1) |
|---|---|---|---|---|---|
| 1 | sGrxS | −194 ±5 | 424 ± 170 | 424 ± 17 | 1.0 ± 0.4 |
| 2 | Grx3(C11U) | −260 ±3 | 2.20 ± 0.52 | 291 ± 36 | 132 ± 27 |
| 3 | Grx3(C14U) | −275 ±5 | 0.72 ± 0.29 | 81 ± 17 | 113 ± 40 |
| 4 | Grx3(C11U-C14U) | −309 ± 3 | 0.05 ± 0.01 | 467 ± 81 | 9140 ± 2040 |
| 5 | sGrxS | −197a | |||
| 6 | wt-Grx3 | −198b | |||
| 7 | wt-Grx1 | −233b | |||
| 8 | wt-Trx1 | −270b | |||
Redox potential of sGrx3 was confirmed by the wt-Grx1/sGrx3 equilibria; using end-point analysis after reaching equilibrium.
Data taken from ref. 7.
| Eq. 1 |
| Eq. 2 |
Discussion
Design of seleno-Grx3 analogs
Since most of the known selenoproteins are redox enzymes that operate via reversible formation of selenenylsulfide bonds, it is critical to understand the effect of an active site S/Se substitution on the enzyme’s redox potential and kinetics. Specifically, the redox potential is a reflection of the relative rates of the selenol/disulfide exchange reaction. Depending on the position of Se in the enzyme and the direction of the reaction, it can act as a nucleophile, a central atom or as a leaving group (Scheme 3). Although small molecule studies have suggested that selenolates are both stronger nucleophiles and better leaving groups than the corresponding thiolates, it is not clear to what extent these intrinsic reactivities are maintained within the enzyme environment and how they affect enzyme kinetics. Grx3 was chosen as a model oxidoreductase for this study due to its intermediate redox potential within the Trx superfamily.7. Both mono-Sec Grx3 analogs containing either CXXU or UXXC, and the diselenide analog with the UXXU motif were examined. In contrast to the Sec derivatives, the synthesis of sGrx3 was found to be more challenging because formation of the disulfide bond does not occur readily under air in this protein. In order to push this variant to the fully oxidized state, we added 1 equiv of GSSG. The redox potential of sGrx3 (−194 mV) was found to be consistent within experimental error to that reported of wt-Grx3 (Table 1, entries 1 and 6).7 This result was confirmed by the wt-Grx1/sGrx3 equilibria (−197 mV; Tablet, entry 5), using the end-point analysis.7
Scheme 3.

General mechanism of the redox exchange between Trx and Grx3 analogs.
Redox kinetics
The equilibration reactions between the oxidized Grx3 analogs and reduced Trx were monitored by HPLC analysis of acid-quenched samples. Fitting the kinetic data (Figure 3) to the developed rate equation provided the second-order rate constants k1, and k−1, (Eq. 1) and Keq, which enabled determination of the redox potentials. Interestingly, the k1, values of all analogs were found to be largely independent of the Cys/Sec substitution at position 11 and/or 14 (Table 1, Scheme 2). This observation suggests that within the protein environment, sulfur and selenium behave similarly when taking the role of either a central atom or a leaving group (Scheme 3). In contrast, the k−1 values were found to be remarkably sensitive to the Cys/Sec substitutions over a range of four orders of magnitude, with sGrx3 being the slowest and Grx3(C11U-C14U) being the fastest enzyme (Table 1). This observation is consistent with previous studies on CXXC mutants of Trx in which the second-order rate constants of reduction of oxidized Trx analogs by DTT were found to be the primary determinant of altered redox potential.27 The sGrx3/Trx thiol/disulfide exchange reaction involves a two-step mechanism (Scheme 3), and it seems likely that the Sec analogs utilize a similar mechanism. The significant increase in k−1 observed in the seleno-Grx3 analogs is consistent with the greater nucleophilicity of the selenolate anion in comparison with that of the thiolate.56 Interestingly, the effects of Sec on k−1 are largely additive, in terms of free energy, with either Grx3(C11U) or Grx3(C14U) exhibiting k−1 with approximately ~100-fold greater than that of sGrx3. The double mutant, Grx3(C11U-C14U), exhibited k−1 that was ~9000-fold higher than that of sGrx3.
Single Sec incorporation into Grx3
The two mono-Sec analogs, Grx3(C11U) and Grx3(C14U), contain a Sec residue in the N and C-terminal position of the 11CXX14C motif. While Cys11 is solvent exposed and exhibits a suppressed pKa, Cys14 is largely hidden from solvent and has a higher pKa.57,58 In reduced Grx3 the upper limit for the pKa of Cys11 (pKa < 5.5) and the lower limit for pKa Cys14 (pKa, > 10.5) were determined experimentally by direct NMR titration.58 Since Grx3 unfolds at pH values below 5.5 and above 10.5, precise values were not measurable. However, the actual pKa’s are expected to be at least 1 unit further away from these limits,58 and clearly demonstrate that Cys11 is largely in the anionic form and Cys14 is neutral at physiological pH.58 Inspection of the dithiol mechanism (Scheme 3) shows that both residues act as a nucleophile in different steps of the catalytic mechanism. Since the pKa, of Sec is three units lower than that of Cys, it is expected that Sec 11 would be largely deprotonated while Sec14 would still be in the neutral selenol form at neutral pH.59 Despite the different protonation states of these residues, the position of the Sec residue does not have a large effect on the redox potential of the selenenylsulfide bond in the context of the Grx3 active site. The redox potentials of Grx3(C11U) (−260 mV; Table 1, entry 2) and Grx3(C14U) (−275 mV; Table 1, entry 3) are within 15 mV of each other and are similar to the most reducing member in the thiol/disulfide oxidoreductase family in E. coli, Trx (−270 mV; Table 1, entry 8). Although both mono-Sec analogs have greatly accelerated back reactions (Scheme 2), Grx3(C14U) shows a 5-fold decrease in the forward reaction as well. This suggests that Sec14 is a worse leaving group (k1′) in the context of Grx3, possibly due to destabilization of the selenolate group in the hydrophobic environment surrounding residue 14. This effect is not seen in the forward reaction of Grx3(C11U) where Sec11 is the leaving group for K1″, since this position is solvent exposed. Although it might seem surprising that Sec exerts its primary effect on redox potential by acting as a potent nucleophile rather than as a leaving group, this relationship is required in order to change the redox equilibrium. In order to decrease the redox potential by ~70 mV, the nucleophilicity of Sec must be increased by ~2.5 orders of magnitude more than the enhancement, if any, of Sec as a leaving group (Scheme 3). If both rates were equally enhanced, no change in redox potential would be observed.
Natural selenoenzymes
The redox potentials of the mono-Sec Grx3 analogs provide insight into other selenenylsulfide bonds in enzymes. For example, a family of eukaryotic selenoproteins has been identified with an active site sequence that is analogous to Grx3(C14U). The NMR structure of two of these proteins, Sep15 and Se1M, revealed that they represent a new Trx-like protein family containing active-site motifs of CGU and CGGU, respectively.23 Although the oxidized form of Sep15 contains a selenenylsulfide bond, it was not possible to express wt-Sep15 in sufficient quantities for redox analysis, so the redox potential of its disulfide analog, CGC, was determined instead (−225 mV).23 It was predicted that the redox potential of wt-Sep15 (CGU) would be similar to that of the CGC analog. In contrast, our results suggest that substitution of Cys by Sec significantly lowers the redox potential, by an average of 73 mV for the two mono-Sec analogs of Grx3. Conferring these properties to wt-Sep15 would suggest the redox potential is in the range of −300 mV. Recently, it has been shown that the selenoenzyme methionine-R-sulfoxide reductase (MsrB1) forms a selenenylsulfide bond in its catalytic cycle. Although this bond was reported to be reduced directly by Trx,8 the redox potential of MsrB1 is still unknown. Our results demonstrate that selenenylsulfide bonds in enzymes are in rapid equilibrium with Trx and can be tuned to be even more oxidizing than Trx. Thioredoxin reductase (TrxR) contains a C-terminal selenenylsulfide bond,6,11 which is reduced directly by a pair of Cys residues located near the N-terminus of this enzyme.6,11 This suggests that the redox potential of this disulfide is not significantly lower than that of the selenenylsulfide.60 Furthermore, a recently discovered ResA from B. subtilis that contains the CXXC motif, achieves a low redox potential (−340 mV)61 without requiring Sec substitution. ResA was suggested to be the protein responsible for reducing oxidized cytochrome c550.62 Finally, the UXXC motif has been found in the N-terminal domain of two structurally homologous proteins: the human selenoprotein P (Se1P)63 and the zebrafish zSe1Pa.10 While in Se1P this motif exhibited glutathione peroxidase (GPx) activity,64 zSe1Pa is thought to have selenol/disulfide oxidoreductase activity.10 The Grx3(C11U) analog suggests that zSe1Pa should have a redox potential in the vicinity of Trx, and future studies will be directed toward the ability of synthetic seleno-Grx3 analogs to act as peroxidases.
The diselenide enzyme, Grx3(C11U-C14U), has a low redox potential and fast kinetics
Although several natural selenoproteins that contain multiple Sec residues have been reported,65 the presence of diselenide bonds in natural proteins has not yet been established. Therefore, the synthetic diselenide analog, Grx3(C11U-C14U), provides an interesting opportunity to study this bond within a protein environment. Grx3(C11U-C14U) was found to have a redox potential of −309 mV (Table 1, entry 4), which is 115 mV lower than that of sGrx3, corresponding to a change of 5.5 kcal/mol in redox potential. This result is consistent with the redox potential of Trx(C32U-C35U), which was previously estimated to be somewhere between Trx (−270 mV) and DTT (−323 mV),37 and with the monoselenol selenocystamine (−348 mV)54 but is significantly higher than previous diselenide-peptide studies (−380 mV).40 Although the redox potential of the diselenide analog, Grx3(C11U-C14U), is lower than that of Trx, at equilibrium ~19% of this analog is in the reduced form (Figure 2D), which suggests that Trx can act as an effective reductant for diselenide bonds in proteins. The ~9000-fold increase in k−1 for the diselenide, combined with a physiologically compatible redox potential, suggests that diselenide selenoenzymes may have a role in biological catalysis.
Selenocysteine as a tool for protein engineering
The relative stability of selenenylsulfide bonds compared to diselenide bonds has generated significant interest in protein engineering. The low redox potential of diselenide bonds could stabilize proteins towards reduction as shown with the disulfide rich peptides apamin and d-conotoxin ImI.66,42 Although the redox potential of Grx3(C11U-C14U) (−309 mV) is significantly below that of the two mixed selenenylsulfide analogs (−260 mV and −275 mV), the sum of two selenenylsulfide bonds compared to a disulfide (−194 mV) and a diselenide (−309 mV) would be slightly in favor of the two mixed selenenylsulfide bonds by 16 mV per bond, a trend also seen in peptide studies,40 and consistent with small molecule selenenylsulfide bonds that do not disproportionate into disulfides and diselenides.67,68 Relative stability of these bonds is clearly dependent on the context of the protein structure and the pH of the aqueous media. The incorporation of Sec residues into proteins could increase the kinetics of disulfide exchange as found in small molecule systems.54 In addition, diselenides can be formed selectively at low pH42 and may be kinetically trapped from exchange under monothiol reducing environments.54 However, the near additive redox potentials of the seleno-Grx3 analogs suggest that caution should be taken when incorporating Sec to direct the formation of native and non-native disulfide bonds in proteins.
Conclusions
The synthesis of a complete set of Sec variants in the active site disulfide of glutaredoxin 3 provides insights into the catalytic machinery of selenoenzyme. In the context of an active site, single Sec substitutions significantly lower the redox potential of the active site to a level similar to that of thioredoxin. Although diselenide bonds have not yet been observed in selenoenzymes, we find that active site diselenides can have low but biologically compatible redox potentials, suggesting a possible role for diselenides in selenoenzyme catalysis. Significantly, the effects of Sec on the reaction kinetics suggest that the difference in nucleophilicity between selenolate and thiolate groups could provide the bulk of the rate enhancement observed in many selenoenzymes. As a result, protein engineering with Sec may enhance the rate of numerous enzymes that utilize nucleophilic cysteine residues.
Experimental Section
Materials and Methods
Buffers for kinetic measurements were prepared using de-ionized water (MilliQ). KH2PO4 and K2HPO4 were purchased from Fisher Biotech. Recombinant E. coli Trx1 and Grx1 were purchased from Promega Corporation (Madison, WI) and American Diagnostica Inc. (Greenwich, CT), respectively. Deuterated solvents (DMSO-d6 and CDCl3) were purchased from Aldrich Chem. Co.
Protein sequence and design
The amino acid sequence of the wild-type Grx3 is:
1ANVEIYTKET 11CPY11CHRAKAL LSSKGVSFQE LPIDGN37A38AKR
EE43MIKRSGRT51TVPQIFIDAQ HIGG65CDDLYA LDARGGLDPL L82K
The sGrx3 protein used in this work contains substitutions: Met43Nle (norleucine) and Cys65Tyr to prevent undesired oxidative side reactions.33 In addition, Ala38Cys was used for NCL and was subsequently alkylated with iodoacetamide to give Ala38Cys(S-CH2CONH2). The seleno-Grx3 analogs contained the above-mentioned substitutions with additional Sec at position 11 and/or 14.
Synthesis of N-Boc-L-Se(4-methylbenzyl)Sec-OH
N-Boc-L-Ser(OTos)-OMe, 3 was synthesized according to van der Donk48 using N-Boc-L-Ser-OMe, 2.69
bis-(4-Methylbenzyl)-diselenide, 4
Following the procedure of Tian et al.52 elemental selenium (7.1 g, 90 mmol), water (20 mL), DMF (160 mL) and 4-methylbenzaldehyde (3.6 g, 30 mmol) were added to a 250 mL three-nicked flask equipped with a stirrer, condenser and gas bubbler. The mixture was stirred at 100 °C under CO for 7 h, then was stirred overnight under air to complete the oxidative dimerization. Water (100 mL) was added and the mixture was filtered through a silica-gel pad, washed with Et2O and then extracted with Et2O. The organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure to yield bis-(4-methylbenzyl)-diselenide, 4 (10 g, 88%) in the form of a yellow-orange solid. 1H-NMR (300 MHz, CDCl3) δ 7.07 (s, 8H), 3.8 (s, 4H), 2.3 (s, 6H).
N-Boc-L-Se(4-methylbenzyl)Sec-OMe, 6
Following the procedure of van der Donk,48 compound 4 (24 g, 64.72 mmol) and N-Boc-L-Ser(OTos)-OMe, 3 (15.05 g, 40.3 mmol) were reacted and purified by column chromatography to give 6 (10 g, 64%). 1H-NMR (300 MHz, CDCl3) δ 7.14 (d, J = 8.1 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H), 5.27 (d, J = 6.9 Hz, 1H), 4.6 (m, 1H), 3.74 (s, 2H), 3.72 (s, 3H) 2.87 (t, J = 4.5 Hz, 2H), 2.3 (s, 3H), 1.43 (s, 9H). MS: [MNa]+ = 410.
N-Boc-L-Se(4-methylbenzyl)Sec-OH, 7
Compound 6 was hydrolyzed by mixing it under argon with trimethyltin hydroxide53 (7 equiv) in degassed 1,2-dichloroethane at 80 °C for 3 h. Purification by column chromatography over silica-gel afforded 7 (95%) in the form of white powder. 1H-NMR (500 MHz, CDCl3) δ 7.14 (d, J = 8 Hz, 2H), 7.06(d, J= 8 Hz, 2H), 5.28 (bd, J = 4.5 Hz, 1H),4.58 (bd, J = 3 Hz, 1H), 3.76 (s, 2H), 2.91 (bs, 2H), 2.29 (s, 3H), 1.43 (s, 9H). MS: [MH]+ = 374, [MNa]+ = 396.
Peptide Synthesis
All peptides were prepared by machine-assisted solid-phase peptide synthesis (SPPS), typically on a 0.2 mmol scale using the in situ neutralization/HCTU activation procedure for Boc-SPPS.70 The peptide coupling was carried out with 11-fold excess (except for norleucine and Sec, which were used in 3-fold excess) of activated amino acid for 20 min. The trityl protecting group of the TAMPAL-Pam resin was first removed by a mixture of TFA:triisopropylsilane:H2O, 95:2.5:2.5, and then transferred to the machine for the synthesis of the thioester peptides as described above. The Cys-peptide Grx3(Cys38-Lys82) was synthesized using the Boc-Lys(2ClZ)-OCH2-Pam resin as described above.
Sec-coupling
The coupling of the Boc-Sec(4-MeBzl)-OH was carried out manually using a DIC/HOBt activation method, using Sec (0.5 mmol in 2 mL of 50% DCM/DMF) and activated with DIC (0.5 mmol) in the presence of HOBt (0.52 mmol) at 0 °C for 5 min. The Boc group on resin-bound peptide was deprotected, neutralized with DIEA (2×1 min) and washed with DMF. The activated Sec was then added to the resin and the mixture was kept at room temperature for 1 h. Upon completion of the polypeptides assembly it was deprotected and cleaved from the resin by treatment of the dry peptide-resin (~300–400 mg) with 10–15 ml HF and ~10% anisole for 1 h at 0 °C. The crude peptide products were precipitated and washed with cold anhydrous ether, dissolved in aqueous acetonitrile and immediately purified by preparative, reversed-phase HPLC.
Conformationally assisted ligation
Preparation of all Grx3 analogs via native chemical ligation was carried out under folding conditions.51 The progress of the reaction was followed by analytical HPLC, indicating that the reaction was complete within 4 h, affording the desired protein in high yields (Figure S1, 40–50% recovered).71 A typical reaction mixture included 10 mg of the thioester-peptide analog (~1.2 equiv.), 10 mg Cys-peptide in 800 μL phosphate buffer (200 mM, pH 7.8, ~3 mM peptide), and 12 μL(1.5% v/v) thiophenol, the ligation was performed at room temperature with periodic vortexing.
Active site oxidation and Cys38 alkylation with iodoacetamide
In the case of Grx3(C11U-C14U-A38C) the resultant diselenide (oxidized form) was found to be stable in the presence of thiophenol in the ligation mixture. At the end of the ligation the excess thiophenol was removed by extraction with Et2O (3×2 mL). Alkylation of Cys38 with iodoacetamide was achieved by addition of excess iodoacetamide (~1000-fold) to the erode ligation mixture to produce Grx3(C11U-C14U-A38Cys(S-CH2CONH2)) or abbreviated Grx3(C11U-C14U), which was purified by semi-preparetive HPLC (41% recovered yield for the two steps).
In the case of the other three analogs, Grx3(A38C), Grx3(C11U-A38C) and Grx3(C14U-A38C), the presence of thiophenol in the ligation mixture resulted in mixtures of the reduced and oxidized forms. Therefore, it was necessary to purify the ligated proteins and oxidize them to produce either the disulfide or the corresponding selenenylsulfide bond. This oxidation was carried out by dissolving each protein (~8 mg) in 100 mL phosphate buffer (200 mM, pH 7.8) and stirring the mixture in an open flask for ~30 min (Figure S2). In the case of Grx3(A38C) it was necessary to add 1 equiv. of oxidized glutathione (GSSG) in order to complete the oxidation process (any dimer formation was separated and recovered). Finally, excess iodoacetamide (~1000-fold) was added and the resultant protein was purified by semi-preparative HPLC.
Equilibration kinetics and redox potential determination
Determining the redox potential was similar to the method of Holmgren,7 by equilibration of equimolar Grx3 analogs (oxidized) and Trx (reduced), which was followed as a function of time. First, the folded proteins were prepared by dissolving 1 mg of each Grx3 analog and 1 mg of Trx in separate tubes using 100 μL argon-degassed potassium phosphate buffer (100 mM, pH 7.07, 1 mM EDTA) and allowing them to stand for 30–60 minutes at room temperature. The equilibrium reactions (5–12 μL typically contained 100–270 μM of each of the two redox-active proteins in a degassed and argon-purged solution of potassium phosphate (100 mM, pH 7.07, 1 mM EDTA). The reduced form of Trx was prepared immediately before use by incubation of the protein (~500 μM in 50 mM dithiothreitol at room temperature for 1 h, followed by extensive centrifugation-dialysis (Amicon Ultra, cutoff 5000 Da, Millipore Corporation, Bedford, MA) by degassed potassium phosphate buffer (8×2 mL). The concentration of each protein was determined by UV (HP 8452A Diode-Array), using the following ε 280 nm values: Trx (ε280 nm = 13700 cm−1M−1); sGrx3 (ε280 nm = 6050 cm−1 M−1); Grx3(C11U) orGrx(C14U) (ε280 nm = 5990 cm−1M−1); Grx3(C11U-C14U) (ε280 nm = 5920 cm−1M−1). The ε280 nm values were calculated using SherpaLite4.0 for Mac.
Redox equilibration of the Grx3 analogs was carried out separately for each time point by mixing the reduced Trx and oxidized Grx3 analog in an Eppendrof tube at room temperature using degassed potassium phosphate buffer. The reaction was stopped after the desired time interval (from 3 sec to 10 min in analogs that contain Sec and to 2 h for the sGrx3) by adding HCl (80 μL, 1 M) followed by immediate HPLC analysis to avoid oxidation of the seleno-Grx3 analogs. The oxidized and reduced forms of Trx72 were well separated by reverse phase HPLC (Varian) on a C18 column (Phenomenex Jupiter 5 μm, 300Å, 150 × 4.6 mm) using a gradient from 35–70% (v/v) buffer B in 25 min at a flow rate of 1 ml/min at room temperature, monitoring at 220 nm. The amounts of oxidized and reduced forms of Trx in the quenched equilibrium mixture were obtained from the chromatograms by peak area integration, each time point is the average of two separate experiments. The data was fit to the second-order rate equation (see SI), which contains a term for a linear background oxidation (only observed for Sec containing analogs). These data are summarized in Table 1. The second-order rate constants (k1 and k−1), as well as the equilibrium constant, Keq(Eq. 1) were calculated by fitting the data to the kinetic model. The difference in redox potential between Trx and each of the Grx3 analog, ΔE, was calculated according to the Nernst equation (Eq. 2), where n is the number of electrons transferred (here n = 2), F is Faraday’s constant (23.04 kcal·mol−1·V−1), R is the gas constant (1.987 cal·K−1·mol−1), and T is the absolute temperature (298 K).
Supplementary Material
Further detailed materials and figures for synthesis, characterization and kinetics are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the Israel-US Binational Science Foundation, the German-Israeli Project Cooperation (DIP) (EK), NIH GM059380 (FED), the Israeli Higher Education Planning and Budgeting Committee and Israel Ministry of Science (NM) and the Skaggs Institute for Chemical Biology for financial support. We also want to thank Dr. John Blankenship for unpublished work on the synthesis and mutagenesis of glutaredoxin, Dr. Michael Churchill for help with the peptide syntheses,
Footnotes
Theresa Tiefenbrunn for discussions, and Dr. Jörg Zimmermann for data fitting.
References
- 1.Flohe L, Gunzler EA, Schock HH. FEBS Lett. 1973;32:132. doi: 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- 2.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Science. 1973;179:588. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 3.Stadtman TC. Ann Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 4.Stadtman TC. Ann NY Acad Sci. 2000;899:399–402. doi: 10.1111/j.1749-6632.2000.tb06203.x. [DOI] [PubMed] [Google Scholar]
- 5.Arnold AP, Tan KS, Rabenstein DL. Inorg Chem. 1986;25:2433. [Google Scholar]
- 6.Zhong L, Arner ESJ, Holmgren A. Proc Natl Acad Sci USA. 2000;97:5854–5859. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Åslund F, Berndt KD, Holmgren A. J Biol Chem. 1997;272:30780–30786. doi: 10.1074/jbc.272.49.30780. [DOI] [PubMed] [Google Scholar]
- 8.Kim H-Y, Gladyshev VN. PloS Biology. 2005;3:2080–2089. doi: 10.1371/journal.pbio.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Ganther HE. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]; b) Becker K. Eur J Biochem. 2000;267:6118–6125. doi: 10.1046/j.1432-1327.2000.01703.x. [DOI] [PubMed] [Google Scholar]
- 10.Kryukov GV, Gladyshev VN. Genes to Cells. 2000;5:1049–1060. doi: 10.1046/j.1365-2443.2000.00392.x. [DOI] [PubMed] [Google Scholar]
- 11.Lee SR, Bar-Noy S, Kwon J, Levine RL, Stadtman TC, Rhee SG. Proc Natl Acad Sci USA. 2000;97:2521–2526. doi: 10.1073/pnas.050579797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH, Jr, Schirmer RH, Arner ESJ. Proc Nat Acad Sci USA. 2003;100:2618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gladyshev VN, Jeang KT, Wootton JC, Hatfield DL. J Biol Chem. 1998;275:8910–8915. doi: 10.1074/jbc.273.15.8910. [DOI] [PubMed] [Google Scholar]
- 14.Korotkov KV, Novoselov SV, Hatfield DL, Gladyshev VN. Mol Cell Biol. 2002;22:1402–1411. doi: 10.1128/mcb.22.5.1402-1411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardikas-Vlamis A, Holmgren A. Methods Enzymol. 2002;347:286–296. doi: 10.1016/s0076-6879(02)47028-0. [DOI] [PubMed] [Google Scholar]
- 16.Eklund H, Cambillau C, Sjoberg BM, Holmgren A, Jornvall H, Hoog JO, Branden CI. EMBOJ. 1984;3:1443–1449. doi: 10.1002/j.1460-2075.1984.tb01994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin JL. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 18.Holmgren A. Structure. 1995;3:239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 19.Nelson JW, Creighton TE. Biochemistry. 1994;33:5974–5983. doi: 10.1021/bi00185a039. [DOI] [PubMed] [Google Scholar]
- 20.Grauschopf U, Winther JR, Korber P, Zander T, Dallinger P, Bardwell JCA. Cell. 1995;83:947–955. doi: 10.1016/0092-8674(95)90210-4. [DOI] [PubMed] [Google Scholar]
- 21.Dyson HJ, Jeng MF, Tennant LL, Slaby I, Lindell M, Cui DS, Kuprin S, Holmgren A. Biochemistry. 1997;36:2622–2636. doi: 10.1021/bi961801a. [DOI] [PubMed] [Google Scholar]
- 22.Jeng MF, Holmgren A, Dyson HJ. Biochemistry. 1995;34:10101–10105. doi: 10.1021/bi00032a001. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson AD, Labunskyy VM, Fomenko DE, Araç D, Chelliah Y, Amezcua CA, Rizo J, Gladyshev VN, Deisenhofer J. J Biol Chem. 2006;281:3536–3543. doi: 10.1074/jbc.M511386200. [DOI] [PubMed] [Google Scholar]
- 24.Krause G, Lundstrom J, Barea JL, Pueyo de la Cuesta C, Holmgren A. J Biol Chem. 1991;266:9494–9500. [PubMed] [Google Scholar]
- 25.Chivers PT, Laboissiere MCA, Raines RT. EMBO J. 1996;16:2659–2667. [PMC free article] [PubMed] [Google Scholar]
- 26.Schultz LW, Chivers PT, Raines RT. Acta Cryst. 1999;55:1533–1538. doi: 10.1107/s0907444999008756. [DOI] [PubMed] [Google Scholar]
- 27.Mössner E, Huber-Wunderlich M, Rietsch A, Beckwith J, Glockshuber R, Åslund F. J Biol Chem. 1999;274:25254–25259. doi: 10.1074/jbc.274.36.25254. [DOI] [PubMed] [Google Scholar]
- 28.21 mV difference in redox potential is equivalent to 1 kcal/mol at room temperature.
- 29.Lin TY, Kim PS. Biochemistry. 1989;28:5282–5287. doi: 10.1021/bi00438a054. [DOI] [PubMed] [Google Scholar]
- 30.Holmgren A. Proc Nat Acad Sci USA. 1976;73:2275–2279. doi: 10.1073/pnas.73.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Åslund F, Ehn B, Miranda-Vizuette A, Pueyo C, Holmgren A. Proc Nat Acad Sci USA. 1994;97:9813–9817. doi: 10.1073/pnas.91.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bushweller JH, Billeter M, Holmgren A, Wüthrich K. J Bio Chem. 1994;255:1585–1597. doi: 10.1006/jmbi.1994.1108. [DOI] [PubMed] [Google Scholar]
- 33.Nordstrand K, Åslund F, Holmgren A, Otting G, Berndt KD. J Mol Biol. 1999;286:541–552. doi: 10.1006/jmbi.1998.2444. [DOI] [PubMed] [Google Scholar]
- 34.Berry MJ, Banu L, Chen YY, Mandel SJ, Kieffer JD, Harney JW, Larsen PR. Nature. 1991;353:273–00276. doi: 10.1038/353273a0. [DOI] [PubMed] [Google Scholar]
- 35.Berry MJ, Banu L, Harney JW, Larsen PR. EMBO J. 1993;12:3315–3322. doi: 10.1002/j.1460-2075.1993.tb06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suppmann S, Persson BC, Böck A. EMBO J. 1999;18:2284–2293. doi: 10.1093/emboj/18.8.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller S, Senn H, Gsell B, Vetter W, Baron C, Böck A. Biochemistry. 1994;33:3404–3412. doi: 10.1021/bi00177a034. [DOI] [PubMed] [Google Scholar]
- 38.Wu ZP, Hilvert D. J Am Chem Soc. 1989;111:4513–4514. [Google Scholar]
- 39.Rajarathnam K, Sykes BD, Dewald B, Baggiolini M, Clark-Lewis I. Biochemistry. 1999;38:7653–7658. doi: 10.1021/bi990033v. [DOI] [PubMed] [Google Scholar]
- 40.Besse D, Siedler F, Diercks T, Kessler H, Moroder L. Angew Chem Int Engl. 1997;36:883–885. [Google Scholar]
- 41.Moroder LJ. Pept Sci. 2005;11:187–214. doi: 10.1002/psc.654. [DOI] [PubMed] [Google Scholar]
- 42.Armishaw CJ, Daly NL, Nevin ST, Adams DJ, Craik DJ, Alewood PP. J Biol Chem. 2006;281:14136–14143. doi: 10.1074/jbc.M512419200. [DOI] [PubMed] [Google Scholar]
- 43.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 44.Dawson PE, Kent SBH. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 45.Dawson PE, Churchill MJ, Ghadiri MR, Kent SBH. J Am Chem Soc. 1997;119:4325–4329. [Google Scholar]
- 46.Hondal RJ, Nilsson BL, Raines RT. J Am Chem Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 47.Ralle M, Berry SM, Nilges MJ, Gieselman MD, Van der Donk WA, Lu Y, Blackburn NJ. J Am Chem Soc. 2004;126:7244–7256. doi: 10.1021/ja031821h. [DOI] [PubMed] [Google Scholar]
- 48.Gieselman MD, Xie L, Van der Donk W. Org Lett. 2001;3:1331–1334. doi: 10.1021/ol015712o. [DOI] [PubMed] [Google Scholar]
- 49.Quaderer R, Sewing A, Hilvert D. Helvetica Chimica Acta. 2001;84:1197–1206. [Google Scholar]
- 50.Eckenroth B, Harris K, Turanov AA, Gladyshev VN, Raines RT, Hondal RJ. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beligere GS, Dawson PE. J Am Chem Soc. 1999;121:6332–6333. [Google Scholar]
- 52.Tian R, Yu Z, Lu S. J Org Chem. 2004;69:4520–4523. doi: 10.1021/jo049733i. [DOI] [PubMed] [Google Scholar]
- 53.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angew Chem Int Ed. 2005;44:2–6. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- 54.Singh R, Whitesides GM. J Org Chem. 1991;56:6931–6933. [Google Scholar]
- 55.Sandberg VA, Kern B, Fuchs JA, Woodward C. Biochemistry. 1991;30:5475–5484. doi: 10.1021/bi00236a021. [DOI] [PubMed] [Google Scholar]
- 56.Johansson L, Gafvelin G, Arnér ESJ. Biochem Biophys Acta. 2005;1726:1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 57.Chivers PT, Prehoda KE, Raines RT. Biochemistry. 1997;36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 58.Nordstrand K, Åslund F, Meunier S, Holmgren A, Otting G, Berndt KD. FEBS Lett. 1999:449, 196–200. doi: 10.1016/s0014-5793(99)00401-9. [DOI] [PubMed] [Google Scholar]
- 59.In principle, the pKa, values of Sec in selenoproteins can be measured by 77Se-NMR, although direct measurement of these values is not possible due to the instability of reduced Grx3 bellow pH 5.
- 60.De Silva V, Woznichak MM, Burns KL, Grant KB, May SW. J Am Chem Soc. 2004;126:2409–2413. doi: 10.1021/ja037294j. [DOI] [PubMed] [Google Scholar]
- 61.Erlendsson LS, Acheson RM, Hederstedt L, Le Brun NE. J Biol Chem. 2003;278:17852–17858. doi: 10.1074/jbc.M300103200. [DOI] [PubMed] [Google Scholar]
- 62.Colbert CL, Wu Q, Erbel PJA, Gardner KH, Deisenhofer J. Proc Natl Acad Sci USA. 2006;103:4410–4415. doi: 10.1073/pnas.0600552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saito Y, Sato N, Hayashi T, Tanaka A, Watanabe Y, Suzuki M, Saito E, Takahashi K. J Biol Chem. 1999;274:2866–2871. doi: 10.1074/jbc.274.5.2866. [DOI] [PubMed] [Google Scholar]
- 64.Saito Y, Sato N, Hirashima M, Takebe G, Nagasawa S, Takahashi K. Biochem J. 2004;381:841–846. doi: 10.1042/BJ20040328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatfield DL, Gladyshev VN. Mol Cell Biol. 2002;22:3565–3576. doi: 10.1128/MCB.22.11.3565-3576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pegoraro S, Fiori S, Cramer J, Rudolph-Bohner S, Moroder L. Protein Sci. 1999;8:1605–1613. doi: 10.1110/ps.8.8.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.du Mont WW, Mugesh G, Wismach C, Jones PG. Angew Chem Int Engl. 2001;40:2486–2488. [PubMed] [Google Scholar]
- 68.Mugesh G, du Mont WW, Wismach C, Jones PG. ChemBioChem. 2002;3:440–447. doi: 10.1002/1439-7633(20020503)3:5<440::AID-CBIC440>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 69.Garner P, Park JM. Org Syn, Coll. 1998;9:300. [Google Scholar]
- 70.Schnolzer M, Alewood PP, Jones A, Alewood D, Kent SBH. Int J Pept Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 71.Under denaturaion conditions (6M GnHCl) the reaction took 16 hrs to give the same yield.
- 72.The two forms of Trx were well separated under the HPLC conditions. The seleno-Grx3 analogs could not be fully separated and the reduced form had partial irreversible adsorption to the reversed phase matrix.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Further detailed materials and figures for synthesis, characterization and kinetics are provided. This material is available free of charge via the Internet at http://pubs.acs.org.