

Abstract
The function of the stacking tryptophan, W290, a second coordination sphere residue in galactose oxidase has been investigated via steady-state kinetics measurements, absorption, CD and EPR spectroscopy, and x -ray crystallography of the W290F, W290G, and W290H variants. Enzymatic turnover is significantly lower in the W290 variants. The Km for D-galactose for W290H is similar to wild type, whereas the Km is greatly elevated in W290G and W290F, suggesting a role for W290 in substrate binding/positioning via the –NH group of the indole ring. Hydrogen bonding between W290 and azide in the wild type-azide crystal structure are consistent with this function. W290 modulates the properties and reactivity of the redox-active tyrosine radical; the Y272 tyrosyl radical in both the W290G and W290H variants have elevated redox potentials and are highly unstable compared to the radical in W290F, which has similar properties to the wild type tyrosyl radical. W290 restricts the accessibility of the Y272 radical site to solvent. Crystal structures show that Y272 is significantly more solvent exposed in W290G variant but that W290F limits solvent access comparable to the wild-type indole side chain. Spectroscopic studies indicate that the Cu(II) ground states in the semi-reduced W290 variants are very similar to that of the wild-type protein. In addition, the electronic structures of W290X-azide complexes the variants are also closely similar to the wild type electronic structure. Azide binding and azide-mediated proton uptake by Y495 are perturbed in the variants, indicating that tryptophan also modulates the function of the catalytic base (Y495) in the wild-type enzyme. Thus, W290 plays multiple critical roles in enzyme catalysis, affecting substrate binding, the tyrosyl radical redox potential and stability, and the axial tyrosine function.
Over the past twenty years, there has been a growing appreciation for the catalytic utility of protein-derived free radical cofactors in enzymes (1–3). Free radical chemistry is harnessed to catalyze bond activation and molecular rearrangements in a wide variety of enzymes including ribonucleotide reductase (4–7), DNA photolyase (8), cytochrome c peroxidase (9), pyruvate-formate lyase (10), lysine-2,3-aminomutase (11), prostaglandin H synthase (12), glyoxal oxidase (13), and galactose oxidase (14).
It is becoming increasingly clear that the environment of a protein-based radical can play a significant role in modulating its properties and reactivity (15–18). The case of E. coli. ribonucleotide reductase is especially pertinent where alteration of the hydrophobic environment of the tyrosyl radical by mutation of second coordination sphere residues dramatically lowers radical stability (19;20). Additionally, substantial evidence now indicates that second coordination shell effects in metalloproteins may profoundly influence the reactivity and electronic structure of active-site metal ions while weak interactions (such as hydrogen bonds) have been strategically manipulated to control reactivity in proteins and model complexes (21–28).
We have initiated a systematic investigation of the influence of the active site microenvironment on the structure and reactivity of the tyrosyl radical (Y•) in the copper-radical enzyme, galactose oxidase (29;30). A notable feature of the Cu(II)-Y• active site is the presence of the so-called “stacking tryptophan”, W290 (Figure 1). As presented herein, our results indicate that the second coordination sphere residue W290 has significant effects on the generation and reactivity of the tyrosyl radical (Y272) in galactose oxidase, and consequently, the catalytic activity of the enzyme.
Figure 1.
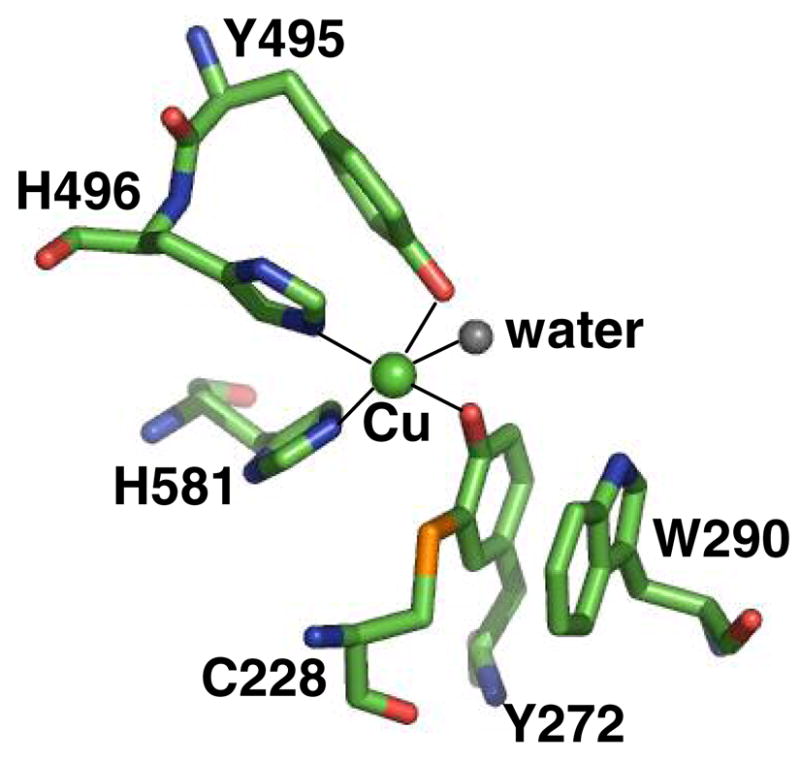
Active site of wild type galactose oxidase at pH 7.0 (1GOG.pdb). The copper is shown as a green sphere and the ligand bonds to the copper are shown as solid lines. Figure produced using Insight (Accelrys, CA).
Galactose oxidase (secreted by Fusarium graminearum) contains a mononuclear copper ion coordinated to a post-translationally modified, cross-linked cysteine-tyrosine radical cofactor
(1).
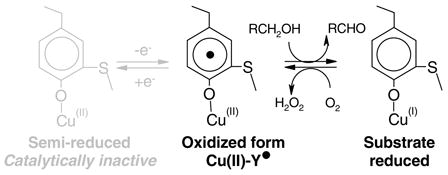
(Figure 1) (31). A growing number of enzymes have been found to have cofactors derived from post-translationally modified amino acids (32). Galactose oxidase catalyzes the stereospecific(33) two-electron oxidation of primary alcohol to the corresponding aldehyde, reducing O2 to hydrogen peroxide (Equation 1). The enzyme has KM values in the millimolar range (30;34) and a broad substrate range (35;36). The precise biological roles of galactose oxidase and related enzymes are not known, but may be associated with peroxide formation (37;38).
The structure of galactose oxidase has been determined by x-ray crystallography to 1.7 Å resolution (31;39) and the chemistry of its active site studied extensively. Y272, the site of the free radical in the oxidized form of galactose oxidase (40), is modified by a thioether crosslink to C228, and W290 “stacks” over this unit (Fig. 1). In model systems thioether substitution of the tyrosyl radical stabilizes the one-electron oxidized state by ~ 540 mV relative to an unsubstituted tyrosyl radical (2;41). The coordinated H2O can be displaced by acetate and azide or by alcohol substrates, suggesting that this is the site of substrate binding (42). The axial ligand Y495 may act as a base to accept a proton from the bound alcohol substrate, activating the alcohol for oxidation and dissociating from the copper ion (43;44).
Previous studies had suggested that W290 influences the redox properties and reactivity of the tyrosyl radical, and may play other roles in catalysis. The W290H tyrosyl radical redox potential is 730 mV (45) compared to 430 mV in wild type enzyme (41), and the radical of W290H appears unstable at pH 7.0 (29;30). Modeling studies suggested that W290 may be involved in binding D-galactose, and perhaps in stabilizing the transition state for oxidation of this substrate (39;46). W290 might also modulate the reactivity of the catalytic base, Y495 (47).
The present paper seeks to define the role(s) of W290 in galactose oxidase by comparing the properties of the wild type enzyme to three W290 mutational variants – W290F, W290H, and W290G. The crystal structures of these variant proteins have been determined, and the reactivity and electronic structure defined through spectroscopic and kinetic methodologies. The roles of W290 are discussed in the light of these new findings.
Experimental Procedures (Materials and Methods)
Enzyme purification
Wild type, W290H, and W290G Fusarium graminearum galactose oxidase were purified from the Aspergillus nidulans over-expression system (30). Purity and molecular weight were confirmed by SDS-PAGE. Wild type and W290F galactose oxidase were alternatively isolated via a Pichia pastoris expression system (48;49). Galactose oxidase purified from the yeast expression system had essentially indistinguishable kinetic and structural properties from the enzyme isolated from the fungal expression system. Protein concentrations were calculated using ε280 of 104900 M−1cm−1 (50). The concentrations of W290 variants were corrected by multiplying by 16/15 to account for the lowered tryptophan content. Thioether bond formation was assessed by SDS-PAGE analysis, taking advantage of the anomalous migration characteristic of the protein with the tyrosine-cysteine crosslink (30).
Copper analysis by atomic absorption spectrometry
The copper content of each galactose oxidase protein was analyzed using a Buck Scientific Model 210VGP atomic absorption spectrophotometer. Protein samples were prepared at a concentration of 1 mg/mL using 50 mM potassium phosphate, pH 7.0 as a background control.
Enzyme kinetics
Specific activity was determined as described previously via detection of H2O2 using a coupled assay (30). Suitably diluted enzyme was added to 1 mL of assay buffer containing 600 mM D-galactose, 0.8 mM ABTS, and 20 U/ml horseradish peroxidase (HRP) in 100 mM sodium phosphate, pH 7.0. The absorbance change at 415 nm was recorded on an HP8453 spectrophotometer at 25 °C. Specific activity was calculated from the rate of production of the ABTS radical cation (ε415 31300 M−1cm−1) assuming two molecules of the ABTS radical cation are produced from every molecule of hydrogen peroxide (48). One unit of specific activity (U) of galactose oxidase corresponds to 1 μmole H2O2 produced min−1 mg−1.
kcat and Km determination
kcat and Km for D-galactose were determined as previously described (30;49) via coupled specific activity assays containing HRP and ABTS, performed over a D-galactose concentration range of 2.5 mM to 907.5 mM. A substrate-depleted assay mixture was supplemented with D-galactose to the required concentration. The amount of protein present in each assay was: wild type, 1.16 × 10−7 μmoles, W290F, 9.46 × 10−6 μmoles, W290G, 6.8 × 10−5 μmoles, and W290H, 2.88 × 10−4 μmoles. Non-linear curve fitting to the Michaelis-Menten equation was used to determine Km and kcat (OriginLab, MA).
Kinetic parameters for 2-methylene-1,3-propanediol (2MP) were determined via oxygen consumption assays (0.680 mL) using a Clark-type oxygen electrode (Instech, PA). Reaction buffer (15 U/mL horseradish peroxidase in 100 mM potassium phosphate, pH 7.0) was added to the reaction chamber, followed by substrate (2 mM to 500 mM) and finally enzyme. The amounts of protein present in these assays were as follows; wild type, 7.6 × 10−12 moles, W290F, 6.9 × 10−12 moles, W290G, 5.13 × 10−9 moles and W290H, 9.44 × 10−9 moles. kcat and Km were determined as above.
X-ray crystallographic data collection and structure determination
Crystals of W290F and W290G were grown from protein as isolated, using hanging drop vapor diffusion from 1–2 M (NH4)2SO4 and 0.1 M sodium acetate at pH 4.0–5.0. To prepare the azide complex, wild type galactose oxidase was dialyzed into 50 mM PIPES, pH 7.0 for two hours, before addition of 20 mM sodium diethyldithiocarbamate (DDC). The copper chelator DDC promotes the formation of orthorhombic crystals, which have more favorable crystal packing for substrate soaking experiments. The copper-free protein was crystallized in 13.5 % PEG 8000, 200 mM calcium acetate, 100 mM MES, pH 6.3. The crystals grew to full size (approximately 400μm by 75μm) in two to three weeks, and were copper-loaded by adding 250 mM copper acetate to the crystallization mother liquor for four hours. After washing with mother liquor to remove excess copper acetate, a suitable crystal was transferred to mother liquor containing 10 mM azide, before transfer to a cryoprotectant comprising mother liquor, 10 mM azide and 20 % PEG 400 followed by flash cooling in liquid nitrogen. Crystals of W290F and W290G were briefly transferred to cryoprotectant, comprising mother liquor and 25% glycerol, before flash cooling in liquid nitrogen. Diffraction data for all crystals were collected at 100 K on station 9.6 at the Daresbury Synchrotron Radiation Source (SRS), using an ADSC quantum IV CCD detector.
Diffraction image data for W290F and the azide complex were integrated using MOSFLM (51) and processed with programs from the CCP4 suite (52). W290F crystals diffracted only to 2.8 Å but were isomorphous with the original galactose oxidase structure (39), and phase information was obtained from the wild type galactose oxidase model (1GOG) with F290 modeled as alanine. Following rigid body, positional and B-factor refinement with CNS, water molecules were added, F290 was built into the model, and refinement continued until R = 17.7 % and Rfree = 23.3 %. The wild type azide complex crystal diffracted to 1.8 Å and was isomorphous with a previously solved orthorhombic wild-type structure (53). An azide ligand was added to this model at the copper site, and only minor changes to the protein and water molecules were needed before refinement to R = 19.2 % and Rfree = 20.9 %. Coordination distances to copper were subjected to weak restraints to standard values in all structures.
W290G crystallized in a space group P6522, with a very long c axis, requiring the detector to be moved further from the crystal, limiting the resolution of the data to 2.2 Å. The diffraction images were integrated using the HKL suite (54), scaled and merged in SCALEPACK and subsequently processed with CCP4. The structure of another variant, Y495K, that had been solved in the same crystal form (55) was used as a starting model, and refinement proceeded in a similar way to W290F to R = 19.1 % and Rfree = 22.7 %. Data collection, processing and refinement statistics are shown in Table 1.
Table 1.
Crystallographic statistics for W290F and W290G galactose oxidase variants and wild type-azide complex
| protein | W290G | W290F | wild type-N3 |
|---|---|---|---|
| Space group | P6522 | C2 | P212121 |
| unit cell (Å) | a = 89.88 | a = 97.77 | a = 59.31 |
| b = 89.88 | b = 88.89 | b = 89.25 | |
| c = 415.33 | c = 86.19 | c = 134.37 | |
| β = 117.9° | |||
| resolution (Å) | 10 - 2.2 | 30 - 2.8 | 39 - 1.8 |
| wavelength (Å) | 0.864 | 0.864 | 0.864 |
| observed reflections | 33161 | 42956 | 682200 |
| unique reflections | 12672 | 14432 | 65987 |
| completeness (%) | 64.6 | 89.3 | 97.0 |
| Rmerge (%) | 7.0 | 8.4 | 4.7 |
| Rcryst (%) | 19.1 | 17.7 | 19.2 |
| Rfree (%) | 22.7 | 23.3 | 20.9 |
| number of atoms | 5341 | 5096 | 5373 |
| Rms deviation | |||
| bond length (Å) | 0.013 | 0.010 | 0.011 |
| bond angle (°) | 1.60 | 1.55 | 1.62 |
All structures were superimposed using LSQMAN (56) to fit Cα atoms of the refined models for W290G, W290F, W290H, and the azide complex to the wild type structure (1GOF). Solvent contact surface area (Å2 on the van der Waals surface of protein atoms contacted by a sphere of 1.4 Å radius) for galactose oxidase proteins were calculated using AREAIMOL (52), utilizing the Lee and Richards method (57). Hydrogens, water and non-protein atoms, but not the copper ion, were excluded from the calculation. Solvent/protein contact surface representations were produced using PyMOL (DeLano Scientific, San Carlos, CA). This is the surface traced out by the surface of a water atom (van der Waals radius 1.4 Å) in contact with the protein molecule represented by atomic spheres with van der Waals radii.
Generation of oxidized and semi-reduced galactose oxidase
As isolated, galactose oxidase is a mixture of the oxidized and semi-reduced forms (14) so treatment with redox agents is required to obtain homogenous redox states of the enzyme prior to spectroscopic experiments. Fully-activated enzyme samples (Cu(II)-Y•) were prepared by oxidizing galactose oxidase (2 mg) with either a 500-fold excess of potassium ferricyanide (E0′ 424 mV) (58) for wild type and W290F, or a 12 -fold excess of cesium octacyanomolybdate (E0′ 892 mV) (59) (8CNMO) for W290G and W290H variants. 8CNMO was synthesized as previously described (29). The concentration of 8CNMO solutions, prepared immediately before use in double deionized water, was determined using ε390 1339 M−1cm−1. Enzymatically-inactive semi-reduced galactose oxidase (Cu(II)-Y) was prepared by reduction with a 500-fold excess of potassium ferrocyanide. Enzyme samples were in 100 mM potassium phosphate, pH 7.0. The oxidant or reductant was rapidly removed by gel filtration using a 10 mL Bio-Gel P-6DG column equilibrated with either 100 mM sodium phosphate, pH 7.0 or 50 mM sodium acetate, pH 4.5. Typically, the concentration of eluted protein was 15–25 μM. The time from the addition of oxidant or reductant to data collection was consistently between 2 and 3 minutes.
Tyrosyl radical decay
Oxidized galactose oxidase was prepared as described above. Following the removal of excess oxidant by gel filtration, the stability of the tyrosyl radical was measured by the decrease in absorbance at 445 nm (pH 7.0) or 455 nm (pH 4.5). The rate of radical decay was determined using non-linear curve fitting methods (OriginLab, MA) with the following equations: wild type, W290H, and W290G: y = Ao exp(−kt) + A∞ (mono-phasic fit) and W290F: y = A1 exp(−k1t) + A2 exp(−k2t) + A∞ (bi-phasic fit).
Spectroscopy of semi-reduced and oxidized galactose oxidase
Optical absorption spectra were recorded using a HP8453 or a Cary 6000I spectrophotometer. Circular dichroism (CD) spectra were recorded on a Jasco J-710 spectropolarimeter using either a cylindrical cell or a masked 1 ml rectangular quartz cell (path length 1 cm). Baseline adjustments were made by subtracting the buffer CD spectrum. X-band EPR spectra were recorded on a Bruker EMX EPR spectrometer at 10 K using an Oxford cryostat. Semi-reduced enzyme samples (200 μl) in 100 mM potassium phosphate buffer, pH 7.0 in quartz EPR tubes were flash frozen in liquid nitrogen prior to insertion into the cryostat. Spectral baseline correction was applied using WinEPR and the spectra were simulated using SimFonia (Bruker, MA).
Interactions with azide
Oxidized or semi-reduced galactose oxidase was prepared as described above. After recording the optical and CD spectra of the enzyme, sodium azide was added to a final concentration of 5 mM and the optical and CD spectra were recorded immediately. Azide dissociation constants were determined by titration of semi-reduced proteins with aliquots of freshly prepared 50 mM sodium azide in 50 mM potassium phosphate buffer, pH 7.0. Dissociation constants were determined from plots of (Aobs-Ao) versus azide concentration via non-linear curve fitting using a hyperbolic function (OriginLab, MA), where A is the absorbance corresponding to the ligand-to-metal charge-transfer band N3− → Cu (II) occurring in the range 370 nm to 383 nm. Solvent proton uptake by galactose oxidase upon azide binding was determined via thymol blue-mediated titrations of the “as purified” proteins at pH 8.0 (43;44). All solutions were CO2-free. Galactose oxidase (80 μM) and thymol blue (80 μM) in 50 mM NaCl (final volume of 1 mL) were transferred to a 3 mL gas-tight cuvette containing a micro-stir bar (25 °C). Small aliquots (5–10 μL) of 1 mM NaOH were added until the change in absorbance at 600 nm became linear. The uptake of protons was then measured by the change in absorbance at 600 nm after the addition of two aliquots of sodium azide. The concentration of azide added was based on the KD for azide previously determined for each protein ([N3−] added =10 KD).
Results
Biochemical properties of the mutational variant proteins
W290F, W290G, and W290H galactose oxidase were isolated in a pure form as determined by SDS-PAGE. Each W290 variant migrated as a single band with an anomalous electrophoretic mobility (apparent Mr ~ 65 kDa) similar to the mature wild type galactose oxidase, and in contrast to the mobility expected for a protein identical in size to the wild-type protein but lacking the C228–Y272 thioether bond (Mr ~ 68 kDa) (30). Therefore the SDS-PAGE results indicate that the thioether bond is completely formed in the W290 variants. Copper analysis of the wild type and W290 variants confirmed full copper loading: wild type, 1.2 ± 0.08; W290F, 0.96 ± 0.09; W290G, 1.0 ± 0.06 and W290H, 1.3 ± 0.1 mol Cu/mol protein. Wild type and W290F galactose oxidase were oxidized to Cu(II)-Y• by potassium ferricyanide (E0′ 424 mV), whereas oxidation to W290H-Y• and W290G-Y• required cesium octacyanomolybdate (E0′ 892 mV). Potassium ferrocyanide treatment was sufficient to reduce all the W290 variants to the Cu(II)-Y form.
Kinetic Properties
Using D-galactose as substrate, the three W290 variants displayed significantly lower specific activities (by factors of 3.3–720, Table 2) compared to wild type galactose oxidase. Wild type and W290H showed saturation kinetics, with kcat/Km for W290H 1000-fold lower than wild type, whereas the activity of W290F and W290G exhibited an almost linear dependence on substrate concentration and could not be saturated with galactose. Values for kcat and Km could be reliably determined only for wild type and W290H; approximate values were estimated for W290F and W290G obtained using the same data fitting method (Table 2).
Table 2.
Kinetic parameters for wild type and W290 variant forms of galactose oxidases
| Specific activity (U/mg) | Km (mM) | kcat (s−1) | kcat/Km (M−1 s−1) | |
|---|---|---|---|---|
| D-galactose | ||||
| Wild type | 321 ± 6.7 | 82 ± 15.6 | 503 ± 16.2 | 6374 ± 709 |
| W290F | 96.6 ± 4.3 | 2950 ± 473.4 | 371 ± 43.0 | 129 ± 14 |
| W290G | 1.09 ± 0.04 | 1686 ± 457.3 | 1.66 ± 0.28 | 1.0 ± 0.2 |
| W290H | 0.445 ± 0.02 | 45 ± 4.5 | 0.24 ± 0.004 | 5.4 ± 0.3 |
| 2-methylene-1,3-propanediol | ||||
| Wild type | 221 ± 23 | 57 ± 4.1 | 283 ± 7.0 | 4965 ± 377 |
| W290F | 149 ± 6 | 29 ± 3.6 | 166 ± 6.4 | 5800 ± 763 |
| W290G | 0.005 ± 0.001 | ND | ND | ND |
| W290H | No measurable activity | |||
ND, not determined
Activities with 2-methylene-1,3-propanediol (2MP) were also examined. Based on modeling analysis, 2MP is unlikely to hydrogen bond to W290, as predicted for galactose binding (39;46), allowing the impact of this possible interaction to be assessed. Kinetic parameters are shown in Table 2. Note that turnover of 2MP by W290H could not be detected, and that saturation kinetics were observed for only for the wild type enzyme and the W290F variant. It should be noted that the low activities of the W290G and W290H variants may be due, in part, to the difficulty of generating the tyrosyl radical under the direct assay conditions, owing to their elevated redox potentials.
Crystal structures of W290G, W290F, wild type-azide complex with comparison to wild type and W290H
Crystallographic data for W290G, W290F, and the wild type-azide complex are shown in Table 1. All refined well, but the more limited resolution of W290 F, and lower completeness for W290G, render these structures less accurate than the wild type-azide complex, W290H (30), and wild type structures (39). Least-squares superposition of the Cα of W290G, W290F and the azide complex to the wild-type structure (1GOF) give rms deviations of 0.37, 0.25 and 0.33 Å respectively, showing the structures exhibit no significant overall changes in the structures. Notably, all the structures indicate that the thioether bond is fully formed and that copper is present at high occupancy, consistent with the SDS-PAGE and atomic absorption data reported above. As previously reported, the histidine ring in W290H superimposes well on the position of the 5-membered ring of the tryptophan side chain (30). Additionally, the H290 ring nitrogen atom hydrogen bonds to the copper-bound acetate (2.92 Å, Figure 2A), suggesting the imidazole -NεH group would be available for interaction with substrates, as proposed for the stacking tryptophan W290. H290 also occupies an alternative flipped (about χ2) conformation where Nε points away and interacts with a water molecule. This result suggests that H290 displays greater mobility compared to W290. W290F lacks a ring –NH group, which precludes hydrogen bonding to coordinated acetate. Acetate is observed in the W290F structure but interacts with R330 (Figure 2B). The phenyl ring of F290 shields the Y272-C228 radical site about as effectively as the indole ring of W290 and more completely than histidine. The absence of the side chain in the W290G variant permits a molecule of the cryoprotectant glycerol to bind, which partly occupies the position of the tryptophan side chain in the wild type enzyme. The bound glycerol also encroaches on the site of the acetate ion, which has been replaced by a water molecule (Figure 2C). In the W290 variants, the axial tyrosine-to-copper distance is not significantly perturbed compared to the wild type structure (Table 3) (39).
Figure 2.
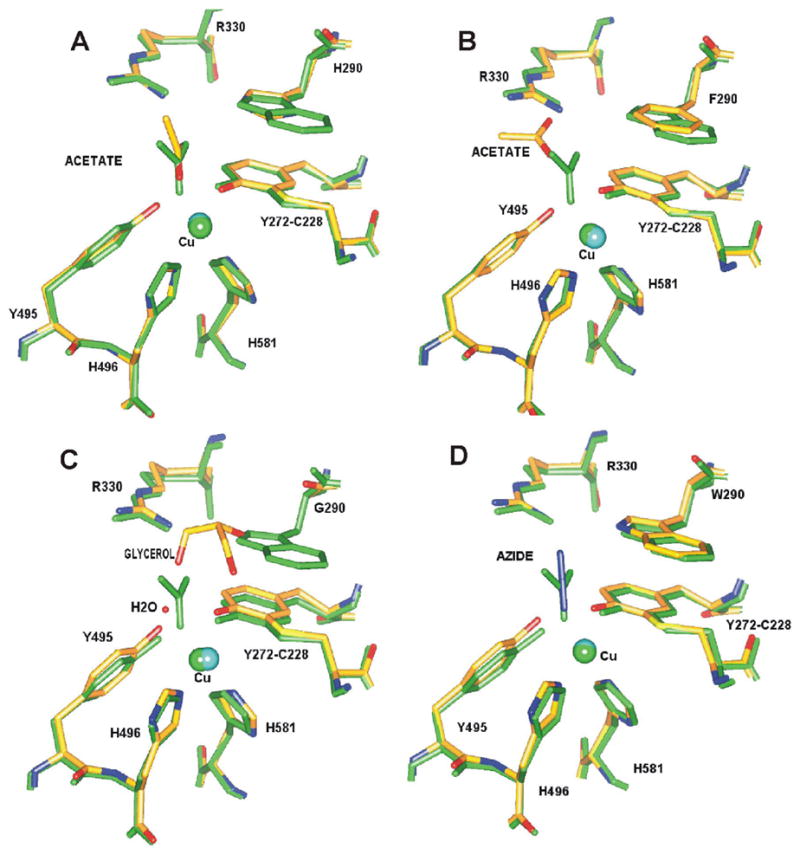
Active site of the W290 variants superimposed with wild type galactose oxidase (A) W290H (B) W290F (C) W290G and (D) wild-type azide complex. The structure of wild-type galactose oxidase is shown in green and that of the variant or azide structure is shown superimposed in atom-based color. The positions of the exogenous ligands in each structure are also shown. The figure was generated using SPOCK (79).
Table 3.
Crystallographic copper-ligand distances in galactose oxidase¶
| Temperature (K) | Y272-Cu (Å) | Y495-Cu (Å) | Exogenous ligand-Cu (Å, ligand) | |
|---|---|---|---|---|
| Wild type, pH 4.5 | 293 | 1. 9 | 2.7 | 2.3, acetate |
| Wild type, pH 7.0 | 293 | 1.9 | 2.6 | 2.8, water |
| W290H | 293 | 1.8 | 2.5 | 2.7, acetate |
| W290F | 100 | 2.1 | 2.7 | 4.1, acetate |
| W290G | 100 | 2.1 | 2.8 | 3.2, water |
| Wild type-azide | 100 | 1.9 | 2.9 | 2.5, azide |
Errors in the coordination distances are difficult to estimate, but are likely to be at least 0.1Å for atoms in Y272 and Y495, and larger for water, azide, and acetate.
Replacement of W290 has implications for the solvent accessibility of the active site. In the wild-type enzyme, the radical site is protected from solvent by the hydrophobic indole side chain, and this is also true for W290F, since the phenyl ring is strongly hydrophobic and almost as large. In W290G, however, the absence of the side chain leaves Y272 exposed to solvent. This can be clearly seen in Figure 3, which shows a view of the solvent-contact surfaces of the wild-type and variants, looking into the active site from the solvent. While solvent accessibility to the Y272-C228 cofactor in wild-type (Figure 3A) and W290F (Figure 3B) are broadly similar, W290G shows a dramatic opening of the cleft above the copper (Figure 3C), allowing solvent direct access to Y272, and this is where glycerol is observed to bind in the structure. H290 is calculated to shield Y272-C228 (Figure 3D) more substantially than anticipated considering the instability of the W290H tyrosyl radical (vide infra).
Figure 3.
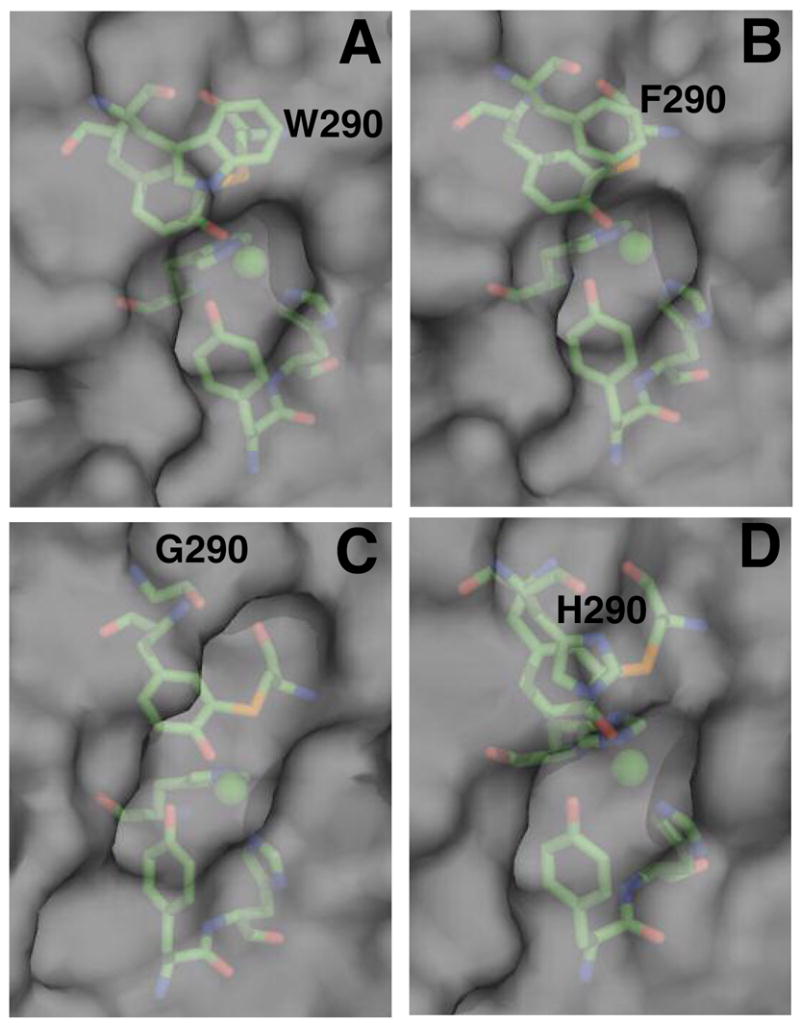
Solvent accessible contact surfaces of (A) wild-type (B) W290F (C) W290G and (D) W290H. The copper ligands are shown with the solvent/protein contact surface represented in gray. Contact surface area for [C228-Y272] in wild type (1GOF.pdb), 1.69 Å2; W290F, 2.02 Å2; W290G, 9.28 Å2; and W290H, 2.42 Å2. The figure was produced using Pymol (DeLano Scientific, San Carlos, CA).
In the wild type-azide complex (Figure 2D), N3− directly coordinates to the copper ion in bent, end-on geometry, with a copper-nitrogen distance of 2.5 Å and angle 121°, well within the range displayed by Cu(II)-N3− complexes of other copper-containing proteins. The terminal nitrogen atom of N3− hydrogen bonds (3.25 Å) to W290-Nε1, which may influence azide binding to the wild type enzyme. Bond distances to the equatorial and axial tyrosines (Table 3) are consistent with an increase of the Cu(II)-Y495 bond in the azide complex, as previously observed (43). Only small variations in the coordination geometry are evident in the various structures, and these are mostly below the significance level at the current resolution.
Stability of the tyrosyl radical
Rates for the Cu(II)-Y• → Cu(II)-Y reaction in the W290 variant proteins varied widely (Table 4). At pH = 7, W290F-Y• had comparable stability (k = 0.2 hr−1) to wild type (k = 0.172 hr−1), whereas W290G decayed more rapidly (k = 2.72 hr−1). The tyrosyl radical in W290H was the least stable, decaying rapidly at pH 7.0 (k = 16.88 hr−1).
Table 4.
Tyrosyl radical decay rates for wild type galactose oxidase and W290 variants
| Decay rate (hr−1) | ||
|---|---|---|
| Phosphate buffer, pH 7.0 | Acetate buffer, pH 4.5 | |
| Wild type | 0.172 ± 0.0017 | 3.95×10−4 ± 1.0×10−4 |
| W290F | 0.20 ± 0.02 | 1.06×10−5 ± 2.7×10−4 |
| W290G | 2.72 ± 0.16 | 0.372 ± 0.13 |
| W290H | 16.88 ± 1.656 | 1.908 ± 0.216 |
While the kinetics of radical decay in wild type, W290G, and W290H were monophasic (Figure 4 and Table 4), biphasic kinetics were observed for W290F (fast phase 2.35 hr−1; slow phase 0.2 hr−1). The decay rate of the second phase for W290F-Y• is comparable to that of wild type, and represents 65 % of the total absorbance change. The chemical nature of the initial fast phase has not yet been identified. Radical lifetimes were extended by rapid buffer exchange from pH 7.0 to pH 4.5. Although the tyrosyl radical in W290G and W290H is stabilized at pH 4.5, the decay is still faster than wild type at pH 7.0. The W290F radical appears to be slightly more stable than wild type galactose oxidase at pH 4.5. It should be noted that in addition to a pH shift, the acidic buffer also contains acetate that can bind to the copper ion.
Figure 4.
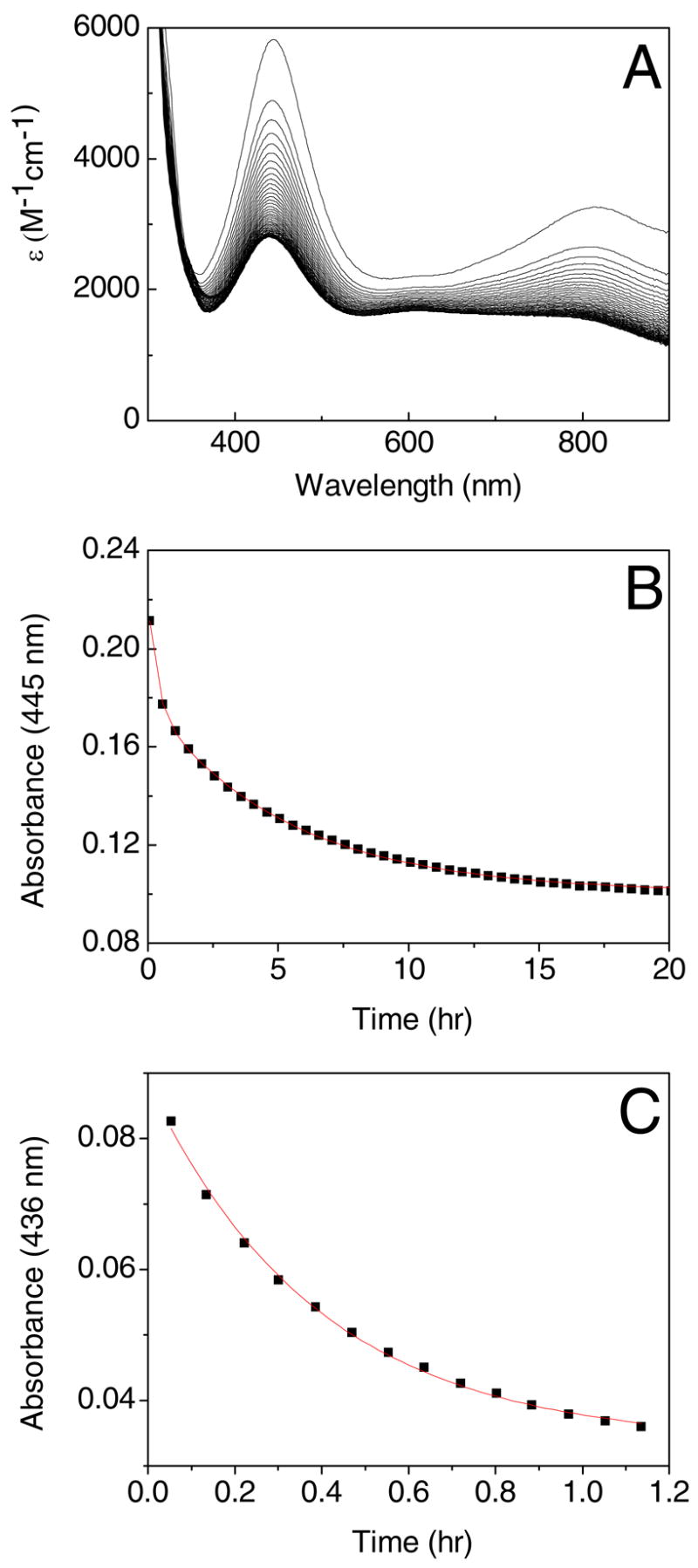
Optical spectra recorded at 30 minute intervals showing (A) the decay of the tyrosyl radical in W290F in 100 mM potassium phosphate, pH 7.0 and (B) the change in A445 versus time. The kinetics of tyrosyl radical decay in W290G in 100 mM potassium phosphate, pH 7.0 is shown in (C).
Spectroscopy of oxidized and semi-reduced W290 variants
As isolated, both the wild type and W290F proteins display thermochromic behavior (color changes from blue-green at 298 K to dusky-pink at 193 K). In contrast, W290G and W290H were purple-pink and orange respectively at both temperatures. Note that ‘as isolated’ wild type and W290F proteins are mixtures of the oxidized and semi-reduced states, whereas the W290G and W290H variants are probably in the semi-reduced state exclusively, owing to their higher tyrosyl redox potentials.
Optical spectra for the oxidized protein states
Wild type Cu(II) -Y• has a distinctive electronic absorption spectrum, with two intense bands in the visible/near-infrared region: a peak at 445 nm attributed to overlapping phenolate → Cu(II) ligand-to-metal charge transfer (LMCT) and Y• π → π* transitions; and an intense low-energy transition at 810 nm, assigned as a ligand-to-ligand (LLCT) charge transfer transition (60), or a π → π* transition of the radical (61). The optical spectra of the oxidized W290 variants are comparable to the wild type enzyme (Figure 5, Table 5). The spectra of W290F-Y• are most similar to that of wild type protein, whereas the energies of the characteristic electronic transitions are perturbed to a greater extent in W290G and W290H. Note that the smaller extinction coefficients of the W290G bands at 437 nm and 825 nm may be attributable, at least in part, to the lower stability of this variant. Given the rapid decay of oxidized W290H, only the band at ~455 nm could be reliably detected; Sykes and coworkers observed the transition at 828 nm in a transient kinetic experiment (45).
Figure 5.
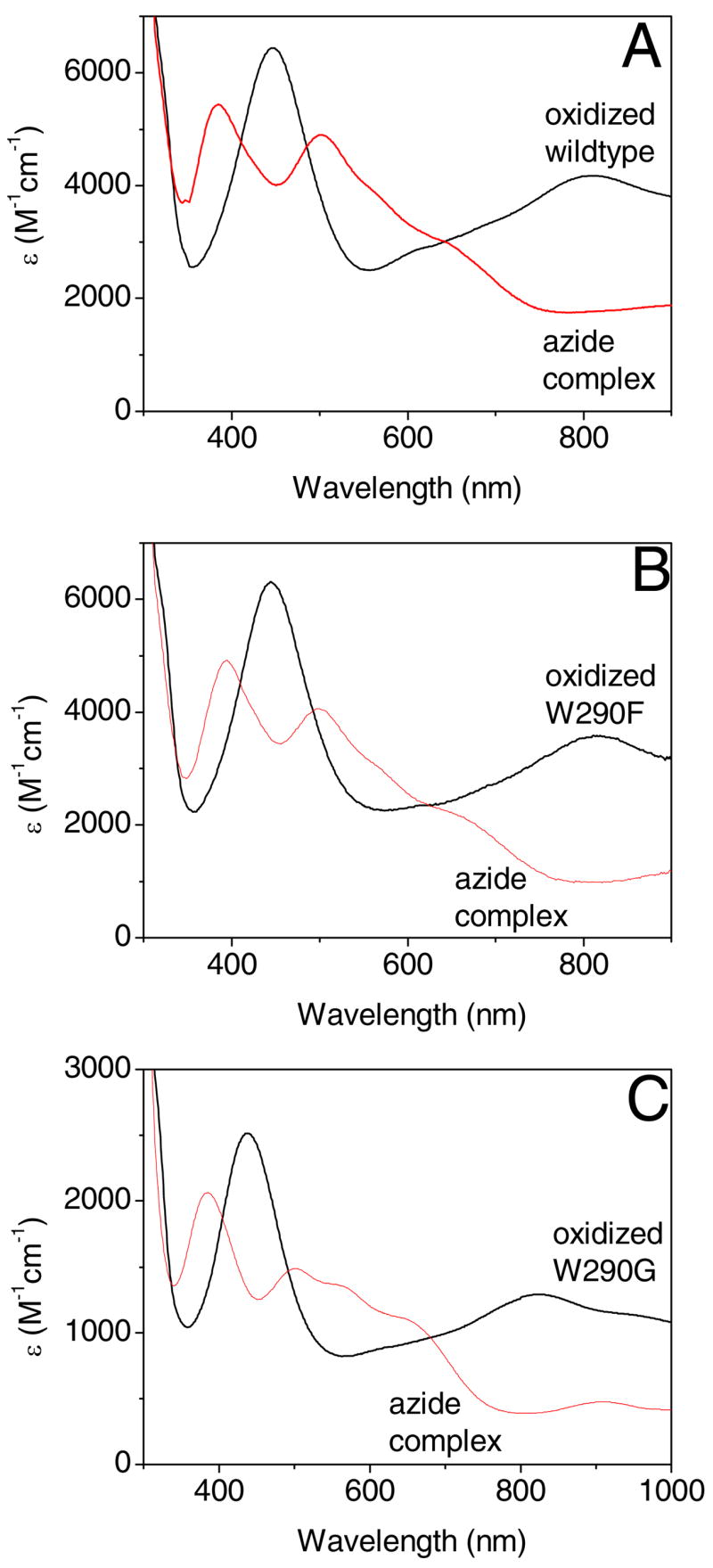
Optical spectra of oxidized galactose oxidase (black) and azide complexes (red) in 100 mM potassium phosphate, pH 7.0 for wild type (A), W290F (B), and W290G galactose oxidase (C).
Table 5.
Comparison of optical and CD spectra of wild type and W290 variants of galactose oxidase
| Wild type | W290F | W290G | W290H | |||||
|---|---|---|---|---|---|---|---|---|
| Optical | λ (nm) | E (M−1cm−1) | λ (nm) | E (M−1cm−1) | λ (nm) | ε (M−1cm−1) | λ (nm) | E (M−1cm−1) |
| Cu(II)-Y• | 445 | 6436 | 445 | 6311 | 437 | 1290 | ||
| 810 | 4133 | 815 | 3582 | 825 | 2515 | |||
| Cu(II)-Y•- N3− | 385 | 5437 | 394 | 4920 | 384 | 2062 | ||
| 500 | 4894 | 500 | 4054 | 500 | 1486 | |||
| 570 | 3993 | 567 | 1348 | |||||
| 650 | 2945 | 665 | 2121 | 660 | 1070 | |||
| 900 | 1782 | 910 | 474 | |||||
| Cu(II)-Y | 441 | 887 | 430 | 1257 | 493 | 2029 | 480 | 1395 |
| 630 | 1112 | 610 | 1425 | 630 | 914 | |||
| Cu(II)-Y- N3− | 380 | 2049 | 385 | 2396 | 370 | 3593 | 370 | 2239 |
| 565 | 736 | 552 | 1035 | 510 | 1940 | 535 | 951 | |
| 775 | 539 | 745 | 352 | 720 | 397 | |||
| CD | λ (nm) | Δε (M−1cm−1) | λ (nm) | Δε (M−1cm−1) | λ (nm) | Δε (M−1cm−1) | λ (nm) | Δε (M−1cm−1) |
|
|
||||||||
| Cu(II)-Y | 314 | 4.79 | 325 | 1.62 | 320 | 1.85 | 318 | 2.23 |
| 365 | −0.14 | 358 | −0.9 | 359 | 0.008 | |||
| 422 | 0.97 | 420 | 1.27 | 399 | 1.47 | 400 | 1.16 | |
| 510 | 0.42 | 515 | 0.42 | 497 | 0.83 | 495 | 0.73 | |
| 625 | −2.89 | 625 | −4.40 | 606 | −3.47 | 600 | −3.03 | |
| 785 | 0.69 | 745 | 1.44 | 752 | 2.12 | |||
| Cu(II)-Y-N3− | 320 | 0.87 | 335 | 0.027 | 355 | 0.006 | ||
| 412 | −1.48 | 404 | −2.06 | 407 | −0.78 | 395 | −1.30 | |
| 555 | −3.32 | 560 | −3.14 | 511 | −1.82 | 530 | −1.67 | |
| 597 | −1.42 | |||||||
| 750 | 1.56 | 740 | 1.24 | 722 | 0.83 | 720 | 1.03 | |
Optical and CD spectra for the semi-reduced protein states
The electronic transitions of the semi-reduced enzymes are generally less intense than those associated with the radical-copper site. Two bands are observed in the absorption spectrum of wild type enzyme at 441 nm (assigned as a phenolate → Cu(II) LMCT transition) and 630 nm (mixed copper ligand-field transition and LMCT) (40;43;62). Spectra of the W290 variants are generally similar (Figure 6) to those of the wild type enzyme, especially W290F. The exception is W290G, with the most prominent ligand-field absorption band observed at ~ 500 nm, reminiscent of the low temperature or anion-bound forms of semi-reduced galactose oxidase (63;64).
Figure 6.
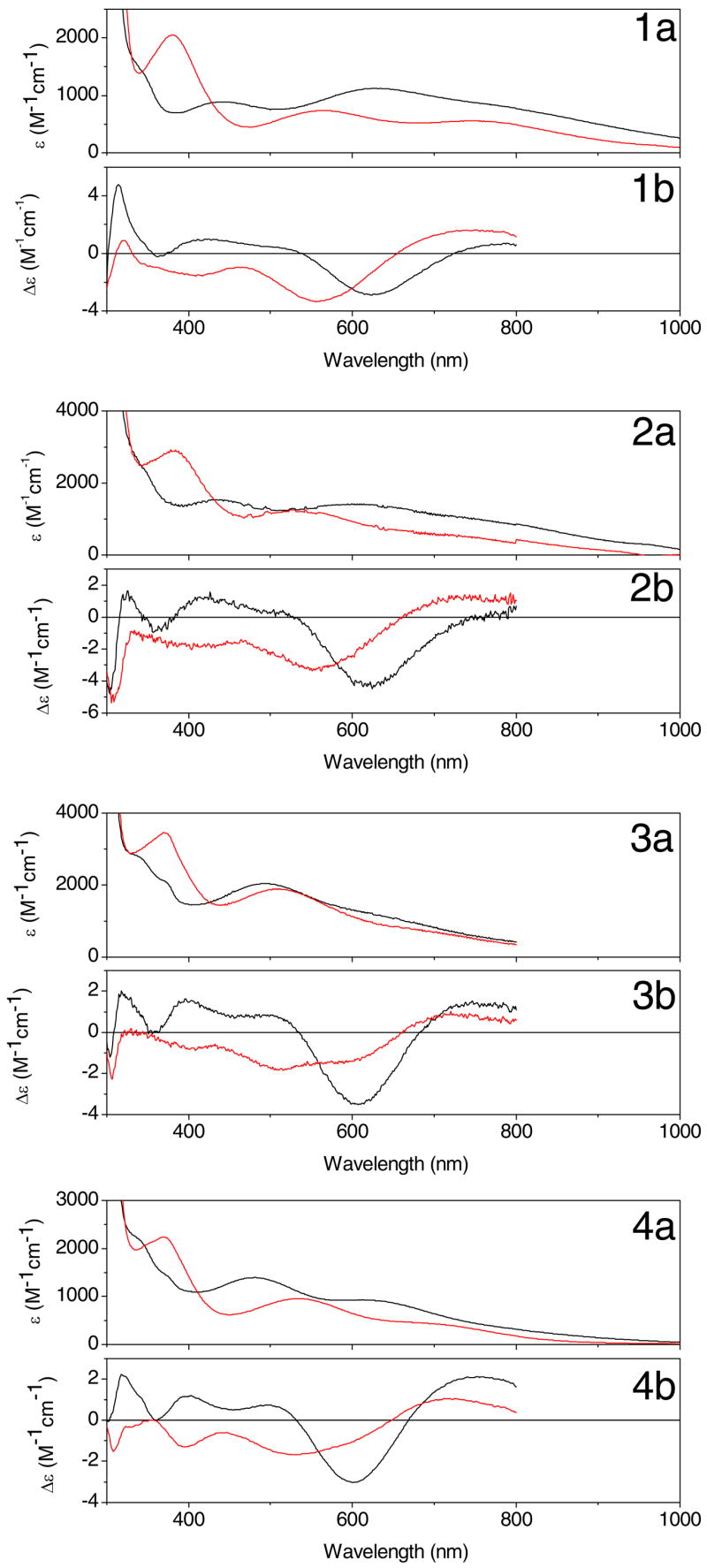
Optical (a) and CD (b) spectra of semi-reduced galactose oxidase (black) and azide complexes (red) in 100 mM potassium phosphate, pH 7.0 for wild type (1), W290F (2), W290G (3), and W290H galactose oxidase (4).
As is frequently the case, the different selection rules for CD spectroscopy permits additional electronic transitions to be resolved (65). Remarkably, the CD spectra are more similar among the proteins than the absorption spectra, indicating that the electronic structure of the Cu(II) center is not grossly perturbed by second coordination shell variations at W290. More specifically, the most prominent CD band at 625 nm has been assigned as a 2B1 → 2B2 (dx2−y2 → dxy) transition, assuming effective C4v symmetry for the Cu(II) complex (43;66). This assignment predicts additional ligand field transitions to both higher and lower energy, which is consistent with the CD spectra. Note the positive CD feature in the semi-reduced spectra at ~ 314–325 nm, which is assigned as an N(π) imidazole → Cu(II) LMCT arising from the histidine ligands.
Inspection of Table 5 shows that the three Cu(II) ligand field transitions (at 510, 625, and 785 nm in wild type) vary in energy by approximately 670 – 790 cm−1 (or 4 – 5 %) among the W290 proteins. Clearly, variation of the second coordination sphere at W290 produces a modest but measurable effect on the Cu(II) electronic structure. The properties of the azide complexes (see below) provide additional evidence for this effect.
EPR spectroscopy
X-band EPR spectra of the semi-reduced W290 variants at 10 K (data not shown) are closely similar, as is evident from the simulated g⊥, g|| and A|| parameters presented in Table 6. The g⊥, g|| and A|| values correspond to an axial type II copper center, in a tetragonal coordination geometry with a dx2−y2 ground state (67). Except for a somewhat decreased A|| seen for W290G, the parameters for the three W290 variants are closely similar to those obtained for the wild-type protein, indicating that the electronic ground state is not significantly affected by variations in the second coordination sphere at W290 at 10 K. Collectively, the electronic and magnetic data are consistent with a common first coordination sphere for Cu(II) in the four proteins and indicate that variations in the second coordination shell at W290 have a measurable, but relatively minor, impact on the electronic structure of the Cu(II) center. Variability in equatorial copper coordination (water, acetate or no ligand) seen in the crystal structures (Figure 2) likely result from the different crystallographic buffer conditions used.
Table 6.
X-band EPR simulation parameters for wild type and W290 variants of galactose oxidase at 10 K
| g|| | g⊥ | A|| (MHz) | ⊥ linewidth (Gauss) | || linewidth (Gauss) | |
|---|---|---|---|---|---|
| Wild type | 2.274 | 2.0570 | 518 | 60 | 50 |
| W290F | 2.270 | 2.0565 | 510 | 58 | 40 |
| W290G | 2.274 | 2.0570 | 488 | 60 | 50 |
| W290H | 2.274 | 2.0574 | 518 | 58 | 50 |
Azide Binding
Azide binding to either oxidized or semi-reduced galactose oxidasegenerates an absorbance band at ~ 380 nm attributed to a N3− → Cu(II) LMCT band (Figures 5 and 6, summarized in Table 5). The energy and intensities of the N3− → Cu(II) LMCT transitions are consistent with equatorial coordination of the anion in all cases, as observed in the crystal structure of the azide complex of the wild type enzyme. Collectively, the shifts in the energy of the N3− → Cu(II) LMCT transition induced by the substitution at W290 are relatively minor, spanning ~ 600 cm−1 (~ 2% shift compared to wild type enzyme) in the oxidized complexes, and ~ 700 cm−1 (~ 3%) in the semi-reduced complexes. Loss of the ~800 nm band in the azide complexes of the oxidized proteins may reflect protonation (and dissociation from copper) of Y495 induced by azide binding (Figure 5), as detailed previously (43). The band at 500 nm may be assigned, in part, as a π → π* radical transition, indicative of the persistence of the tyrosyl radical in the W290 variants upon anion binding (68).
At first inspection, the CD spectra for the semi-reduced azide complexes are quite similar, with perturbations of the Cu(II) ligand-field bands on azide binding of comparable magnitude. However, there is some variation in the energy and intensity of the CD bands (Figure 6 and Table 5). As anticipated, the CD for W290F•N3− is the most similar to that of the wild type complex. The persistence of a weak negative CD feature around 597 nm (Δε = −1.47 M−1cm−1) in the spectrum of W290G•N3− suggests the presence of uncomplexed enzyme, consistent with the lower affinity of the variant for azide (vide infra). The shift of the 625 nm band to higher energy in the wild type enzyme has been interpreted as a change in copper geometry from square pyramidal to tetragonal initiated by the protonation/dissociation of the axial tyrosine ligand by either anion complexation, low pH, or low temperature (64). This shift of the ligand field band to higher energy is also evident in the spectra of the variants.
Cupric–azide dissociation constants (KD) were determined using semi-reduced protein in 50 mM potassium phosphate buffer, pH 7.0. The KD values are: W290H, 0.130 ± 0.006 mM < wild type, 0.146 ± 0.007 mM < W290F, 0.730 ± 0.006 mM < W290G, 1.69 ± .057 mM. Clearly, variations in the second coordination sphere significantly perturb azide binding to Cu(II), with the largest perturbation (W290G) resulting in an order of magnitude decrease in affinity for the azide anion. Azide-binding induces dissociation of the axial tyrosinate (Y495) and proton uptake. The H+-uptake stoichiometries were: wild type protein, 0.86 ± 0.003, in agreement with previous reports (43;44); W290H, 0.57 ± 0.01 > W290F, 0.41 ± 0.009 > W290G, 0.24 ± 0.006 proton/mole. According to the proposed mechanism of proton uptake (63), these data suggest that the pKa of the axial tyrosine (Y495) has been perturbed significantly by side chain variation in the W290 position. This finding concurs with the previous assignment of a pKa of 6.9 to the protonation of Y495 in the W290H variant, compared to a pKa of 7.9 in the wild type enzyme (45).
Discussion
As discussed in the following sections, our data demonstrate that mutation of W290 in the second coordination shell of the copper active-site in galactose oxidase produces substantial changes in some enzyme characteristics (radical stability, kinetic parameters), while only slightly perturbing others (geometry and ground state electronic structure). Structural, spectroscopic, and kinetic analyses establish that the predominant factors are likely to be altered solvent accessibility of the [Cu(II)-Y•] site, although subtle second-shell electronic effects may also contribute to modulating reactivity. An additional effect is that loss of the W290 ring –NH apparently decreases the affinity for D-galactose.
Structural characteristics and active site biochemical properties of the mutational variants
There are no substantial changes in structure between the wild type and W290X variants (Fig. 2), apart from the change in residue at position 290. The crystal structures also established unambiguously that the thioether bond in the [C228-Y272] cofactor is present in all the variants, substantiating the SDS-PAGE analysis (30). A key structural change during processing of the precursor protein is a 6.3 Å movement of the W290-Cα that moves this residue into its stacking location over the thioether bond (69). Our data clearly show that the side chain of W290 is not absolutely required for this structural reorganization. Apparently the indole side chain of W290 is not obligatory for the processing steps leading to thioether bond formation, although further study would be needed to establish whether the rates of these processing steps are affected. Furthermore, the copper analysis results establish that W290 is not essential for metal incorporation. The results from copper analysis were supported by the protein crystallography, which revealed strong electron density at the metal site in all variants. Consequently, altered catalytic properties of the W290 variants compared to wild-type enzyme are not due to sub-stoichiometric copper incorporation or incomplete processing.
However, the resolution of the structures, especially for W290F and W290G, do not allow evaluation of perturbations in the bonding and electronic structure, such as electronic coupling between Cu(II) and the singly-occupied orbital on Y•, which requires a higher degree of accuracy. Spectroscopic data are more informative in this regard.
Tyrosyl radical generation and stability
Previous work (45) has established the redox potential for the [Cu(II)-Y•]/[Cu(II)-Y] couple to be ~ 730 mV for W290H. We observed that a strong oxidant, cesium octacyanomolybdate (E0′ 892mv), is required to generate the activated states of W290G (and W290H) whereas a weaker oxidant, potassium ferricyanide (E0′ 424mv), was sufficient to oxidize the wild-type and W290F proteins. Remarkably, substitution of H or G at W290 has a larger effect on the cofactor tyrosine (Y272) redox potential than does removal of the coordinating copper, which increases the redox potential only to 570 mV (70). Thus it appears that hydrophobic aromatic residues lead to lower redox potentials whereas smaller polar residues or absence of a side chain lead to higher redox potentials. These results indicate that W290 plays a significant role in controlling the redox potential of the free radical site. Model systems (71) had not revealed this effect, which is clearly dependent on the chemical nature of the stacking residue at the 290 position.
The stabilities of the radical site in wild type and W290F are comparable at pH 7, and the decay rates are a factor of ten- to a thousand-fold slower than for W290G and W290H, suggesting that a hydrophobic residue at position 290 increases radical stability and lowers E0′. EPR experiments (14;67;72;73) indicate that spin density is not delocalized over the W290 in the wild type enzyme, but this does not preclude a n electronic stabilizing effect from a stacking aromatic ring (74). It is reasonable to suppose that a radical site exposed to solvent (e.g. the W290G variant) would be less stable than a site protected by a hydrophobic, aromatic “lid”. Solvent contact surface representations show that Y272 in the W290F and W290H variants appears slightly more exposed than in the wild type, and that W290G provides little protection for the radical (Figure 3). The unexpectedly low solvent contact surface area to [C228-Y272] calculated for W290H (2.42 Å2) does not appear to correlate with the highly unstable character of the tyrosyl radical in this variant, which is less stable than the tyrosyl radical in W290G. However, H290 apparently occupies two conformations in the crystal structure, consistent with increased side chain mobility; together with its increased polarity and H-bonding capability these characteristics of H290 may enhance solvent accessibility, which could partly account for the accelerated decay of W290H-Y•. Alternatively, instability of the tyrosyl radical in W290H may reflect enhancement of specific radical decay pathways by the imidazole ring.
It is striking that the stability of the radical site in wild type and W290F increases dramatically (3–4 orders of magnitude) when the pH is lowered from pH 7.0 to pH 4.5 (Table 4). At pH 4.5 the red absorption band (at 810 nm) of oxidized galactose oxidase is lost, possibly resulting from dissociation of Y495 from the copper ion (13;29). Enhanced radical stability at pH 4.5 may result from disruption of an electronic interaction between Y495 and Y272•, mediated by Cu(II), perhaps by abolishing a radical decay pathway via Y495. Notably, the radical in both W290G and W290H also displays increased stability at pH 4.5, but by approximately one order of magnitude. This attenuated difference in stability may reflect partial dissociation of Y495 at neutral pH for the latter variants (vide infra). At pH 4.5 the exogenous copper ligand is acetate from the buffer, and we cannot fully discount the possibility that this may also contribute to the increased stability of the radical at this pH despite the observation that addition of acetate at pH 7.0 did not improve the stability of the radical in the W290H variant.
Sequence alignments suggest that the stacking tryptophan of galactose oxidase may be replaced by histidine in glyoxal oxidase. While the sequence similarity is less than 20 %, the key active site residues are conserved (13). The glyoxal oxidase radical has a redox potential of 0.64 V (galactose oxidase 0.4 V) (41) and is 43-fold less stable than the galactose oxidase radical (13;75). These differences between galactose oxidase and glyoxal oxidase further illustrate the modulating effects of the residue stacked over [C228-Y272•].
Kinetic properties
As immediately evident from Table 2, substitution at W290 has substantial effects on galactose oxidase catalysis. Although the catalytic ability of the W290F variant is reduced, it is clearly the most competent catalyst among the variants, whereas W290G and W290H are severely compromised. Further, the Km for D-galactose in W290F is strikingly elevated (> 1000 mM) compared to wild type (82 mM), implying a role for W290 in binding D-galactose, probably by hydrogen bonding. Indirect evidence for the role of W290 in substrate binding may be inferred by a hydrogen bond between W290 and the acetate copper ligand (2.84 Å from W290-Nε1 to acetate-O, 1GOF.pdb). In contrast, the nearest acetate to the copper ion in the W290F crystal structure is 4.08 Å away, and is hydrogen bonded to R330. A closer correspondence in Km values for 2-methylene-1,3-propanediol as substrate, where such H-bonding to W290 is not possible, supports this inference. M odel building studies (39;46;76) have also predicted a H-bonding interaction between W290 and D-galactose. The value of kcat for W290F (371 s−1) is comparable in magnitude to wild type (503 s−1) reinforcing the conclusion that W290F can be a competent catalyst when the substrate concentration is high.
Although the specific activity of W290H with D-galactose as substrate is very low, the Km (45 mM) is more favorable than wild type. The observation of a H-bond between H290 and coordinated acetate (2.92 Å, similar to that observed for the wild-type enzyme) supports the hypothesis that H-bonding from position 290 is important to D-galactose binding. The low specific activity in W290H may be attributed to the protein being predominantly in the enzymatically inactive semi-reduced state. Although the presence of peroxidase in the coupled assay (see Experimental Section) is sufficient to support oxidization of the wild-type enzyme and W290F, the elevated redox potential of the free radical site in W290H precludes this. Note that the value of Km determined for W290H is an accurate estimate since, unlike kcat, it does not require that the enzyme be fully activated. W290G also displays a low specific activity that may be attributed to the high redox potential required to activate this site.
Spectroscopic studies on the W290 variants
Thermochromic behavior of wild type galactose oxidase has been modeled as an equilibrium involving a proton transfer from Cu(II)-H2O to Y495, leading to dissociation of Y495 from copper at low temperature (63). Investigation of the thermochromic properties of the ‘as isolated’ W290 variants (largely in the semi-reduced state) suggests that the axial ligand Y495 is coordinated to copper in W290F at 298 K whereas Y495 is mostly dissociated from copper in W290G and W290H. Such an outcome is relevant to the interpretation of the spectroscopic results and the proton uptake results discussed below. Note that for crystal structures based on data collected at 100 K, the Cu(II)–O(Y495) distance is anticipated to reflect at least partial dissociation.
The spectroscopic data establish that the perturbations of the electronic structure of the galactose oxidase active site, induced by side chain variation at position 290, are relatively modest, in both the semi-reduced state and the catalytically important oxidized state. The EPR spectra for the semi-reduced forms of the wild type and W290 variants are essentially identical and indicate that there are no major differences in the geometry of the copper centers at 10 K. At this temperature, Y495 is expected to be completely dissociated for all samples, such that the EPR parameters reflect only equatorial bonding and a tetragonal geometry, which must then be essentially identical for the wild type enzyme and the W290 variants.
Interactions with azide
Azide coordination mimics the binding behavior of galactose oxidase substrates (42;43) and thus serves as a model for this reaction. The crystal structure of the azide complex (Figure 2D) establishes that the ligand coordinates in an equatorial position, with the expected bent, end-on geometry, and that Y495 has moved to a longer distance from the copper. The terminal nitrogen atom of the azide anion is hydrogen bonded to W290, further suggesting a role for the residue in substrate binding. Electronic spectra of the azide complexes, for both the semi-reduced and oxidized states, of wild type and W290 variants confirm equatorial azide coordination. In the oxidized complex, absence of the red band at ~810 nm would be consistent with protonation of Y495 and its dissociation from copper.
Despite the close similarities in the electronic structures of the azide complexes of the wild-type enzyme and the W290 variants, Keq of azide binding to Cu(II) in the semi-reduced forms of the proteins is substantially altered by the conservative variations in the second coordination sphere. Once again, the most straightforward interpretation of this effect is that mutation of the residue at position 290 alters properties of the active-site microenvironment, e.g. solvent accessibility and polarity, which perturbs the thermodynamics for azide binding.
Conclusions
The data presented herein demonstrate that, for a series of structurally-characterized derivatives, conservative mutations of a single second-coordination shell residue can produce significant changes in critical aspects of active-site reactivity. Such a result is hardly surprising, but the particular mechanisms that modulate the reactivity of the Cu(II) - radical site in galactose oxidase are interesting and illustrative. Most obviously, the dramatic increase in the Km for D-galactose in W290F is strong evidence that W290 is involved in recognition and binding of some substrates. It can be argued that the relatively similar magnitudes for the Km for D-galactose in wild type and W290H (as compared to the other variants) indicates that hydrogen bonding, at least for D-galactose, between substrates and W290 is important. W290 is one of several residues in the active site of galactose oxidase that can hydrogen bond to sugar substrates and the totality of these hydrogen bonding interactions govern the stereo-specificity of the enzyme (46;49;77;78).
The detailed comparisons among the W290 mutational variants and the wild type enzyme (see also ref. 45) provide compelling evidence that W290 plays an important role in lowering the redox potential from that normally required for generation of tyrosine radical at Y272. Moreover, W290 clearly contributes to the stability of the active-site tyrosine radical; it is likely that both electronic and steric effects associated with the stacking of the indole ring with the cross-linked tyrosine–cysteine cofactor contribute. W290 may also modulate the reactivity of Y495, which has been implicated to function as the active-site base in turnover. This effect may be a consequence of the role of W290 in the active-site microenvironment, and may also be evident in the impact of mutational variations at position 290 on the thermodynamics of azide binding to Cu(II). These findings have potentially important implications for modeling and theoretical studies of the galactose oxidase active site: perturbations of weak interactions modulated by W290 have substantial impacts on reactivity without major perturbations of the electronic structure and bonding of the Cu(II) and its ligands.
Finally, W290F has comparable characteristics to wild type with respect to radical generation and stability, but is much less competent with regard to D-galactose recognition and binding. W290H displays similar D-galactose affinity as the wild type enzyme, while lacking the thermodynamic and kinetic stabilization of the radical. Therefore, the choice of tryptophan at position 290 may best satisfy necessary reactivity constraints. It is not surprising that mutations to W290 are pleiotropic, since the active site of the enzyme is a delicately poised and optimized environment, and changing any one of amino acids in the active site would affect this balance.
Acknowledgments
We thank Mark R. Parsons for assistance with processing the crystallographic data for the W290F variant. We are also grateful for facilities pro vided by the BBSRC funded North of England Structural Biology Centre (NESBIC). NA and SJF thank the BBSRC for studentship awards. KM thanks the Egyptian Government for studentship funding.
Abbreviations and textual footnotes
- Wild type
wild type galactose oxidase
- W290F
W290F galactose oxidase
- W290G
W290G galactose oxidase
- W290H
W290H galactose oxidase
- 2MP
2-methylene-1,3-propanediol
- 8CNMO
cesium octacyanomolybdate
- LMCT
ligand-to-metal-charge transfer
- LLCT
ligand-to-ligand-charge transfer
- ABTS
2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid), diammonium salt
- HRP
horseradish peroxidase
References
- 1.Stubbe JA. Protein radical involvement in biological catalysis. Annu Rev Biochem. 1989;58:257–285. doi: 10.1146/annurev.bi.58.070189.001353. [DOI] [PubMed] [Google Scholar]
- 2.Stubbe J, van der Donk WA. Protein radicals in enzyme catalysis. Chemical Reviews. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 3.Frey PA. Radical mechanisms of enzymatic catalysis. Annu Rev Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 4.Reichard P, Ehrenberg A. Ribonucleotide reductase--a radical enzyme. Science. 1983;221:514–519. doi: 10.1126/science.6306767. [DOI] [PubMed] [Google Scholar]
- 5.Eklund H, Uhlin U, Farnegardh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Biol. 2001;77:177–268. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
- 6.Stubbe J, Nocera DG, Yee CS, Chang MC. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 7.Kolberg M, Logan DT, Bleifuss G, Potsch S, Sjoberg BM, Graslund A, Lubitz W, Lassmann G, Lendzian F. A new tyrosyl radical on Phe208 as ligand to the diiron center in Escherichia coli ribonucleotide reductase, mutant R2-Y122H. Combined x-ray diffraction and EPR/ENDOR studies. J Biol Chem. 2005;280:11233–11246. doi: 10.1074/jbc.M414634200. [DOI] [PubMed] [Google Scholar]
- 8.Sancar A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev. 2003;103:2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 9.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science. 1989;245:738–740. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 10.Knappe J, Wagner AF. Stable glycyl radical from pyruvate formate-lyase and ribonucleotide reductase (III) Adv Protein Chem. 2001;58:277–315. doi: 10.1016/s0065-3233(01)58007-9. [DOI] [PubMed] [Google Scholar]
- 11.Ballinger MD, Reed GH, Frey PA. An organic radical in the lysine 2,3-aminomutase reaction. Biochemistry. 1992;31:949–953. doi: 10.1021/bi00119a001. [DOI] [PubMed] [Google Scholar]
- 12.Tsai A, Kulmacz RJ. Tyrosyl radicals in prostaglandin H synthase-1 and -2. Prostaglandins Other Lipid Mediat. 2000;62:231–254. doi: 10.1016/s0090-6980(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 13.Whittaker MM, Kersten PJ, Nakamura N, Sanders-Loehr J, Schweizer ES, Whittaker JW. Glyoxal oxidase from Phanerochaete chrysosporium is a new radical-copper oxidase. J Biol Chem. 1996;271:681–687. doi: 10.1074/jbc.271.2.681. [DOI] [PubMed] [Google Scholar]
- 14.Whittaker MM, Whittaker JW. A Tyrosine-derived free radical in apogalactose oxidase. J Biol Chem. 1990;265:9610–9613. [PubMed] [Google Scholar]
- 15.Kim SY, Barry BA. The protein environment surrounding tyrosyl radicals D′ and Z′ in photosystem II: A difference Fourier-transform infrared spectroscopic study. Biophys J. 1998;74:2588–2600. doi: 10.1016/S0006-3495(98)77965-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narvaez AJ, Kalman L, LoBrutto R, Allen JP, Williams JC. Influence of the protein environment on the properties of a tyrosyl radical in reaction centers from Rhodobacter sphaeroides. Biochemistry. 2002;41:15253–15258. doi: 10.1021/bi0264566. [DOI] [PubMed] [Google Scholar]
- 17.Barrows TP, Bhaskar B, Poulos TL. Electrostatic control of the tryptophan radical in cytochrome c peroxidase. Biochemistry. 2004;43:8826–8834. doi: 10.1021/bi049531g. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Boada M, Ruiz-Duenas FJ, Pogni R, Basosi R, Choinowski T, Martinez MJ, Piontek K, Martinez AT. Versatile peroxidase oxidation of high redox potential aromatic compounds: site-directed mutagenesis, spectroscopic and crystallographic investigation of three long-range electron transfer pathways. J Mol Biol. 2005;354:385–402. doi: 10.1016/j.jmb.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 19.Gerez C, Elleingand E, Kauppi B, Eklund H, Fontecave M. Reactivity of the tyrosyl radical of Escherichia coli ribonucleotide reductase - Control by the protein. Eur J Biochem. 1997;249:401–407. doi: 10.1111/j.1432-1033.1997.t01-2-00401.x. [DOI] [PubMed] [Google Scholar]
- 20.Ormo M, Regnstrom K, Wang Z, Que L, Jr, Sahlin M, Sjoberg BM. Residues important for radical stability in ribonucleotide reductase from Escherichia coli. J Biol Chem. 1995;270:6570–6576. doi: 10.1074/jbc.270.12.6570. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Zheng H, Groce SL, Lipscomb JD. Basis for specificity in methane monooxygenase and related non-heme iron-containing biological oxidation catalysts. Journal of Molecular Catalysis A. 2006;251:54–65. [Google Scholar]
- 22.Kohzuma T, Inoue T, Yoshizaki F, Sasakawa Y, Onodera K, Nagatomo S, Kitagawa T, Uzawa S, Isobe Y, Sugimura Y, Gotowda M, Kai Y. The structure and unusual pH dependence of plastocyanin from the fern Dryopteris crassirhizoma. The protonation of an active site histidine is hindered by pi-pi interactions. J Biol Chem. 1999;274:11817–11823. doi: 10.1074/jbc.274.17.11817. [DOI] [PubMed] [Google Scholar]
- 23.Chen K, Tilley GJ, Sridhar V, Prasad GS, Stout CD, Armstrong FA, Burgess BK. Alteration of the reduction potential of the [4Fe-4S](2+/+) cluster of Azotobacter vinelandii ferredoxin I. J Biol Chem. 1999;274:36479–36487. doi: 10.1074/jbc.274.51.36479. [DOI] [PubMed] [Google Scholar]
- 24.Xie J, Yikilmaz E, Miller AF, Brunold TC. Second-sphere contributions to substrate-analogue binding in iron(III) superoxide dismutase. J Amer Chem Soc. 2002;124:3769–3774. doi: 10.1021/ja016254h. [DOI] [PubMed] [Google Scholar]
- 25.Zoidakis J, Sam M, Volner A, Han A, Vu K, Abu-Omar MM. Role of the second coordination sphere residue tyrosine 179 in substrate affinity and catalytic activity of phenylalanine hydroxylase. J Biol Inorg Chem. 2004;9:289–296. doi: 10.1007/s00775-004-0527-2. [DOI] [PubMed] [Google Scholar]
- 26.Miranda FF, Kolberg M, Andersson KK, Geraldes CF, Martinez A. The active site residue tyrosine 325 influences iron binding and coupling efficiency in human phenylalanine hydroxylase. J Inorg Biochem. 2005;99:1320–1328. doi: 10.1016/j.jinorgbio.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Tomchick DR, Phan P, Cymborowski M, Minor W, Holman TR. Structural and functional characterization of second-coordination sphere mutants of soybean lipoxygenase-1. Biochemistry. 2001;40:7509–7517. doi: 10.1021/bi002893d. [DOI] [PubMed] [Google Scholar]
- 28.Borovik AS. Bioinspired hydrogen bond motifs in ligand design: the role of noncovalent interactions in metal ion mediated activation of dioxygen. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 29.Rogers MS, Knowles PF, Baron AJ, McPherson MJ, Dooley DM. Characterization of the active site of galactose oxidase and its active site mutational variants Y495F/H/K and W290H by circular dichroism spectroscopy. Inorganica Chimica Acta. 1998;275–276:175–181. [Google Scholar]
- 30.Baron AJ, Stevens C, Wilmot C, Seneviratne KD, Blakeley V, Dooley DM, Phillips SEV, Knowles PF, McPherson MJ. Structure and mechanism of galactose oxidase. The free radical site. J Biol Chem. 1994;269:25095–25105. [PubMed] [Google Scholar]
- 31.Ito N, Phillips SE, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KD, Knowles PF. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 32.Okeley NM, van der Donk WA. Novel cofactors via post-translational modifications of enzyme active sites. Chemistry and Biology. 2000;7:159–171. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 33.Amaral D, Bernstein L, Morse D, Horecker BL. Galactose oxidase of Polyporus circinatus: a copper enzyme. J Biol Chem. 1963;238:2281–2284. [PubMed] [Google Scholar]
- 34.Sun L, Petrounia IP, Yagasaki M, Bandara G, Arnold FH. Expression and stabilization of galactose oxidase in Escherichia coli by directed evolution. Protein Eng. 2001;14:699–704. doi: 10.1093/protein/14.9.699. [DOI] [PubMed] [Google Scholar]
- 35.Kosman DJ. Galactose Oxidase. In: Lontie R, editor. Copper Proteins and Copper Enzymes. CRC Press; Boca Raton, FL: 1984. pp. 1–26. [Google Scholar]
- 36.Avigad G, Amaral D, Asensio C, Horecker BL. The D-galactose oxidase of Polyporus circinatus. J Biol Chem. 1962;237:2736–2743. [PubMed] [Google Scholar]
- 37.Silakowski B, Ehret H, Schairer HU. fbfB, a gene encoding a putative galactose oxidase, is involved in Stigmatella aurantiaca fruiting body formation. J Bacteriol. 1998;180:1241–1247. doi: 10.1128/jb.180.5.1241-1247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leuthner B, Aichinger C, Oehmen E, Koopmann E, Muller O, Muller P, Kahmann R, Bolker M, Schreier PH. A H2O2-producing glyoxal oxidase is required for filamentous growth and pathogenicity in Ustilago maydis. Mol Genet Genomics. 2005;272:639–650. doi: 10.1007/s00438-004-1085-6. [DOI] [PubMed] [Google Scholar]
- 39.Ito N, Phillips SEV, Yadav KDS, Knowles PF. Crystal structure of a free radical enzyme, galactose oxidase. J Mol Biol. 1994;238:794–814. doi: 10.1006/jmbi.1994.1335. [DOI] [PubMed] [Google Scholar]
- 40.Whittaker MM, Whittaker JW. The active site of galactose oxidase. J Biol Chem. 1988;263:6074–6080. [PubMed] [Google Scholar]
- 41.Johnson JM, Halsall HB, Heineman WR. Redox activation of galactose oxidase: thin-layer electrochemical study. Biochemistry. 1985;24:1579–1585. doi: 10.1021/bi00328a001. [DOI] [PubMed] [Google Scholar]
- 42.Knowles PF, Brown RD, Koenig SH, Wang S, Scott RA, McGuirl MA, Brown DE, Dooley DM. Spectroscopic studies of the active site of galactose oxidase. Inorg Chem. 1995;34:3895–3902. [Google Scholar]
- 43.Whittaker MM, Whittaker JW. Ligand interactions with galactose oxidase: Mechanistic insights. Biophys J. 1993;64:762–772. doi: 10.1016/S0006-3495(93)81437-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reynolds MP, Baron AJ, Wilmot CW, Vinecombe E, Stevens C, Phillips SEV, Knowles PF, McPherson MJ. Structure and mechanism of galactose oxidase: Catalytic role of tyrosine 495. J Biol Inorg Chem. 1997;2:327–335. [Google Scholar]
- 45.Saysell CG, Barna T, Borman CD, Baron AJ, McPherson MJ, Sykes AG. Properties of the Trp290His variant of Fusarium NRRL 2903 galactose oxidase: interactions of the GOasesemi state with different buffers, its redox activity and ability to bind azide. JBIC. 1997;2:702–709. [Google Scholar]
- 46.Wachter RM, Branchaud BP. Molecular modeling studies on oxidation of hexopyranoses by galactose oxidase. An active site topology apparently designed to catalyze radical reactions, either concerted or stepwise. J Amer Chem Soc. 1996;118:2782–2789. [Google Scholar]
- 47.Whittaker MM, Duncan WR, Whittaker JW. Synthesis, structure and properties of a model for galactose oxidase. Inorganic Chemistry. 1996;35:382–386. doi: 10.1021/ic951116c. [DOI] [PubMed] [Google Scholar]
- 48.McPherson MJ, Mark R, Parsons Spooner RK, Wilmot CM. Handbook of Metalloproteins. Wiley; Chichester, New York: 2001. Galactose oxidase. [Google Scholar]
- 49.Deacon SE, Mahmoud K, Spooner RK, Firbank SJ, Knowles PF, Phillips SE, McPherson MJ. Enhanced fructose oxidase activity in a galactose oxidase variant. Chembiochem. 2004;5:972–979. doi: 10.1002/cbic.200300810. [DOI] [PubMed] [Google Scholar]
- 50.Kosman DJ, Ettinger MJ, Weiner RE, Massaro EJ. The molecular properties of the copper enzyme galactose oxidase. Arch Biochem Biophys. 1974;165:456–467. doi: 10.1016/0003-9861(74)90271-9. [DOI] [PubMed] [Google Scholar]
- 51.Leslie AGW. Joint CCP4+ESF-EAMCB Newsletter on Protein. Cyrstallography. 1992;26 [Google Scholar]
- 52.Collaborative Computational Project, N. Acta Crystallogr, Sect D: Biol Crytallogr. 1994;50:760–763. [Google Scholar]
- 53.Firbank SJ. PhD Thesis. University of Leeds; 2002. Structural investigation of processing and catalysis in galactose oxidase. [Google Scholar]
- 54.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 55.Vinecombe E. PhD Thesis. Univeristy of Leeds; 1999. X-ray crystallographic studies on copper-containing oxidases. [Google Scholar]
- 56.Kleywegt GJ, Jones TA. CCP4/ESF-EACBM Newsletter on Protein. Crystallogrpahy. 1994;31:9–14. [Google Scholar]
- 57.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 58.Hawkridge FM, Kuwana T. Indirect coulometric titration of biological electron transport components. Anal Chem. 1973;46:1021–1026. doi: 10.1021/ac60329a038. [DOI] [PubMed] [Google Scholar]
- 59.Kolthoff IM, Tomsicek WJ. The oxidation potential of the system potassium molybdocyanide-molybdocyanide and the effect of neutral salts on the potential. J Phys Chem. 1936;40:247–255. [Google Scholar]
- 60.McGlashen ML, Eads DD, Spiro TG, Whittaker JW. Resonance raman spectroscopy of galactose oxidase: a new interpretation based on model compound free radical spectra. J Phys Chem. 1995;99:4918–4922. [Google Scholar]
- 61.Rokhsana D, Dooley DM, Szilagyi RK. Structure of the oxidized active site of galactose oxidase from realistic in silico models. J Am Chem Soc. 2006;128:15550–15551. doi: 10.1021/ja062702f. [DOI] [PubMed] [Google Scholar]
- 62.Whittaker JW. Spectroscopic studies of galactose oxidase. In: Klinman JP, editor. Redox-active Amino Acids in Biology. Academic Press; San Diego, CA: 1995. pp. 262–278. [Google Scholar]
- 63.Whittaker MM, Ekberg CA, Peterson J, Sendova MS, Day EP, Whittaker JW. Spectroscopic and magnetochemical studies on the active site copper complex in galactose oxidase. Journal of Molecular Catalysis B: Enzymatic. 2000;8:3–15. [Google Scholar]
- 64.Whittaker JW. Galactose oxidase. Adv Protein Chem. 2002;60:1–49. doi: 10.1016/s0065-3233(02)60050-6. [DOI] [PubMed] [Google Scholar]
- 65.Johnson MK. Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism. University Science Books; Sausalito: 2000. [Google Scholar]
- 66.Whittaker JW. The free radical-coupled copper active site of glactose oxidase. In: Sigel H, Sigel A, editors. Metalloenzymes involving amino acid residues and related radicals. Marcel Dekker; New York: 1994. [Google Scholar]
- 67.Peisach J, Blumberg WE. Structural implications derived from the analysis of electron paramagnetic resonance spectra of natural and artificial copper proteins. Arch Biochem Biophys. 1974;165:691–708. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 68.Whittaker MM, DeVito VL, Asher SA, Whittaker JW. Resonance Raman evidence for tyrosine involvement in the radical site of galactose oxidase. J Biol Chem. 1989;264:7104–7106. [PubMed] [Google Scholar]
- 69.Firbank SJ, Rogers MS, Wilmot CM, Dooley DM, Halcrow MA, Knowles PF, McPherson MJ, Phillips SEV. Crystal structure of the precursor of galactose oxidase: An unusual self-processing enzyme. Proc Natl Acad Sci USA. 2001;98:12932–12937. doi: 10.1073/pnas.231463798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wright C, Sykes AG. Interconversion of CuI and CuII forms of galactose oxidase: comparison of reduction potentials. J Inorg Biochem. 2001;85:237–243. doi: 10.1016/s0162-0134(01)00214-8. [DOI] [PubMed] [Google Scholar]
- 71.Halcrow MA. Dissecting an enzyme - Model compounds for the galactose oxidase radical site. Heteroatom Chemistry. 2002;13:494–500. [Google Scholar]
- 72.Gerfen GJ, Bellew BF, Griffin RG, Singel DJ, Ekberg CA, Whittaker JW. High-frequency electron paramagnetic resonance spectroscopy of the apogalactose oxidase radical. J Phys Chem. 1996;100:16739–16748. [Google Scholar]
- 73.Babcock GT, El-Deeb MK, Sandusky PO, Whittaker MM, Whittaker JW. Electron paramagnetic resonance and electron nuclear double resonance spectroscopies of the radical site in galactose oxidase and of thioether-substituted phenol model compounds. J Am Chem Soc. 1992;114:3727–3734. [Google Scholar]
- 74.Rogers MS, Dooley DM. Posttranslationally modified tyrosines from galactose oxidase and cytochrome c oxidase. Adv Protein Chem. 2001;58:387–436. doi: 10.1016/s0065-3233(01)58009-2. [DOI] [PubMed] [Google Scholar]
- 75.Whittaker MM, Ballou DP, Whittaker JW. Kinetic isotope effects as probes of the mechanism of galactose oxidase. Biochemistry. 1998;37:8426–8436. doi: 10.1021/bi980328t. [DOI] [PubMed] [Google Scholar]
- 76.Whittaker JW. The radical chemistry of galactose oxidase. Arch Biochem Biophys. 2005;433:227–239. doi: 10.1016/j.abb.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 77.Ito N, Philips SE, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KD, Knowles PF. Three-dimensional structure of galactose oxidase: an enzyme with a built-in secondary cofactor. Faraday Discussions. 1992;93:75–84. doi: 10.1039/fd9929300075. [DOI] [PubMed] [Google Scholar]
- 78.Sun L, Bulter T, Alcalde M, Petrounia IP, Arnold FH. Modification of galactose oxidase to introduce glucose 6-oxidase activity. Chem BioChem. 2002:781–783. doi: 10.1002/1439-7633(20020802)3:8<781::AID-CBIC781>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 79.Christopher JA. The Center for Macromolecular Design. Texas A&M University; College Station, TX: 1998. SPOCK: The structural properties observation and calculation kit. [Google Scholar]