

Abstract
PYK2 is a Ca2+-dependent, nonreceptor protein tyrosine kinase that is involved in the induction of left ventricular hypertrophy (LVH) and its transition to heart failure. We and others have previously investigated PYK2’s function in vitro using cultured neonatal and adult rat ventricular myocytes as model systems. However, the function of PYK2 in the in vivo adult heart remains unclear. Here we evaluate the effect of PYK2 inhibition following myocardial infarction (MI) using adenoviral (Adv) overexpression of the C-terminal domain of PYK2, known as CRNK. First we demonstrate that CRNK functions as a dominant-negative inhibitor of PYK2-dependent signaling, presumably by displacing PYK2 from focal adhesions and costameres. Then, male Sprague-Dawley rats (~300g) underwent permanent left anterior descending coronary artery ligation. One wk post-MI, either Adv-GFP (n=34) or Adv-CRNK (n=28) was administered (1010 pfu, 0.1ml) via catheter-based, Optison®-mediated gene transfer. LV structure and function were evaluated by echocardiography 1 and 3wk after gene transfer, and LV tissue was analyzed by real-time RT-PCR and Western blotting. CRNK overexpression was readily detected by Western blotting 1wk following gene transfer. Adv-CRNK improved overall survival (P=0.03; Logrank Test) and LV fractional shortening (23±2% vs. 31±2% for Adv-GFP vs. Adv-CRNK infected animals, respectively; P<0.05). Whereas MI hearts exhibited increased β-, and decreased α-myosin heavy chain (MHC) mRNA expression characteristic of LVH, Adv-CRNK reversed the MHC isoenzyme switch (3.3±1.4 fold increase in αMHC; 0.4±0.1 fold decrease in βMHC; P<0.05 for both). In summary, CRNK gene transfer improves survival, increases LV function, and alters MHC gene expression suggesting an attenuation of LV remodeling post-MI.
Keywords: signal transduction, heart failure, PYK2
INTRODUCTION
Segmental loss of viable myocardium following myocardial infarction (MI) activates cardiomyocyte signal transduction pathways leading to pathological left ventricular hypertrophy (LVH). Pathological LVH is often associated with hemodynamic overload and decreased systolic and/or diastolic performance, which can ultimately lead to the clinical syndrome of heart failure (HF). The progressive structural and functional alterations in both the infarcted and noninfarcted LV myocardium that occur following irreversible ischemic injury are referred to as post-MI ventricular remodeling (1). Interventions that block the maladaptive cellular signaling leading to ventricular remodeling may also be useful in preventing or attenuating loss of ventricular performance seen in HF.
The Ca2+-dependent, nonreceptor protein tyrosine kinase PYK2 has been previously implicated in cardiomyocyte cell signaling pathways leading to LVH and HF (2–6). PYK2 is a member of the focal adhesion kinase (FAK) family of nonreceptor PTKs. PYK2 coordinates integrin-, Ca2+-, and PKC-dependent signal transduction in a number of tissues (for review, see (7)). PYK2 expression and phosphorylation are regulated by intracellular Ca2+ and protein kinase C (PKC) in cardiomyocytes and vascular smooth muscle cells (8–10). As examined in various cell lines, PYK2 serves as an “activatable” scaffolding protein, and transduces signals from G-protein coupled receptors to the mitogen-activated protein kinases (MAPK) and the phosphoinositol-3-kinase (PI-(3)K)-PDK1-Akt signaling pathway depending upon which adaptor proteins bind to the phosphorylated enzyme (6, 11–13). PYK2 has also been shown to link a variety of stressful stimuli, including Ca2+ overload, UV irradiation, and H2O2 and TNF-α treatment to MAPK and Akt activation in many cell types.
Like FAK, the function of PYK2 is regulated by an endogenously expressed inhibitor, known as PYK2-Related Non-Kinase (PRNK) (14), also known as Cell Adhesion Kinase-β-Related Non-Kinase (CRNK) (15). CRNK consists of the C-terminal portion of PYK2, containing the focal adhesion targeting sequence, but lacking the N-terminal autophosphorylation site and kinase domain. CRNK is structurally analogous to FAK-Related Non-Kinase (FRNK), the autonomously expressed C-terminal domain of FAK. CRNK transcripts are generated by either alternative splicing of the PYK2 hnRNA, or, as is the case of FRNK, by utilization of an intronic promoter (16). CRNK is expressed at relatively high levels in the brain and other organs (14). When ectopically expressed, CRNK can inhibit PYK2 (but not FAK) tyrosine autophosphorylation at Y402, presumably by displacing PYK2 from its cytoskeleton-binding sites (15). Thus CRNK, like its structurally homologous polypeptide FRNK, has been used as a tool to specifically inhibit PYK2-dependent signal transduction (17–20).
In this study we evaluated the effect of PYK2 inhibition using adenoviral (Adv)-mediated overexpression of CRNK in cultured cardiomyocytes and in and in vivo model of post-MI ventricular remodeling. Data are presented to indicate that CRNK prevents PYK2 autophosphorylation and downstream signaling in cultured heart cells. Furthermore, CRNK gene transfer in vivo improves survival, increases LV function, and alters MHC gene expression suggesting an attenuation of LV remodeling post-MI.
METHODS
Reagents
PC-1 tissue culture medium was obtained from BioWhittaker (Walkersville, MD). Dulbecco's Modified Eagle Medium (DMEM) and Medium 199 were obtained from Gibco BRL (Grand Island, NY). Anti-FLAG M2 and anti-sarcomeric α-actinin monoclonal antibodies (mAb) were obtained from Sigma (St. Louis, MO). Rhodamine-conjugated goat anti-mouse IgG and FITC-congugated phalloidin were obtained from Molecular Probes (Eugene, OR). Phosphospecific PYK2-Y402, phospho-specific Akt-S473, and Akt polyclonal Ab (pAb) were purchased from Cell Signaling Technology (Danvers, MA). N-terminal PYK2 pAb was obtained from BioLegend (San Diego, CA). C-terminal PYK2/CRNK mAb was obtained BD Transduction Laboratories (San Jose, CA). Horseradish peroxidase conjugated goat anti-rabbit and goat antimouse IgG were obtained from BioRad (Hercules, CA). Real-time RT-PCR reagents were obtained from GE Healthcare Biosciences (Piscataway, NJ), and Invitrogen (Carlsbad, CA). cDNA primers and probes were obtained from Integrated DNA Technologies, Inc. (Coralville, IA). All other reagents were of the highest grade commercially available and were obtained from Sigma, and Baxter S/P (McGaw Park, IL).
Cell culture
Animals used in these experiments were handled in accordance with the Guiding Principles in the Care and Use of Animals, approved by the Council of the American Physiological Society. Neonatal rat ventricular myocytes (NRVM) were isolated from the hearts of 2-day old Sprague-Dawley rats by collagenase digestion, as previously described (21). Cells were pre-plated for 1h in serum-free PC-1 medium to reduce nonmyocyte contamination. The nonadherent NRVM were then plated at a density of 1600 cells per mm2 onto collagen-coated chamberslides or 60mm dishes, and left undisturbed in a 5% CO2 incubator for 36h. Unattached cells were removed by aspiration, washed twice in HBSS, and the attached cells were maintained in a solution of DMEM/Medium 199 (4:1) containing antibiotic/antimycotic solution. NRVM were infected (24h) with replication-defective adenoviruses (Adv) diluted in DMEM/Medium 199. Medium was then replaced with virus-free DMEM/Medium 199, and the cells stimulated with agonists, or cultured for an additional 24h. Adult rat ventricular myocytes (ARVM) were isolated according to Westfall et al. (22). with modification as described previously (9). Myocytes were then either fixed and permeabilized, or plated in PC-1 medium onto laminincoated (15 µg/ml) chamberslides for 60 min in a 5% CO2 incubator. Unattached myocytes were removed and attached myocytes were maintained in PC-1 medium, and infected with Adv for 24h.
Adenoviral constructs
A replication-deficient Adv encoding FLAG-tagged, human CRNK (Adv-CRNK) was kindly provided by Dr. Andrey Sorokin, Medical College of Wisconsin (18). An Adv expressing GFP (Adv-GFP) was used to control for nonspecific effects of Adv infection (23). An Adv expressing nuclear-encoded β-galactosidase (Adv-neβgal; kindly provided by Dr. MK Rundell, Northwestern University) was used in some in vivo gene transfer experiments to demonstrate the regions of ventricular myocardium transduced by adenoviral infection. The multiplicity of viral infection (moi) was determined by dilution assay in HEK293 cells grown in 96 well clusters.
Immunolocalization
NRVM and ARVM were fixed and permeabilized as previously described (9, 24). Myocytes were stained with anti-FLAG mAb followed by rhodamine-conjugated goat anti-mouse IgG, and counterstained with FITC-phalloidin. Fluorescently-labeled cells were then viewed using a Zeiss LSM 510 laser scanning confocal microscope.
Western blotting
Cultured cells and tissue samples were homogenized in lysis buffer (25), and equal amounts of extracted proteins (50–150µg) were separated by SDS-PAGE and Western blotting on 10% polyacrylamide gels. Primary antibody binding was detected with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibodies, and visualized by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL). Developed films were then scanned on a HP Deskjet 4890 Scanner, and band intensity was quantified using UN-SCAN-IT Gel Software, Ver. 6.1 (Silk Scientific, Orem UT).
Cellular compositon
For the analysis of NRVM hypertrophy, cellular protein and DNA were quantitatively scraped from dishes in 0.2N perchloric acid and collected by centrifugation (10,000g, 10min). The precipitate was re-dissolved by incubation (60°C, 20min) in 0.3N KOH. Aliquots were used for analysis of total protein by the Lowry method with crystalline human serum albumin as standard, and for DNA using 33258 Hoechst dye and salmon sperm DNA as standard (26).
mRNA analysis
Total cellular RNA was isolated from NRVM using the RNeasy Mini Kit (Qiagen, Inc., Valencia, CA) or from LV tissue by the method of Chomczynski and Sacchi (27). RNA was quantified by absorbance at 260 nm and its integrity was determined by examining the 28S and 18S rRNA bands in ethidium bromide-stained agarose gels. SERCA2, ANF, αMHC, and βMHC mRNAs were then analyzed by real-time RT-PCR, as previously described (5, 28). The mixture consisted of 1µL of sample cDNA, 21µL DEPC water, 25µL Platinum Quantitative PCR SuperMix-UDG, and 3µL of a primer/dual-labeled probe combination specific for each gene of interest (Table 1). TaqMan® Rodent GAPDH or 18S rRNA Control Reagents were obtained from Applied Biosystems, (Foster City, CA) and used to normalize mRNA expression levels. Probes were labeled at the 5’ end with 6-FAM and at the 3’ end with BHQ-1. Specific gene primers were 10µM, and probe was 1µM. Rodent GAPDH and 18S rRNA primers were 10µM, and probe was 5µM. PCR amplification was performed by cycling between 95°C (15 sec) and 60°C (60 sec) for 45 cycles, using the 6-FAM fluorophore for quantification. All samples were run in triplicate, and the results were averaged. Data were analyzed as previously described (28).
Table 1.
Primers and Probes Used for Real-Time RT-PCR
| cDNA | Sense Primer | Antisense Primer | Probe |
|---|---|---|---|
| SERCA2 | 5' TCT GTC ATT CGG GAG TGG GG 3' | 5' GCC CAC ACA GCC AAC GAA AG 3' | 5' TGG CCA CTC ATG ACA ACC CG 3' |
| ANF | 5' CTT GCG GTG TGT CAC ACA GC 3' | 5' GGG AGA GGT AAG GCC TCA CT 3' | 5' CCT CCT GGA GCT GCA GCT TC 3' |
| αMHC | 5' ACA GAG TGC TTC GTG CCT GAT 3' | 5' CGA ATT TCG GAG GGT TCT GC 3' | 5' ACA GTC ACC GTC TTG CCG TTT TCA GT 3' |
| βMHC | 5' GCT ACC CAA CCC TAA GGA TGC 3' | 5' TCT GCC TAA GGT GCT GTT TCA 3' | 5' TGT GAA GCC CTG AGA CCT GGA GCC 3' |
Probes were labeled at the 5’ end with 6-FAM and at the 3’ end with BHQ-1.
Coronary artery ligation
Adult male Sprague-Dawley rats weighing ~300g were anesthetized with ketamine (70mg/kg IM) and xylazine (10mg/kg IM), intubated, and placed on a heated operating table. Ligation of the left anterior descending coronary artery was then performed as described by Pfeffer et al. (29). Sham-operated animals underwent surgery without placement of the coronary ligature.
M-mode and 2-D echocardiography
Transthoracic 2-D guided, M-mode echocardiography was performed on anesthetized rats using an Acuson 15L8 Microson High Resolution Transducer and an Acuson Sequoia C256 Echocardiography System (Siemens Medical Solutions USA; Malvern, PA). Images were analyzed in real-time, or saved onto 630mbyte magneto-optical discs for off-line analysis.
Adenoviral gene transfer
A modification of a catheter-based, Optison®-mediated gene transfer technique (30) was used to administer Adv-neβgal, Adv-GFP or Adv-CRNK to the myocardium of normal rats, and rats 1wk after coronary artery ligation. Briefly, animals were anesthetized with ketamine/xylazine, and ventilated with O2 administered via a conical facial mask. A right carotid artery cutdown was used to insert a custom-designed, 2.5F double-lumen, 8 mm long × 4 mm diameter balloon catheter (Tyshak Mini Pediatric Valvuloplasty Catheter; NuMED, Inc., Hopkinton, NY) into the ascending aorta. With the tip of the catheter positioned under echocardiographic guidance exactly 3mm above the aortic valve, the balloon was inflated, thus completely occluding the proximal aorta. Acetylcholine (1.5µg in 100µl saline) was injected to transiently arrest the heart and induce coronary vasodilation, followed immediately by a mixture of Adv (~1010 pfu in 100µl) + Optison® echocontrast reagent (100µl) and then 1 ml of saline flush. The balloon was deflated, and the 15L8 ultrasound transducer was positioned over the heart. The mechanical index of the transducer was raised to 1.9, and the injected micobubbles were disrupted over the next 5min. Finally, the catheter was removed, the carotid artery was tied off and the incision closed, and the animal allowed to recover.
Tissue fixation and β-galactosidase staining
Following induction of general anesthesia, the heart was removed, rinsed in ice-cold Tyrode’s solution and the ascending aorta was cannulated with PE-290 tubing. The coronary circulation was pressure-perfusion flushed (5min, 25°C) with Tyrode’s at 80mm Hg and then fixed with Tyrode’s solution containing 0.2% (v/v) gluteraldehyde (10min, 25°C). Hearts were embedded into OCT Compound (Sakura Finetek, Torrence, CA), frozen to −20°C, and sectioned in the short axis to obtain 14µm thick sections. Sections were stained for β-galactosidase with X-gal (Takara Bio USA, Madison, WI) in a humidified chamber at 37°C for 2h in the dark, according to manufacturer’s instructions. Specimens were washed 3 times with PBS, counterstained with eosin solution (Sigma; St. Louis, MO) for 30 sec, and washed three times with PBS. Images were obtained with an Olympus microscope fitted with a Nikon digital camera at 300x and 600x magnification.
Flow cytometry
Freshly isolated ARVM were fixed in 4% paraformaldehyde and permeabilized with 0.1% saponin. Cells were then stained with anti-sarcomeric α-actinin monoclonal antibody, followed by Alexa Fluor® goat anti-mouse IgG to distinguish the myocyte vs. nonmyocyte population. Flow cytometry was performed using a BD FACSCanto Flow Cytometer running FACSDiva software.
Data analysis
Results were expressed as means±SEM. Normality was assessed using the Kolmogorov-Smirnov test. Data were compared using one-way ANOVA followed by Student-Newman-Keuls test, one-way ANOVA on Ranks followed by Dunn’s test, or unpaired t-test, where appropriate. Differences among means were considered significant at P<0.05. Data were analyzed using SigmaPlot for Windows, Ver. 9.0 (Systat Software, San Jose, CA).
RESULTS
CRNK overexpression and localization in NRVM and ARVM
Initial experiments were conducted in cultured cardiomyocytes to analyze FLAG-tagged, CRNK expression, and to verify its function as an inhibitor of PYK2-dependent signal transduction. NRVM and ARVM were infected (5 or 100moi, respectively; 24h) with Adv-CRNK and the transgene was localized by immunocytochemistry with anti-FLAG monoclonal antibody (1:10) and confocal microscopy. Of note, CRNK expression did not appear to affect cell morphology, nor did it cause cell detachment over this time period. As seen in Figure 1A, CRNK was found in a reticular pattern throughout the cytoplasm of NRVM, but was excluded from the nucleus. In contrast, CRNK was predominantly localized to rib-like structures adjacent to the sarcoplasmic membrane of ARVM, consistent with the appearance of costameres (Figure 1B). The difference in CRNK localization between NRVM and ARVM was reminiscent of the difference in PYK2 localization between neonatal and adult cardiomyocytes observed in our previous studies (5, 9).
Figure 1. Immunolocalization of CRNK in neonatal and adult cardiomyocytes.
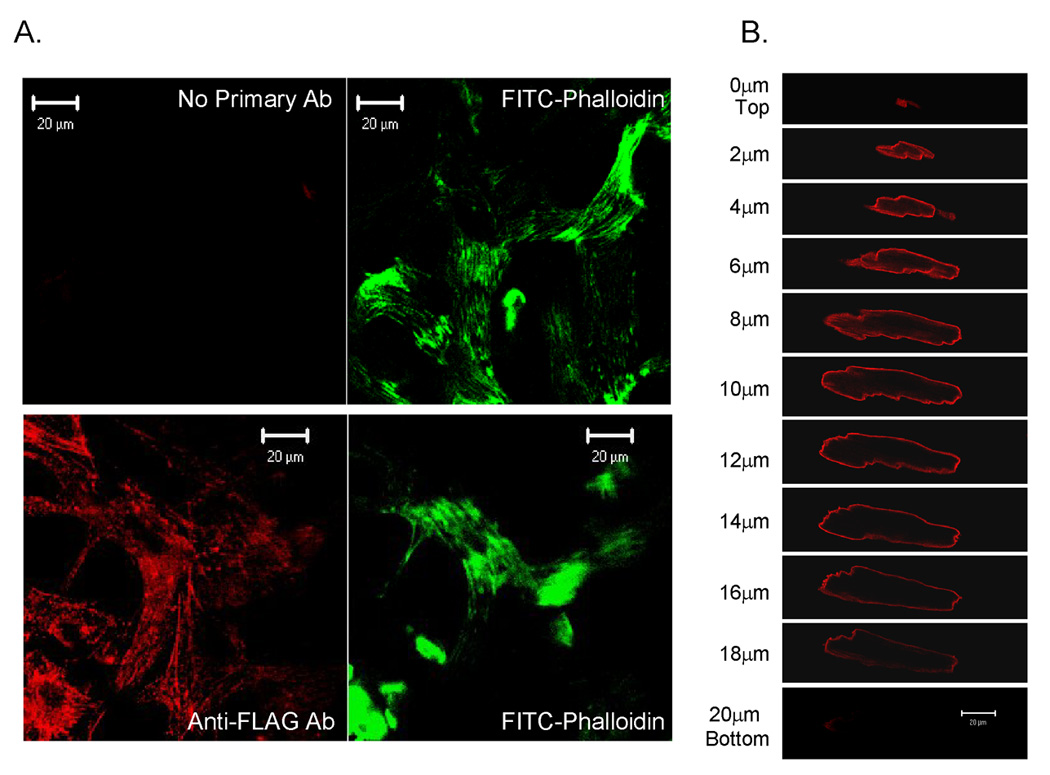
In Panel A, NRVM grown on Permanox® chamberslides were infected with Adv-CRNK (5moi, 24h). Cells were then fixed, permeabilized, stained with anti-FLAG monoclonal antibody (1:10) followed by rhodamine-conjugated goat anti-mouse secondary antibody, and counterstained with FITC-conjugated phalloidin to detect actin filaments. Control chamberslides were handled in an identical fashion, except that the anti-FLAG antibody was omitted (No Primary Ab). Cells were then viewed with a confocal microscope. In Panel B, ARVM were infected (100moi, 24h) with Adv-CRNK. Cells were then fixed, permeabilized and stained with anti-FLAG antibody followed by rhodamine-conjugated goat-antimouse secondary antibody. Cells were then optically sectioned (1µm) using the confocal microscope.
CRNK overexpression inhibits PYK2-dependent signal transduction
We next examined whether CRNK inhibited cardiomyocyte PYK2 activation in response to known activators of the kinase (2, 6, 9, 31, 32). As seen in Figure 2A, NRVM infected with a control adenovirus (Adv-GFP) expressed little endogenous CRNK, but contained readily detectable amounts of PYK2 that was phosphorylated at Y402 under basal conditions. Endothelin-1 (ET; 100nM, 10min) and H202 (100µM, 10min) further increased PYK2 phosphorylation 2–3 fold in cells expressing GFP. In contrast, CRNK overexpression significantly reduced basal PYK2 phosphorylation, and inhibited the increase in PYK2 phosphorylation induced by ET and H2O2. The quantitative analysis of 6–8 Western blotting experiments is depicted in Figure 2B.
Figure 2. CRNK overexpression inhibits basal, and endothelin-and H2O2-induced PYK2 phosphorylation.
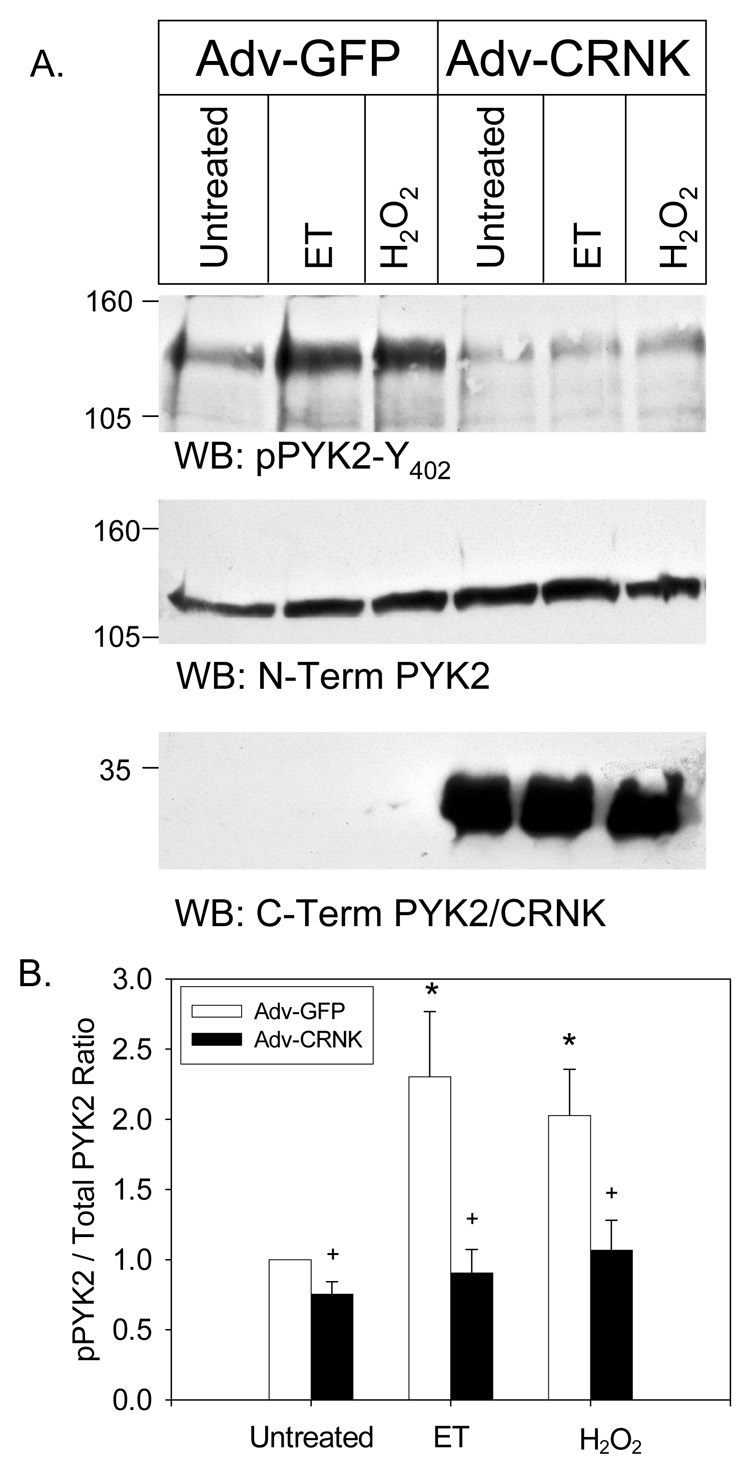
In Panel A, NRVM were infected (5moi, 24h) with either Adv-GFP or Adv-CRNK. Cells were then left untreated, or stimulated (10min) with endothelin-1 (ET, 100nM) or H2O2 (100µM). Equal amounts of extracted cellular protein (50–150µg) were then separated by SDS-PAGE and Western blotting (WB). The position of molecular weight standards is indicated to the left of each blot. In Panel B, the quantitative analysis of 6–8 experiments is depicted. *P<0.05 vs. untreated, Adv-GFP infected cells; +P<0.05 for Adv-GFP vs. Adv-CRNK for each treatment group.
When phosphorylated at Y402, PYK2 serves as a scaffolding protein for the PYK2-and Src-dependent tyrosine phosphorylation of PDK1 in vascular smooth muscle cells (VSMC) (13). PYK2 co-localizes with PDK1 and paxillin in VSMC focal adhesions, and PDK1 phosphorylation is required for downstream activation of Akt in response to angiotensin II (13). A similar PYK2-dependent signaling pathway is required for phenylephrine (PE)-induced activation of Akt in NRVM, although this pathway appears to be tonically inhibited by novel PKCs in cardiomyocytes (6). Therefore, we examined whether CRNK overexpression inhibited ET- or H2O2-induced Akt activation in NRVM. As seen in Figure 3A, Akt was phosphorylated at S473 under basal conditions in NRVM infected with Adv-GFP. H2O2 (but not ET) significantly increased Akt phosphorylation 2–3 fold. In contrast, CRNK overexpression reduced basal Akt phosphorylation, and also reduced Akt phosphorylation in ET-and H2O2-treated NRVM. The quantitative analysis of 6 Western blotting experiments is depicted in Figure 3B.
Figure 3. CRNK overexpression inhibits basal and H2O2-induced Akt phosphorylation.
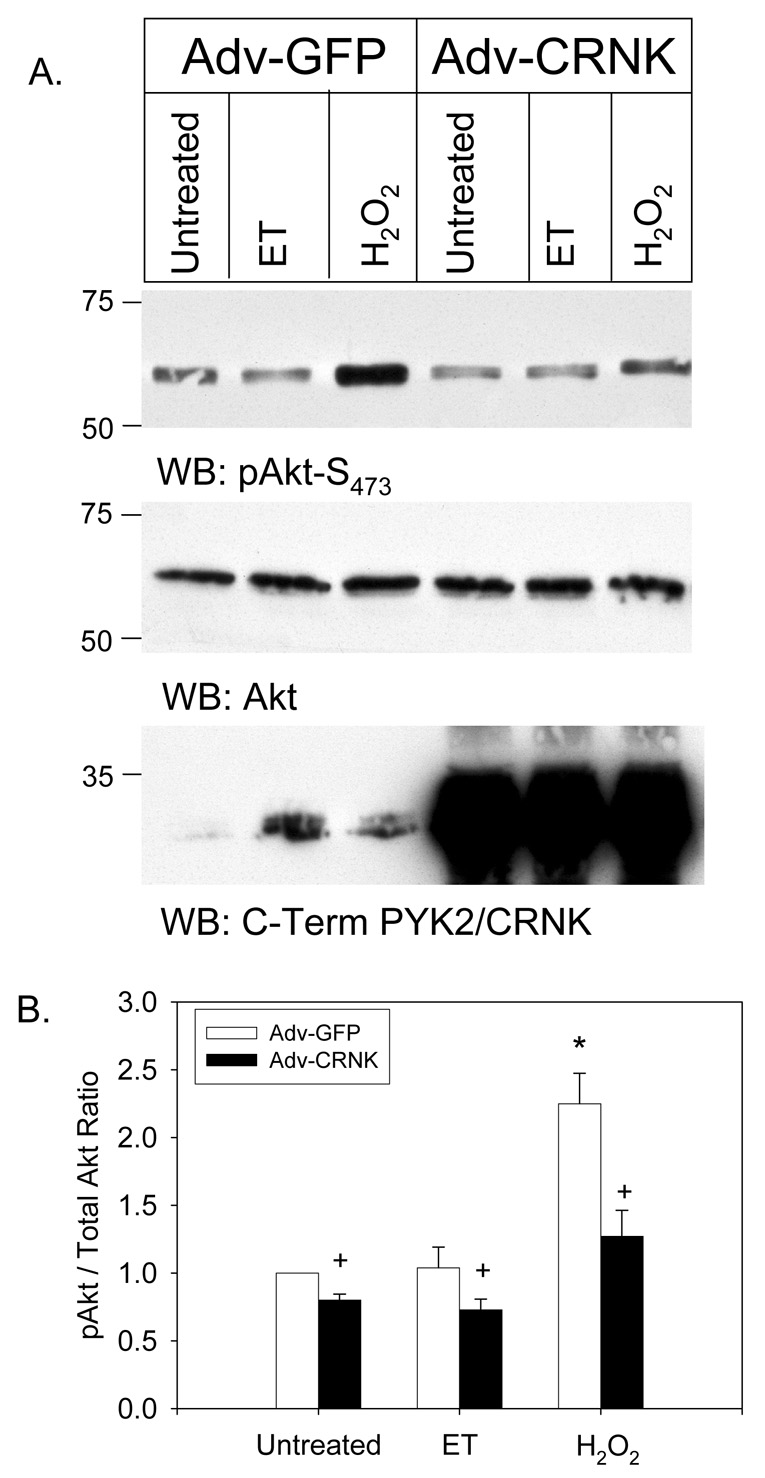
In Panel A, NRVM were infected (5moi, 24h) with either Adv-GFP or Adv-CRNK. Cells were then left untreated, or stimulated (10min) with endothelin-1 (ET, 100nM) or H2O2 (100µM). Equal amounts of extracted cellular protein (50µg) were then separated by SDS-PAGE and Western blotting (WB). The position of molecular weight standards is indicated to the left of each blot. In Panel B, the quantitative analysis of 6 experiments is depicted. *P<0.05 vs. untreated, Adv-GFP infected cells; +P<0.05 for Adv-GFP vs. Adv-CRNK for each treatment group.
CRNK overexpression alters hypertrophic gene expression in NRVM
PYK2 has been implicated in signaling various aspects of pathological hypertrophy in cultured cardiomyocytes and the intact heart, including alterations in gene expression, increases in cell size and protein synthesis, the induction of apoptosis, and the production of reactive oxygen species in response to PE and ET stimulation (2–6, 9, 31–33). Therefore, we tested whether CRNK overexpression prevented NRVM hypertrophy (as measured by total protein/DNA ratio) or altered the hypertrophic gene program in these cultured cells. As seen in Figure 4A, CRNK overexpression (5moi, 48h) had no effect on total protein/DNA ratio as compared to uninfected NRVM, or NRVM infected with Adv-GFP. CRNK also did not prevent the increase in protein/DNA ratio elicited by treatment with phorbol myristate acetate (PMA; 200nM, 48h). However, CRNK overexpression increased SERCA2 mRNA levels, in keeping with PYK2’s role in regulating SERCA2 gene transcription in NRVM (Figure 4B) (5). Furthermore, CRNK significantly reduced ANF mRNA levels, suggesting a role for PYK2 in mediating the PKC and/or Ca2+ dependence of ANF gene transcription in NRVM (33, 34). CRNK overexpression also appeared to affect MHC gene expression, although the observed increase in αMHC mRNA levels did not achieve statistical significance.
Figure 4. CRNK overexpression alters hypertrophic gene expression in NRVM.
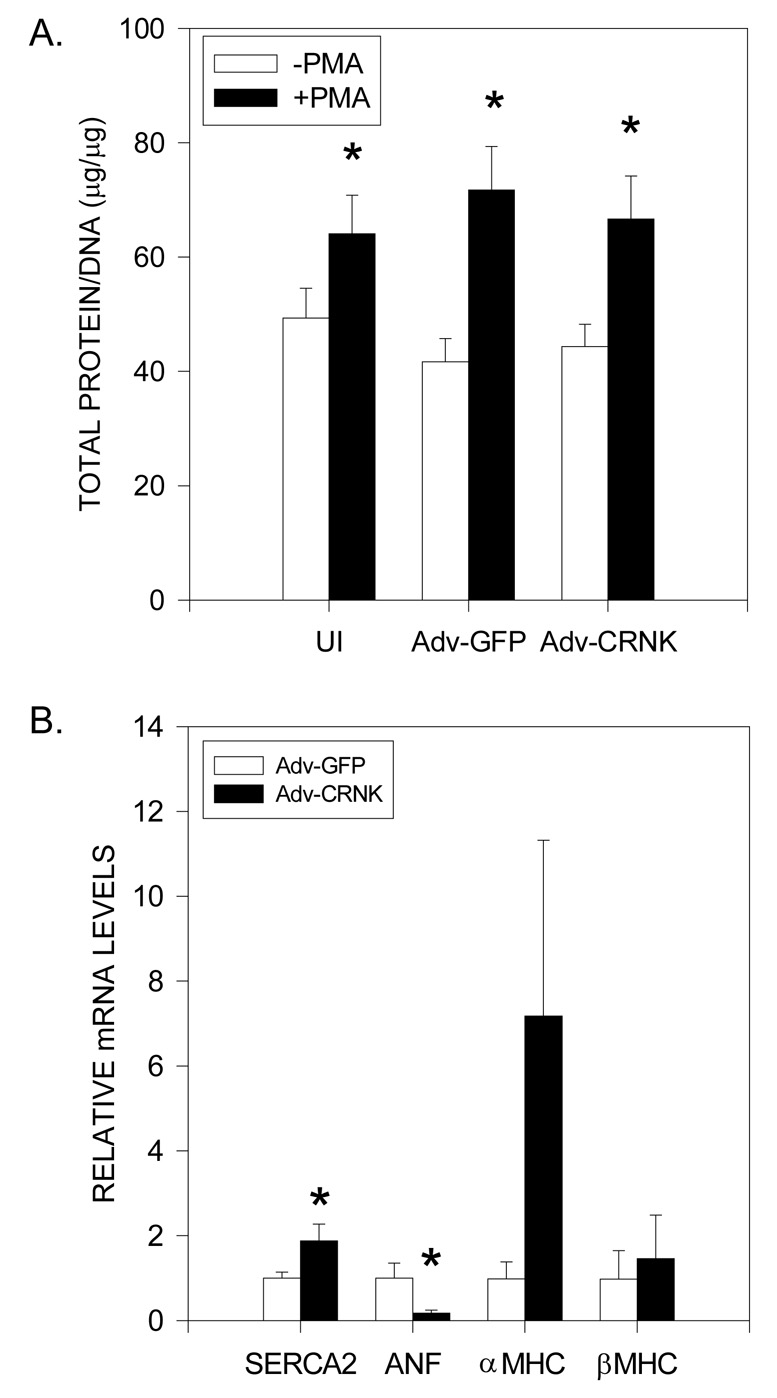
In Panel A, NRVM were maintained in control medium (UI), or infected (5moi, 24h) with either Adv-GFP or Adv-CRNK. Cells were then left untreated, or treated with phorbol myristate acetate (PMA, 200nM) for an additional 24h. Total protein and DNA were analyzed in cell extracts following perchloric acid precipitation. Data are the mean±SEM for 7 experiments. *P<0.05 vs. untreated cells in each group. In Panel B, NRVM were in infected (5moi, 24h) with either Adv-GFP or Adv-CRNK. Cell extracts were then analyzed for SERCA2, ANF, αMHC, and βMHC mRNA levels (relative to GAPDH mRNA) by real-time RT-PCR. *P<0.05 vs. Adv-GFP infected cells for each gene of interest.
Adenoviral gene transfer of GFP and CRNK into normal hearts
Having determined that CRNK overexpression resulted in inhibition of PYK2-dependent signal transduction in cultured cardiomyocytes, we used a modification of a catheter-based, Optison®-mediated gene transfer technique (30) to introduce adenoviral vectors into normal adult ventricular myocardium. First, we used Adv-neβgal to define the regions of myocardium transduced by the Optison-mediated gene transfer technique. As seen in Figure 5, injection of ~1010 pfu of Adv-neβgal into the aortic root of anesthetized Sprague-Dawley rats produced scattered regions of nuclear β-galactosidase expression 3–4 days after gene transfer. Transgene expression occurred throughout the anterior as well as posterior walls of the normal LV. High power images indicated that both cardiomyocytes and vascular endothelial cells expressed the transgene. Then, ~1010 pfu of Adv-GFP (n=16) or Adv-CRNK (n=12) was injected using the identical catheter-based technique. Five days after gene transfer of Adv-GFP, peak fluorescence in the myocyte (α-actinin positive) and non-myocyte (α-actinin negative) cell populations was assessed by flow cytometry, and compared to the background fluorescence of cells isolated from an uninfected heart. Myocytes showed a large shift in peak fluorescence in the cells isolated from the Adv-GFP infected LV myocardium (Figure 6A). Interestingly, we also observed a significant shift in the peak fluorescence in the α-actinin negative, non-myocyte population following Adv-GFP gene transfer (Figure 6B), again indicating that both myocytes and nonmyocytes were infected and expressing the transgene. Phase-contrast and epifluorescent microscopy of freshly isolated cells 5d after gene transfer revealed that ~5–10% of the adult myocytes were expressing GFP (Figure 6C). LV tissue homogenates from 3 Adv-GFP and 3 Adv-CRNK infected hearts were then analyzed for CRNK expression by Western blotting. As seen in Figure 6D, only trace amounts of CRNK were found in LV tissue samples derived from animals receiving Adv-GFP 1wk after gene transfer. However, LV CRNK protein levels were substantially increased in animals receiving Adv-CRNK. Transgene expression was readily detectable by Western blotting in the septum, and LV and RV free walls at both the apex and base of the heart. Furthermore, CRNK overexpression was still detectable up to 2wk after gene transfer, but was undetectable at 3wk (data not shown). Of note, CRNK overexpression did not appear to affect animal survival. In fact, Adv-CRNK modestly increased LV weight/body weight ratio, and also modestly increased fractional shortening as determined by echocardiography 1wk after gene transfer (Figures 6E and 6F).
Figure 5. Adenoviral gene transfer of nuclear-encoded β-galactosidase into normal hearts.
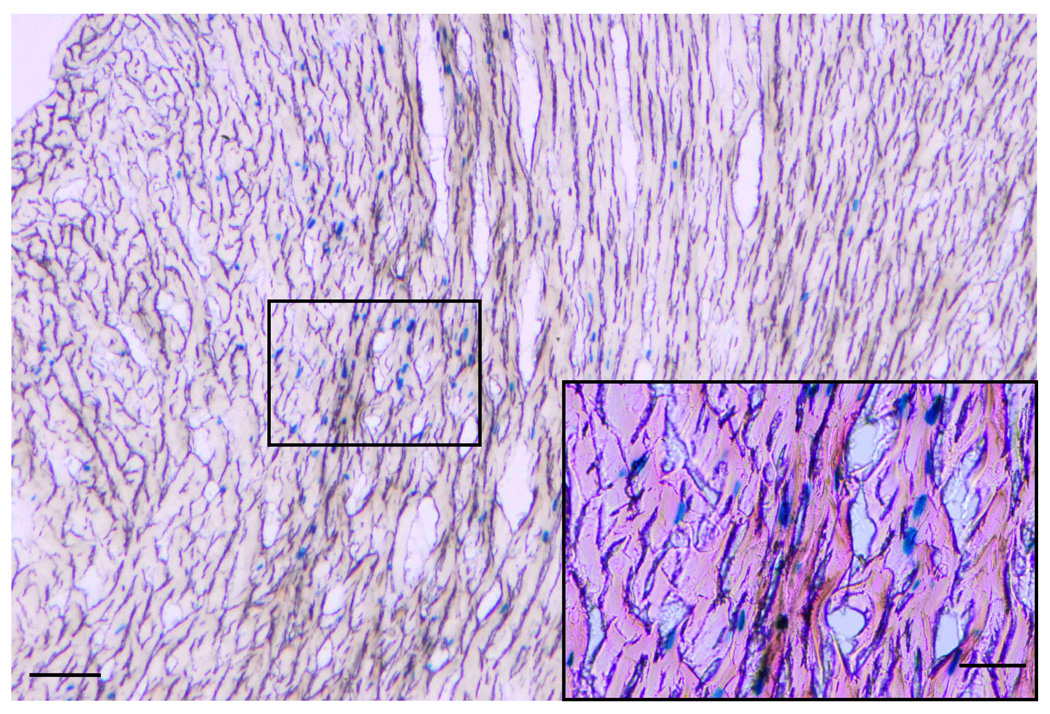
Adv-neβgal (~1010 pfu) was injected by an Optison-mediated gene transfer technique into the aortic root of anesthetized rats in order to define the regions of myocardium transduced by the procedure. Following gluteraldehyde perfusion of whole hearts 3−4 days after gene transfer, tissue sections (14µm thick) were processed to detect β-galactosidase activity by X-gal staining. As is evident from this section of the LV posterior wall, scattered regions of nuclear β-galactosidase expression was detected throughout the full thickness of the myocardium. A high power image (box and insert) indicated that both cardiomyocytes and vascular endothelial cells expressed the transgene. Bar = 25µm.
Figure 6. Adenoviral gene transfer of GFP and CRNK in normal hearts.
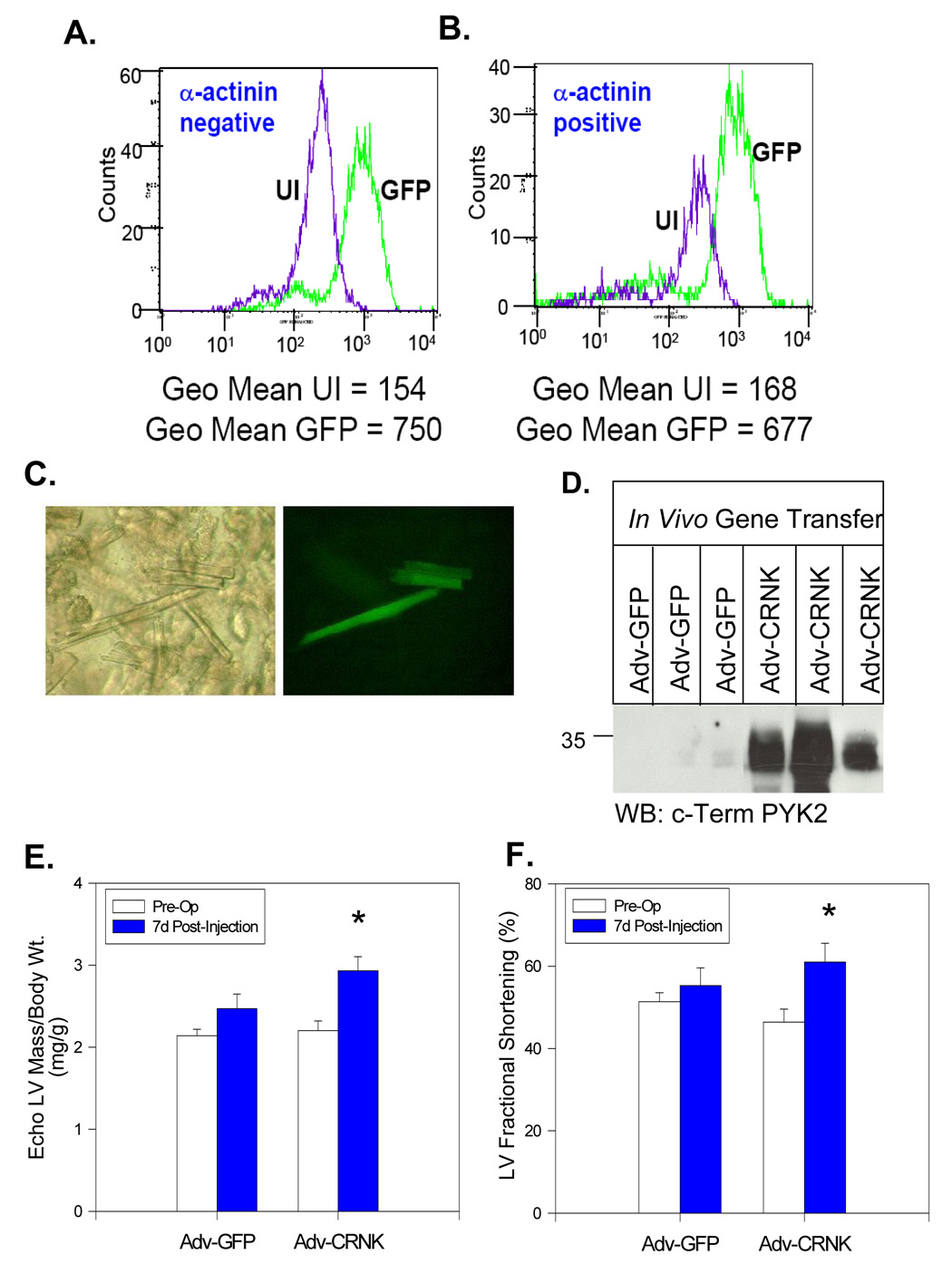
Peak fluorescence of GFP in the myocyte and non-myocyte cell populations was assessed by flow cytometry. in ventricular cells isolated from an uninfected heart, and a GFP-expressing heart 5 days following adenoviral gene transfer. Myocytes (α-actinin postitive cells) showed a large shift in peak GFP fluorescence in the cells isolated from the Adv-GFP infected heart as compared to the cells from the uninfected heart (Panel B). Interestingly, in the non-myocyte population (α-actinin negative), there was also a significant increase in the peak fluorescence in cells isolated from the GFP heart (Panel A). In Panel C, freshly isolated cells were examined by phase contrast microscopy (left) and by epifluorescent microscopy (right). In Panel D, ventricular tissue homogenates derived from the LV apex of 3 rats infected with Adv-GFP and 3 rats infected with Adv-CRNK were compared by SDS-PAGE and Western blotting 1 wk after gene transfer. In Panels E and F, pre-operative and 7-d post-operative echo-derived LV/body weight ratios and LV fractional shortening were compared in animals infected with Adv-GFP (n=16) and animals infected with Adv-CRNK (n=12). *P<0.05 for pre-vs. post-operative measurements for each treatment group.
Alterations in LV structure, function and gene expression during post-infarction LV remodeling
Adult male rats were then subjected to either sham surgery (n=39) or left anterior descending coronary artery ligation (n=49). Approximately 25% of the rats died within the first 24h after sham or infarct surgery. However, after the immediate post-operative period, all of the sham-operated animals survived, whereas 43 of 49 infarct animals survived the entire 5wk observation period. M-mode and 2-D echocardiography was performed at 5d and at 5wk (i.e., immediately prior to euthanasia of the surviving animals). As seen in Figure 7A, representative M-mode and 2-D echocardiographic images demonstrated anterior wall thinning and increased systolic and diastolic LV chamber dimensions as early as 5d after coronary artery ligation. Thereafter, LV end-diastolic dimension progressively increased over time, consistent with post-MI LV remodeling (Figure 7B). Fractional shortening was also significantly reduced at 5d, and remained depressed for the 5wk observation period (Figure 7B). Gene expression analysis conducted using a portion of the non-infarcted LV tissue from a subset of infarct animals (Figure 7C) demonstrated the typical pattern of hypertrophic gene expression associated with pathological LVH. The significant alterations in SERCA2, ANF, αMHC and βMHC mRNA levels (relative to sham-operated controls) were consistent with the progressive increase in echo-derived LV/body weight ratio following coronary artery ligation (Figure 7B).
Figure 7. Alterations in LV structure, function and gene expression during post-myocardial infarction LV remodeling.
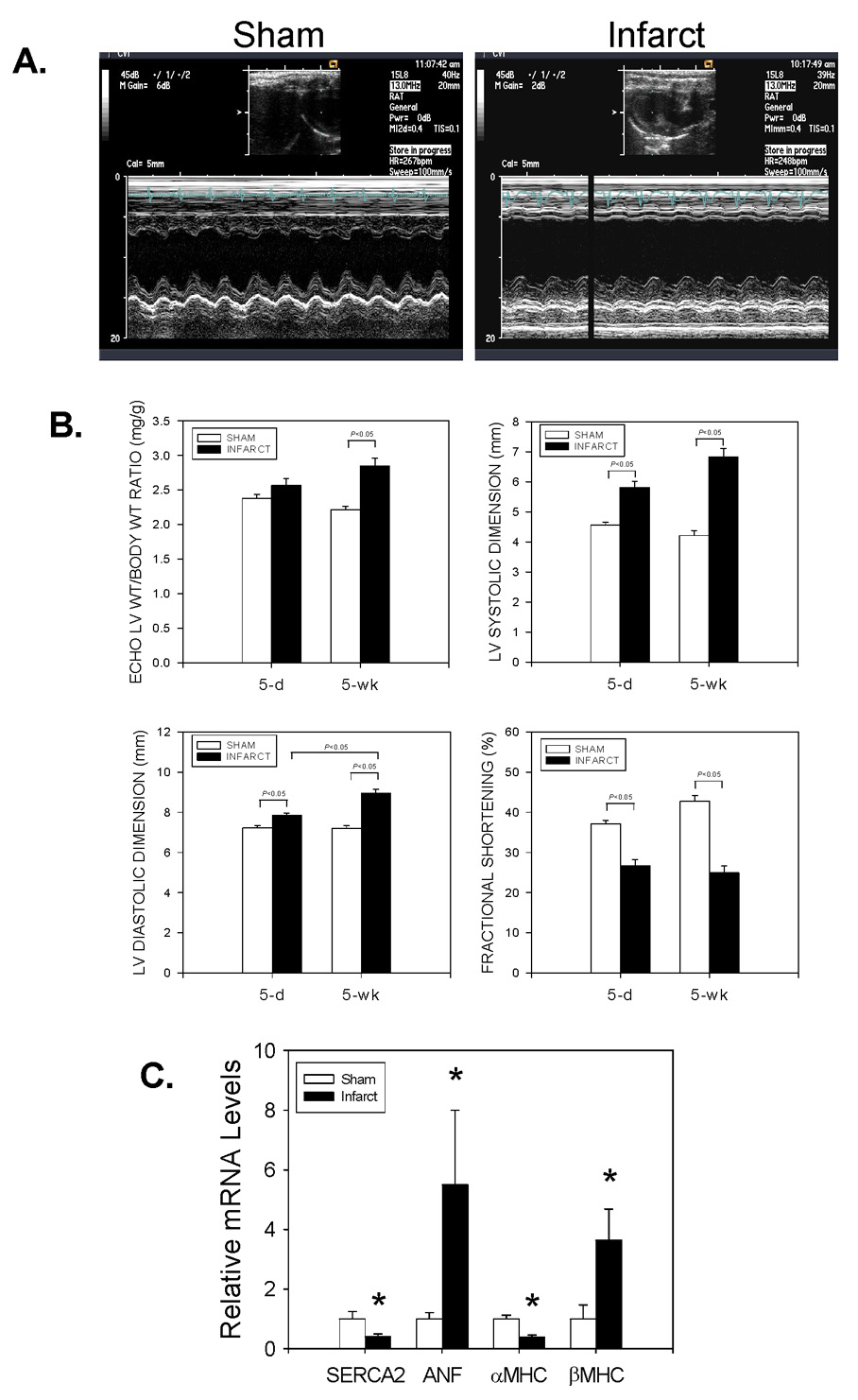
In Panel A, representative M-mode echocardiograms from Sham-operated and Infarct animals 5 days after surgery are depicted. In Panel B, echo-derived LV/body weight ratios, LV systolic dimensions, LV diastolic dimensions, and LV fractional shortening measurements from Sham-operated (n=39) and Infarct (n=49) animals at 5d and 5wk after surgery are depicted. In Panel C, LV tissue extracts from Sham-operated (n=11) and Infarct (n=8) animals 5wks after surgery were analyzed for SERCA2, ANF, αMHC, and βMHC mRNA levels (relative to 18S rRNA) by real-time RT-PCR. *P<0.05 vs. Sham-operated for each gene of interest.
Distribution of transgene following gene transfer into rats with coronary artery ligation
As seen in Figure 8, Optison-mediated gene transfer of Adv-neβgal into rats 3–4 days after coronary artery ligation revealed scattered transgene expression throughout the noninfarted septum and posterior wall, and at the border zone of the anterolateral wall infarct. No transgene was detected within the infarcted tissue; however, rare, nuclear β-galactosidase activity was detected in the rim of viable, epicardial muscle immediately adjacent to the infarct.
Figure 8. Distribution of nuclear-encoded β-galactosidase following gene transfer into rats following coronary artery ligation.
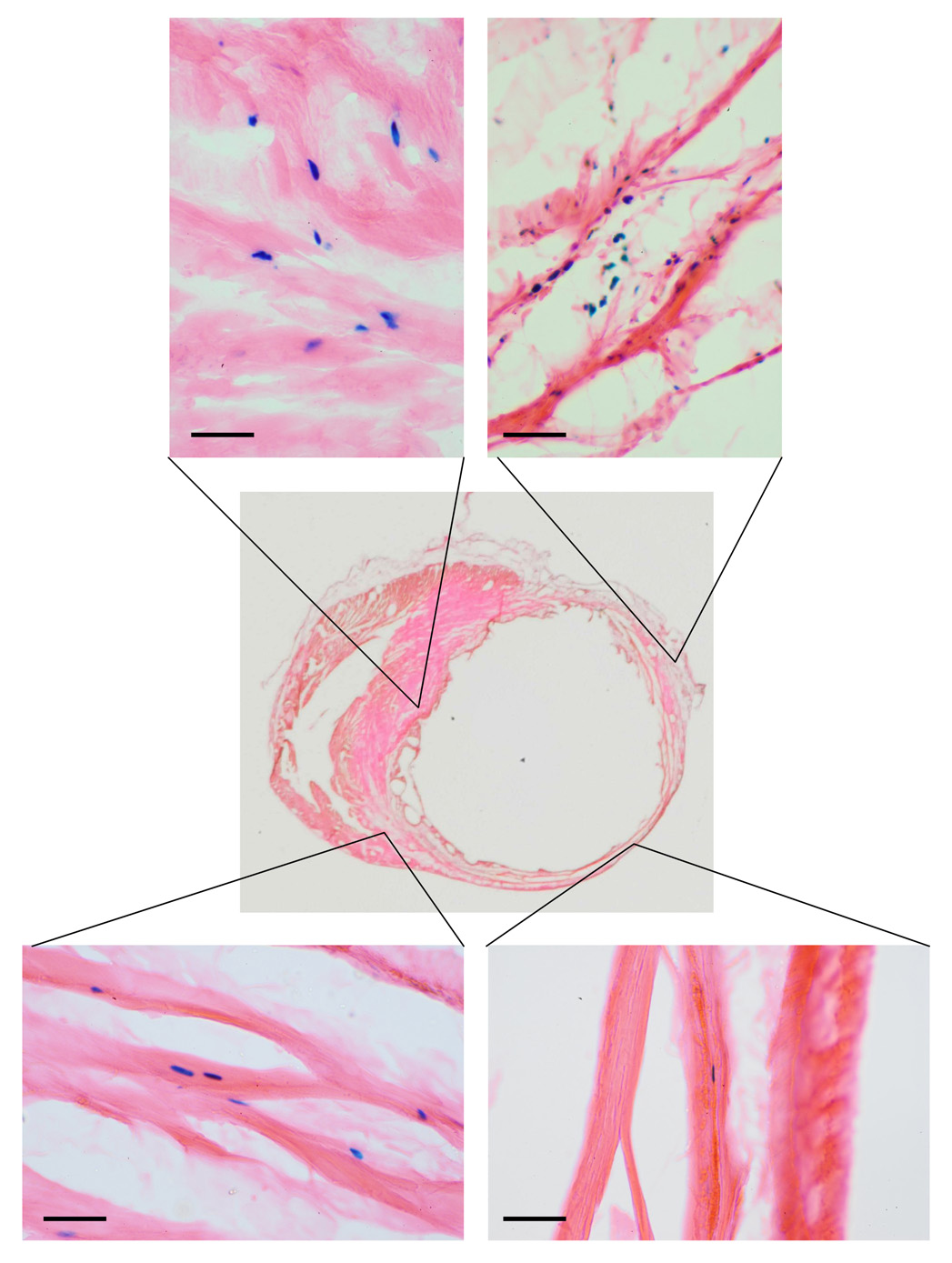
Optison-mediated gene transfer of Adv-neβgal (~1010 pfu) was performed 7d after ligation of the left anterior descending coronary artery. After removing the heart 3d later, perfusion-fixation, tissue sectioning (14µm thick sections), and X-gal staining was performed to identify regions of myocardium transduced by the procedure. As is evident from the figure, X-gal staining revealed scattered nuclear β-galactosidase activity throughout the noninfarted septum and posterior wall, and at the border zone of this large, anterolateral wall infarct. No transgene was detected within the infarcted tissue; however, rare, nuclear β-galactosidase activity was detected in the rim of viable, epicardial muscle immediately adjacent to the infarct. Bar = 25µm.
Adenoviral gene transfer of Adv-GFP and Adv-CRNK after myocardial infarction
As depicted in the gene transfer protocol schematically outlined in Figure 9A, another group of rats was then subjected to coronary artery ligation, and 1wk later, received either Adv-GFP (n=34) or Adv-CRNK (n=28). Surviving animals were then subjected to general anesthesia, echocardiography, and euthanasia at either 2 or 4wk following infarct surgery (i.e., 1 or 3wk after gene transfer). Survival curves for Adv-GFP and Adv-CRNK animals were generated by the method of Kaplan and Meier, and compared using the Logrank test. As seen in Figure 9B, animals receiving Adv-CRNK demonstrated a significant survival advantage over animals receiving Adv-GFP, especially within the first 7d after gene transfer. The improved survival of Adv-CRNK animals was associated with significantly improved LV fractional shortening at both 2 and 4wk after infarct surgery (i.e., 1 and 3wk after gene transfer) (Figure 9C). Differences in function and survival were not due to differences in initial infarct size between the two groups, as LV fractional shortening at the time of gene transfer were similar (28.7±1.9% vs. 28.7±1.5% for Adv-GFP vs. Adv-CRNK; P=0.98). Indeed, fractional shortening in the surviving Adv-GFP infected rats decreased during the first week after gene transfer (i.e., between 1wk and 2wk post-MI), whereas animals receiving Adv-CRNK slightly improved (−6.2±2.9% vs. +2.7±2.6% change in fractional shortening between 1wk and 2wk echocardiograms; P=0.034; Figure 9D). Gene expression analysis revealed that the functional improvement in rats receiving Adv-CRNK was associated with a significant decrease in MHC mRNA levels 1wk after gene transfer (Figure 10A). The reduction in βMHC mRNA persisted, and was also associated with a significant increase in αMHC mRNA levels 3wk after gene transfer as compared to animals receiving Adv-GFP (Figure 10B).
Figure 9. Adenoviral gene transfer of Adv-GFP and Adv-CRNK after myocardial infarction.
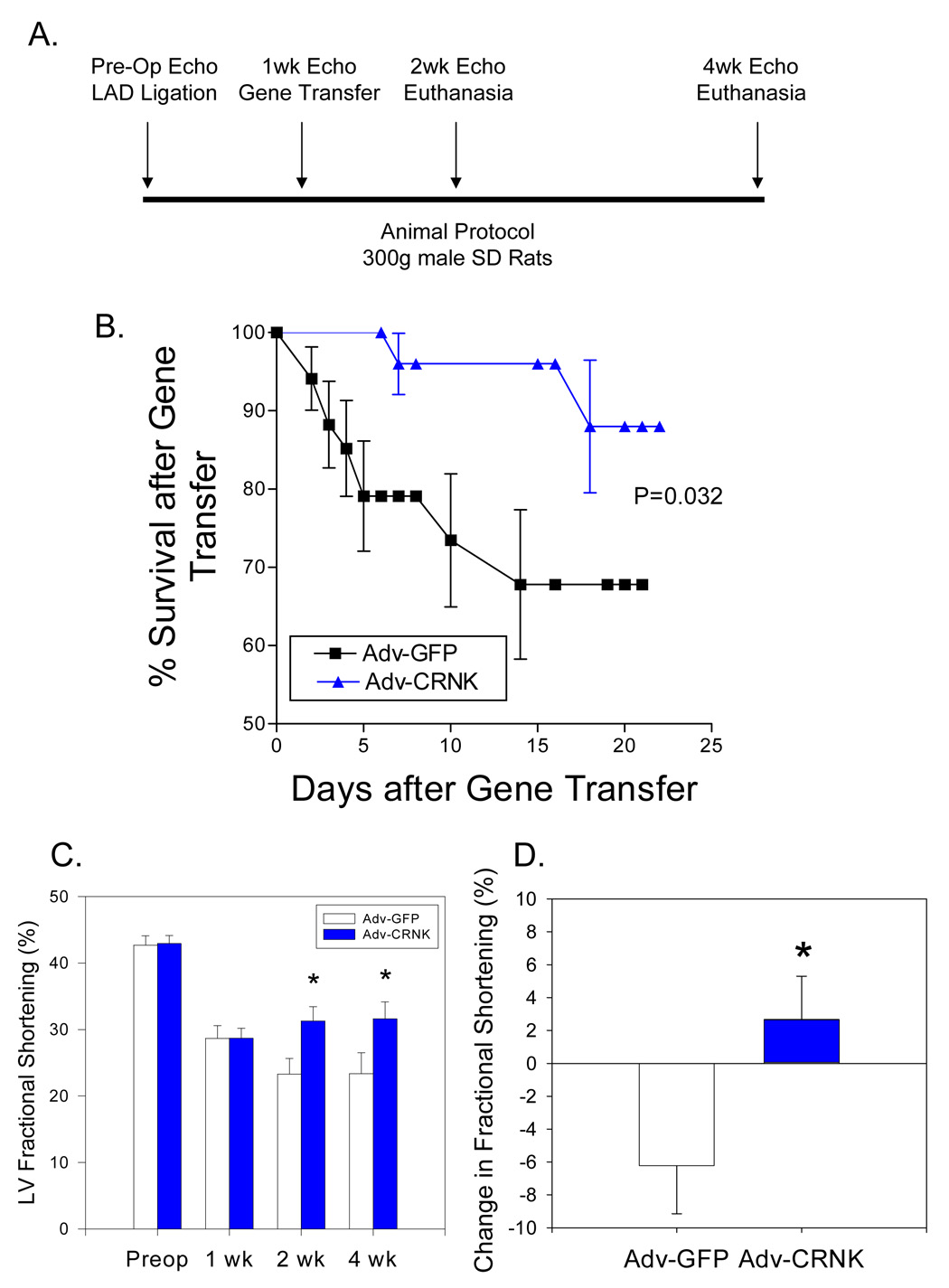
In Panel A, the gene transfer protocol is schematically outlined. Rats were subjected to coronary artery ligation and gene transfer with ~1010 pfu of either Adv-GFP (n=34) or Adv-CRNK (n=28). Surviving animals were then subjected to general anesthesia, echocardiography, and euthanasia at either 2 or 4 wks following infarct surgery (i.e., 1 or 3 wks after gene transfer). In Panel B, survival curves for Adv-GFP and Adv-CRNK animals were generated by the method of Kaplan and Meier, and compared using the log-rank test (P=0.032 for Adv-GFP vs. Adv-CRNK). In Panel C, LV fractional shortening measurements were compared in Adv-GFP vs. Adv-CRNK infected rats each time point in the gene transfer protocol. In Panel D, the change in fractional shortening between the 1wk and 2wk echocardiograms of Adv-GFP and Adv-CRNK infected rats are depicted. *P<0.05 vs. Adv-GFP at each time point examined.
Figure 10. CRNK overexpression alters hypertrophic gene expression during post-MI ventricular remodeling.
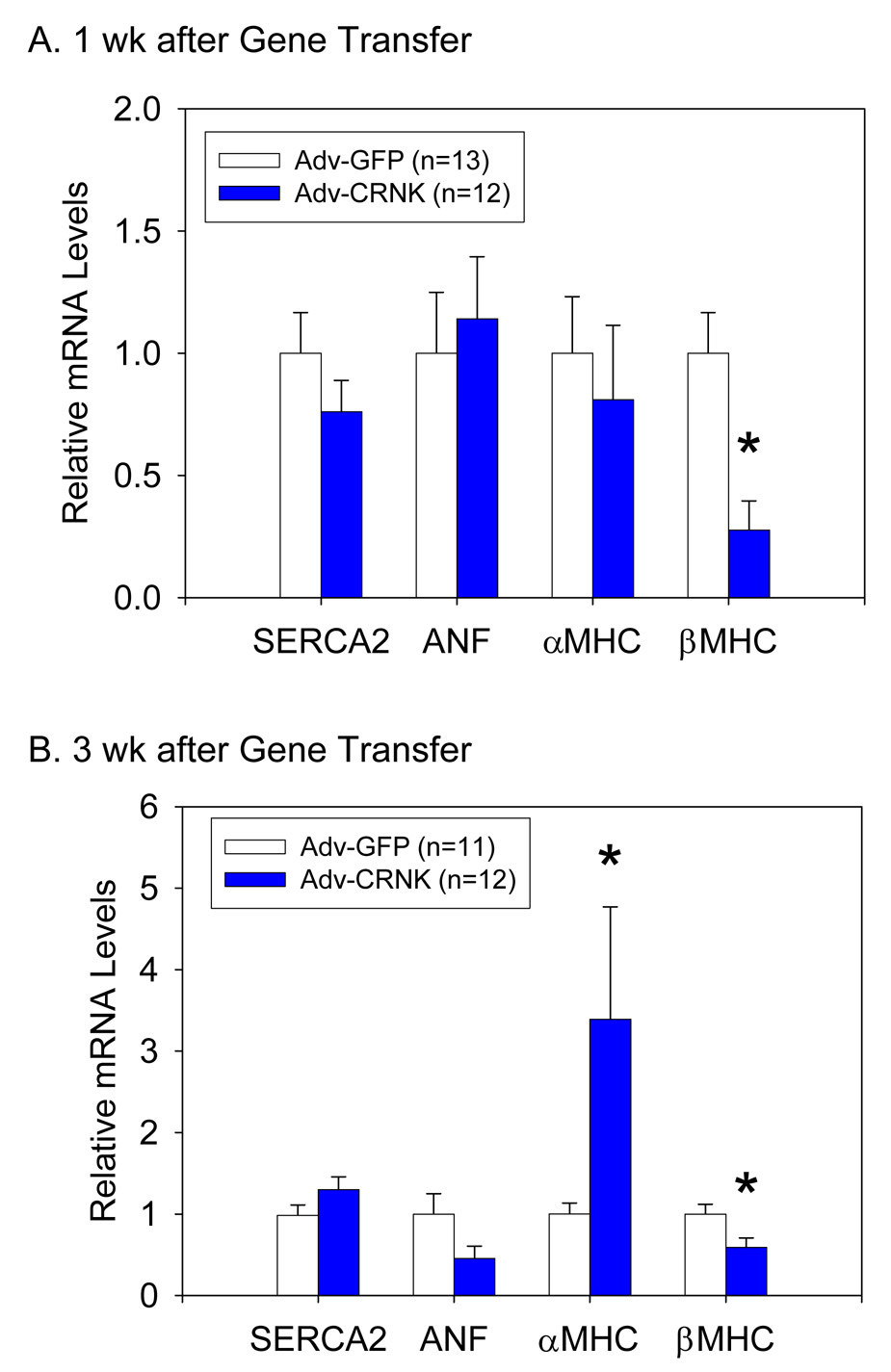
LV tissue extracts were analyzed for SERCA2, ANF, αMHC, and βMHC mRNA levels (relative to 18S rRNA) by real-time RT-PCR 1wk (Panel A) and 3wk (Panel B) after gene transfer with Adv-GFP or Adv-CRNK. *P<0.05 vs. Adv-GFP infected tissue for each gene of interest.
DISCUSSION
In this report, adenoviral gene transfer was used to overexpress CRNK, the C-terminal domain of PYK2, in cultured cardiomyocytes and in the intact heart. CRNK overexpression reduced basal PYK2 phosphorylation, and interfered with PYK2-dependent activation of Akt in response to the pro-apoptotic agonist H2O2. Furthermore, CRNK overexpression improved LV function and reversed some of the gene expression changes associated with post-MI LV remodeling. Thus, the results of these experiments help to define the role of PYK2-dependent signal transduction in mediating some of the adverse effects of PYK2 activation and overexpression that accompanies LVH and HF (2, 4).
Like its homolog FAK, PYK2 contains a C-terminal focal adhesion targeting sequence that directs PYK2 to cardiomyocyte focal adhesions and costameres. Once localized, PYK2 undergoes autophosphorylation at Y402, providing a docking site for Src-family protein tyrosine kinases to bind to, and phosphorylate PYK2 at other sites. Thus, PYK2 functions as a scaffold for the assembly of other signaling kinases and adaptor proteins within cardiomyocyte focal adhesions and costameres. In contrast to FAK, PYK2 appears to direct downstream signals to either pro-or anti-apoptotic signaling cascades depending on which molecules bind to the phosphorylated kinase. For instance, Melendez et al. (3) previously demonstrated that overexpression of wildtype PYK2 promoted cardiomyocyte apoptosis, perhaps via the preferential activation of JNKs (3, 5, 6, 32) and direct activation of the mitochondrial apoptotic machinery (35). PYK2-dependent apoptosis could be suppressed by prior overexpression of paxillin, which binds to the C-terminal domain of PYK2 (14, 36) and prevented JNK activation (3). Similarly, Guo et al. (6) recently demonstrated that PYK2 forms a signaling complex with PDK1 and paxillin, which when formed in the presence of a JNK inhibitor, promoted cell survival via preferential activation of Akt. Our results indicating that CRNK overexpression suppressed H2O2-induced Akt phosphorylation are consistent with their results, as CRNK likely displaced endogenous PYK2 from the complex, and interfered with downstream signaling to Akt. However, CRNK overexpression alone did not appear to induce apoptosis or prevent cardiomyocyte hypertrophy, but rather reversed some of the PYK2-dependent alterations in gene expression observed in pathological LVH and HF (5, 33).
Our results confirm earlier studies (15, 17–20) and indicate that CRNK indeed functions as a dominant-negative inhibitor of PYK2 in cardiomyocytes. CRNK prevented PYK2 autophosphorylation at Y402 in response to both ET and H2O2 treatment of NRVM. CRNK, like its homolog FRNK, appears to mediate its inhibitory effect by preventing PYK2 localization and autophosphorylation in response to a variety of extracellular signals. However, CRNK overexpression does not prevent FAK activation, presumably due to C-terminal sequence differences between FAK and PYK2 (15). Conversely, FRNK overexpression can prevent both FAK and PYK2 activation in a variety of cell types, including cardiomyocytes (23). Thus it is conceivable that some of the reported effects of FRNK overexpression are mediated by inhibition of PYK2, rather than FAK.
A novel aspect of this report was the modification of a previously described, catheter-based gene transfer method (30) to administer a potentially “therapeutic” adenovirus via selective injection into the coronary circulation. We showed that this catheter-based technique caused adenovirally mediated transgene expression in both the cardiomyocyte and nonmuscle cell populations of the rat heart, and induced the robust expression of a potent, polypeptide inhibitor of an important signaling kinase. CRNK overexpression clearly preserved LV function, and provided a significant survival advantage over animals receiving Adv-GFP, especially within the first 7 days after gene transfer. In fact, we believe that the beneficial effects of CRNK overexpression may have been underestimated, as a substantial number of animals receiving Adv-GFP did not survive and were therefore excluded from subsequent analysis of cardiac structure and function.
Nevertheless, the mechanism(s) responsible for this survival advantage remain unclear. Unlike other agents, CRNK overexpression did not appear to prevent the progressive LV dilatation associated with post-MI ventricular remodeling, as LV end-diastolic dimension was not significantly different between animals receiving Adv-GFP or Adv-CRNK at any time point in the gene transfer protocol (data not shown). Although βMHC mRNA levels were significantly reduced within 1 wk of CRNK gene transfer, it is unclear whether this was the effect of, rather than the cause of the improved LV function. Furthermore, the significant increase in αMHC mRNA was observed at time when transgene expression was undetectable (i.e., 3 wks after gene transfer). As CRNK was overexpressed long after the onset of myocardial necrosis, it seems unlikely that the transgene prevented further ischemic injury by augmenting signaling to cardiomyocyte cell survival pathways (6). As indicated above, CRNK overexpression altered the hypertrophic gene program in the hemodynamically overloaded, noninfarcted LV myocardium. This hemodynamic overload likely stimulated integrin-dependent signal transduction at costameric sites within the cardiomyocyte population of the heart (37). However, it is also conceivable that the inhibitor blocked some of the adverse effects of the gene transfer technique that occurred in the coronary endothelium. Van Buul et al. (20) have shown that CRNK overexpression prevented the PYK2-dependent, tyrosine phosphorylation of vascular endothelial (VE)-cadherin in response to the addition of a VE-cadherin-blocking blocking antibody (cl75) added to the medium of cultured human vascular endothelial cells. CRNK also markedly attenuated the increase in endothelial permeability induced by cl75. As increased vascular permeability is required for efficient adenoviral gene transfer (38), CRNK overexpression in coronary microvascular endothelial cells may have promoted the restoration of barrier function, and improved coronary perfusion during the post-gene transfer period. It is also conceivable that the gene transfer technique itself induced cardiomyocyte apoptosis, and CRNK (but not GFP) reduced the loss of muscle cells, thus accounting for its beneficial effects on fractional shortening and survival. However, neither Beeri et al. (30) nor Bekeredjian et al. (39) reported any significant structural or functional effects of the microbubble-ultrasound technique in delivering plasmids or adenoviruses via coronary artery injection, other than an increase in myocardial stiffness. Additional studies will be necessary, however, to determine the primary site of action of CRNK, and to ascertain the mechanisms responsible for PYK2-dependent eterioration of LV performance post MI.
ACKNOWLEDGMENTS
These studies were supported by NIH RO1 grant HL63711, and a grant to the Cardiovascular Institute from the Ralph and Marian Falk Trust for Medical Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling-concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 2.Bayer AL, Heidkamp MC, Patel N, Porter MJ, Engman SJ, Samarel AM. PYK2 expression and phosphorylation increases in pressure overload-induced left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2002;283:H695–H706. doi: 10.1152/ajpheart.00021.2002. [DOI] [PubMed] [Google Scholar]
- 3.Melendez J, Turner C, Avraham H, Steinberg SF, Schaefer E, Sussman MA. Cardiomyocyte apoptosis triggered by RAFTK/pyk2 via Src kinase is antagonized by paxillin. J Biol Chem. 2004;279:53516–53523. doi: 10.1074/jbc.M408475200. [DOI] [PubMed] [Google Scholar]
- 4.Melendez J, Welch S, Schaefer E, Moravec CS, Avraham S, Avraham H, Sussman MA. Activation of pyk2/related focal adhesion tyrosine kinase and focal adhesion kinase in cardiac remodeling. J Biol Chem. 2002;277:45203–45210. doi: 10.1074/jbc.M204886200. [DOI] [PubMed] [Google Scholar]
- 5.Heidkamp MC, Scully BT, Vijayan K, Engman SJ, Szotek EL, Samarel AM. PYK2 regulates SERCA2 gene expression in neonatal rat ventricular myocytes. Am J Physiol Cell Physiol. 2005;289:C471–C482. doi: 10.1152/ajpcell.00130.2005. [DOI] [PubMed] [Google Scholar]
- 6.Guo J, Sabri A, Elouardighi H, Rybin V, Steinberg SF. α1-adrenergic receptors activate AKT via a Pyk2/PDK-1 pathway that is tonically inhibited by novel protein kinase C isoforms in cardiomyocytes. Circ Res. 2006;99:1367–1375. doi: 10.1161/01.RES.0000252830.01581.fd. [DOI] [PubMed] [Google Scholar]
- 7.Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000;12:123–133. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 8.Sabri A, Govindarajan G, Griffin TM, Byron KL, Samarel AM, Lucchesi PA. Calcium- and protein kinase C-dependent activation of the tyrosine kinase PYK2 by angiotensin II in vascular smooth muscle. Circ Res. 1998;83:841–851. doi: 10.1161/01.res.83.8.841. [DOI] [PubMed] [Google Scholar]
- 9.Bayer AL, Ferguson AG, Lucchesi PA, Samarel AM. Pyk2 expression and phosphorylation in neonatal and adult cardiomyocytes. J Mol Cell Cardiol. 2001;33:1017–1030. doi: 10.1006/jmcc.2001.1369. [DOI] [PubMed] [Google Scholar]
- 10.Bayer AL, Heidkamp MC, Howes AL, Heller Brown J, Byron KL, Samarel AM. Protein kinase Cε-dependent activation of proline-rich tyrosine kinase 2 in neonatal rat ventricular myocytes. J Mol Cell Cardiol. 2003;35:1121–1133. doi: 10.1016/s0022-2828(03)00228-1. [DOI] [PubMed] [Google Scholar]
- 11.Blaukat A, Ivankovic-Dikic I, Gronroos E, Dolfi F, Tokiwa G, Vuori K, Dikic I. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J Biol Chem. 1999;274:14893–14901. doi: 10.1074/jbc.274.21.14893. [DOI] [PubMed] [Google Scholar]
- 12.Pandey P, Avraham S, Kumar S, Nakazawa A, Place A, Ghanem L, Rana A, Kumar V, Majumder PK, Avraham H, Davis RJ, Kharbanda S. Activation of p38 mitogen-activated protein kinase by PYK2/related adhesion focal tyrosine kinase-dependent mechanism. J Biol Chem. 1999;274:10140–10144. doi: 10.1074/jbc.274.15.10140. [DOI] [PubMed] [Google Scholar]
- 13.Taniyama Y, Weber DS, Rocic P, Hilenski L, Akers ML, Park J, Hemmings BA, Alexander RW, Griendling KK. Pyk2- and Src-dependent tyrosine phosphorylation of PDK1 regulates focal adhesions. Mol Cell Biol. 2003;23:8019–8029. doi: 10.1128/MCB.23.22.8019-8029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong WC, Macklem M, Parsons JT. Expression and characterization of splice variants of PYK2, a focal adhesion kinase-related protein. J Cell Sci. 1998;111:1981–1991. doi: 10.1242/jcs.111.14.1981. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Dy RC, Cance WG, Graves LM, Earp HS. Interactions between two cytoskeleton-associated tyrosine kinases: calcium-dependent tyrosine kinase and focal adhesion tyrosine kinase. J Biol Chem. 1999;274:8917–8924. doi: 10.1074/jbc.274.13.8917. [DOI] [PubMed] [Google Scholar]
- 16.Hayasaka H, Martin KH, Hershey ED, Parsons JT. Disruption of FRNK expression by gene targeting of the intronic promoter within the focal adhesion kinase gene. J Cell Biochem. 2007 doi: 10.1002/jcb.21329. in press. [DOI] [PubMed] [Google Scholar]
- 17.Park JG, Bose A, Leszyk J, Czech MP. PYK2 as a mediator of endothelin-1/Gα11 signaling to GLUT4 glucose transporters. J Biol Chem. 2001;276:47751–47754. doi: 10.1074/jbc.C100524200. [DOI] [PubMed] [Google Scholar]
- 18.Sorokin A, Kozlowski P, Graves L, Philip A. Protein-tyrosine kinase Pyk2 mediates endothelin-induced p38MAPK activation in glomerular mesangial cells. J Biol Chem. 2001;276:21521–21528. doi: 10.1074/jbc.M008869200. [DOI] [PubMed] [Google Scholar]
- 19.Watson JM, Harding TW, Golubovskaya V, Morris JS, Hunter D, Li X, Haskill JS, Earp HS. Inhibition of the calcium-dependent tyrosine kinase (CADTK) blocks monocyte spreading and motility. J Biol Chem. 2001;276:3536–3542. doi: 10.1074/jbc.M006916200. [DOI] [PubMed] [Google Scholar]
- 20.van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL. Proline-rich tyrosine kinase 2 (Pyk2) mediates vascular endothelial-cadherin-based cell-cell adhesion by regulating β-catenin tyrosine phosphorylation. J Biol Chem. 2005;280:21129–21136. doi: 10.1074/jbc.M500898200. [DOI] [PubMed] [Google Scholar]
- 21.Samarel AM, Engelmann GL. Contractile activity modulates myosin heavy chain-β expression in neonatal rat heart cells. Am J Physiol. 1991;261:H1067–H1077. doi: 10.1152/ajpheart.1991.261.4.H1067. [DOI] [PubMed] [Google Scholar]
- 22.Westfall MV, Rust EM, Albayya F, Metzger JM. Adenovirus-mediated myofilament gene transfer into adult cardiac myocytes. Methods Cell Biol. 1997;52:307–322. [PubMed] [Google Scholar]
- 23.Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM. GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ Res. 2002;90:1282–1289. doi: 10.1161/01.res.0000023201.41774.ea. [DOI] [PubMed] [Google Scholar]
- 24.Eble DM, Strait JB, Govindarajan G, Lou J, Byron KL, Samarel AM. Endothelin-induced cardiac myocyte hypertrophy: role for focal adhesion kinase. Am J Physiol Heart Circ Physiol. 2000;278:H1695–H1707. doi: 10.1152/ajpheart.2000.278.5.H1695. [DOI] [PubMed] [Google Scholar]
- 25.Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strait JB, Martin JL, Bayer A, Mestril R, Eble DM, Samarel AM. Role of protein kinase C-ε in hypertrophy of cultured neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2001;280:H756–H766. doi: 10.1152/ajpheart.2001.280.2.H756. [DOI] [PubMed] [Google Scholar]
- 27.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 28.Porter MJ, Heidkamp MC, Scully BT, Patel N, Martin JL, Samarel AM. Isoenzyme-selective regulation of SERCA2 gene expression by protein kinase C in neonatal rat ventricular myocytes. Am J Physiol Cell Physiol. 2003;285:C39–C47. doi: 10.1152/ajpcell.00461.2002. [DOI] [PubMed] [Google Scholar]
- 29.Pfeffer JM, Pfeffer MA, Braunwald E. Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ Res. 1985;57:84–95. doi: 10.1161/01.res.57.1.84. [DOI] [PubMed] [Google Scholar]
- 30.Beeri R, Guerrero JL, Supple G, Sullivan S, Levine RA, Hajjar RJ. New efficient catheter-based system for myocardial gene delivery. Circulation. 2002;106:1756–1759. doi: 10.1161/01.cir.0000035240.92015.e4. [DOI] [PubMed] [Google Scholar]
- 31.Kodama H, Fukuda K, Takahashi T, Sano M, Kato T, Tahara S, Hakuno D, Sato T, Manabe T, Konishi F, Ogawa S. Role of EGF Receptor and Pyk2 in endothelin-1-induced ERK activation in rat cardiomyocytes. J Mol Cell Cardiol. 2002;34:139–150. doi: 10.1006/jmcc.2001.1496. [DOI] [PubMed] [Google Scholar]
- 32.Kodama H, Fukuda K, Takahashi E, Tahara S, Tomita Y, Ieda M, Kimura K, Owada KM, Vuori K, Ogawa S. Selective involvement of p130Cas/Crk/Pyk2/c-Src in endothelin-1-induced JNK activation. Hypertension. 2003;41:1372–1379. doi: 10.1161/01.HYP.0000069698.11814.F4. [DOI] [PubMed] [Google Scholar]
- 33.Hirotani S, Higuchi Y, Nishida K, et al. Ca2+-sensitive tyrosine kinase Pyk2/CAKbeta-dependent signaling is essential for G-protein-coupled receptor agonist-induced hypertrophy. J Mol Cell Cardiol. 2004;36:799–807. doi: 10.1016/j.yjmcc.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 34.McDonough PM, Stella SL, Glembotski CC. Involvement of cytoplasmic calcium and protein kinases in the regulation of atrial natriuretic factor secretion by contraction rate and endothelin. J Biol Chem. 1994;269:9466–9472. [PubMed] [Google Scholar]
- 35.Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, Izumo S. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 36.Schaller MD, Sasaki T. Differential signaling by the focal adhesion kinase and cell adhesion kinase β. J Biol Chem. 1997;272:25319–25325. doi: 10.1074/jbc.272.40.25319. [DOI] [PubMed] [Google Scholar]
- 37.Domingos PP, Fonseca PM, Nadruz W, Jr, Franchini KG. Load-induced focal adhesion kinase activation in the myocardium: role of stretch and contractile activity. Am J Physiol Heart Circ Physiol. 2002;282:H556–H564. doi: 10.1152/ajpheart.00534.2001. [DOI] [PubMed] [Google Scholar]
- 38.Hajjar RJ, del Monte F, Matsui T, Rosenzweig A. Prospects for gene therapy for heart failure. Circ Res. 2000;86:616–621. doi: 10.1161/01.res.86.6.616. [DOI] [PubMed] [Google Scholar]
- 39.Bekeredjian R, Chen S, Frenkel PA, Grayburn PA, Shohet RV. Ultrasound-targeted microbubble destruction can repeatedly direct highly specific plasmid expression to the heart. Circulation. 2003;108:1022–1026. doi: 10.1161/01.CIR.0000084535.35435.AE. [DOI] [PubMed] [Google Scholar]