

Abstract
 Recognition imaging microscopy is an analytical technique used to map the topography and chemical identity of specific protein molecules present in complex biological samples. The technique relies on the use of antibodies tethered to the cantilever tip of an AFM probe to detect cognate antigens deposited onto a mica surface. Despite the power of this technique to resolve single molecules with nanometer-scale spacing, the recognition step remains limited by the availability of suitable quality antibodies. Here we report the in vitro selection and recognition imaging of anti-histone H4 aptamers. In addition to identifying aptamers to highly basic proteins, these results suggest that aptamers provide an efficient, cost effective route to highly selective affinity reagents for recognition imaging microscopy.
Recognition imaging microscopy is an analytical technique used to map the topography and chemical identity of specific protein molecules present in complex biological samples. The technique relies on the use of antibodies tethered to the cantilever tip of an AFM probe to detect cognate antigens deposited onto a mica surface. Despite the power of this technique to resolve single molecules with nanometer-scale spacing, the recognition step remains limited by the availability of suitable quality antibodies. Here we report the in vitro selection and recognition imaging of anti-histone H4 aptamers. In addition to identifying aptamers to highly basic proteins, these results suggest that aptamers provide an efficient, cost effective route to highly selective affinity reagents for recognition imaging microscopy.
Recognition imaging microscopy is an analytical technique used to map the topography and chemical identity of specific protein molecules present in complex biological samples.1-3 The technique relies on the use of affinity reagents immobilized to the cantilever tip of an atomic force microscope (AFM) to identify the precise location of a single protein in an aqueous environment. Since surface images can be acquired in near real-time, recognition imaging has been used to study many time dependent processes.1 Despite the ability of this technique to resolve single molecules with nanometer-scale spacing, the recognition step remains limited by the availability of antibodies of suitable quality. In our work on chromatin remodeling we have found that many commercial antibodies developed to recognize DNA binding proteins show batch-to-batch variation in performance and mild to severe cross-reactivity with other proteins.4 To overcome these limitations we initiated an investigation into alternative affinity reagents as antibody mimics in recognition imaging microscopy.5 Here we report the in vitro selection and evaluation of DNA aptamers selected to bind histone H4 tails.
Aptamers are nucleic acid molecules that exhibit antibody-like properties by adopting structures that are complementary in shape and charge to a selected target.6 In contrast to antibodies, aptamers are smaller in size, easier to engineer, and can be generated relatively quickly using standard in vitro selection technologies. Although aptamers have been selected to bind a diverse array of targets with high affinity and specificity,6 some concern remains over their ability to bind highly charged molecules due to the potential for nonspecific binding.7 Recognizing that histones are highly charged proteins that contain many lysine and arginine residues, we wondered whether aptamers could be selected to recognize different histone classes.
To address this question we used an in vitro selection protocol that relies on capillary electrophoresis (CE) to separate functional aptamers from unbound sequences.8 This approach enables binding to occur free in solution, thereby eliminating some of the biases associated with traditional SELEX.6 We began by incubating the histone H4 peptide (1.5 kDa, pI = 12.0) with a nucleic acid library for 1 h at room temperature. The DNA library contained 48 random nucleotide positions flanked on both sides with constant primer binding sites for PCR amplification (5′-GGC GGC GAT GAG GAT GAC-(48N)-ACC ACT GCG TGA CTG CCC-3′). The 5'-end of the DNA was fluorescently labeled with 6-carboxy-fluorescein (FAM) to facilitate detection by laser induced fluorescence. Approximately 10 nL of this mixture was injected onto a neutral coated capillary. Five injections were made for each round of section, and ∼1011 unique DNA sequences were surveyed in round 1 of the selection.
Electrophoresis was performed using an electric field of 526 V/cm in a 57 cm long capillary with an inner diameter of 50 μm. Under these conditions, the unbound DNA migrated faster than the DNA-peptide complex. Functional sequences were recovered by allowing unbound sequences to pass into a waste vial, applying pressure to the column, and collecting the bound DNA in a separate vial. The DNA from each round of selection was amplified by PCR, purified, and made single-stranded by denaturing the PCR product on streptavidin-coated beads. After 4 rounds of selection, a second peak became visible in the CE chromatogram (Fig. 1a), indicating that the pool had become enriched in aptamers with affinity to the H4 peptide.
Figure 1.
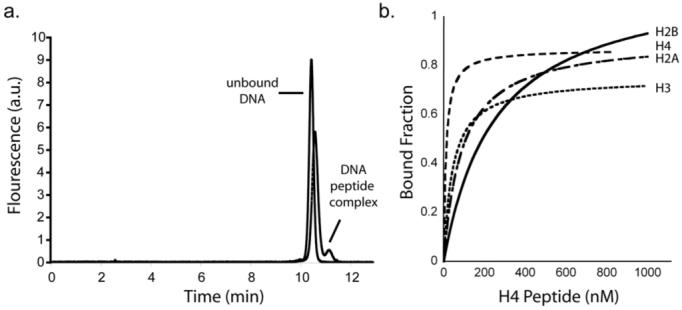
Aptamer enrichment and specificity to different histone classes. (a) Representative chromatograms from rounds 1 and 4 of the selection. The bound fraction is too small to be seen as a distinct peak in round 1, but is clearly visible after round 4. (b) Binding curves were obtained for aptamer 4.15 with the H4, H3, H2A and H2B peptide tail sequences.
Eighteen clones from the output of round 4 were sequenced. Secondary structures for all of the sequences were generated using mFold to calculate the lowest energy structure. Analysis of the different aptamers reveals a common unpaired loop connected by stems. Similar structures have been observed for other protein aptamers.9 Two of these sequences were randomly chosen and assayed for affinity to the H4 peptide using affinity capillary electrophoresis (ACE). Dissociation constants (Kd) for the two aptamers were measured for the H4 peptide sequence. Analysis of the binding curves (Fig. 1b) revealed that both aptamers bound the H4 peptide with Kd's (5-10 nM) similar to a typical antibody.
Next, we evaluated the selectivity of both aptamers for the H4 peptide by measuring their Kd for the peptide tails found in H3, H2A, and H2B type histones. Results from this experiment (Fig. 1b, Table 1) revealed that aptamer 4.15 showed slightly higher selectivity for the H4 peptide than aptamer 4.13 (5-20 versus 2-7 fold selectivity, respectively). The difference in selectivity between the two aptamers is consistent with the notion that higher affinity binding does not necessarily lead to greater specificity.10 In this case, it was discovered that aptamer 4.13, which bound the H4 peptide 2-fold more tightly than aptamer 4.15, was overall less selective for the H4 histone tail than the weaker affinity aptamer.
Table 1.
Affinity and Specificity of the Selected Aptamers.
| H4: GGKGLGKGGAKRHRK H2A: SAPAPKKGSKKAVTK | ||||
|---|---|---|---|---|
| H3: ARTKQTARKSTGGKA H2B: GKQGGKTRAKAKTRS | ||||
|
Kd (nM) |
||||
| Clone | H4 | H3 | H2A | H2B |
| 4.13 | 5.0 ± 2.0 | 12 ± 6.0 | 38 ± 26 | 34 ± 10 |
| 4.15 | 9.0 ± 2.0 | 44 ± 30 | 160 ± 50 | 200 ± 30 |
To evaluate the ability of our in vitro selected aptamers to function as antibody mimics in recognition imaging microscopy, we covalently linked each aptamer to the cantilever tip of our AFM and imaged recombinant histones in solution. To our surprise, aptamer 4.15, which showed high specificity in our solution binding assays, was unable to detect the recombinant histone target. This could be due to differences between the way it was selected (as a free peptide tail) versus its presentation on the protein surface or possibly a problem related to aptamer folding. Aptamer 4.13, however, gave very convincing recognition images. Using this aptamer, we simultaneously acquired topography and recognition images of recombinant H4 histones deposited onto freshly cleaved mica surfaces. The white spots in the topography image (Fig 2a) mark the locations of the protein on the surface, while the circled dark spots in the recognition image (Fig. 2b) denote the locations where a recognition event took place. In this example, 53 out of 62 histone H4 molecules were recognized by the aptamer. This gives a recognition efficiency of ∼85%, which is considerably higher than the 48% efficiency we measured using a commercial H4 antibody (unpublished data). The small number of histone molecules not detected in the recognition scan may reflect the small number of binding sites on the protein that remain occluded due to random adsorption on the mica surface.
Figure 2.

Demonstration of aptamer recognition in recognition imaging microscopy. Topography (a) and recognition images (b) were simultaneously acquired for pure histone H4 protein deposited on a mica surface. Recognition events are shown with circles. In this image-pair, 53 out of 62 histone molecules are recognized by the anti-H4 aptamer 4.13.
To investigate the selectivity of our in vitro selected aptamer for histone H4, we performed a series of recognition imaging experiments against recombinant histone H3, H2A, and H2B. By analyzing ∼250 molecules for each protein target (Table 2), we were able to determine that the H4 aptamer recognized the H4, H3, H2A, and H2B proteins with recognition efficiencies of 80%, 29%, 3%, and 5%, respectively. The low level of recognition to the H2A and H2B proteins correlates with the previous affinity measurements performed on the histone tail sequences. Although it is difficult, based on the current data, to determine the exact contacts between the aptamer and histone, comparison of the different tail sequences reveals a GGX motif that is present twice in H4 tails, once in H3 tails, but absent from the H2A and H2B tail sequences. Given the close correlation between the presence of this motif in the different histone tails and the observed recognition efficiency of aptamer 4.13 for these proteins, it seems likely that this motif is important in aptamer binding.
Table 2.
Recognition Imaging Efficiencies with Aptamer 4.13.
| Recognition Efficiency (%) | ||||
|---|---|---|---|---|
| H4 | H3 | H2A | H2B | |
| Recognitiona | 194 | 65 | 7 | 13 |
| Proteinsb | 242 | 225 | 240 | 275 |
| Efficiencyc | 80% | 29% | 2.9% | 4.7% |
Recognition refers to the total number of recognition events detected by monitoring changes in the tip oscillation amplitude over background.
Proteins denote the total number of protein molecules found in the topographical image.
Efficiency was measured as the total number of recognition events over the total number of proteins found in the image.
In summary, we have found that DNA aptamers represent a viable alternative to traditional antibodies in recognition imaging microscopy. Moreover, this work highlights the observation that aptamers, which are negatively charged molecules, can be selected to bind highly basic proteins displaying many positively charged sidechains. We suggest that this approach could be used to study key epigenetic modifications involved in chromatin remodeling.
Supplementary Material
Acknowledgements
We gratefully acknowledge Prof. Michael Bowser for training L.L. in CE SELEX. This work was supported by grants from the Agilent Foundation to S.M.L, and the National Cancer Institute R21 CA125510 to J.C.C. and S.M.L.
Footnotes
Supporting Information Available: Detailed protocols, sequences, mFold structures, and AFM images are available in the supplementary section. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kienberger F, Ebner A, Bruber HJ, Hinterdorfer P. Acc. Chem. Res. 2006;39:29–36. doi: 10.1021/ar050084m. [DOI] [PubMed] [Google Scholar]
- 2.Stroh C, Wang H, Bash R, Ashcroft B, Nelson J, Gruber H, Lohr D, Lindsay SM, Hinterdorfer P. Proc. Natl. Acad. Sci. 2004;34:12503–12507. doi: 10.1073/pnas.0403538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raab A, Han W, Badt D, Smith-Gill SJ, Lindsay SM, Schindler H, Hinterdorfer P. Nature Biotech. 1999;17:902–905. doi: 10.1038/12898. [DOI] [PubMed] [Google Scholar]
- 4.Bash R, Wang H, Anderson C, Yodh J, Hager G, Lindsay SM, Lohr D. FEBS Lett. 2006;580:4757–4761. doi: 10.1016/j.febslet.2006.06.101. [DOI] [PubMed] [Google Scholar]
- 5.Lin L, Wang H, Liu Y, Yan H, Lindsay SM. Biophys. J. 2006;90:4236–4238. doi: 10.1529/biophysj.105.079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Wilson DS, Szostak JW. Ann. Rev. Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]; (b) Bunka DHJ, Stockley PG. Nature Rev. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- 7.Sazani PL, Larralde R, Szostak JW. J. Am. Chem. Soc. 2004;126:8370–8371. doi: 10.1021/ja049171k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Mendonsa SD, Bowser MT. J. Am. Chem. Soc. 2004;126:20–21. doi: 10.1021/ja037832s. [DOI] [PubMed] [Google Scholar]; (b) Mendonsa SD, Bowser MT. J. Am. Chem. Soc. 2005;127:9382–9383. doi: 10.1021/ja052406n. [DOI] [PubMed] [Google Scholar]
- 9.Bartel DP, Zapp ML, Green MR, Szostak JW. Cell. 1991;67:529–536. doi: 10.1016/0092-8674(91)90527-6. [DOI] [PubMed] [Google Scholar]
- 10.Carothers JM, Oestreich SC, Szostak JW. J. Am. Chem. Soc. 2006;128:7929–7937. doi: 10.1021/ja060952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.