

Abstract
Two major determinants of the transparency of the lens are protein-protein interactions and stability of the crystallins, the structural proteins in the lens. βB2 is the most abundant β-crystallin in the human lens and is important in formation of the complex interactions of lens crystallins. βB2 readily forms a homodimer in vitro, with interacting residues across the monomer-monomer interface conserved among β-crystallins. Due to their long life spans, crystallins undergo an unusually large number of modifications, with deamidation being a major factor. In this study the effects of two potential deamidation sites at the monomer-monomer interface on dimer formation and stability were determined. Glutamic acid substitutions were constructed to mimic the effects of previously reported deamidations at Q162 in the C-terminal domain and at Q70, its N-terminal homologue. The mutants had a nativelike secondary structure similar to that of wild type βB2 with differences in tertiary structure for the double mutant, Q70E/Q162E. Multiangle light scattering and quasi-elastic light scattering experiments showed that dimer formation was not interrupted. In contrast, equilibrium unfolding and refolding in urea showed destabilization of the mutants, with an inflection in the transition of unfolding for the double mutant suggesting a distinct intermediate. These results suggest that deamidation at critical sites destabilizes βB2 and may disrupt the function of βB2 in the lens.
Cataract formation is the leading cause of blindness worldwide and affects an estimated 16 to 20 million people (1). Crystallins are highly soluble structural proteins that comprise 90% of lens proteins (2). The highly ordered, tightly packed crystallins make up the transparent structure of the lens and allow it to focus light onto the retina. These proteins undergo little turnover during the human life span, allowing for accumulation of modifications that may decrease crystallin solubility, alter lens transparency, and diminish vision.
During normal aging, crystallins are modified by truncation, oxidation, deamidation, and disulfide bond formation (3-9). Most notable by two-dimensional gel electrophoresis are trains of deamidated species (3). Each of the major crystallin polypeptides in the lens is deamidated during aging, and most are deamidated at multiple residues. Deamidation introduces a negative charge at physiological pH by replacing an amide with a carboxyl group and causes some isomerization. These changes potentially disrupt protein structure.
Recently, 34 sites of deamidations have been identified in a single cataractous lens (10). Due to the difficulty of detecting a single mass unit change resulting from deamidation, quantitative data associating deamidation with cataract formation is limited. The large numbers of modifications that occur during aging complicate the analysis. However, specific deamidations have been associated with cataractous lenses in γS (11, 12). Interestingly, in Alzheimer’s, Huntington’s, and Parkinson’s diseases the aggregated proteins characteristic of these debilitating diseases are increasingly deamidated (13), although it is not known if deamidation is causative or a correlation in these cases.
The crystal structures for two β-crystallins, βB1 and βB2, have been determined (14-17). β-Crystallin subunits have two domain regions joined by a connecting peptide. Each of these tightly folded domains has two Greek key motifs composed of four β-pleated sheet strands and N- and C-terminal extensions of varying lengths. Although βB1 and βB2 share high sequence homology, differences in the interface and linker regions of the proteins lead to different dimerization properties. Both form domain-swapped dimers where the domain of one subunit interacts with the opposite domain of the second subunit. However, βB1 has an intradomain interface that is not observed in βB2. The two differing conformations have been referred to as “open” and “closed” for βB2 and βB1, respectively (18). Homologous residues in these two crystallins participate in hydrophobic interactions across the domain and subunit interfaces. We have previously reported that replacing a Gln of βB1 at the intradomain interface with a Glu markedly decreased the stability of βB1 without disrupting the dimer (19, 20). Others have determined the contribution of the hydrophobic and ionic interactions to the interface stability with several site-directed alanine mutants in βB2 and in the intradomain interface of γD-crystallin (21-23). In general, mutations at the interface led to decreased stability with the detection of partially unfolded intermediates.
βB2 is thought to play an important role in hetero-oligomer formation in the lens. It is the major β-crystallin in the lens (24) and is the least modified during aging (25). βB2 is also the most soluble of the β-crystallins, remaining soluble during aging (26), and is needed to maintain the solubility of hetero-oligomers during isolation (27). There is a tendency for other β-crystallins to precipitate when separated from βB2 (25, 27). This has led to the proposal of a role for βB2 in maintaining the solubility of other β-crystallins that are heavily modified during aging.
The purpose of this study was to determine the effect of deamidation on the protein-protein interactions and stability of βB2 by introducing charges at the protein-protein interface. The effects of two deamidation sites were measured both independently and in combination. Deamidation at Q162 and Q70 were studied because they were recently found in vivo from a cataractous lens (10, 28). Glutamine162 is located at the C-terminal domain interface and is highly conserved among all the crystallins in the βγ family, suggesting a critical role in crystallin structure. The topologically equivalent site in the N-terminal domain in βB2 is Q70. Glutamines 70 and 162 and two Glu-Arg ion pairs appear to orient the docking of the N-terminal to the C-terminal domains around a hydrophobic cluster of residues (Figure 1, ref 15, 16). Deamidation at both sites was studied to determine the effects of accumulated deamidations. This is the first study addressing the effects of deamidation on βB2 structure.
Figure 1.
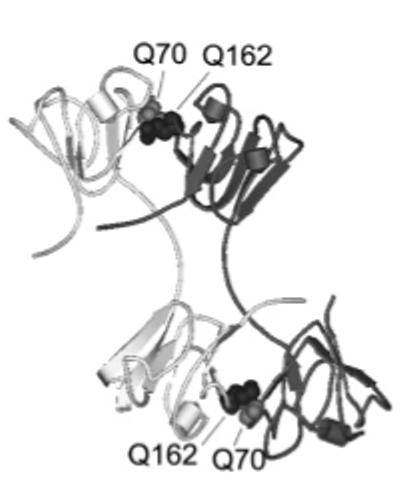
The crystal structure of βB2 shown in ribbon representation with interface glutamines 70 and 162 shown in spacefill (PDB code, 1blb, ref 15, 16).
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant Proteins
Deamidations at residues 70 and 162 were introduced by replacing a glutamine with a glutamic acid by site directed mutagenesis using internal primers containing the desired mutation (Quick Change Mutagenesis, Stratagene, Cedar Creek, TX) as previously described for βB1 (19). The forward primers contained the sequence TAA TAC GAC TCA CTA TAG GG and T GGA CTG GAG TAC CTG CTG to generate βB2 Q70E and Q162E, respectively (GenBank accession No. L10035). The expression plasmid, a pET 3a, containing the wild type (WT) βB2 sequence, was kindly provided by Drs. Nicolette Lubsen (University of Nijmegen) and Orval Bateman (University of London). The Q70E/Q162E mutant was made with the primer introducing the Q162E mutation into the vector containing the Q70E mutation. Plasmids were sequenced and mutagenesis was confirmed (Nevada Genomics Center). Recombinant human βB2 was expressed as previously described for βB1 in ref 19 using the Escherichia coli strain BL21(DE3) (Novagen, Madison, WI). Cells were lysed by repeated freeze/thaw in chromatography buffer appropriate for first step chromatography for each protein.
All βB2 proteins were purified from E. coli protein by successive ion-exchange chromatography. Cell lysates from WT βB2, Q70E, and Q162E were applied to a MacroPrep DEAE anion exchange column (BioRad, Hercules, CA) equilibrated in a Tris buffer (pH 8.0) for the first step of purification and a DEAE Sepharose Fast Flow anion exchange column (Amersham Biosciences, Piscataway, NJ) equilibrated in a Tris buffer (pH 7.4) for the second step. Cell lysate from Q70E/Q162E culture was applied to a SP Sepharose cation exchange column (Amersham Biosciences) equilibrated in a phosphate buffer (pH 5.9) for the first step and a MacroPrep DEAE anion exchange column equilibrated in a Tris buffer (pH 7.4) for the second step. A 20 mM Tris buffer with 1 mM EGTA, 0.16 mM EDTA, and 1 mM DTT of was used for anion exchange columns and a phosphate buffer of 6.6 mM Na2HPO4, 6.6 mM KH2PO4, 23.0 mM KCl, 0.16 mM EDTA, 1 mM EGTA, and 1 mM DTT was used for the cation exchange chromatography. Proteins were eluted with a 0-500 mM NaCl gradient. Mass spectrometry was performed to confirm the deamidation sites as previously described (29).
Circular Dichroism Measurements
Circular dichroism spectra in the far- and near-UV range were obtained using a JASCO J500-A spectropolarimeter (JASCO, Easton, MD). Samples were dialyzed into 5 mM NaH2PO4, 5 mM Na2HPO4 (pH 6.8), containing 100 mM NaF, and measured in a 0.01-cm cell for far-UV and in a 1-cm cell for near-UV at 20 °C. Spectra were measured at 0.1 nm resolution and averaged over 30 scans. WT βB2, Q70E, and Q70E/Q162E were analyzed in triplicate, and Q162E was analyzed in duplicate. Protein concentrations between 0.1 and 0.5 mg/mL were determined by amino acid analysis. Estimates of secondary structures were made using the variable selection method (30).
Associative Behavior of Expressed Proteins
Using a multiangle light scattering (MALS) instrument (miniDAWN, Wyatt Technology, Santa Barbara, CA), the association state of βB2 was observed based on the measured molar masses. The instrument was placed in line with a UV detector and a quasi-elastic light scattering detector (QELS, Wyatt Technology) while performing size-exclusion chromatography (SEC). SEC was performed with a Superose 12 10/300 GL column (Amersham Biosciences) equilibrated in buffer containing 29 mM Na2HPO4, 29 mM KH2PO4, 100 mM KCL, 0.7 mM EDTA, 1 mM DTT (pH 6.8) with a flow rate of 0.4 mL/min. Samples were concentrated to between 0.3 and 15 mg/mL using 10 000 MWCO cellulose centrifugal spin filters (Amicon, Millipore, Billerica, MA), filtered to remove particulate matter, and 50 μL injected. Samples were analyzed at each concentration step to avoid dilution effects.
MALS experiments were performed as previously described (29). Software provided by the manufacturer (ASTRA IV, Wyatt Technology) was used to calculate the weight-averaged molar mass of a species in a very narrow eluting volume every 2-5 s from the intensity of the scattered light according to Rayleigh light scattering principles. Concentration was determined from the UV absorbance at 280 nm and a molar extinction coefficient for βB2 of 40 210 (M-1 cm-1). An in-line refractive index detector (Opti-lab, Wyatt Technology) was also used to determine concentration. The molar masses using either method agreed within 5%.
Signals from the UV and MALS detector were normalized using bovine serum albumin. Monodisperse regions under the peak were analyzed. The weight-averaged molar masses (Mw) were reported based on the concentration of the recovered protein and the average protein concentration for the peak area analyzed. The peak area analyzed was approximately 75%. The uncertainty, due to noise in the signal, was generally below 2%.
Size of Expressed Proteins
QELS was used to measure the translational diffusion constant (DT). The QELS detector was attached to the 90° angle detector of the MALS instrument. For a single species undergoing translational diffusion, the autocorrelation functions are expected to follow a simple exponential function and the data were fit to this model with software provided by the manufacturer. The software reported the radius of hydration assuming a spherical conformation (RH). The frictional coefficient, f, was determined from this value using Stokes’s law, f = 6πηRH, where η is the viscosity of the solvent. The frictional coefficient for βB2 was compared to the theoretical frictional coefficient of βB2, fo, in a spherical conformation and the predicted axial ratio determined (31).
Stability of Expressed Proteins
Stability of deamidated mutants relative to WT βB2 was measured by fluorescence spectrometry during protein unfolding in urea. A stock solution of 8 M urea in phosphate buffer was made according to the method of Pace (32). An Abbe refractometer was used to measure the urea concentration (VEE GEE Scientific, Inc., Kirkland, WA). The phosphate buffer (pH 7.0) contained 50 mM Na2HPO4,50mMNaH2PO4, 5 mM DTT, and 2 mM EDTA. Experiments were also performed with 300 mM (NH4)2SO4 to stabilize the double mutant (33). Proteins at 1 μM were incubated in urea for 24 h at 21 °C. Protein concentrations were determined by absorbance at 280 nm, and a calculated extinction coefficient of 1.72. For refolding experiments, proteins were incubated in 6 M urea for 5 h, then diluted to the desired urea concentration and incubated for a total of 24 h. Initial timed fluorescence experiments determined that the proteins had completely unfolded in 6 M urea after 5 h.
Fluorescence measurements were made on a Photon Technology International QM-2000-7 spectrofluorimeter using the manufacturer’s supplied software, FeliX (Photon Technology International, Lawrenceville, NJ). Emission spectra were recorded between 300 and 400 nm with an excitation wavelength at 283 nm and repeated at 295 nm. Slit widths were set to 2 nm. Emission spectra were corrected for the buffer signal. The percent of unfolded protein was calculated either from the normalized intensities at 320 nm or the ratios of the 360 to 320 intensities (FI 360/320) and fit to a two-state model by the methods of Pace (32) or a three-state model according to the methods of Flaugh et al. (22) and Clark (34). Curve fitting was performed using Kaleidagraph software (Version 4.0, Synergy Software, Reading, PA) in order to calculate ΔG°s for a three-state model (two-step transition). Initial parameters were provided, and best fit of the averaged data was chosen by the smallest distribution of residuals. The following three-state model was employed:
| (1) |
| (2) |
| (3) |
where Y is the observed FI 360/320 nm signal, YN and YU are the intercepts of the native and unfolded baselines, SN and SU are the slopes of the native and unfolded baselines, and YI is the signal of the intermediate. Additionally, m1 and ΔG1 are the m value and ΔG° for the native to intermediate transition, and m2 and ΔG2 are the m value and ΔG° for the intermediate to unfolded transition. T is the temperature in kelvins, and R is the gas constant in units kcal mol-1 K-1 (34).
Stability of Heterodimers
Heterodimers were formed according to previously published methods (35-37). Equal molar amounts of recombinant proteins were mixed, unfolded in 6 M urea overnight, and then refolded by removing the urea by exhaustive dialysis for 24 h into phosphate buffer (pH 7.0) containing 50 mM Na2HPO4, 50 mM NaH2PO4, 5 mM DTT, and 2 mM EDTA. Samples were then unfolded in 0, 1.5, or 2.0 M urea in the same buffer, and the fluorescence emission was measured as described above. These urea concentrations were chosen because these concentrations gave the greatest differences between WT and the mutants during unfolding.
Electrophoresis
Electrophoresis was performed using precast, 1.0 mm thick 8 × 8 cm, polyacrylamide NuPAGE 10% Bis-Tris gels (Invitrogen, Carlsbad, CA). Proteins were visualized by staining with SimplyBlue SafeStain (Invitrogen).
RESULTS
The Effect of Deamidation on the Structure of βB2-Crystallin
In order to study the effect of deamidation on the structure of βB2-crystallin, deamidation was simulated by replacing the amino acid residues glutaminyl with glutamyl by site directed mutagenesis. Protein expression yields were notably lower for only the double mutant, which also required a different purification protocol. The presence of Glu at residues 70, 162, and at both residues in the double mutant was confirmed by mass spectrometry. Representative spectra of the parent ion isotopic distributions and the MS/MS fragment ions used to deduce the sequence for the peptide containing Q162E are shown in Figure 2A and Figure 2B. The proteins were purified to >98% homogeneity (Figure 3).
Figure 2.

Mass spectra of peptide 160-167 from recombinant βB2-crystallin containing a Q162E mutation. High-resolution zoom scan (A) of the isotopic distribution of the parent ion and tandem MS/MS spectra (B) used to determine the deamidation site location. The parent ion of peptide 160-167 was singly charged with a measured monoisotopic m/z of 964.6 (unmodified peptide monoisotopic m/z is calculated to be 963.5). The asterisk indicates the site of deamidation.
Figure 3.

Gel electrophoresis of WT (lane 1), Q70E (lane 2), Q162E (lane 3), and Q70E/Q162E (lane 4) βB2. One microgram of each protein was visualized with Coomassie stain on a 1.0 mm thick, 10% Bis/Tris gel.
The circular dichroic absorption in the far-UV showed characteristics typical for β-crystallins with a minimum at ∼218 nm (19, 32) (Figure 4A). The similar spectra and minima for all proteins suggest similar secondary structure. Differences in ellipticity were greatest between the wild type (WT) and double mutant (Table 1). The near-UV CD spectra were similar between WT and the single mutants, whereas the spectrum for the double mutant clearly differed from that for the WT, indicating differences in tertiary structure (Figure 4B).
Figure 4.

(A) Far-UV and (B) near-UV circular dichroism of WT (blue), Q70E (red), Q162E (purple), and Q70E/Q162E (green) βB2. Samples contained 10 mM sodium phosphate and 100 mM NaF (pH 6.8) and were measured in a 0.01-cm cell for far-UV and in a 1-cm cell for near-UV at 20 °C. Spectra shown are representative of experiments performed in triplicate.
Table 1.
Secondary Structure Composition of WT, Q70E, Q162E, and Q70E/Q162E βB2 as Determined by the Variable Selection Method (30)
| percent of structure |
|||||
|---|---|---|---|---|---|
| mutant | helix | antiparallel | parallel | turn | other |
| WT βB2 | 21 | 27 | 9 | 15 | 28 |
| Q70E | 23 | 22 | 8 | 18 | 29 |
| Q162E | 22 | 21 | 9 | 21 | 27 |
| Q70E/Q162E | 22 | 22 | 6 | 20 | 30 |
Fluorescence spectra of WT and the mutants were measured using an excitation wavelength of 283 nm (Figure 5). There was a similar emission maximum between 327 and 330 nm without urea and red-shift to 350 nm after unfolding in urea for WT and the single mutants. The double mutant had a slight shift in emission maximum compared to WT that was more pronounced after 24 h. The shape of the emission scan for the Q162E mutant was extended to the right of the peak. The significance of this is not known. The circular dichroism and fluorescence spectroscopy suggest that introducing a single charge at the interface did not disrupt the overall fold of the protein. In contrast, introducing both charges may have disrupted the tertiary structure and to a lesser extent the overall fold of the protein.
Figure 5.
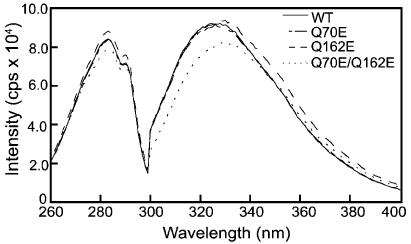
Excitation and emission spectra of WT, Q70E, Q162E, and Q70E/Q162E βB2. Excitation spectra were measured from 240 to 300 nm with emission wavelength at 336 nm. Emission spectra were measured from 300 to 400 nm with excitation at 283 nm. Proteins were analyzed at 1 μM in 100 mM sodium phosphate, 5 mM DTT, 2 mM EDTA, and 300 mM (NH4)2SO4 (pH 7.0).
Associative Behavior of Expressed Proteins
Introducing a negative charge across the interface of the βB2 dimer did not alter dimer formation. Both singly deamidated mutants and the double mutant formed dimers with the same molar mass and size as WT βB2 at all concentrations tested (Figure 6A and Figure 6B). A wide range of concentrations was used in order to facilitate aggregation at high concentrations and dissociation at low concentrations. The double mutant, Q70E/Q162E, was not analyzed at the higher concentrations because it tended to precipitate during concentration. There were no differences in elution behavior of the dimers.
Figure 6.

Molar mass (A) and radius of hydration (B) of the WT (×), Q70E (△), Q162E (□), and Q70E/Q162E (◇) βB2 determined by SEC in line with MALS or QELS. Column was equilibrated in 58 mM Na/K phosphate, 100 mM KCl, 0.7 mM EDTA, 1 mM DTT (pH 6.8) with a flow rate of 0.4 mL/min. Predicted molar mass for WT βB2 dimer is 46.6 kDa. Error bars are standard deviations, N = 3.
Taking an RH for βB2 as 2.8 nm with a weight-averaged molar mass, Mw, of 45 kDa (Figure 6) gave a calculated frictional coefficient, f, of 4.7 × 10-11 kg/s. The ratio of this f to that predicted for a sphere of the same Mw, fo, would give a f/fo of 1.1 and a predicted axial ratio of 2-3. The overall shape of the dimer for all mutants more closely approximated that of a sphere than what was previously reported for the βB1 dimer with a predicted axial ratio of 4-5 (29). βB2 is a more compact dimer than βB1, even with an extended linker. This can likely be attributed to the shorter N-terminal extension on βB2.
Stability of Expressed Proteins
In order to determine whether or not deamidation altered the stability of the βB2 dimer, the relative stabilities of the mutants were compared to WT βB2 during unfolding in urea. The double mutant was found to have a slight shift in wavelength, even in the absence of urea (Figure 5). Therefore, a high salt buffer (including 100 mM phosphate and 300 mM (NH4)2SO4) was initially used to facilitate comparison of all four proteins (33). In this buffer all proteins had approximately the same absorbance and emission at 1 μM (Figure 5). An excitation wavelength of 283 was chosen to monitor global unfolding of the proteins and to compare with previously published results for both βB2 and βB1 (19, 33). The data were analyzed by using the ratio of intensities of 360 to 320 nm (23, 34), in order to include differences in the shapes of the emission scans noted in Figure 5. Using these fluorescence intensities, a sigmoidal unfolding curve with a transition midpoint of ∼2.9 M urea was observed for WT βB2 (Figure 7) similar to that which others have reported (21, 33, 38).
Figure 7.

Unfolding of WT (×), Q70E (△), Q162E (□), and Q70E/Q162E (◇) βB2 in 0.0-5.0 M urea at 283 nm excitation. Proteins were analyzed at 1 μM concentration in the same buffer as in Figure 5. Samples were excited at 283 nm and emission intensities recorded as described in the text. Data are shown as the ratio of fluorescence intensities at 360/320 nm (FI 360/320 nm).
The cooperativity of unfolding was diminished for the Q70E, Q162E, and Q70/162E mutants (Figure 7), as reflected in the differences in slopes of the transitions compared to WT. The midpoints of the equilibrium unfolding/refolding transitions for the mutants were similar to WT when a two-state model of native to unfolded protein was assumed. However, this did not reflect differences in the unfolding curves between 0.75 and 2.5 M urea (Figure 7). In contrast to WT, there was a slight inflection in the transition of unfolding of Q70E/Q162E at ∼1.75 M urea, indicating a two-step transition with the first step occurring between 0.5 and 1.5 M urea (Figure 8). This mutation-induced shift to lower urea concentrations in the unfolding equilibrium indicates decreased stability of all the mutants, with a stable intermediate for the double mutant.
Figure 8.

Equilibrium unfolding and refolding of Q70E/Q162E βB2 in 0.0-5.0 M urea as described in Figure 7. Curve is fit with method from Clark et al. (34).
Since a plateau was present in the unfolding of Q70/162E βB2, the free energy was calculated assuming a three-state model (22, 34, Table 2). While the data appeared to fit the curves, the actual ΔG° calculations were difficult to solve without large error in one of the two transitions (Table 2). Quantitative analysis was hindered by the inability to set the pre- and post-transitions for two transitions when only a single transition was apparent. Nonetheless, apparent ΔG°s were calculated for comparisons. For the double mutant, the ΔG°s for both transitions of unfolding were lower than those estimated for WT, with the greatest difference in the first transition.
Table 2.
Equilibrium Unfolding Free Energy for WT, Q70E, Q162E, and Q70E/Q162E βB2
| transition 1 |
transition 2 |
|||||
|---|---|---|---|---|---|---|
| protein | ΔG° a | errorb | ΔΔG° c | ΔG° a | errorb | ΔΔG° c |
| WT βB2 | 7.2 | 0.4 | 7.8 | 5.4 | ||
| Q70E | 2.5 | 1.6 | -4.7 | 3.8 | 0.7 | -3.9 |
| Q162E | 2.9 | 0.6 | -4.3 | 8.0 | 0.5 | 0.2 |
| Q70E/Q162E | 3.7 | 2.0 | -3.5 | 4.6 | 0.7 | -3.1 |
Apparent free energy, ΔG° (kcal/mol), of unfolding based on urea curve.
Error of fit to determine ΔG° value, as reported by Kaleida-graph software.
in units of kcal/mol.
Since assuming a two-state model did not account for an intermediate and assuming a three-state model resulted in poor fits of the data, comparisons were made using the midpoint of the first transition where the greatest differences were seen. The inflection point seen for the double mutant was used to evaluate the comparative post-transition values of the first transition for all the proteins (Figure 8). This midpoint for WT was ∼2.25 M urea and was decreased for the single mutants to ∼1.75 M urea, with a further decrease to ∼1.25 M urea for the double mutant. The two single mutants had similar midpoints, but differed in their slopes with the greatest difference between WT and Q70E.
Results indicated that unfolding is reversible for all four proteins, and Q70E/Q162E appears to refold via an intermediate similar to that observed during unfolding. At urea concentrations <1 M urea, there was no sharp increase in intensity as would have been expected from light-scattering aggregates. The shift in wavelength during unfolding was also reversed during refolding, including for Q70E/Q162E (data not shown). The high salt in the buffer may have prevented formation of aggregates or intermediates detected by other researchers (33).
To determine whether or not greater differences could be observed, the unfolding of βB2 was compared at excitation wavelengths of 283 and 295 nm (data not shown). The tryptophan fluorescence intensities at 295 nm were used to follow changes in the microenvironment during unfolding. βB2 has five tryptophans, three of which are buried, one is partially buried, and one is exposed (37). Unfolding was also analyzed using intensities at 320 nm, rather than the ratio of intensities at 360/320. Similar m and C1/2 values were obtained by both of these methods. However, the slight inflection of the transition was not as apparent using these parameters for analysis. The reason for this is not known, but has also been observed by Sathish et al. (21).
A high salt buffer previously used to stabilize the expressed N-terminal domain was also used in our system to stabilize the double mutant (33). In order to determine whether or not the high salt buffer masked the effects of the introduced charges, experiments were repeated in the absence of (NH4)2SO4. First, it was determined by MALS that the proteins formed dimers under these conditions (Figure 9). A 50 μL injection of 1 mg/mL of each protein was analyzed by SEC-MALS in the same mobile phase as the fluorescence experiments. All proteins formed a dimer with molar masses between 42 and 44 kDa, although the molar masses were slightly lower at the tailing edge of the peak (Figure 9). All four proteins eluted ∼0.3 mL later in this buffer than in the “stabilizing” buffer, suggesting a column interaction. During chromatography, proteins eluted in 1 mL with concentrations under the peak ranging from 20 to 27 μg/mL or about 1 μM. Therefore, proteins formed dimers in the new buffer at 1 μM, the concentrations used to measure unfolding. A shift in the fluorescence spectrum for the double mutant was present in this buffer, even without urea, and all four proteins unfolded in lower urea without changing the pattern of unfolding (Figure 10). Experiments were repeated in a low ionic strength buffer of 10 mM phosphate with similar results. Because WT βB2 also unfolded at lower urea concentrations, the differences among all four proteins were only slightly increased in this buffer. The ΔG°s were not calculated because a true pretransition was not seen for the double mutant.
Figure 9.

Chromatogram of molar masses of recombinant WT (blue), Q70E (red), Q162E (purple), and Q70E/Q162E (green) βB2 determined by SEC-MALS in 100 mM sodium phosphate, 5 mM DTT, and 2 mM EDTA (pH 7.0). The line tracing represents the signal from the UV detector. The individual data points represent the molar masses in a narrowly eluting volume. A 50 μL sample of ∼1 mg/mL was analyzed for each protein.
Figure 10.

Unfolding of WT (×), Q70E (△), Q162E (□), and Q70E/Q162E (◇) βB2 in 0.0-5.0 M urea at 283 nm excitation. Proteins were analyzed at 1 μM concentration in the same buffer as Figure 9.
A 50:50 mixture of WT and Q70E, Q162E, or Q70E/Q162E resulted in a greater shift in wavelength than with WT alone at either 1.5 or 2 M urea (data shown for WT:Q162E at 1 μM, Figure 11). The shift in wavelength of the mixtures was intermediate between that of the WT and individual mutants alone.
Figure 11.

Fluorescence emission spectra of mixed WT:Q162E showing the decrease in intensity and shift in maximum wavelength. WT (-), mixed WT:Q162E (- - -), and Q162E (- - -) βB2 in 2.0 M urea at 283 nm excitation. Proteins were analyzed at 1 μM concentration in the same buffer as Figure 9.
DISCUSSION
Effects of Deamidation on βB2 Structure
Both Q70 and Q162 lie at the interface of the βB2 dimer and participate in H-bonds with residues of the opposite domain from the partner subunit in the crystal structure (16). Q70 interacts with L164 and F176, and Q162 interacts with V72 (16). Simulating deamidation at these sites did not alter dimer formation, with changes in the overall structure observed most notably for the double mutant.
Our findings are consistent with several other studies probing the interface interactions (21, 23, 39). Replacing hydrophobic or ionic residues at the monomer-monomer interface in βB2 or the domain-domain interface in γD or γB with alanines resulted in only minor changes to the overall fold of the protein, and did not disrupt dimer formation in βB2. In γD, mutating the homologous Gln mutated in this study did not alter the nativelike structure of γD (22). However, a mutation observed in congenital cataracts eliminating 51 residues from the C-terminal domain of βB2 was enough to disrupt dimer formation (40). Together, these results indicate that mutating specific interface residues in βB2 alone may not be enough to alter the dimer formation of βB2.
Despite the absence of changes detected by MALS and QELS, changes in secondary and tertiary structure, particularly of the doubly deamidated mutant, while not dissociating the dimer, led to differences in stability.
Effects of Deamidation on βB2 Stability
Deamidation decreased the stability of βB2-crystallin. Deamidation at one or both of the homologous sites, Q70 in the N-terminal domain or Q162 in the C-terminal domain of βB2, shifted the equilibrium of unfolding preferentially in the early segment of the unfolding curves. This was associated with a loss of cooperativity, which was more apparent for the Q70E mutant.
The unfolding curve of WT βB2 did not show a discrete intermediate, previously seen with a low ionic strength buffer (41) or when GdnHCl was used as the denaturant (42, 43). The monophasic nature of the unfolding curve for WT suggests a highly cooperative unfolding from the native to the unfolded protein or a spectroscopically silent transition from native to intermediate or intermediate to unfolded protein. The nature of the intermediate for the double mutant was not determined in this study, but it can be inferred from previous reports that βB2 unfolds in a two-step transition (33, 42, 43). The unfolding of rat βB2 is dominated by simultaneous dissociation of the dimer and unfolding of the N-terminal domain followed by the unfolding of the C-terminal domain (33). Therefore, the intermediate state is most likely populated by monomers with an unfolded N-terminal domain. Alternatively, the inflection in unfolding of the double mutant may have been due to aggregation.
We have previously reported similar results for homologous deamidations in βB1. Simulating deamidation in the C-terminal domain at Q204 in βB1 homologous to Q162 in βB2 was associated with a decrease in stability of the N-terminal domain (20).
Single mutations in β/γ-crystallins have generally led to changes in stability. For example, mutating either of the domain Gln at Q54 or Q143 in γD decreased the apparent free energy (ΔG°) by 0.2 to 0.5 kcal/mol (22). These decreases in stability may be enough to alter crystallin interactions in the lens. In another study, a marginally destabilized βB2 mutant, βB2 L164A, bound to R-crystallin, indicating formation of an intermediate that was not detected in the unfolding curve (21). Even minor changes in stability may be enough to alter crystallin-crystallin interactions.
When both interface Gln residues were replaced with charged residues in βB2, there was a further decrease in stability that could be explained by the observed effect on the tertiary structure and appearance of a possible intermediate. Likewise, in γD when both interface Gln residues were mutated, the difference in ΔG°s increased to 1.4 kcal/mol and a distinct intermediate was seen in the unfolding curve (22). Several other sites of deamidation in βB2 have been detected in vivo (8, 10). It may be that either a single strategically located deamidation or an accumulation of deamidation at several sites may lead to detrimental changes in the lens.
A greater percentage of the possible deamidation sites have been reported in βB1 and βA3 than in βB2 (8, 10, 28). This is consistent with other reports that βB2 is more resistant to modifications than the other crystallins (25). Preliminary data suggest that the extent of deamidation may be greater in the insoluble than the soluble proteins. The deamidation levels appear to be between 5% and 15%. The effects of this amount of deamidation of βB2 in the complex mixture of other modified crystallins in vivo are not known.
In summary, deamidation in the major lens β-crystallin, βB2, altered protein stability and structure. Glutamine residues 70 and 162 in βB2 are located at the monomer-monomer interface in the dimer (16). As a result of deamidation at these key sites, newly introduced charges on the side-chain groups decreased protein stability without dissociating the dimer. Deamidation also led to changes in secondary and tertiary structure, particularly of the doubly deamidated mutant. These changes may facilitate further modifications and may contribute to insolubilization of βB2, although these studies do not rule out the possibility that other lens modifications such as oxidation and disulfide bond formation are principally responsible for insolubilization (10, 44). Deamidation may also alter the function of βB2 in vivo by compromising the ability of βB2 to maintain the solubility of other more modified β-crystallins. Alternatively, changes in structure may facilitate the interaction of βB2 with other β-crystallins in the complex mixture in the lens (41). Future studies will explore the interactions between the deamidated proteins described here and other β-crystallins.
ACKNOWLEDGMENT
We thank Hans Peter Bächinger, Larry David, Phillip A. Wilmarth, Mike Harms, Julie Cherry, and Ted Brandon for expert technical help and discussions.
Footnotes
Funding provided by the National Institutes of Health, National Eye Institute, Grant Reference No. R01-EY012239 (K.J.L.), Oregon Lions Sight and Hearing Foundation grant (K.J.L.), Shriners Hospitals Shared Facilities Grant, and core grant EY 10572.
REFERENCES
- 1.Thylefors BNA, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull. W.H.O. 1995;73:115–121. [PMC free article] [PubMed] [Google Scholar]
- 2.Bloemendal H. The vertebrate eye lens. Science. 1977;197:127–138. doi: 10.1126/science.877544. [DOI] [PubMed] [Google Scholar]
- 3.Lampi KJ, Ma Z, Hanson SRA, Azuma M, Shih M, Shearer TR, Smith DL, Smith JB, David LL. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp. Eye Res. 1998;67(1):31–43. doi: 10.1006/exer.1998.0481. [DOI] [PubMed] [Google Scholar]
- 4.Lund AL, Smith JB, Smith DL, David LL. Modifications of the water-insoluble human lens α-crystallins. Exp. Eye Res. 1996;63:662–672. doi: 10.1006/exer.1996.0160. [DOI] [PubMed] [Google Scholar]
- 5.Hanson SR, Smith DL, Smith JB. Deamidation and disulfide bonding in human lens γ-crystallins. Exp. Eye Res. 1998;67:301–312. doi: 10.1006/exer.1998.0530. [DOI] [PubMed] [Google Scholar]
- 6.Hanson SR, Hasan A, Smith DL, Smith JB. The major in vivo modifications of the human water-insoluble lens crystallins are disulfide bonds, deamidation, methionine oxidation and backbone cleavage. Exp. Eye Res. 2000;71:195–207. doi: 10.1006/exer.2000.0868. [DOI] [PubMed] [Google Scholar]
- 7.Takemoto L, Boyle D. Deamidation of specific glutamine residues from αA crystallin during aging of the human lens. Biochemistry. 1998;37:13681–13685. doi: 10.1021/bi981542k. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Smith DL, Smith JB. Human β-crystallins modified by backbone cleavage, deamidation, and oxidation are prone to associate. Exp. Eye Res. 2003;77:259–272. doi: 10.1016/s0014-4835(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 9.Groenen PJ, van Dongen MJ, Voorter CE, Bloemendal H, de Jong WW. Age-dependent deamidation of αB-crystallin. FEBS Lett. 1993;322:69–72. doi: 10.1016/0014-5793(93)81113-e. [DOI] [PubMed] [Google Scholar]
- 10.Searle BC, Dasari S, Wilmarth PA, Turner M, Reddy AP, David LL, Nagalla SR. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J. Proteome Res. 2005;4(2):546–554. doi: 10.1021/pr049781j. [DOI] [PubMed] [Google Scholar]
- 11.Lapko VN, Purkiss AG, Smith DL, Smith JB. Deamidation in human γS-crystallin from cataractous lenses is influenced by surface exposure. Biochemistry. 2002;41:8638–8648. doi: 10.1021/bi015924t. [DOI] [PubMed] [Google Scholar]
- 12.Takemoto L, Boyle D. Increased deamidation of asparagine during human senile cataractogenesis. Mol. Vision. 2000;6:164–168. [PubMed] [Google Scholar]
- 13.Robinson NE, Robinson AB. Molecular Clocks. Althouse Press; Cave Junction, OR: 2004. pp. 270–273. Chapter 15. [Google Scholar]
- 14.Van Montfort R, Bateman O, Lubsen N, Slingsby C. Crystal structure of truncated human βB1-crystallin. Protein Sci. 2003;12:2606–2612. doi: 10.1110/ps.03265903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bax B, Lapatto R, Nalini V, Driessen H, Lindley PF, Mahadevan D, Blundell TL, Slingsby C. X-Ray analysis of βB2-crystallin and evolution of oligomeric lens proteins. Nature. 1990;347:776–780. doi: 10.1038/347776a0. [DOI] [PubMed] [Google Scholar]
- 16.Lapatto R, Nalini V, Bax B, Driessen H, Lindley PF, Blundell TL, Slingsby C. High-resolution structure of an oligomeric eye lens β-crystallin. Loops, arches, linkers, and interfaces in βB2 dimer compared to a monomeric γ-crystallin. J. Mol. Biol. 1991;222:1067–1083. doi: 10.1016/0022-2836(91)90594-v. [DOI] [PubMed] [Google Scholar]
- 17.Nalini V, Bax B, Driessen H, Lindley PF, Blundell TL, Slingsby C. Close packing of an oligomeric eye lens β-crystallin induces loss of symmetry and ordering of sequence extensions. J. Mol. Biol. 1994;236:1250–1258. doi: 10.1016/0022-2836(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 18.Sergeev YV, Hejtmancik JF, Wingfield PT. Energetics of domain-domain interactions and entropy driven association of β-Crystallins. Biochemistry. 2004;43:415–424. doi: 10.1021/bi034617f. [DOI] [PubMed] [Google Scholar]
- 19.Lampi KJ, Oxford JT, Bachinger HP, Shearer TR, David LL, Kapfer DM. Deamidation of human βB1 alters the elongated structure of the dimer. Exp. Eye Res. 2001;72:279–288. doi: 10.1006/exer.2000.0950. [DOI] [PubMed] [Google Scholar]
- 20.Kim YH, Kapfer DM, Boekhorst J, Lubsen NH, Bachinger HP, Shearer TR, David LL, Feix B, Lampi KJ. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, βB1-crystallin. Biochemistry. 2002;41:14076–14084. doi: 10.1021/bi026288h. [DOI] [PubMed] [Google Scholar]
- 21.Sathish HA, Koteiche HA, Mchourab HS. Binding of destabilized βB2-crystallin mutants to α-Crystallin. J. Biol. Chem. 2004;279:16425–16432. doi: 10.1074/jbc.M313402200. [DOI] [PubMed] [Google Scholar]
- 22.Flaugh S, Kosinski-Collins M, King J. Interdomain side-chain interactions in human γD crystallin influencing folding and stability. Protein Sci. 2005;14:2030–2043. doi: 10.1110/ps.051460505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flaugh S, Kosinski-Collins M, King J. Contributions of hydrophobic domain interface interactions to the folding and stability of human γD-crystallin. Protein Sci. 2005;14:569–581. doi: 10.1110/ps.041111405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lampi KJ, Ma Z, Shih M, Shearer TR, Smith JB, Smith DL, David LL. Sequence analysis of βA3, βB3, and βA4 crystallins completes the identification of the major proteins in young human lens. J. Biol. Chem. 1997;272:2268–2275. doi: 10.1074/jbc.272.4.2268. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, David L, Smith D, Smith J. Resistance of human βB2-crystallin to in vivo modification. Exp. Eye Res. 2001;73:203–211. doi: 10.1006/exer.2001.1023. [DOI] [PubMed] [Google Scholar]
- 26.Feng J, Smith DL, Smith JB. Human lens β-crystallin stability. J. Biol. Chem. 2000;275(16):11585–11590. doi: 10.1074/jbc.275.16.11585. [DOI] [PubMed] [Google Scholar]
- 27.Bateman OA, Slingsby C. Structural studies on β H-crystallin from bovine eye lens. Exp. Eye Res. 1992;55(1):127–33. doi: 10.1016/0014-4835(92)90100-7. [DOI] [PubMed] [Google Scholar]
- 28.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post - translational modifications by blind search of mass spectra. Nat. Biotechnol. 2005;23(12):1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 29.Harms MJ, Wilmarth PA, Kapfer DM, Steel EA, David LL, Bachinger HP, Lampi KJ. Laser light-scattering evidence for an altered association of βB1-crystallin deamidated in the connecting peptide. Protein Sci. 2004;13(3):678–686. doi: 10.1110/ps.03427504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Compton LA, Mathews CK, Johnson WC., Jr. The conformation of T4 bacteriophage dihydrofolate reductase from circular dichroism. J. Biol. Chem. 1987;262:13039–13043. [PubMed] [Google Scholar]
- 31.Cantor CR, Schimmel PR. Biophysical Chemistry. W.H. Freeman; San Francisco: 1980. [Google Scholar]
- 32.Pace CN, Scholtz JM. In: Protein Structure: A practical approach. 2nd ed. Creighton TE, editor. IRL Press; Oxford, U.K.: 2002. pp. 299–320. [Google Scholar]
- 33.Wieligmann K, Mayr E, Jaenicke R. Folding and self-assembly of the domains of βB2-crystallin from rat eye lens. J. Mol. Biol. 1999;286:989–994. doi: 10.1006/jmbi.1999.2554. [DOI] [PubMed] [Google Scholar]
- 34.Clark AC, Sinclair JF, Baldwin TO. Folding of bacterial luciferase involves a non-native heterodimeric intermediate in equilibrium with the native enzyme and the unfolded subunits. J. Biol. Chem. 1993;268:10773–10779. [PubMed] [Google Scholar]
- 35.Slingsby C, Bateman OA. Quaternary interactions in eye lens β-crystallins: Basic and acidic subunits of β-crystalliins favor heterologous association. Biochemistry. 1990;29:6592–6599. doi: 10.1021/bi00480a007. [DOI] [PubMed] [Google Scholar]
- 36.Slingsby C, Bateman OA. Formation and crystallization of the eye lens heterodimer βB2-βB3-crystallin. Exp. Eye Res. 1994;58:761–764. doi: 10.1006/exer.1994.1073. [DOI] [PubMed] [Google Scholar]
- 37.Bateman OA, Sarra R, van Genesen ST, Kappe G, Lubsen NH, Slingsby C. The stability of human acidic β-crystallin oligomers and hetero-oligomers. Exp. Eye Res. 2003;77:409–422. doi: 10.1016/s0014-4835(03)00173-8. [DOI] [PubMed] [Google Scholar]
- 38.Jaenicke R, Slingsby C. Lens crystallins and their microbial homologs: structure, stability, and function. Crit. Rev. Biochem. Mol. Biol. 2001;36:435–499. doi: 10.1080/20014091074237. [DOI] [PubMed] [Google Scholar]
- 39.Palme S, Slingsby C, Jaenicke R. Mutational analysis of hydrophobic domain interactions in γB-crystallin from bovine eye lens. Protein Sci. 1997;6(7):1529–1536. doi: 10.1002/pro.5560060717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu BF, Liang JJ. Interaction and biophysical properties of human lens Q155* βB2-crystallin mutant. Mol. Vision. 2005;11:321–327. [PubMed] [Google Scholar]
- 41.MacDonald J, Purkiss A, Smith M, Evans P, Goodfellow J, Slingsby C. Unfolding crystallins: The destabilizing role of a β-hairpin cysteine in βB2-crystallin by simulation and experiment. Protein Sci. 2005;14:1282–1292. doi: 10.1110/ps.041227805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu L, Liang J. Unfolding of human lens recombinant βB2- and γC-crystallins. J. Struct. Biol. 2002;139:191–198. doi: 10.1016/s1047-8477(02)00545-2. [DOI] [PubMed] [Google Scholar]
- 43.Fu L, Liang J. Spectroscopic analysis of lens recombinant βB2- and γC-crystallin. Mol. Vision. 2001;7:178–183. [PubMed] [Google Scholar]
- 44.Takemoto LJ. Disulfide bond formation of cysteine-37 and cysteine-66 of βB2 crystallin during cataractogenesis of the human lens. Exp. Eye Res. 1997;64:609–614. doi: 10.1006/exer.1996.0247. [DOI] [PubMed] [Google Scholar]