

Abstract
Cholesterol is predicted to associate more strongly with the outer than the inner leaflet of plasma membrane bilayers based on the relative in vitro affinities of their phospholipids. Complex formation with the high affinity species (especially saturated sphingomyelins) is said to reduce the chemical activity (escape potential or fugacity) of the sterol. We therefore tested the hypothesis that scrambling the sidedness of plasma membrane phospholipids of intact cells will increase the chemical activity of outer surface cholesterol. Upon activating the plasma membrane scramblase in intact human red cells by introducing ionomycin to raise cytoplasmic Ca++, phosphatidylserine became exposed and, concomitantly, the chemical activity of exofacial cholesterol was increased. (This was gauged by its susceptibility to cholesterol oxidase and its rate of transfer to cyclodextrin.) Similar behavior was observed in human fibroblasts. Two other treatments known to activate cell surface cholesterol (namely, exposure to glutaraldehyde and to low ionic strength buffer) also brought phosphatidylserine to the cell surface but by a Ca++-independent mechanism. Given that phospholipid scrambling is important in blood coagulation and apoptosis, the concomitant activation of cell surface cholesterol could contribute to these and other pathophysiological signaling processes.
It has been observed that increasing the cholesterol in the mammalian PM1 beyond the physiological level evokes a sharp rise in its susceptibility to attack by CO and in its transfer both to extracellular cyclodextrin and the endoplasmic reticulum (1, 2). These findings have been explained by the following hypothesis (2, 3): Cholesterol forms complexes of varied strength and stoichiometry with different bilayer PLs. The complexed sterol has a lower chemical activity (escape potential or fugacity) than uncomplexed (free) cholesterol; for example, that added in excess of the binding capacity of the PL. It is the high chemical activity of the uncomplexed sterol that accounts for its increased interaction with CO and cyclodextrin. Physiologically, the high fugacity of excess cholesterol provides a homeostatic mechanism that keeps the PM sterol level near the capacity of its PL partners, ~0.8 mol per mol phospholipid (4). [Note that the concept of cholesterol chemical activity of interest here differs distinctly from that of active cholesterol, which refers to the attributes of biologically functional sterols (5).]
Cholesterol equilibrates rapidly between the two leaflets of the PM bilayer; however, its transbilayer distribution is debated (6). The transverse distribution of the PM PLs is asymmetrical, with a preponderance of PC and SM in the exofacial leaflet and anionic PL, mostly PE and PS, in the cytoplasmic leaflet (7). Furthermore, the chains of PC and SM tend to be more saturated than those of PE and PS, suggesting that the outer and inner leaflets of the PM bilayer have different physicochemical characteristics (8-10). In particular, evidence from model systems suggests that the exofacial PLs might interact more strongly with sterols than the endofacial species (11, 12). According to the hypothesis outlined above (2, 3), the activity of cholesterol in the inner leaflet might then be higher than that in the outer leaflet.
While the activity of membrane cholesterol is modulated by various intercalators, such as octanol, diglycerides, ceramides and lysophosphatidylcholine (13), the premise that the chemical activity of cholesterol in biological membranes is affected by neighboring PLs is in need of further substantiation. Given the aforementioned evidence of significant asymmetry in the PM bilayer, we reasoned that randomizing the PL distribution between the two leaflets should increase the activity of cholesterol at the cell surface, as reflected by a rise in sterol reactivity to CO and cyclodextrin. Two early observations gave indirect support for this premise. First, cholesterol is a far better substrate for CO in artificial vesicles composed of PL of the endofacial type than of the exofacial type (14). Second, the susceptibility of red cell surface cholesterol to CO is greatly increased in disrupted membranes (15). It was suggested at that time that translocated inner surface PLs could create a more favorable environment for the action of CO on the cholesterol in the outer leaflet of the ghosts.
To test this hypothesis in a biological context, we have now examined the effect of relaxing the PL asymmetry of the plasma membrane bilayer on the activity of surface cholesterol in human red cells and fibroblasts. We used the ionophore ionomycin to raise the level of cytoplasmic Ca++. Intracellular Ca++ serves to activate PM scramblase which then randomizes the PLs between the leaflets (16). We observed that this simple treatment caused a rapid increase in the apparent activity of PM cholesterol, thus confirming our hypothesis. We then asked whether phospholipid scrambling might also be the mechanism for the observed potentiation of CO attack by glutaraldehyde and by pre-incubation of cells in low ionic strength iso-osmotic buffer (15, 17, 18). This hypothesis was also confirmed.
EXPERIMENTAL PROCEDURES
Materials
Human red blood cells were obtained from normal volunteers and washed three times with 10 volumes of HBS or PBS. Human foreskin fibroblasts were cultured as described (4). 1α,2α-[3H]Cholesterol was from Amersham Biosciences. Ionomycin, TNBS, DIDS, CO (Streptomyces sp) and MBCD were all from Sigma. Amplex Red was from Molecular Probes. FITC-annexin V was obtained from BD Biosciences. Agents were delivered in buffer or in ethanol, the final concentration of which was always ≤ 2%.
Methods
Transfer of [3H]cholesterol from red cells to MBCD-cholesterol complexes was carried out as described (13). To gauge the action of CO, untreated and treated cells were washed, lysed in 5 mM NaPi (pH 8), the ghosts washed twice in the lysis buffer, and the oxidation of cholesterol determined as the disappearance of input cholesterol using Amplex Red. In experiments using glutaraldehyde-fixed cells, cholesterol oxidation was determined as the appearance of the product, cholest-4-en-3-one (cholestenone), using HPLC. The amount of ionomycin and CO was optimized for the various experiments. FITC-Annexin V binding was determined by flow cytometry according to the specifications of BD Biosciences.
RESULTS
Activation of PM cholesterol by cytoplasmic calcium
Human red cells are normally not susceptible to CO; however, oxidation can be elicited by a variety of non-disruptive perturbations (15). Conceivably, those treatments could act by bringing endofacial lipids to the outer surface. We examined this possibility by promoting the mixing of the PLs across the bilayer. This was done by activating membrane scramblase with elevated intracellular Ca++ (16). As seen in Fig. 1, as little as 0.02 μM of the ionophore, ionomycin, substantially increased the oxidation of cholesterol in intact red cells from its near-zero baseline. Complete oxidation was achieved with 0.05 μM ionomycin in the presence of 1 mM CaCl2. In this and all other cases, 1 mM EGTA abolished the effect of ionomycin (not shown). The effect of treating the cells with ionomycin plus Ca++, as judged by the induced oxidation of PM cholesterol by CO, began immediately, had a half-time of ~4 min and was complete within 15 min (Fig. 2). The rapidity of this effect is consistent with the kinetics of PL translocation by activated scramblases rather than a more complex intracellular process (16). (We frequently observed partial and variable hemolysis of the red cells late in CO time courses but only as a consequence of cholesterol oxidation. This effect does not change the interpretation of the data.)
FIGURE 1.

Effect of ionomycin on the oxidation of red cell cholesterol by cholesterol oxidase. Aliquots of washed and pelleted cells (5 μl bearing ~4.8 μg cholesterol) were preincubated for 5 min at 37 °C in 0.1 ml (final) HBS containing 1 mM CaCl2. Ionomycin was added and the cells were incubated for 5 min at 37 °C. Cholesterol oxidase (0.2 IU) was added and the mixtures were incubated for 30 min at 37 °C. The cells were washed and their residual cholesterol determined by Amplex Red analysis of their ghost membranes. Note that in this experiment, lipid scrambling was not inhibited and presumably continued throughout the 30 min oxidation step.
FIGURE 2.

Kinetics of activation of red cell cholesterol by PL scrambling. Aliquots of washed and pelleted cells (5 μl bearing ~4.8 μg cholesterol) were preincubated for 5 min at 37 °C in 0.1 ml (final) HBS containing 1 mM CaCl2. Ionomycin was added to 1 μM and the cells incubated at 37 °C for the times indicated. The cells were washed in HBS containing 1 mM EGTA to arrest scrambling and then incubated 100 μl HBS with 0.2 IU cholesterol oxidase at 37 °C for 30 min. The cells were washed and their residual cholesterol determined by Amplex Red analysis of their ghost membranes. (The first time point, taken at 10 sec, verifies that EGTA instantaneously inhibited the ionomycin effect.) The data were fit to a first order function with a half-time of 2.7 min.
To examine whether the action of ionomycin was in fact related to lipid scrambling and not to its simple intercalation into the membrane, we tested the effect of A23187, a chemically-unrelated Ca++ ionophore, and two monovalent ionophores, monensin and nigericin. While A23187 gave the same Ca++-dependent result as ionomycin, the monovalent ionophores had no effect with or without Ca++ (not shown).
Another indicator of membrane cholesterol activity is its rate of transfer to an aqueous acceptor, MBCD (2, 13, 19). We therefore tested the effects of increased cytoplasmic Ca++ on this process. Red cells were pre-equilibrated with an MBCD-cholesterol exchange partner so that the movement of subsequently-introduced [3H]cholesterol from the cells to the acceptor could be followed without mass transfer. Fig. 3 shows that ionomycin stimulated the rate of [3H]cholesterol transfer 1.9-fold; in two other experiments, the stimulation was 1.9- and 2.1-fold. The presence of 1 mM EGTA abolished the acceleration of sterol transfer by ionomycin (not shown).
FIGURE 3.
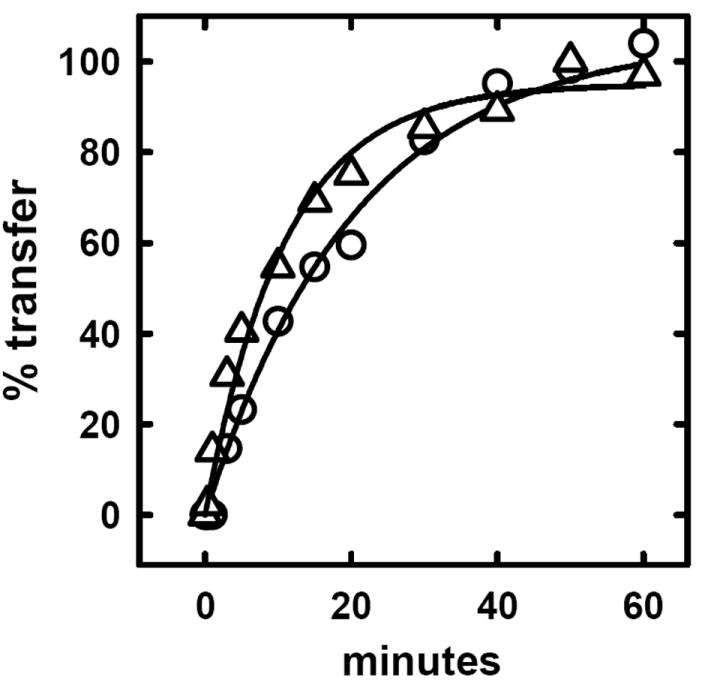
Effect of scrambling on the rate of transfer of cholesterol from red cells to cyclodextrin. As described previously (13), pelleted cells (160 μl) were brought to 16 ml in HBS containing 14.4 mg MBCD-cyclodextrin plus 1.6 mg cholesterol and pre-equilibrated for 10 min at room temperature. The cells and supernatant were separated by centrifugation, and both fractions saved. The cells were pulse labeled with [3H]cholesterol and washed. Two 60 μl aliquots of packed cell were then incubated for 15 min at 37 °C in 6 ml HBS containing 1 mM CaCl2 plus either the solvent alone (0.2% ethanol) or 1 μM ionomycin. The cells were pelleted and resuspended to 6 ml in the cyclodextrin-cholesterol mixture with which they had been previously equilibrated. The transfer of label to the supernatant at 10 °C was followed by periodically pelleting the cells; the supernatants were counted and compared to the unspun input. The data were fit to a first order expression with 100% transfer as the final (plateau) value. (O) Minus ionomycin; half time = 14 min. (Δ) plus ionomycin; half time = 7.5 min.
Phospholipid scrambling by cytoplasmic Ca++
We used annexin V, a high-affinity ligand for PS, to verify that the ionomycin-Ca++ treatment was in fact stimulating PL scrambling (16). We observed that preincubation with 1 μM ionomycin plus 1 mM CaCl2 as described above caused more than half of the cells to bind FITC-annexin V strongly (Fig. 4A). In the absence of Ca++, the fraction of ionomycin-treated cells stained by annexin V was less than 1.5%.
FIGURE 4.
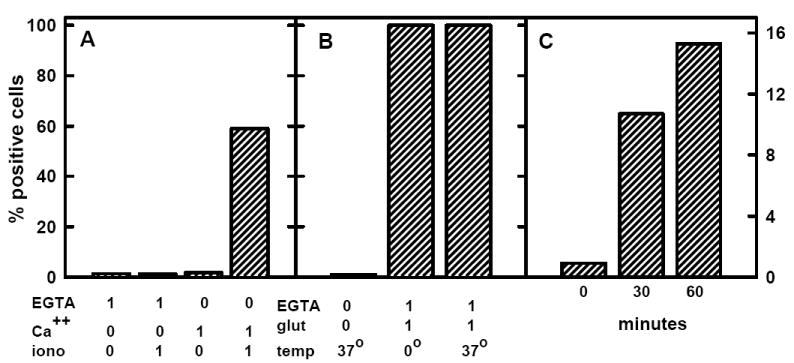
Annexin V binding to red cells. Panel A: Red cells were washed in HBS, suspended to 0.2 μl/ml (~2 × 106 cells/ml) in HBS containing 0 or 1 mM EGTA, 0 or 1 mM CaCl2 and 0 or 1 μM ionomycin and incubated 30 min at 37 °C. The cells then were washed and analyzed for the binding of FITC-annexin V. Panel B: As in panel A, except that 0 or 1% w/v glutaraldehyde was the agent and the 30 min incubation was performed at 0 or 37 °C. Panel C: Red cells were washed and 3 aliquots suspended to 0.2 μl/ml (~2 × 106 cells/ml) in 310 mM sucrose-5 mM histidine (pH 7.5). (This iso-osmotic buffer does lyse the cells or cause the leakage of hemoglobin.) The suspensions were incubated at 37 °C for the times indicated prior to chilling, washing and analysis. [Note that the full scale for panels A and B is 100% and for panel C is 16%.]
Blocking cholesterol activation with inhibitors of scramblase
We tested whether cytoplasmic Ca++ was activating PM cholesterol through PL scrambling or by some other mechanism. For this purpose, we tested the effect of two scramblase inhibitors, TNBS and DIDS, on the activation of red blood cell surface cholesterol (20). Both agents are membrane-impermeable, so that their action here could be assumed to be directed at the outer leaflet of the membrane. Using a concentration range previously found to inhibit scramblase (20), we found that both agents substantially reduced the ability of ionomycin plus Ca++ to evoke CO attack on PM cholesterol (Fig. 5).
FIGURE 5.

Effect of scramblase inhibitors on ionomycin-Ca++ promoted oxidation of red cell cholesterol. Washed cells (80 μl) were incubated for 30 min at 37 °C in 0.72 ml PBS, 25 μM DIDS or 1.5 mM TNBS, all containing 0.1 mM EGTA. The cells were washed and 56 μl aliquots (bearing ~54 μg cholesterol) incubated at 37 °C in 0.96 ml PBS containing 4 μM ionomycin plus 0.1 mM CaCl2. The reaction was stopped after 7 min by the addition of EGTA to 1 mM and the mixtures incubated with 0.2 IU cholesterol oxidase at 37 °C for 30 min. The cells were washed and their residual cholesterol determined by Amplex Red analysis of their ghost membranes.
Phospholipid scrambling by glutaraldehyde
We tested the hypothesis that the enhanced susceptibility of PM cholesterol in cells fixed with glutaraldehyde was mediated by the randomization of the transverse distribution of bilayer phospholipids. First, we verified the stimulatory effect of glutaraldehyde on the action of CO on red cells (Fig. 6). (Control experiments showed that the removal of ambient Ca++ with 1 mM EGTA did not block the stimulation of cholesterol oxidation by glutaraldehyde.) Then, we showed that glutaraldehyde stimulated the exchange-transfer of [3H]cholesterol from red cells to the acceptor, MBCD-cholesterol, a second indicator of enhanced cholesterol fugacity (Fig. 7). The stimulation was nearly 3-fold in these experiments. Finally, we demonstrated that treatment with glutaraldehyde greatly increased the appearance of phosphatidylserine at the surface of intact red cells in a Ca++-independent fashion (Fig. 4B).
FIGURE 6.
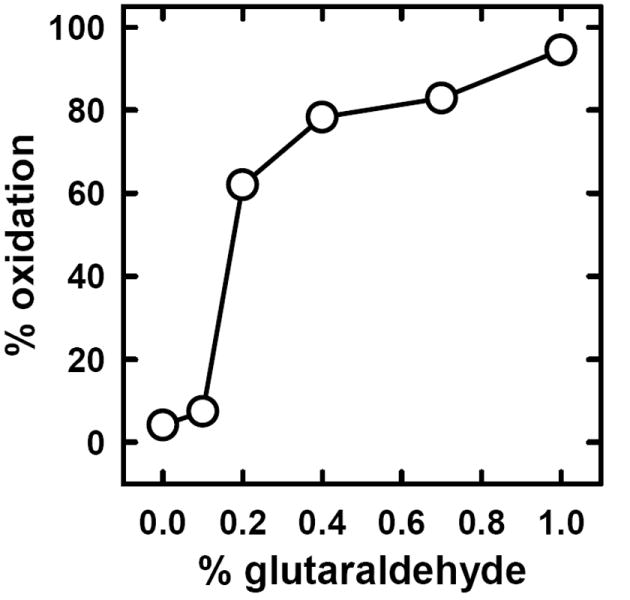
Effect of glutaraldehyde on the oxidation of red cell cholesterol by cholesterol oxidase. Washed cells (7 μl) were incubated for 10 min on ice in 100 μl chilled PBS containing 0 to 1% w/v glutaraldehyde. The cells were washed free of glutaraldehyde and incubated for 30 min at 37 °C in 0.1 ml PBS containing 0.3 IU cholesterol oxidase. The cells were washed and oxidation of their cholesterol determined by HPLC.
FIGURE 7.
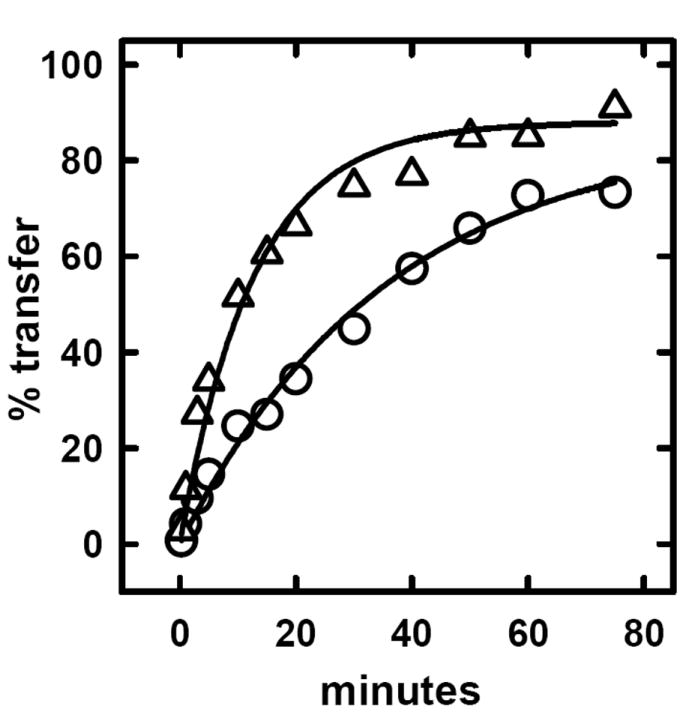
Effect of glutaraldehyde on the rate of transfer of cholesterol from red cells to cyclodextrin. The protocol was exactly as described in the legend to Fig. 3 except that one of two aliquots of labeled red cells was incubated for 10 min on ice with 1% w/v glutaraldehyde and washed before being transferred back to the cyclodextrin-cholesterol mixture with which the cells had been previously equilibrated. The exit of label to the supernatant was then followed at 10 °C. The data were fit to first order expressions. Half times of transfer were 24.6 min for the control (O) and 8.7 min for the glutaraldehyde-treated cells (Δ).
Phospholipid scrambling by exposure to low ionic strength
Because lengthy pre-incubation of intact red cells in iso-osmotic, low ionic strength buffer induces a lagged (accelerating) development of susceptibility to cholesterol oxidation (15), we examined whether this pre-treatment likewise acted by scrambling PM PLs. In support of this hypothesis, we found that incubation in low ionic strength buffer evoked the appearance of PS at the surface of the red cells in the absence of Ca++ (Fig. 4C).
Activation of fibroblast PM cholesterol by cytoplasmic calcium
We confirmed our observations on red cells using cultured human fibroblasts. As shown in Fig. 8, the susceptibility of fibroblast surface cholesterol to CO was greatly increased by ionomycin in a Ca++-dependent fashion, just as in red cells.
FIGURE 8.

Dependence of oxidation of fibroblast cholesterol on Ca++ and ionomycin. Cultured fibroblasts were dissociated from their flasks (3), washed and resuspended in HBS. Aliquots (containing ~2 μg cholesterol) were suspended in a final volume of 500 μl containing 0 or 1 mM CaCl2, 0 or 1 mM EGTA and 0 or 1 μM ionomycin. The mixtures were incubated for 10 min at 37 °C, 2 IU cholesterol oxidase was added and the incubation continued for 20 min at 37 °C. The oxidation of cholesterol was then determined by HPLC.
DISCUSSION
The principal finding of this study is that three kinds of perturbations that induce the redistribution of PM PLs across the bilayer also cause an increase in the activity (escape potential) of the cholesterol at the outer surface. The first of these perturbants is ionomycin. It seems very likely that the action of the ionomycin was mediated by PL scrambling (see Fig. 4A). First, ionomycin plus Ca++ caused the appearance of PS at the exofacial surface. The specific binding of annexin V to this inner-leaflet PL at the outer surface of intact cells is a well-established indicator of the redistribution of PM PLs and signifies the randomization of PL across the bilayer (16). Second, its effect was Ca++-dependent. Third, Ca++ had no effect without ionomycin; thus, Ca++ acts within the intracellular space, consistent with a scramblase effect (16). Fourth, two inhibitors of that transport protein (20) substantially block the effect of introducing Ca++ into the cytoplasm (Fig. 5). Fifth, another Ca++ ionophore, A23187, acted just like ionomycin; in contrast, nigericin and monensin, monovalent ionophores of a similar chemical form, had no effect on the accessibility of cholesterol to CO either in the presence or the absence of Ca++.
Sixth, the other two perturbants, glutaraldehyde and low ionic strength buffers, also caused PS redistribution (Fig. 4B and C). These data strongly suggest that PL scrambling was necessary and sufficient to activate cell surface cholesterol in these experiments but that scramblase and its activators were not. That the ability of glutaraldehyde to promote the susceptibility of PM cholesterol to CO (Fig. 6) involves an increase in the activity of the sterol is confirmed by its ability to also stimulate the transfer of the membrane sterol to cyclodextrin (Fig. 7). Unlike the first two perturbants, however, pre-incubation in low ionic strength buffer (90 min at 37 °C) did not appreciably enhance the rate of transfer of [3H]cholesterol to cyclodextrin (not shown). This negative result is consistent with the modest effect this treatment had on bilayer PL scrambling compared to ionomycin-Ca++ and to glutaraldehyde (Figs. 1 and 4). Presumably, exposure to low ionic strength increased the activity of surface cholesterol enough to be detected by the very sensitive CO susceptibility test but not by the less-sensitive [3H]cholesterol transfer assay. Although the molecular basis for the activation of exofacial PM cholesterol by cytoplasmic Ca++, glutaraldehyde and exposure to low ionic strength needs further elucidation, it is nevertheless parsimonious to suppose that their shared ability to bring PS to the outer leaflet is not a coincidence but, rather, mechanistic.
Free bilayer cholesterol molecules, like other lipids, have at least three kinds of activity. One is the potential to diffuse and distribute laterally in plane. The second is the potential to diffuse and distribute across the midplane (flip-flop). The third is the propensity of bilayer lipid molecules to transiently project into the aqueous phase, driven by thermal motion. While the lateral and transverse distributions of cholesterol are apparently each in rapid diffusional equilibrium, the tendencies of the sterol population at each leaflet to project into the neighboring aqueous space do not come to equilibrium but rather remain different from one another. The transfer of membrane cholesterol to aqueous acceptors such as cyclodextrin appears to depend on this bobbing motion (6). In particular, it is during these transient projections that sterol molecules are captured by collision with the acceptors. Fruitful encounters with CO would presumably require (and therefore report on) the same fleeting exposure (2, 13). We therefore assume that the form of cholesterol activity measured by both CO assays and the transfer to MBCD is its bobbing frequency. This basic molecular mechanism presumably is also required for the passive transfer of sterols to physiologic acceptors, including lipid binding proteins or lipoproteins in both the cytoplasm and the extracellular space (21).
The activity of membrane cholesterol is influenced strongly by its lipid environment (3, 13). A natural constraint would be that imposed by the association of the sterol with PLs (19, 22). If so, the activity of free cholesterol—that not bound by PL partners—will greatly surpass that of sterol molecules in complexes, leading to their increased interactions with exogenous probes. This is why both cholesterol oxidation and transfer to cyclodextrin increase sharply when the abundance of the sterol exceeds a stoichiometric equivalence with PL ligands and when the sterol is displaced from PL complexes by amphipaths (1-3, 13).
As discussed in the Introduction, data from model lipid systems support the prediction that stronger sterol complexes exist at the outer than the inner leaflet due to the asymmetric distribution of PM PLs. In that case, exofacial cholesterol should have a lower bobbing frequency or fugacity than that at the cytoplasmic surface. Consistent with this hypothesis is the demonstration in synthetic vesicles that PLs characteristic of the endofacial leaflet of PMs foster CO attack while the exofacial species inhibit it (14). Furthermore, the introduction of lysophosphatidylcholine (a membrane-intercalating PC analog with affinity for cholesterol) inhibits the action of CO on membrane cholesterol, while lysophosphatidylethanolamine and lysophosphatidylserine do not (15). It has been demonstrated that PLs characteristic of the inner leaflet have far less ability to associate with cholesterol in segregated domains (rafts) than the PL identified with the outer leaflet (11). Since such phase separation appears to be driven by complexation of the sterol with favorable PL species (19), these findings provide further evidence for differential interactions of cholesterol with the PLs in the two bilayer leaflets; hence, asymmetrical sterol fugacity. The implication of our findings that cholesterol is not strongly associated with the PL in the inner leaflet also bears on the question of domain (raft) formation at the cytoplasmic surface (11).
Thus, our results give strong support to the premise that the activity of the cell surface sterol reflects its lipid environment. Our findings also greatly strengthen the supposition that, because of the compositional asymmetry of the PM bilayer, the cholesterol in the exofacial leaflet is more strongly (completely) associated or complexed with the PL than that at the inner leaflet. There is, however, an alternative explanation for our data. Briefly, the low activity of cholesterol in membranes rich in PC and SM has been attributed to the ability of their large head groups to shelter the sterols from contact with water (23). Sterols in molar excess of the vicinal PLs—or sterols among PLs with small head groups—would not enjoy such protection and would have a higher activity. While both this “umbrella” model and that based on varied weak chemical complexes among sterols and PLs (19, 22) are compatible with our results, the latter provides a more satisfying formulation. In either case, relaxing the asymmetry between the two bilayer leaflets would be expected to decrease the sequestration of exofacial cholesterol by PL molecules and cause a corresponding increase its activity. This is our finding.
It is conceivable that scrambling activates cell surface cholesterol by increasing its abundance relative to the phospholipids in the outer leaflet. For example, there could be unequal exchange of PL across the scrambled bilayer. But even tiny alterations in the relative areas of the two lipid leaflets should change the shape of the red cells by a bilayer couple mechanism (24). That the biconcave disk shape was unaffected by the treatment of red cells with ionomycin plus Ca++ makes it more likely that the observed increase in the activity of their cholesterol reflects its new, low-affinity PL environment.
The activity of PM cholesterol is important in cholesterol homeostasis in nucleated cells. It has been suggested that the fugacity of the fraction of PM sterol in excess of the physiological threshold set by the capacity of the PLs quickly drives the sterol to the cell interior (1, 2, 25). There, it continually sets and resets the sterol level in the ER which, in turn, helps to govern the activity of manifold regulatory pathways in the ER. In particular, transient increments in ER cholesterol can rapidly reduce cell cholesterol levels through interaction with at least two cholesterol-regulated systems. These are the esterification of cholesterol via acyl-cholesterol acyltransferase and the proteolysis of hydroxymethylglutaryl-CoA reductase, leading to a prompt decrease in the rate of cholesterol biosynthesis (2). In addition, the expression of multiple genes for sterol accretion is adjusted by an ER sterol-sensing protein system (21, 26).
The scrambling of PM PLs in response to elevated cytoplasmic Ca++ might also increase the content of PC and SM in the cytoplasmic leaflet of the PM and thereby reduce the activity of endofacial PM cholesterol. This could lower the level of ER cholesterol in equilibrium with the PM (3). We were unable to test this hypothesis due to the deterioration of fibroblasts treated with ionomycin plus CaCl2. Furthermore, high cytoplasmic Ca++ could evoke so many varied responses that any particular outcome from such experiments would not carry much weight. Along these lines, it has been shown that excess cholesterol loading in macrophages promotes the release of ER Ca++ to the cytosol, thereby stimulating their apoptosis (27). Since elevated cytosolic Ca++ promotes PM PL scrambling, it should reduce the activity of cholesterol in the cytoplasmic leaflet of the PM. If this were to lead to reduced ER cholesterol, PM lipid scrambling might counter or buffer the release of ER Ca++ and, therefore, mitigate apoptosis.
Among the functions of the asymmetrical distribution of lipids in the PM bilayer is the information it conveys regarding the state of the cell. In particular, the scrambling of that asymmetry (in response to elevated cytoplasmic Ca++, for example) serves as a powerful signal for blood coagulation and apoptosis (16). It will be important to learn if the concomitant increase in cholesterol activity at the surface of the scrambled PM contributes to these and/or other pathophysiological processes. As one example, it has recently been reported that cholesterol itself may increase phospholipid randomization by scramblase (28) which, our data would suggest, could create a positive feedback loop of scramblase activation.
Acknowledgments
We thank Jeff Martinson for help with the annexin V analysis of lipid scrambling.
Footnotes
This work was supported in part by National Institutes of Health grant HL 28448.
The abbreviations used are: CO, cholesterol oxidase; DIDS, 4,4’-Diisothiocyanostilbene-2,2’-Disulfonic Acid; ER, endoplasmic reticulum; HBS, 150 mM NaCl-5 mM histidine, pH 7.5; MBCD, methyl-β-cyclodextrin; PBS, 150 mM NaCl-5 mM NaPi, pH 7.5; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PL, phospholipid; PS, phosphatidylserine; PM, PM; SM, sphingomyelin; TNBS, trinitrobenzenesulfonic acid.
References
- 1.Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;40:2264–70. [PubMed] [Google Scholar]
- 2.Lange Y, Ye J, Steck TL. How cholesterol homeostasis is regulated by plasma membrane cholesterol in excess of phospholipids. Proc Natl Acad Sci U S A. 2004;101:11664–7. doi: 10.1073/pnas.0404766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radhakrishnan A, McConnell HM. Chemical activity of cholesterol in membranes. Biochemistry. 2000;39:8119–24. doi: 10.1021/bi0005097. [DOI] [PubMed] [Google Scholar]
- 4.Lange Y, Swaisgood MH, Ramos BV, Steck TL. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J Biol Chem. 1989;264:3786–93. [PubMed] [Google Scholar]
- 5.Barenholz Y. Cholesterol and other membrane active sterols: from membrane evolution to “rafts”. Prog Lipid Res. 2002;41:1–5. doi: 10.1016/s0163-7827(01)00016-9. [DOI] [PubMed] [Google Scholar]
- 6.Steck TL, Ye J, Lange Y. Probing red cell membrane cholesterol movement with cyclodextrin. Biophys J. 2002;83:2118–25. doi: 10.1016/S0006-3495(02)73972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinn PJ. Plasma membrane phospholipid asymmetry. Subcell Biochem. 2002;36:39–60. doi: 10.1007/0-306-47931-1_3. [DOI] [PubMed] [Google Scholar]
- 8.Hullin F, Bossant MJ, Salem N., Jr Aminophospholipid molecular species asymmetry in the human erythrocyte plasma membrane. Biochim Biophys Acta. 1991;1061:15–25. doi: 10.1016/0005-2736(91)90263-8. [DOI] [PubMed] [Google Scholar]
- 9.Ohvo-Rekila H, Ramstedt B, Leppimaki P, Slotte JP. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41:66–97. doi: 10.1016/s0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- 10.Silvius JR. Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim Biophys Acta. 2003;1610:174–83. doi: 10.1016/s0005-2736(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 11.Wang TY, Silvius JR. Cholesterol does not induce segregation of liquid-ordered domains in bilayers modeling the inner leaflet of the plasma membrane. Biophys J. 2001;81:2762–73. doi: 10.1016/S0006-3495(01)75919-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leventis R, Silvius JR. Use of cyclodextrins to monitor transbilayer movement and differential lipid affinities of cholesterol. Biophys J. 2001;81:2257–67. doi: 10.1016/S0006-3495(01)75873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lange Y, Ye J, Steck TL. Activation of membrane cholesterol by displacement from phospholipids. J Biol Chem. 2005;280:36126–31. doi: 10.1074/jbc.M507149200. [DOI] [PubMed] [Google Scholar]
- 14.Patzer EJ, Wagner RR, Barenholz Y. Cholesterol oxidase as a probe for studying membrane organisation. Nature. 1978;274:394–5. doi: 10.1038/274394a0. [DOI] [PubMed] [Google Scholar]
- 15.Lange Y, Matthies H, Steck TL. Cholesterol oxidase susceptibility of the red cell membrane. Biochim Biophys Acta. 1984;769:551–62. doi: 10.1016/0005-2736(84)90053-1. [DOI] [PubMed] [Google Scholar]
- 16.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–88. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 17.el Yandouzi EH, Zlatkine P, Moll G, Le Grimellec C. Cholesterol distribution in renal epithelial cells LLC-PK1 as determined by cholesterol oxidase: evidence that glutaraldehyde fixation masks plasma membrane cholesterol pools. Biochemistry. 1994;33:2329–34. doi: 10.1021/bi00174a046. [DOI] [PubMed] [Google Scholar]
- 18.Lange Y, Ramos BV. Analysis of the distribution of cholesterol in the intact cell. J Biol Chem. 1983;258:15130–4. [PubMed] [Google Scholar]
- 19.McConnell HM, Radhakrishnan A. Condensed complexes of cholesterol and phospholipids. Biochim Biophys Acta. 2003;1610:159–73. doi: 10.1016/s0005-2736(03)00015-4. [DOI] [PubMed] [Google Scholar]
- 20.Kamp D, Sieberg T, Haest CW. Inhibition and stimulation of phospholipid scrambling activity. Consequences for lipid asymmetry, echinocytosis, and microvesiculation of erythrocytes. Biochemistry. 2001;40:9438–46. doi: 10.1021/bi0107492. [DOI] [PubMed] [Google Scholar]
- 21.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol Sensing, Trafficking, and Esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 22.Radhakrishnan A, McConnell H. Condensed complexes in vesicles containing cholesterol and phospholipids. Proc Natl Acad Sci U S A. 2005;102:12662–6. doi: 10.1073/pnas.0506043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Feigenson GW. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys J. 1999;76:2142–57. doi: 10.1016/S0006-3495(99)77369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim HWG, Wortis M, Mukhopadhyay R. Stomatocyte-discocyte-echinocyte sequence of the human red blood cell: evidence for the bilayer- couple hypothesis from membrane mechanics. Proc Natl Acad Sci U S A. 2002;99:16766–9. doi: 10.1073/pnas.202617299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan DP, Ohvo-Rekila H, Baumann NA, Beh CT, Menon AK. Sterol trafficking between the endoplasmic reticulum and plasma membrane in yeast. Biochem Soc Trans. 2006;34:356–8. doi: 10.1042/BST0340356. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 27.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–92. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 28.Damek-Poprawa M, Golub E, Otis L, Harrison G, Phillips C, Boesze-Battaglia K. Chondrocytes utilize a cholesterol-dependent lipid translocator to externalize phosphatidylserine. Biochemistry. 2006;45:3325–36. doi: 10.1021/bi0515927. [DOI] [PMC free article] [PubMed] [Google Scholar]