

Abstract
Superoxide reductase (SOR) is a non-heme iron enzyme which reduces superoxide to peroxide at a diffusion-controlled rate. Sulfur K-edge x-ray absorption spectroscopy (XAS) is used to investigate the ground state electronic structure of the resting high-spin and CN− bound low-spin FeIII forms of the 1Fe SOR from Pyrococcus furiosus. A computational model with constrained Imidazole rings (necessary for reproducing spin states), H-bonding interaction to the thiolate (necessary for reproducing Fe-S bond covalency of the high-spin and low-spin forms) and H-bonding to axial ligand (necessary to reproduce the ground state of the low-spin form) was developed and then used to investigate the enzymatic reaction mechanism. Reaction of the resting ferrous site with superoxide and protonation leading to a high-spin FeIII-OOH species and its subsequent protonation resulting in H2O2 release is calculated to be the most energetically favorable reaction pathway. Our results suggest that the thiolate acts as a covalent anionic ligand. Replacing the thiolate with a neutral non-covalent ligand makes protonation very endothermic and greatly raises the reduction potential. The covalent nature of the thiolate weakens the FeIII bond to the proximal Oxygen of this hydroperoxo species, which raises its pKa by an additional 5 log units relative to a primarily anionic ligand facilitating its protonation. A comparison with Cytochrome P450 indicates that the stronger equatorial ligand field from the porphyrin results in a low-spin FeIII-OOH species which would not be capable of efficient H2O2 release due to a spin-crossing barrier associated with formation of a high spin 5C FeIII product. Additionally, the presence of the di-anionic porphyrin π ring in Cytochrome P450 allows O-O heterolysis forming a FeIV-oxo porphyrin radical species, which is calculated to be extremely unfavorable for the non-heme SOR ligand environment. Finally the 5C FeIII site which results from the product release at the end of the O2− reduction cycle is calculated to be capable of reacting with a second O2− resulting in superoxide dismutase (SOD) activity. However in contrast to FeSOD the 5C FeIII site of SOR which is more positive is calculated to have a high affinity for the binding a 6th anionic ligand which would inhibit its SOD activity.
Keywords: Superoxide Reductatse, S K-edge XAS, DFT, Reaction Mechanism
INTRODUCTION
The enzyme superoxide reductase (SOR) is found in microaerophilic and strictly anaerobic microorganisms and is capable of reducing superoxide to peroxide.1, 2, 3, 4,5 Though its in vivo function is not completely understood, it is thought to provide a mechanism for combating oxidative stress in anaerobes, which typically lack the classical superoxide dismutases (SOD), by depleting superoxide concentration via direct reduction to hydrogen peroxide, rather than by a dismutation reaction which produces H2O and O2. 6 Thus, unlike SOD, SOR does not accumulate dioxygen in these high anoxic microorganisms. The crystal structure of the resting oxidized form of the 1Fe-SOR from Pyrococcus furiosus (Fig. 1) shows that the ferric active site has a non-heme iron coordinated to four histidines in the equatorial plane and a cysteine and glutamate in the trans-axial positions.7 There are two peptide N–H---SCys H-bonds to the axial cysteine ligand. Both 1Fe and 2Fe SOR’s have been characterized and found to have similar active site structures and reactivities.8, 9 The second Fe in the 2Fe SOR is in a rubredoxin-like Fe(SCys)4 site for which the function is unknown as eliminating it by mutation does not affect the reactivity of this enzyme.9
Fig. 1.
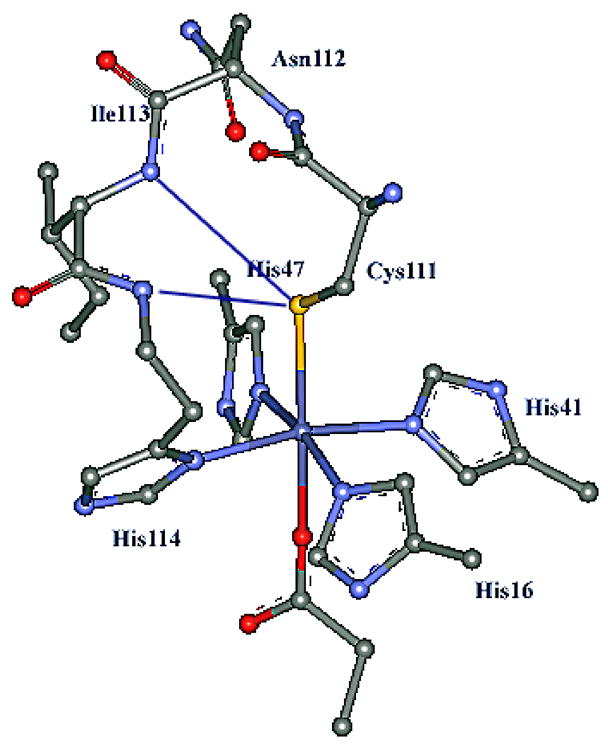
Active site of SOR from P. furiosus at 1.7 Å resolution (pdb:1DQI).7 Residues Ile113 and Asn112 are in the vicinity of the active site and proposed to H-bond to the Cys111 thiolate.
The resting oxidized form of 1Fe P. furiosus SOR has been spectroscopically characterized in detail by Clay et. al.10, 11 It has a high-spin ground state and an intense CysS− → FeIII charge transfer (CT) band at 15100 cm−1 (ε ~2500 M−1cm−1) which gives it its characteristic blue color. The EPR spectrum of the high-spin resting state indicated that it is quite axial (E/D = 0.06) and VTVH MCD indicated that a trans NHis-Fe-NHis axis defined the unique direction of the ZFS tensor and not the trans SCys-Fe-OGlu axis.10 This site is solvent exposed and binds exogenous ligands such as N3− and OH− in place of the glutamate. Binding CN− converts it to a low-spin site with an intense CysS− → FeIII CT at 14600 cm−1 (ε ~2700 M−1cm−1).10
The reaction mechanism of SOR, involving transfer of an electron and two protons to superoxide to form hydrogen peroxide, has been studied in detail using kinetic and spectroscopic techniques.12, 13, 14, 15, 16, 17,18 Using kinetic data, Nivière et al. and Kurtz et al. have proposed the presence of at least one FeIII intermediate (henceforth referred to as intermediate I, characterized by a CT band at 600 nm) in the reaction. The formation of the intermediate was diffusion controlled and had no pH dependence or deuterium isotope effect and thus was proposed to be an FeIII-μ2-O22− species. The decay of this intermediate involved a diffusion controlled protonation from its pH dependence and a deuterium isotope effect of 2. Lys 14 has been identified as a participating 2nd sphere residue (mutation of this residue decreases the turnover rate by a factor of 20–30) and has been proposed to assist superoxide binding to the active site.19, 20 Though no intermediate has been trapped with the physiological substrate superoxide, Olivier et. al. have trapped and characterized (using EPR, resonance Raman, Mössbauer spectroscopy and DFT calculations) a high-spin (S=5/2) side-on η2-O22−-FeIII species by addition of H2O2 to the resting FeII form of the enzyme.21, 22 The relevance of such species in the turnover cycle of SOR is still an issue.23
A computational study has used DFT to investigate the ground state and has considered possible intermediates along the reaction pathway.17 The study successfully reproduced most of the geometric parameters and spin state of the experimentally observed high spin ferrous and ferric ground state and concluded that a low-spin FeIII-OOH is the best candidate for the Intermediate I. A subsequent protonation driven H2O2 release from this low-spin FeIII-OOH was proposed, though the energetic feasibility of such steps were not evaluated.17 Kovacs et. al. 24 reported a high-spin and Kovacs et. al. 25 and Goldberg et. al. 26 reported low-spin FeIII-OOH(R) intermediates upon reacting FeII SOR model complexes with superoxide or alkyl peroxide in protic solvents. The former study reported weak Fe-O and high O-O stretches in the rRaman spectrum which correlated with the facile rate of dissociation of H2O2 from the FeIII-OOH intermediate in presence of weak acids e.g. methanol.24
The presence of an axial cysteine ligand distinguishes the SOR active site from all other catalytic non-heme Fe enzymes. The role of this thiolate ligand is currently an open issue. The weak Fe-O bond in the high-spin FeIII-OOH model by Kovacs et. al. requires a strong trans effect of the axial thiolate.24 The trans effect of the thiolate has also been proposed to affect NO binding to this active site.27 The thiolate may also play a major role in controlling the reduction potential of the active site.23 Alternatively, there is some similarity between the SOR active site and the active site of cytochrome P450 where the four porphyrin Nitrogens are in the equatorial plane and there is an axial cysteine ligand.28 However, these sites differ drastically in reactivity. SOR releases hydrogen peroxide via proton assisted heterolytic cleavage of the Fe-O bond and P450 activates O2 to oxidize substrates via a low-spin FeIII-OOH intermediate which protonates at the distal Oxygen to heterolytically cleave the O-O bond to form Compound I.29, 30 Also, in spite of having a favorable redox potential (+250 mV), the SOR active site is incapable of performing SOD activity.31 The reduced SOR (FeII form) will reduce O2− to H2O2, but the oxidized SOR (FeIII form) will not oxidize O2− to O2.23 This is crucial in maintaining low oxidative stress in anaerobes that are proposed to use SOR to deplete the superoxide concentration in vivo. However FeSOD with Nitrogen and Oxygen based ligands can perform both reactions. The lack of an open co-ordination site has been suggested to be a possible reason for this23 although the origin of the affinity for this 6th ligand binding or the feasibility of a SOD reaction in a SOR active site has not been evaluated. Thus it is important to understand the role of the axial thiolate ligand and the lack of heme in determining the reactivity of SOR.
Sulfur K-edge X-Ray Absorption Spectroscopy (S K-edge XAS) is a relatively new spectroscopic method that has been used to study metal-Sulfur (M-S) bonding and how it is affected by the protein in various Cu-S and Fe-S electron transfer (ET) active sites.32,33,34 The S K edge refers to the ionization of Sulfur 1s (S1s) orbital. This is a ligand based transition and thus this technique does not suffer from the complications that arise from the interaction of the core hole with the metal 3d manifold in the final state.35 The, lower-energy pre-edge transitions (pre-edge) to unoccupied metal 3d (M3d) orbitals gain intensity through Sulfur 3p (S3p) mixing due to covalency and because the Sulfur 1s→3p transition is electric dipole allowed. Thus the S K pre-edge intensity is a direct experimental probe of M-S bond covalency. Recently, we have used this method to investigate the active site structure and bonding in a series of P450 model complexes to quantitatively understand the effect of H-bonding on Fe-thiolate bond covalency and redox potential.36
In the present study we use S K-edge XAS to quantitatively define the Fe-S bond covalency in the resting high-spin and CN− bound low-spin ferric forms of SOR from P. furiosus. We use the S K-edge results in addition to other well defined experimental parameters, e.g. geometry, spin states and Fe-S bond covalency etc., to develop a computational model for the resting ferric SOR active site that required inclusion of H-bonding to the axial thiolate ligand. The S K-edge XAS data on the low-spin CN− bound form are then used to evaluate computational models for this spin state change and further expand the model to include H-bonding to the exchangeable axial ligand. The model that best fits these experimental data is then used to computationally investigate the proposed mechanism of SOR and evaluate possible intermediates. The role of the trans axial thiolate and H-bonding to it in the SOR reaction are computationally evaluated. The FeIII-OOH species in the SOR site is compared to that in P450 (compound 0). The role of the non-heme vs heme ligand in difference in their reactivity is evaluated. A hypothetical SOD reaction cycle is constructed for the SOR active site and compared to that for the Fe-SOD active site to obtain insight into the lack of SOD reactivity of SOR.
EXPERIMENTAL DETAILS
Sample Preparation
The recombinant form of P. furiosus SOR was obtained from E. coli as described in ref. 10. Absorption at 660nm (ε = 2500 M−1cm−1)10 indicated that 85% of the protein was loaded with Fe. The protein solutions (in 100 mM phosphate buffer having pH 7.2–7.3) were pre-equilibrated in a water-saturated He atmosphere for ~1 h to minimize bubble formation in the sample cell. The protein samples was oxidized or reduced before experiments by using a ~3–4-fold excess of sodium hexachloroiridate or ascorbic acid solution in the same buffer as the protein solution. The CN− bound form was generated by adding 40 equivalent of CN− to a pre-oxidized sample at pH 9.0. The complete conversion to the low-spin form was confirmed by room temperature absorption and EPR spectroscopy at 8K (Fig. S1, supplementary information). Samples were loaded via a syringe into a Pt-plated Al block sample holder sealed in front using a 6.3 μm polypropylene window and maintained at a constant temperature of 4°C during data collection using a controlled flow of N2 gas pre-cooled by liq. N2 passing through an internal channel in the Al block.
Data Collection and Reduction
XAS data were measured at the Stanford Synchrotron Radiation Laboratory using the 54-pole wiggler beam line 6–2. Details of the experimental configuration for low energy studies have been described previously.37 The energy calibration, data reduction and error analysis follow the same approach described previously.38
Fitting Procedures
Pre-edge features were fit using pseudo-Voigt line shapes (sums of Lorentzian and Gaussian functions) using the program PEAKFIT. This line shape is appropriate as the experimental features are expected to be a convolution of a Lorentzian transition envelope39 and a Gaussian line shape imposed by the beam line optics.40 A fixed 1:1 ratio of Lorentzian to Gaussian contribution successfully reproduced the pre-edge features. The rising edges were also fit with pseudo-Voigt line shapes. Fitting requirements included reproducing the data and its second derivative, using a minimum number of peaks. Fits were performed using single peaks to simulate the pre-edge feature of each component with a half-width of ~0.9 eV. The intensity of a pre-edge feature (peak area) represents the sum of the intensity of all the pseudo-Voigt peaks which were needed to fit the feature in a given fit. The fitted intensities were converted to %S3p character using the pre-edge feature of plastocyanin as a reference (where 1.27 units of intensity, obtained using this program, corresponded to 38% S3p character).41 Note that the 85% loading of the active site was accounted for by renormalization of the data. This is because the free cysteines that are not bound to iron will contribute to the rising edge but not to the pre-edge.
DFT Calculations
All calculations were performed on dual Intel Xeon workstations using the ADF 2004.01 and the Gaussian 03 package.42 The different computational models used in this study are described and evaluated in the results section. The geometries were optimized using either a) the meta GGA BP86 functional available in the ADF package using a TZP basis set on all atoms or b) with B3LYP method using 6-311G* basis sets on Fe, S, N, O atoms and 6-31G* basis set on C and H atoms using the Gaussian 03 program.43 The default convergence criteria in both software were used during optimization. For the Gaussian optimized geometries the energies were obtained from single point calculations using 6-311G* basis-set on all atoms. The stability of the ground states was tested using the “stable” command in the Gaussian program. Frequency calculations were performed on the optimized geometries to ensure that they had no imaginary mode. Only the larger H-bonded models having 7 frozen coordinates had 4–5 <30 cm−1 imaginary modes involving these frozen co-ordinates. For calculating the SOD reaction, the co-ordinates from the X-tal structure of Fe-SOD from P. shermanii (pdbid:1BSM) were used for the starting structure.44 The α carbons of the histidine and the glutamate residues were frozen during optimization. All other geometric parameters were optimized. Mulliken45 population analyses were performed using PYMOLYZE46 program and the molecular orbitals were plotted using Molden ver. 4.1 with a contour value of 0.05. For solvent corrections, experimental free energies of solvation were used for all small ions (since the site is solvent exposed) and a PCM model47 with an epsilon of 4.0 was applied to the active site models.48 The fragment dissociation energies (bond energies) were computed by comparing the energies of the full molecule to the sum of the energies of the fragments. To check for basis set supposition error we have estimated the energies of the two weakly endothermic key protonation steps with a larger 6-311+g** basis set and found less than 2 Kcal difference in the calculated energies.
RESULTS
A. S K-edge XAS
The S K-edge XAS data for the resting oxidized (blue), CN− bound oxidized (red) and reduced (black) forms of SOR enzymes are shown in Fig. 2. The XAS data of the resting oxidized species show two distinct pre-edge features at 2469.3 eV and 2470.5 eV and a rising edge transition at 2472.8 eV. The two pre-edge features correspond to S1s transitions to the t2 and e half occupied molecular orbitals of the resting high spin ferric ground state. These features can be quantified by fits to the experimental data to have 0.49 and 0.33 units of normalized intensity. These correspond to 15±2% and 10±2% S3p character in the singly occupied d-π (t2) and the d-σ (e) orbitals in the d5 resting oxidized high-spin ground state. The CN− bound low-spin form (Fig. 2, red) also exhibits t2 and e transitions at 2469.5 eV and 2470.7 eV with 0.50 and 0.48 units of intensity, respectively.49 These correspond to 13±2% and 15±3% S3p character mixed into the singly occupied t2 π and unoccupied e σ orbitals, respectively, of the low-spin d5 ferric state. The significant intensity in the low-energy peak indicates that the single d-π t2 hole is oriented along the Fe-S bond. Although one may have expected the e intensity to go up in the low-spin form (as both α and β Fe-S d-σ orbitals are unoccupied in the low spin state relative to one β d-σ orbital in the high spin state and the Fe-S bond length is shorter) the intensity actually goes down. However for low-spin FeIII the assignment of the pre-edge features is complicated by multiplet effects in the final state. The S1s→Fe3d transition produces an L1t25e1 excited state configuration (where L = ligand hole) which produces two 2A excited states (the t25e1 configuration produces a singlet and triplet state which couple to the ligand hole). These wavefunctions were obtained as linear combinations of the parent microstates using lowering operators.50, 51 The 2A state originating from the parent dn+1 triplet configuration has three times the intensity of the 2A originating from the dn+1 singlet parent configuration. The observed pre-edge feature at 2470.5 eV can be assigned as the transition to the lower 2A state (originating from the triplet dn+1 parent) with 3/4th of the intensity while the higher energy multiplet should be shifted up in energy (estimated ~1 eV from DFT)52 and obscured by overlap with the C-S σ* transition.53 Corrected for this multiplet shift of intensity the total intensity for the e transition in the low-spin state is 20%.
Figure 2.

S K-edge XAS of superoxide reductase in its reduced (black), resting oxidized (blue) and CN− bound oxidized (red) forms. The 2nd derivatives of the data are shown in dotted lines. The FeII pre-edge feature is indicated by the black arrow.
The data for the resting reduced form show an intense S1s→ SC-S σ* transition at 2473.6 eV. The S1s→ FeII3d pre-edge transition appears as a weak feature in the 2nd derivative of the data54 at 2472.1 eV (Fig. 2, arrow on dotted black line) overlapping with the stronger feature at 2473.6 eV.
B. DFT Modeling
a. High-Spin resting
The active site was modeled using the crystal structure of 1Fe SOR (pdb: 1DQI)7 with four neutral imidazole rings (representing the coordinated histidines), an axial methyl thiolate (representing cysteine) and an axial acetate (representing glutamate) (fig. 3A). Geometry optimized DFT calculations were performed using BP86 and B3LYP methods on four sets of models (B, C, D and E) presented in figure 3. The relevant optimized parameters are given in Table 1. Full optimization of the active site using BP86 with no constraints led to structure B (Fig. 3) where the metal-ligand bond lengths were in good agreement with the published solution EXAFS results (Fig. 3A), and the 1.7 Å resolution crystal structure.10, 7 However, the optimized low-spin state was found to be more stable by 11.2 Kcal/mol (Table 1, row 1). This indicates that the full optimization, which does not include the constraints imposed by protein environment, does not correctly describe the ground state at the BP86 level. This could be either a result of the simplified model that does not account for the constraints imposed on an active site by the protein backbone or an artifact of using a pure density functional, which generally stabilizes a low-spin state.55
Figure 3.
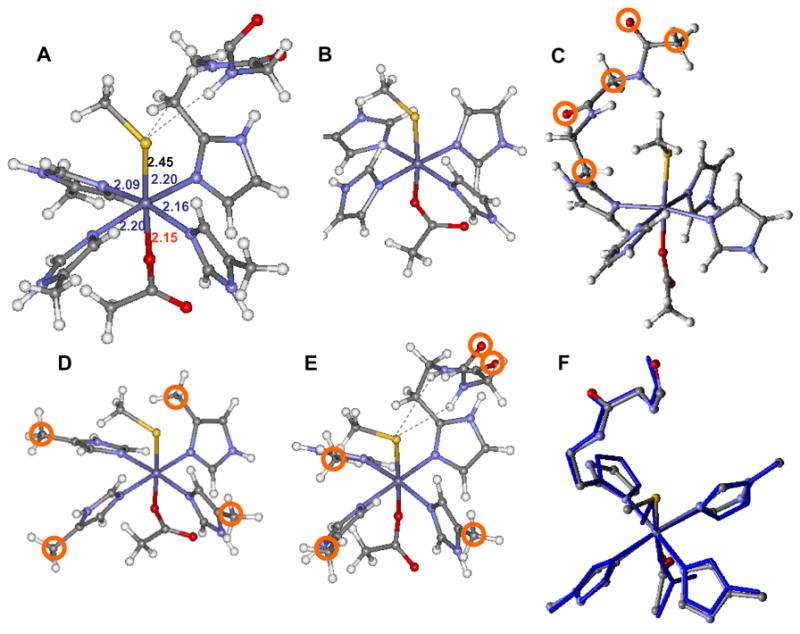
The crystal structure of the active site of resting oxidized SOR (A), the unconstrained model (B), H-bonded only model (C)60, α carbon constrained model (D), H-bonded and α-carbon constrained model (E) and overlay between the optimized model E (in blue) and X-tal structure (atom colored) from 1DQI (F). The constrained atoms are circled in the respective structures. Bond lengths of the protein crystal structure are given in Å and are color coded as Fe-S, Fe-N and Fe-O.
Table 1.
Calculated geometric parameters and energy difference of models B, C, D and E using BP86 and B3LYP methods.
| Method | Model | Fe-Sa | Fe-N1a,b | Fe-N2a,b | Fe-Oa | C-S-Fec | N-Sa | S- -H-Nc | dΔEHS-LS |
|---|---|---|---|---|---|---|---|---|---|
| X-Ray | 1DQI | 2.46 | 2.12 | 2.20 | 2.15 | 117.0 | 3.50, 3.72 | 131.7, 165.1 | |
| EXAFS | 2.36 | 2.17 | 2.07 | 2.15 | |||||
| BP86 | B-HS | 2.39 | 2.18 | 2.22 | 2.03 | 111.5 | +11.2 | ||
| B-LS | 2.21 | 2.00 | 2.03 | 2.03 | 113.0 | ||||
| C-HS | 2.42 | 2.20 | 2.22 | 1.97 | 121.4 | 3.62, 3.82 | 112.8, 140.9 | +9.0 | |
| C-LS | 2.20 | 2.01 | 2.02 | 1.98 | 114.7 | 3.69, 3.97 | 107.2, 137.1 | ||
| D-HS | 2.39 | 2.15 | 2.20 | 2.03 | 120.1 | +4.5 | |||
| D-LS | 2.23 | 2.05 | 2.06 | 2.00 | 124.4 | ||||
| B3LYP | D-HS | 2.43 | 2.18 | 2.18 | 2.01 | 109.9 | −20.0 | ||
| D-LS | 2.31 | 2.02 | 2.03 | 1.98 | 109.6 | ||||
| E-HS | 2.43 | 2.15 | 2.20 | 1.95 | 111.4 | 3.65, 3.90 | 126.0, 144.5 | −6.8 | |
| E-LS | 2.28 | 2.07 | 2.04 | 1.97 | 113.0 | 3.81, 4.03 | 121.8, 143.6 |
Units Å
T he Fe-N distances are grouped together representing long and short Fe-N bonds.
Units degrees
Units Kcal/mol
Two conserved H-bonds to the axial CysS− (see Fig. 1) from the amide N-H’s of His114 and the Ile113 residues are present in the active site of SOR that are absent in model B. These H-bonds can affect the covalency of the axial Fe-S bond and may be an important determinant of the spin state. Thus the two H-bonds were also included in the model and optimized (the carbonyl Oxygen of the H-bonding amides were frozen in space during optimization as these are strongly H-bonded to 2nd sphere residues, see Fig. 3C). The optimized bond-lengths and bond angles of the high-spin and low-spin states are give in Table 1, Column 3 and 4, respectively. The H-bonding to CysS causes ~0.03 Å elongation of the Fe-S bond length in the optimized structures for both spin sates. Also the H-bonding interaction is weaker in the low-spin state as indicated by the longer N-S heteroatom distances and less linear N-H---S angles (Table 1). This is because the Fe-S bond is more covalent in the low-spin state which shifts electron density away from the thiolate and weakens the H-bonding interaction. Note that even in this model the optimized structure of the low-spin state shows significant shortening of the Fe-NHis bond length relative to the high-spin state (like in model B) and this results in a 9 Kcal/mol more stable low-spin state. This implies that though the H-bonding weakens the Fe-S bond, it does not result in a high-spin ground state for this model of SOR.
In the crystal structure of the enzyme the co-ordinated Histidine residues are involved in direct or indirect (via bridging water molecules) H-bonds from peptide residues (e.g. pro42). These H-bonding interactions will oppose the shortening of Fe-NHis bond lengths observed in these unconstrained calculations that lead to a low-spin ground state. To simulate this tethering of the Histidine residues the active site model was expanded to include the α carbon of the four histidine rings that were frozen during optimization (Fig. 3D). Though the resulting geometry is generally in good agreement with the solution EXAFS results (Table 1, row 3), the low-spin ground state is still more stable; albeit, the energy difference between the spin states in this model is only 5 Kcal/mol relative to 11.2 Kcal/mol in the unconstrained model without the H-bond (Fig. 3B).56, 57
DFT calculations were performed on the model C using the hybrid B3LYP method, which is reportedly less covalent than a pure functional and thus favors the high-spin state.58 In this α carbon constrained model (Fig. 3C) the optimized high-spin ground state is more stable than the optimized low-spin ground state by 20 Kcal/mol, thus reproducing the experimentally observed high-spin ground state of resting six-coordinate SOR active site.59 The optimized Fe-S distances in B3LYP are ~0.04Å longer than those obtained from BP86 calculations. This is a consequence of over estimation of Fe-S bond covalency by a pure functional.58 However, this model does not reproduce one short N-Fe-N (Fe-NHis 2.09 Å, 2.16 Å) and one long N-Fe-N (Fe-NHis 2.20 Å and 2.20 Å) axis as observed in the active site structure. Though this geometric feature may not contribute to reactivity, it serves as another parameter to test the accuracy of these calculations.10 As a final step we included the H-bonding (which affected the Fe-S bond length) and the α carbon constraints on the histidines (which affected the spin state) in the same model (Fig. 3 E). This B3LYP optimized structure reproduces the experimental Fe-S bond length and has one short N1-Fe-N1 axis ((Fe-N1 2.17Å and 2.14 Å) and one long N2-Fe-N2 axis ((Fe-N2 2.20 Å and 2.19 Å) and the high spin state is 6.8 Kcal/mol more stable. Note that the H-bonding residue is covalently linked to one of the histidine ligands with the longer Fe-NHis bond (Fig. 3E). This suggests that this Fe-NHis bond elongation may be a result of steric interaction between the H-bonding arm and the histidine imidazole ring. We considered the possibility of a H2O molecule H-bonded to the Glu residue since the active site is solvent exposed (Table 1, F) and there is direct Raman evidence for a solvent interaction in the low-spin CN− form (vide infra). However, no significant change in geometry was observed other than a ~ 0.04 Å elongation of the Fe-O Glu bond upon H-bonding.
Electronic structure
The MO diagrams (spin unrestricted unoccupied β orbitals from the B3LYP calculations) of the resting six-coordinate ferric site without (left) and with (right) H-bonding show a pseudo-octahedral ligand field (Fig. 4, left). In both models there are two Fe-S bonding interactions. The lowest energy t2 orbital forms a π bond with the out of C-S-Fe plane CysS3p orbital while the highest energy e orbital has a pseudo-σ interaction with the in plane S3p orbital (Fig. 4, Fe-S π and Fe-S σ). 61 In the non-H-bonded models these %S3p covalencies are calculated to be 28% (π) and 15% (σ). These are much higher than the experimental values (15% and 10%, respectively), which suggests the H-bonding interaction in the SOR active site might have a significant effect on the S-Fe bond covalency. Indeed in the H-bonding model, the calculated %S3p character in these orbitals are 17% and 12%, in reasonable agreement with the experimental values.62, 63 The decrease in Fe-S bond covalency is mainly in the π bonding orbital consistent with the orientation of the H-bonds (Fig. 3 A). It is also worth noting that the total calculated %S3p using the BP86 method on the high-spin H-bonded model (Fig. 3 C) are ~39–40% which are again much higher than the experimental value of 25% (15% π + 10% σ). This reflects the over estimation of Fe-S bonding by a pure functional and further indicates that B3LYP is better for describing the exchange in this system.
Figure 4.
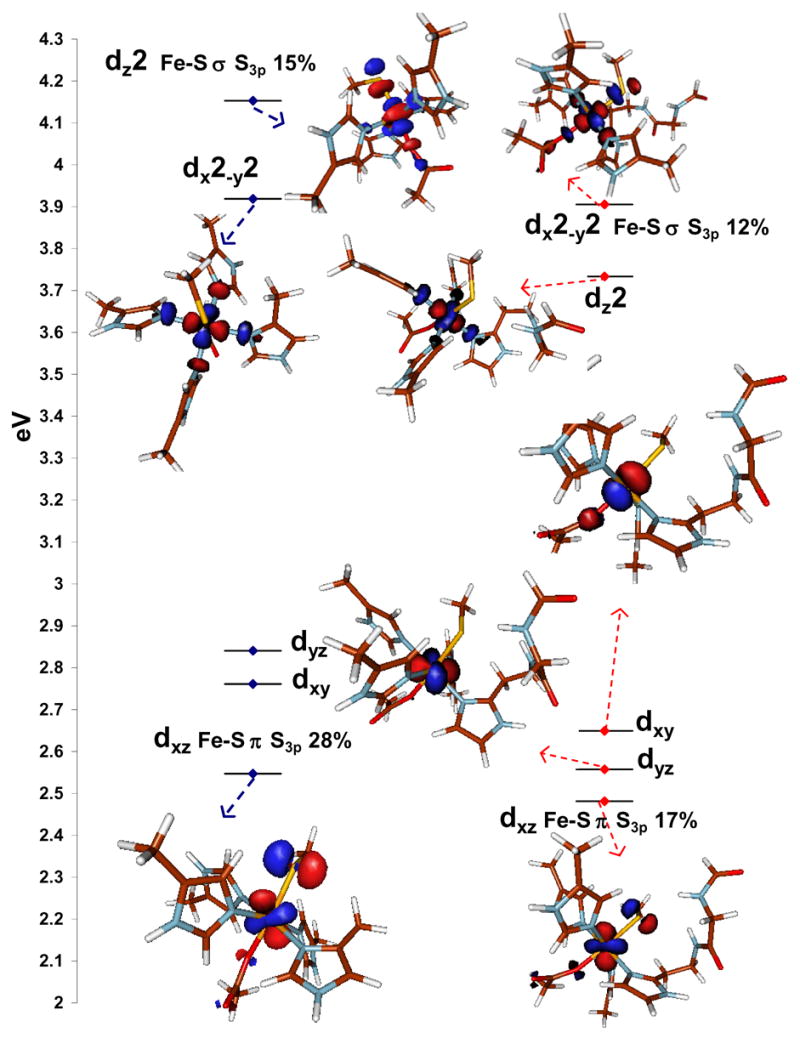
The MO diagrams of the resting six-coordinate high-spin FeIII active site without, (left) and with (right) H-bonding interactions. The S3p co-efficients are indicated in text. Only the β contours are shown. The dyz and dxy contours of the model without H-bonding are not shown to avoid crowding.
b. Low-spin CN− adduct
The CN− bound low-spin ferric active site was modeled with the α carbon constraint that proved important to reproduce the correct ground state for the resting FeIII site (Fig. 3D). The geometry optimized calculations on the CN− bound form of model 3D indicates than the low-spin form is more stable by 90 Kcal/mol consistent with experiments. Thus these α carbon constraints were successful in reproducing the experimentally observed spin states of both the resting and the CN− bound forms. Further the H-bonds to the thiolate were included (model E) as they were crucial in reproducing the experimental covalency of the resting oxidized form.
A recent study using vibrational spectroscopy and pulsed EPR techniques by Clay et. al. has indicated the presence of at least three CN− bound forms in solution; One linear and two bent conformations having Fe-C-N angles of 175°, 131° and 125°, respectively.20 From 2H2O isotope effect observed on one set of C-N vibrations, it was proposed that the linear conformation is H-bonded to a solvent molecule.20 Thus in addition to the α-carbon constraints and H-bonding interactions that were necessary to reproduce the ground state spin and Fe-S covalency of the high-spin model, an H-bond to the axial CN− ligand from a H2O molecule was also included in the LS model. Both the optimized geometries (E and E+H2O, Table 2) have a linear Fe-CN unit and have shorter Fe-S and Fe-N bond lengths relative to the resting form consistent with the low-spin state of the CN− form. The optimized geometries with and without the H2O H-bonded to the CN− shows that H-bonding leads to an elongation of the Fe-CN bond by 0.02 Å, which is compensated by 0.05 Å shortening of the Fe-S bond (Table 2, E and E + H2O). This suggests that H-bonding to CN− weakens the Fe-CN bond which is compensated by strengthening of the trans Fe-S bond. These interactions determine the orientation of the single d-π hole in this low-spin FeIII center (vide infra).
Table 2.
Optimized bond distance and angles of linear, bent and H-bonded CN− models using B3LYP
| Fe- S | Fe-N1 | Fe-N2 | Fe-C | C-N | Fe-S-C | Fe-C-N | N-S | S- -H-N | |
|---|---|---|---|---|---|---|---|---|---|
| E | 2.42 | 2.03 | 2.06 | 1.95 | 1.17 | 113.1 | 171.6 | 3.72, 3.97 | 126.5, 146.1 |
| E + H2O | 2.37 | 2.03 | 2.06 | 1.97 | 1.17 | 115.3 | 174.6 | 3.74, 4.02 | 125.6, 145.9 |
The MO diagrams of CN− bound L.S. models E and E + H2O (H-bonded to thiolate and H-bonded to thiolate and CN−, Fig. 5 left and right, respectively) show a normal low-spin t25 configuration with one singly occupied β t2 hole and two pairs (α+β) of un-occupied e orbitals (only three β orbitals shown in Fig. 5). However, there is a very significant difference in their electronic structure. The t2 hole of the low-spin FeIII center is localized in the in-plane dxy orbital in the H-bonded to thiolate model E, while it is in the dxz (which is the Fe-S d-π antibonding) orbital in model E + H2O. The description of model E is in contrast to the experimentally observed low-lying S→ Fe CT, the low energy t2 pre-edge feature in the S K-edge XAS and recently published ENDOR data all of which indicate that the t2 hole in the CN− bound state is in the Fe-S d-π orbital.20 The nature of the t2 hole is determined by an interplay between the stabilization of the dxz, dyz orbitals by back bonding to the CN π* orbitals and the destabilization of the dyz orbital by the donation of the S3p π orbital. In the case of the linear CN− model with H-bonding to the thiolate (E), the back bonding from the dxz, dyz orbitals along the Fe-CN axis into the two CN− π* orbitals dominates which results in localization of the t2 hole on the dxy orbital in the Fe-N4 equatorial plane. H-bonding from the solvent molecule to the CN− (Fig. 5, E + H2O) weakens this π backbonding interaction with one of the two d-π orbitals (evident from the decreased Fe3d mixing in one of the two π* orbital from 3.2% to 1%) and the t2 hole is now localized in the antibonding Fe-S d-π dxz orbital. Note that of the two possible d-π orbitals along the Fe-CN bond, the Fe-S d-π orbital is higher in energy due to its π* nature which localizes the t2 hole in this orbital. Note that single point calculations on bent Fe-CN models (as indicated by the Raman data) also show weakening of Fe-CN backbonding and similar localization of the t2 hole in the d-π dxz orbital (the effect of bending and H-bonding has an orientation dependence, supplemental information page S1).
Figure 5.
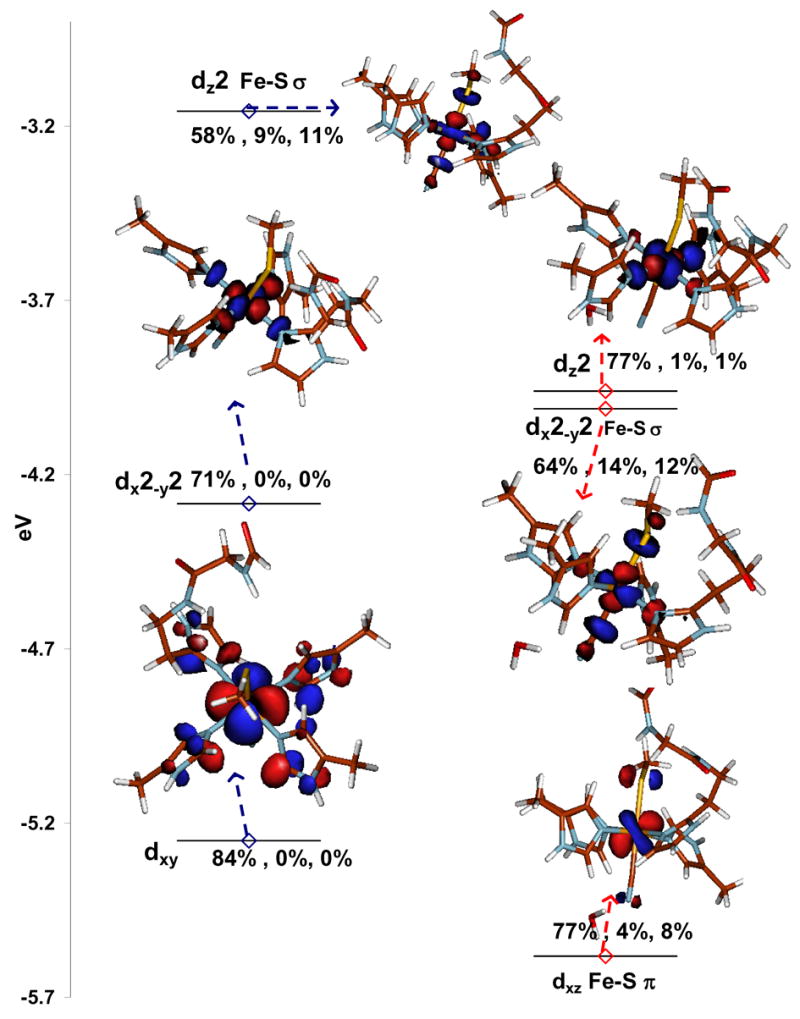
β contours of the E (left) and E +H2O (right) models of the linear Fe-CN bound LS forms. The two α unoccupied contours are not shown for clarity. The designations of the orbitals are written next to them and the %Fe3d, CN2p, S3p are given as well.
A net result of these interactions is that the t2 hole in the low-spin CN− bound form has S3p mixing consistent with the low-energy feature in S K-edge XAS and the presence of a low-energy CT at 14000 cm−1.10 The %S3p mixing in the t2 hole in the E + H2O and the bent Fe-CN models are calculated to be 8% and 11%, respectively, which are lower than the experimentally observed 13% intensity in the 2469.5 eV feature in Fig. 2. The total %S3p in the unoccupied d orbitals is calculated to be 35% (11% d-π+24% d-σ (α+β)) for both the linear and bent models which is distributed over one d-π and two dn+1 d-σ final states as presented in Section A. The experimental total S3p is estimated to be 33% (13% d-π and 20% d-σ).64 Note that the Fe-S bond may be expected to get more covalent in its low-spin state relative to high-spin state but the very strong trans CN− ligand in the low-spin state weakens it. This is provides an explanation for the result that the S3p co-efficient in the d-σ orbital remains the same (12%) between the high (Fig. 4 and Fig. 5 right hand side) and the low-spin states. However the total covalency of the Fe-S bond in the low-spin form is 35% (11% d-π + 2×12% d-σ) which is more than that of the high-spin form (25%) because the d-σ orbital is unoccupied in the low-spin state while it is half-occupied in the high-spin state. While this computational model provides a good qualitative description of the t2 hole, the Fe-S d-π covalency is under-estimated. Note that the nature of this t2 hole is strongly dependent on the modeling of the CN− fragment (as described above) and the estimation of the π Fe-S covalency may be improved by expanding the model. However this is not essential for the properties investigated in the following sections.
The pre-edge of the resting FeII form could not be resolved due to its overlap with the rising edge. However, the geometric parameters of the calculated model (Fe-S =2.35 Å, Fe-N=2.20 Å) agrees reasonably well with the published EXAFS (Fe-S =2.37 Å, Fe-N =2.16 Å) data. The Fe-S bond covalency is calculated to be 14% which is similar to the Fe-S bond covalency reported for a FeII(SR)42− model complex.65
Overall the computational results indicate that the α-carbon constrained and H-bonded (to both thiolate and the trans axial ligand) model using the B3LYP hybrid functional reproduces the experimentally observed electronic and geometric parameters for the resting high-spin and its conversion to the CN− bound low-spin state. A different modeling approach was used in Ref. 17. Though neither the α carbon constraints nor the H-bonding interactions were included, this model reproduced the high-spin resting state using BP86 functional and a DN** basis set.66 In another study the BP86 functional and 6-311g* basis set was used to describe the low-spin CN− bound form.67 Though this model gave the correct spin state and localization of the single t2 hole, it predicts a low-spin ground state for the high-spin resting form and over estimates the Fe-S bond covalency in both high-spin and low-spin forms (which we find is significantly reduced upon inclusion of H-bonding and HF exchange in the computational models) that are both well reproduced by the model used here.
C. Reaction mechanism
In this section we use the experimentally calibrated computational model developed above to evaluate the reaction mechanism commonly invoked for SOR23 which involves reduction by one-electron and double protonation of superoxide anion to release H2O2. Fig. 6 presents the results of the H-bonded thiolate model which also includes H-bonding to the exchangeable axial ligand from a solvent H2O molecule i.e. the E+H2O model in section B (the non- H-bonded model is given in Scheme S1, Supplementary Information). One intermediate (intermediate I) has been unambiguously reported in literature by several groups which has the characteristics of a ferric species.13,23 The possible intermediates involved in the reaction mechanism were calculated in Ref. 17 using a non H-bonded model although the energies of individual steps were not evaluated and a low spin FeIII-OOH was assigned as intermediate I. Evaluating the energies of the individual steps of the mechanism and identifying possible intermediates (including spin states) will be the main focus of this section. The relevant geometric and ground state parameters are listed in Table 3.
Figure 6.
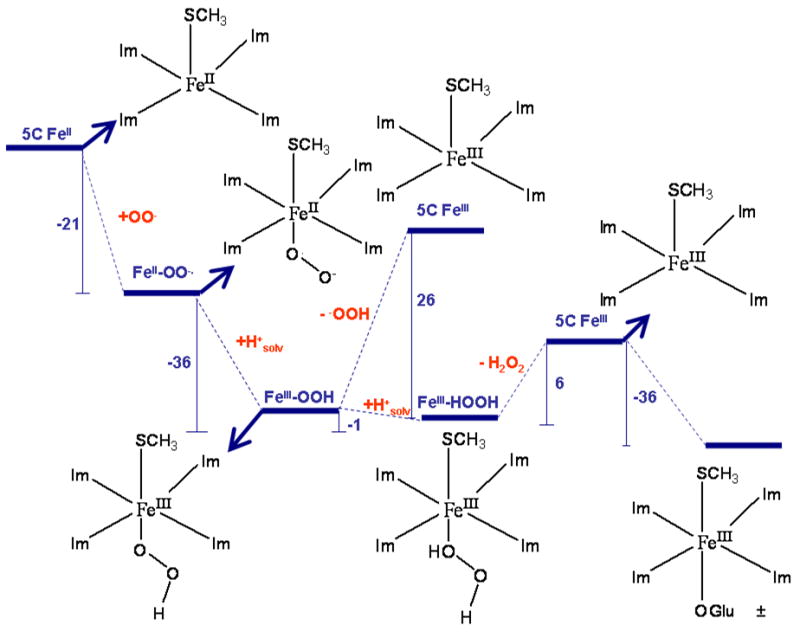
Reaction mechanism of SOR with H-bonding to the CysS− included. The numbers represent ΔE of individual steps (ΔE calculated as Eproducts - Ereactants) and were calculated using a PCM model (ε = 4.0) and are given in Kcal/mol. Solvation energy of 260 Kcal/mole was used for a proton (H+solv) in this scheme.68 The computational model used for this calculation is described in Figure 3E. The H-bonds to thiolate and the axial ligand are not shown for clarity. ± In the E47A mutant where the Glu is replaced by Ala, the axial ligand has been shown to be OH− using rR.72 The binding of a OH− from water is calculated to be exothermic by −17 Kcal/mole.
Table 3.
Results of DFT calculations on H-bonded models relevant to the mechanism
| Spin | Fe-S (Å) | Fe- O (Å) | O-O (Å) | Fe-N (Å) | Fe-O-O (0) | SS1 | S Fe1 | SO1 | qS1 | qFe1 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Resting FeII | S=2 | 2.35 | 2.21 | 2.21 | 0.14 | 3.71 | −0.66 | 1.36 | ||||
| FeIII 5C | S=5/2 | 2.26 | 2.14 | 2.13 | 0.53 | 4.08 | −0.37 | 1.61 | ||||
| FeII-O2− | S=5/2 | 2.63 | 2.16 | 1.33 | 2.24 | 2.18 | 123 | 0.03 | 3.78 | 0.5,0.5 | −0.79 | 1.54 |
| FeIII-OOH | S=5/2 | 2.45 | 1.95 | 1.44 | 2.19 | 2.17 | 123 | 0.24 | 4.15 | 0.27 | −0.59 | 1.73 |
| FeIII-OOH | S=1/2 | 2.41 | 1.81 | 1.45 | 2.07 | 2.00 | 118 | −0.05 | 0.99 | 0.11 | −0.50 | 1.47 |
| FeIII-H2O2 | S=5/2 | 2.29 | 2.75 | 1.45 | 2.15 | 2.15 | 125 | 0.53 | 4.10 | −0.38 | 1.71 | |
| Resting FeIII | S=5/2 | 2.43 | 1.95 | 2.2 | 2.16 | 0.31 | 4.19 | −0.53 | 1.76 | |||
Mulliken spin densities and charges.
The first step involves attack by the superoxide anion at the open co-ordination position of the resting FeII to form an inner-sphere complex. The Δ E of the reaction is −21 Kcal/mol. This species is best described as a FeII-superoxide complex as the O-O distance is 1.33Å and the spin density on the O2 fragment is 1.0. Thus it is important to note that this step does not involve electron transfer from the FeII center to the bound superoxide ion and hence this species is not likely to be the experimental intermediate I which has spectroscopic characteristics of a FeIII species.23, 13
The next step involves protonation of the bound superoxide to generate a η1-HOO-Fe complex.69 This species is best described as a FeIII-OOH species based on the optimized Fe-O, O-O bond distances and the resultant spin densities on Fe and OOH (Table 3, row 4). The calculated energies of the geometry optimized high-spin and the low-spin states of this model indicate that the former is more stable by 4 Kcal/mol. This is in contrast to the results in Ref. 17 where a low-spin ground state was calculated to be more stable. Thus this computational model that predicts the correct ground state and thiolate covalency of both the resting high-spin and the CN− bound low-spin form of SOR predicts a high-spin ground state of an FeIII-OOH intermediate. The high energy of this protonation step is −35 Kcal/mol (using 260 Kcal/mol for solvated H+) will help overcome any barrier that may be involved in protonating the distal Oxygen of this solvent exposed FeII-O2− adduct. The H+ may be donated by Lys48 or abstracted from bulk solvent as this site is very solvent exposed. Thus the above results favor a high-spin FeIII-OOH as the first stable ferric species formed in the reaction cycle. Note that the large driving force for the formation of this species is consistent with its diffusion controlled rate of formation. Further the favorable protonation energy will help drive the process and explain the lack of a 2H isotope effect in its formation.14
At this point, heterolytic cleavage of the Fe-O bond to generate a five-coordinate high-spin FeIII center and an −OOH ion may be possible. In fact, a potential energy scan of Fe-O bond in the non H-bonded model of this active site results in a heterolytic Fe-O cleavage (with a barrier of +26 Kcal/mol) in contrast to homolytic Fe-O cleavage generating FeII and an •OOH radical found in DFT calculations of high-spin non-heme iron hydroperoxide intermediates with neutral ligands.70 However this step is calculated to be endothermic (for the computational model used here) and will introduce a 26 Kcal/mol energy barrier (ΔE) in the reaction pathway. An alternative energetically favored path involves protonation of the proximal Oxygen of the FeIII-OOH species resulting in a FeIII-O2H2 species. This process is enthalpically favorable (ΔE= −1 Kcal/mol, using a solvated proton) as the resulting high-spin FeIII-O2H2 species is stabilized by strong interaction with the axial H-bonded thiolate (the Fe-S bond shortens from 2.45 Å in FeIII-OOH to 2.29 Å in FeIII-O2H2, Table 3, rows 4 and 6). Note that the favorable enthalpy of protonation does not reflect a greater stability of the FeIII-O2H2 species relative to the FeIII-OOH species (see calculated ΔG’s below). The ΔE for this step is +6.6 Kcal/mol for a model that does not include the H-bonding interaction with the exchangeable axial ligand from the solvent molecule. This interaction had to be included in the model to get reasonable agreement with the S Kedge XAS data on the low-spin CN− bound form and is observed in the crystal structure of this FeIII-OOH species. This additional interaction from a solvent molecule makes the reaction more favorable by ~8 Kcal/mol (6.6 Kcal/mol + 1 Kcal/mol from Fig. 6) by stabilizing the weakly bound H2O2 by H-bonding. The resulting Fe-O2H2 species has a long Fe-O bond (2.7 Å) which facilitates the release of the final H2O2 product. Thus the anionic thiolate ligand weakens the Fe-OOH bond favoring its protonation and cleavage. Note that in Compound 0 of peroxidases the protonation of a proximal Oxygen of a FeIII-OOH species is calculated to have a barrier of 9 Kcal/mol (including ZPE correction).71 However this is still less than the ΔG± of diffusion of a small ion in solution (11–12 Kcal/mol) involved in the formation of the FeII-O2− adduct. The following step is cleavage of the weak Fe-(O2H2) bond which is endothermic by only E = 8 Kcal/mol and this process is associated with a further 0.03 Å decrease in the Fe-S bond length. The small dissociation energy of the H2O2 implies that the axial thiolate also helps avoid product inhibition. It is also possible that the axial glutamate residue displaces this weakly bound H2O2 as its binding is exothermic by −36 Kcal/mol. In the E47A mutant the Glu is replaced by a H2O which binds as HO− from rR studies72 and is also calculated to be exothermic by −17 Kcal/mol. This results in the formation of the 6C FeIII resting form of the enzyme which would be re-reduced in turnover. Note that the presence of the axial glutamate residue competes with superoxide binding to the FeIII site and has been proposed to be an important factor in avoiding superoxide dismutase activity (vide infra).2
Along the calculated reaction pathway, three species could be identified as possible candidates for the observed intermediate I namely the FeII-O2−, the FeIII-OOH and the FeIII-O2H2 species. Though the calculated ΔE’s provide some insight into the favored pathway, the ΔG’s are more useful in evaluating the feasibility of a possible intermediate. The ΔG for the formation of the FeIII-OOH upon protonation of the FeII-O2− is calculated to be −25 Kcal/mol. The large driving force for protonation along with the observation that the deprotonated species is FeII in nature disfavors the FeII-O2− species as the ferric intermediate I. The heterolytic dissociation of the Fe-OOH to FeIII and HOO− has a ΔG of +14 Kcal/mol that will result in an estimated rate constant of 400 s−1 (using the Eyring equation and assuming ΔG± =ΔG for an endothermic process without any additional barrier) which is significantly lower than the experimentally observed rate. Thus this step can not be involved in the reaction pathway. Alternatively, the protonation of this FeIII-OOH species has a ΔG of +5.4 Kcal/mol and the further dissociation of the Fe-O2H2 species have a ΔG of −10 Kcal/mol. The lack of a significant barrier (ΔG± = 9 Kcal/mol)71 and requirement of a H+ for decay of the FeIII-OOH are consistent with the pH dependent diffusion controlled decay of intermediate I.73 Also the observed intermediate has a S→ Fe CT band that is blue shifted to 600nm from 647 nm in the resting enzyme. 14,15 This blue shift of the S→ Fe CT is indicative of a stronger ligand field around the FeIII in the intermediate which is only present in the FeIII-OOH species.74 Thus the high-spin FeIII-OOH species is the likely candidate for intermediate I.
The above mechanism is in general agreement with the mechanisms proposed by Kurtz et. al. and Nivere et. al. but with some differences.14, 15 While Nivière et. al. suggest that the intermediate I is a high-spin side-on FeIII deprotonated peroxide our results here suggest that the deprotonated Fe-O2 adduct is FeII-superoxide in nature. In the DFT calculations a side-on FeIII-μ2-O2 species is only formed in the absence of H-bonding to the thiolate (supporting information Scheme S1). Based on DFT calculated energies Kurtz et. al. favor a low-spin FeIII-OOH as intermediate I17 but we find the high-spin FeIII-OOH species to be more stable by 4 Kcal/mol. Also protonation of a low-spin FeIII-OOH will result in the formation of a high-spin FeIII-O2H2 species and will involve a spin-crossing which will have a barrier. Also the possibility of a 2nd intermediate has been proposed by Nivere et. al. At this point the calculated mechanism in Fig. 6 does not provide sufficient support for the presence of another intermediate.
D. Perturbations to the model: Contributions to reactivity
The presence of a H-bonded thiolate ligand is a unique feature of the SOR active site. The following sections address the effect of the axial ligand on the two key reaction steps; the protonation of the FeIII-OOH species and the reduction of the ferric site generated at the end of the reaction in Fig. 6 for further turnover. The following perturbations will be computationally evaluated: Effect of the axial ligand (H-bonded MeS− to H2O to and F−), and the effect of H-bonding (H-bonded MeS− to MeS−). These calculations use a model which includes only H-bonding to the thiolate and not to the exchangeable axial ligand i.e. model 3E, Fig. 3.
i. Protonation of FeIII-OOH
The calculated Fe-O bond length and the energy required to protonate the proximal Oxygen of the FeIII-OOH species for the different models listed above are given in table 4. Replacing the negatively charged thiolate with a neutral H2O results in a shorter optimized Fe-O bond length (1.89 Å with H2O vs 1.95 Å with a thiolate). The stronger Fe-OOH bond with the trans H2O ligand makes the subsequent protonation of this species to form H2O2 energetically uphill by 43 Kcal/mol (also the heterolytic cleavage into 5C FeIII and −OOH fragments is calculated to be uphill by > 100 Kcal/mol). This is in agreement with the lack of Fe-O bond heterolysis in the 6C high-spin FeIII-OOtBu complex with neutral ligands and facile Fe-O heterolysis observed in the 6C high-spin FeIII-OOH species with an anionic ligand.70,24 Interestingly, substitution of the axial ligand with another anionic ligand like F− results in a 7 Kcal/mol increase in the protonation energy relative to a trans thiolate ligand. This suggests that although the anionic nature of the thiolate ligand plays a dominant role in lowering the protonation energy (by 29 Kcal/mol) relative to a neutral ligand, its covalent nature further assists the protonation by 7 Kcal/mol. H-bonding to the thiolate makes it a weaker donor and results in a process which is 4.2 Kcal/mol more endothermic relative to a non H-bonded thiolate.
Table 4.
Optimized Fe-O bond length and calculated FeIII-OOH protonation enthalpy
| Model | Fe-O (Å) | ΔE (Kcal/mol) |
|---|---|---|
| (MeS)FeIII(His)4OOH H-bonded | 1.95 | + 6.7 |
| (F)FeIII(His)4OOH | 1.95 | + 14.1 |
| (H2O)FeIII(His)4OOH | 1.89 | + 43.0 |
| (MeS)FeIII (His)4OOH | 1.95 | +2.5 |
ii. Redox potential
The SOR active site has a redox potential ~250–300 mV. This process involves a 6C ferric and a 5C ferrous species (i.e. FeIII-Glu + e− → FeII + Glu−) and is essential for the re-reduction of the FeIII during turnover.
The effects of the above perturbations of the axial thiolate ligand on the energy of this redox process are tabulated in Table 5. Note that these are shifts in ionization energies (IE) calculated using a PCM model and not reduction potentials. However these shifts in IE are reasonable approximations for the shifts in reduction potentials as the contributions from entropy, zero point energy and solvation of the axial ligand should be similar between these models. Changing the axial thiolate to a neutral H2O shifts the redox potential by more than +3 V. This is expected as this substitution changes the charge of the model. On substituting the thiolate with an F− the potential shifts more negative by 516 mV which could imply greater stabilization of the FeIII state by F− or a greater stabilization of the FeII state by CysS− ligand. Note that the SOR redox couple includes an increase in co-ordination number upon oxidation. Thus the oxidation process can be divided into two steps; oxidation of a reduced 5C site, and the subsequent co-ordination of the glutamate ligand to the oxidized 5C site. The calculations on a 5C site show that replacing the CysS− with F− shifts this reduction potential only by +44 mV indicating that this ligand change has negligible effect on the relative stabilities of the 5C FeII and the 5C FeIII states. However the F− donates less charge to the Fe and increases its Lewis acidity. This results in an increased 6th ligand binding energy (~13 Kcal/mole = ~560 mV) for the F− bound oxidized site. Thus F− binding results in additional stabilization of the 6C FeIII state, which shifts the reduction potential of the coupled redox process by −516 mV. The H-bonded thiolate has a redox potential which shifted by −137 mV relative to a non H-bonded thiolate ligand. This is in contrast to several past studies where H-bonding has been found to shift the redox potential more “positive” as it stabilizes the increased charge density in the reduced form.36 H-bonding affects the oxidation of a reduced 5C site, and the subsequent coordination of the glutamate ligand to the oxidized 5C site differently. The calculations on a 5C site show that the H-bonding interaction with the thiolate shifts the reduction potential by +250 mV as observed in past studies.36 However the H-bonded thiolate donates less charge to the Fe and increases its Lewis acidity. This results in an increased 6th ligand binding energy (~9 Kcal/mole = ~387 mV) for the H-bonded oxidized site. The net result is that H-bonding shifts the coupled redox process by −137 mV.
Table 5.
Calculated shift of reduction potential upon axial ligand perturbation.
| Model | ΔIEo (mV) |
|---|---|
| (MeS)FeIII(His)4Glu | +137 |
| (MeS)FeIII(His)4Glu H-bonded | 0 |
| (F)FeIII(His)4Glu | −516 |
| (H2O)FeIII(His)4Glu | +3000 |
In summary the anionic nature of the thiolate ligand is necessary to raise the pKa of FeIII-OOH species while keeping its Eo (tuned by the H-bond to the cysteine) in the range required for SOR reduction. Its covalent character of the thiolate Fe bond helps to further weaken the Fe-OOH bond by +7.4 Kcal/mol relative to a non-covalent anionic ligand like F−. This decrease in protonation affinity due to a covalent thiolate corresponds to a ~5 log unit increase in pKa of the FeIII-OOH species.
E. SOR vs SOD
Interestingly, the SOR active site shows very limited SOD activity (0.5–1% of native Fe type SODs) in spite of having a redox potential (+250 mV) comparable to that of the Fe-SOD active site, which is in between those of the O2− → + e− and O2− + 2H+ + e− → H2O2 employed to investigate the oxidation of superoxide by the H-bonded SOR in comparison to a model of the Fe-SOD active site (Fig. 7).76 Elaborate spectroscopic characterization and DFT calculations of Fe-SOD have been reported where the importance of the 2nd sphere residues has been demonstrated.77, 78 We employ a simpler model here consisting of three imidazoles, one acetate and one hydroxide/water and focus mainly on the energies for the individual steps (which are not evaluated elsewhere) as a reference for the SOD activity of the SOR active site. Inner-sphere superoxide binding to FeIII was assumed. The relevant bondlengths and spin densities are given in Table 6. The superoxide binding is exothermic for both SOR and SOD site leading to the formation of a FeIII-OO− complex. The binding energy is higher for SOR (−57 Kcal/mol) than SOD (−29 Kcal/mol) active site. This is largely the result of the two anionic donors present at the SOD active site (−OH and CH3COO−) which makes a third anionic ligand binding less favorable relative to the one anionic donor in SOR (−SMe). The higher shift of electron density from O2− to FeIII in SOR leads to an FeII-O2 like species having a O-O bondlength of 1.28 Å and a spin density of 1.5 on the O2 fragment (Table 6, row 2). Thus facile O2 dissociation is calculated for this species which is only 1 Kcal/mol uphill. Note that this represents the ΔE for the process. The ΔG will be ~12 Kcal/mol down hill as this step involves O2 dissociation. In SOD this step is H+ driven (as FeIII-OH is converted to FeII-OH2 in the product) and very exothermic (−35 Kcal/mol).79 It is important to note that these results indicate that a five coordinate SOR active site is capable of SOD activity.
Figure 7.
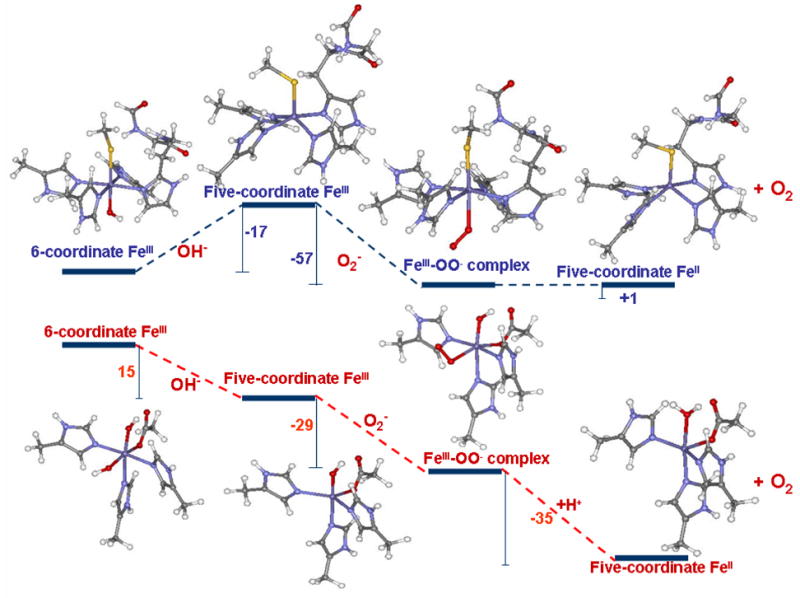
Comparison of energies of superoxide oxidation by SOR (upper panel) and SOD (lower panel) active sites (energies (ΔE) were calculated using a PCM model (ε = 4.0) and are given in Kcal/mol); Solvation energy of 260 Kcal/mol was used for an H+.
Table 6.
Relevant Bond Lengths and Mulliken Spin Densities for Species Involved in SOD cycle.
| Model | Fe-Ltransa | Fe-Oa | O-Oa | SpinFe | SpinO-O | |
|---|---|---|---|---|---|---|
| 5C FeIII | SOR | 2.26 | 4.08 | |||
| SOD | 1.90 | 4.20 | ||||
| FeIII+O2− | SOR | 2.43 | 2.24 | 1.28 | 4.03 | 1.51 |
| SOD | 2.05 | 2.09 | 1.30 | 4.15 | 1.28 | |
| FeII | SOR | 2.35 | 3.71 | |||
| SOD | 2.07 | 3.79 | ||||
| FeIII-OH | SOR | 2.47 | 1.89 | 4.21 | ||
| SOD | 2.06 | 1.88 | 4.24 | |||
Units Å
However while the resting ferric SOD active site is five coordinate, resting SOR is six co-ordinate and the reaction with O2− would require dissociation of the axial glutamate ligand. This dissociation energy (ΔE) for the glutamate residue in the resting SOR active site is calculated to be +36 Kcal/mol. Although a 5C active site is generated during turnover (Fig. 6) one also needs to consider the possibility of H2O binding to form an FeIII-OH species which is kinetically favored over reaction with superoxide ([O2−]=10−6–10−9 M under turnover conditions) as proposed by Kurtz et. al.23 From Fig. 7 the ΔE for FeIII + 2H2O → FeIII-OH + H3O+ for the open five-coordinate FeIII active site in SOR is favorable by −17 Kcal/mol. Alternatively this process is uphill, ΔE = +15 Kcal/mol, for the five-coordinate FeIII site in SOD. This is consistent with the SOD active site remaining five-coordinate in solution80 while the E47A mutant of SOR (this mutant lacks the axial carboxylate ligand) has a bound −OH at physiological pH.72 Thus for SOR but not SOD, O2− binding to the FeIII site requires dissociation of an anionic ligand which is endothermic by at least ~15 Kcal/mol (for OH−, 36 Kcal/mol for Glu), along the reaction co-ordinate (Fig. 7, left). This will effectively inhibit the SOD activity of the SOR active site.
DISCUSSION
The resting high-spin FeIII site of SOR has an axial Fe-SCys bond with two peptide NH---S H-bonding interactions. It’s S K-edge XAS exhibits a large t2-e splitting (1.2 eV) such that the σ and π covalency of this Cysteine thiolate-FeIII can be directly measured (Fig. 2). The %S3p characters (i.e. covalency) in the t2 and e Fe3d orbitals are obtained experimentally to be 15% and 10%, respectively (total 25%). The total Fe-S bond covalency of SOR is comparable to H-bonded heme thiolate models (30%).36 Comparison of the H-bonded thiolate ligand with a neutral (H2O) and an anionic ligand (F−) reveals that the thiolate plays the role of a covalent anionic ligand. The anionic character of the thiolate ligand increases the pKa of the FeIII-OOH intermediate facilitating its protonation by 29 Kcal/mol and tuning down its potential to a physiological range. The covalent character of the thiolate facilitates the reaction by further lowering the protonation energy of the high-spin FeIII-OOH intermediate by 7.5 Kcal/mol relative to a non-covalent anionic donor like F−. The high π covalency of the resting ferric SOR site can also facilitate electron transfer into the active site by providing efficient ligand super-exchange coupling into the protein.81
The mechanism of superoxide reduction by SOR has been found to involve one or more intermediates.23,16 One is proposed to be a high-spin side-on peroxide based on resonance Raman studies.22 We could computationally stabilize a side-on η2-FeIII-peroxo species only in the absence of H-bonding to the thiolate and its protonation (leading to an FeIII-OOH species) was energetically very favorable (Scheme S1). On including the conserved H-bonding residues to the thiolate ligand, which are necessary to reproduce the experimental Fe-S bond covalency, a peroxide level intermediate could be generated only upon protonation. This high spin FeIII-OOH species is consistent with the experimentally observed stronger ligand field in the intermediate (S→ FeIII CT blue shifted by ~1100 cm−1) relative to the resting ground state (HOO− is a stronger donor ligand that GluO−). The formation of this O2− adduct and its further protonation, with a large driving force (which helps overcome barriers), and the decay requiring a H+ (with ΔG± = 9 kcals/mol) are consistent with the diffusion controlled pH independent rate of formation and diffusion controlled pH dependent rate of decay observed experimentally.14 The above mechanism is consistent with recent results from Kitagawa et. al. where a high-spin FeIII-OOH species has been identified as an intermediate in a functional model of SOR.24 It is important to note that with a non-covalent anionic axial ligand like F− in place of the thiolate this step is +14 Kcal/mol uphill which would be detrimental towards the fast kinetics required for this enzyme.
The active site of SOR is similar to the active site of Cytochrome P450 which also has an axial cysteine co-ordination and a equatorial N4 co-ordination. However, the porphyrinato di-anion in the active site of P450 provides a strong equatorial ligand field and an aromatic macro-cycle with an additional 2− charge.23 This produces dramatic differences in the reactivities of the FeIII-OOH intermediate involved in both active sites. The additional negative charge favors the protonation of the FeIII-OOH (Compound 0) species in P450 is more favorable than protonation of SOR. Protonation of the distal Oxygen of compound 0 in P450 leads to an energetically favorable O-O bond heterolysis (ΔE = −40 Kcal/mol) forming Compound I where the FeIII gets oxidized to FeIV and the porphyrin ring gets oxidized to a ligand radical. An analogous O-O bond heterolysis of the FeIII-OOH species in SOR active site results in a FeIV and histidine cation radical species and is calculated to be ~+250 Kcal/mol uphill in energy, clearly demonstrating the necessity of the porphyrin dianion in stabilizing the FeIV-oxo ligand radical species. Protonation of the proximal Oxygen in Compound 0 in P450 leads to the formation of a FeIII-O2H2 species analogous to SOR. However, due to the strong equatorial ligand field of the porphyrin ring, Compound 0 in P450 is low-spin. Thus protonation and elimination of H2O2 from the low-spin Compound 0 which will form the 5C high-spin FeIII state will involve a barrier associated with the forbidden two-electron spin flip. Indeed the elimination of H2O2 in P450 active site is experimentally determined to be <1%.82, 83 However, the high-spin nature of the FeIII-OOH intermediate in SOR enables facile H 2O2 release forming a high-spin product.
An important feature of the SOR active site is the presence of the H-bonded thiolate in the first coordination sphere of Fe trans to the O2− binding site. The H-bonding to the active site causes a significant decrease of the Fe-S bond covalency (decrease in covalency of 11% π and 3% σ in model 3D) which shifts the reduction potential by −137 mV. This negative shift in Eo is opposite to the results in previous studies where positive shifts are observed upon H-bonding. It reflects the net effect of two opposing factors: a) increased stabilization of the 5C FeII site (+250 mV, consistent with previous results)36, and b) increased 6th axial ligand binding affinity (due to increased Lewis acidity) of the 5C FeIII site (−387 mV). A further effect of the H-bonding to the thiolate is in the nature of the FeII + O2− adduct. It can be described as an η1-O2−-FeII species in the presence and an η2-O22−-FeIII species in the absence of H-bonding. The FeII-η1-O2− species has higher net charge on the distal Oxygen and a higher energy HOMO donor orbital that activates it for protonation relative to an FeIII-η2-O22− species where the charge on the peroxide moiety is reduced because of its very covalent charge donation to the FeIII.84 Finally the five-coordinate SOR active site is calculated to be able to perform superoxide oxidation to dioxygen. The lack of SOD activity in SOR enzymes then appears to result from a favorable 6th ligand binding energy of the solvent exposed SOR active site as has been proposed previously by Kurtz et. al..23 The Lewis acidity of the SOR active site, having only one anionic ligand, is such that the FeIII state generated during turnover binds a glutamate ligand or an −OH ligand. Superoxide binding to the 6C FeIII SOR site would entail a dissociative mechanism which is calculated to be endothermic by at least ~15 Kcal/mol that will preclude SOD activity. In contrast, the Fe-SOD active site which has two strong anionic ligands (hydroxide and aspartate) has an unfavorable energy for binding an additional anionic −OH ligand and thus remains five-coordinate at physiological pH’s allowing facile SOD activity.
Supplementary Material
All optimized geometries, MO diagram of the H-bonded CN− bound form and the reaction scheme without H-bonding is available free of charge via the web at http://pucs.acs.org.
Acknowledgments
This research was supported by NIH Grants GM 40392 and NSF-CHE 446304 (E.I.S.), RR-01209 (K.O.H.), GM 60329 (M.W.W.A. and M.K.J), AD has an Evelyn Laing McBain fellowship from Stanford University. AD acknowledges Dr. M. D. Clay, Dr. Andrea Decker and Dr. Diego Del Rio for valuable discussions. SSRL operations are supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and by the Department of Energy, Office of Biological and Environmental Research.
References
- 1.Jenney FE, Jr, Verhagen MFJM, Cui X, Adams MWW. Science. 1999;286:306–309. doi: 10.1126/science.286.5438.306. [DOI] [PubMed] [Google Scholar]
- 2.Kurtz DM, Jr, Coulter ED. J Biol Inorg Chem. 2002;7:653–658. doi: 10.1007/s00775-002-0360-4. [DOI] [PubMed] [Google Scholar]
- 3.Adams MWW, Jenney FE, Jr, Clay MD, Johnson MK. J Biol Inorg Chem. 2002;7:647–652. doi: 10.1007/s00775-002-0359-x. [DOI] [PubMed] [Google Scholar]
- 4.Nivière V, Fontecave M. J Biol Inorg Chem. 2004;9:119–123. doi: 10.1007/s00775-003-0519-7. [DOI] [PubMed] [Google Scholar]
- 5.Abreu IA, Xavier AV, LeGall J, Cabelli DE, Teixeira M. J Biol Inorg Chem. 2002;7:668–674. doi: 10.1007/s00775-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 6.Imlay JA. J Biol Inorg Chem. 2002;7:659–663. doi: 10.1007/s00775-002-0361-3. [DOI] [PubMed] [Google Scholar]
- 7.Yeh AP, Hu Y, Jenney FE, Jr, Adams MW, Rees DC. Biochem. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]
- 8.Coehlo AV, Matias P, Fülop V, Thomson A, Gonzalez A, Carrondo MA. J Biol Inorg Chem. 1997;2:680–689. [Google Scholar]
- 9.Emerson JP, Cabelli DE, Kurtz DM., Jr Proc Natl Acad Sci USA. 2003;100:3802–3807. doi: 10.1073/pnas.0537177100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clay MD, Jenney FE, Jr, Hagedoorn PL, George GN, Adams MWW, Johnson MK. J Am Chem Soc. 2002;124:788–805. doi: 10.1021/ja016889g. [DOI] [PubMed] [Google Scholar]
- 11.Clay MD, Jenney FE, Jr, Noh HJ, Hagedoorn PL, Adams MWW, Johnson MK. Biochem. 2002;41:9833–9841. doi: 10.1021/bi025833b. [DOI] [PubMed] [Google Scholar]
- 12.Coulter ED, Emerson JP, Kurtz DM, Jr, Cabelli DE. J Am Chem Soc. 2000;122:11555–11556. [Google Scholar]
- 13.Nivière V, Lombard M, Fontecave M, Houée-Levin C. FEBS Lett. 2001;497:171–173. doi: 10.1016/s0014-5793(01)02468-1. [DOI] [PubMed] [Google Scholar]
- 14.Emerson JP, Coulter ED, Cabelli DE, Phillips RS, Kurtz DM., Jr Biochem. 2002;41:4348–4357. doi: 10.1021/bi0119159. [DOI] [PubMed] [Google Scholar]
- 15.Nivière V, Asso M, Weill CO, Lombard M, Guigliarelli B, Favaudon V, Houée-Levin C. Biochem. 2004;43:808–818. doi: 10.1021/bi035698i. [DOI] [PubMed] [Google Scholar]
- 16.Lombard M, Touati D, Fontecave M, Nivière V. J Biol Chem. 2000;275:27021–27026. doi: 10.1074/jbc.M004201200. [DOI] [PubMed] [Google Scholar]
- 17.Silaghi-Dumitrescu R, Silaghi-Dumitrescu I, Coulter ED, Kurtz DM., Jr Inorg Chem. 2003;42:446–456. doi: 10.1021/ic025684l. [DOI] [PubMed] [Google Scholar]
- 18.Halfen JA, Moore HL, Fox DC. Inorg Chem. 2002;41:3935–3943. doi: 10.1021/ic025517l. [DOI] [PubMed] [Google Scholar]
- 19.Lombard M, Houée-Levin C, Touati D, Fontecave M, Nivière V. Biochem. 2001;40:5032–5040. doi: 10.1021/bi0023908. [DOI] [PubMed] [Google Scholar]
- 20.Clay MD, Yang TC, Jenney FE, Jr, Kung IY, Cosper CA, Krishnan R, Kurtz DM, Jr, Adams MWW, Hoffman BM, Johnson MK. Biochem. 2006;45:427–438. doi: 10.1021/bi052034v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathé C, Mattioli TA, Horner O, Lombard M, Latour JM, Fontecave M, Nivière V. J Am Chem Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]
- 22.Horner O, Mouesca JM, Oddou JL, Jeandey C, Nivière V, Mattioli TA, Mathé C, Fontecave M, Maldivi P, Bonville P, Halfen JA, Latour JM. Biochem. 2004;43:8815–8825. doi: 10.1021/bi0498151. [DOI] [PubMed] [Google Scholar]
- 23.Kurtz DA. Acc Chem Res. 2004;37:902–908. doi: 10.1021/ar0200091. [DOI] [PubMed] [Google Scholar]
- 24.Kitagawa T, Dey A, Lugo-Mas P, Solomon EI, Kovacs JA. J Am Chem Soc. 2006;128:14448–14449. doi: 10.1021/ja064870d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shearer J, Scarrow RC, Kovacs JA. J Am Chem Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- 26.Krishnamurthy D, Kasper GD, Namuswe F, Kerber WD, Narducci Sarjeant AA, Moenne-Loccoz P, Goldberg DP. J Am Chem Soc. 2006;128:14222–14223. doi: 10.1021/ja064525o. [DOI] [PubMed] [Google Scholar]
- 27.Clay MD, Cosper CA, Jenney FE, Adams MWW, Johnson MK. PNAS. 2003;100:3796–3801. doi: 10.1073/pnas.0636858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulos TL, Finzel BC, Howard AJ. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 29.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 30.Akhtar M, Calder MR, Corina DL, Wright JN. Biochem J. 1982;201:569–580. doi: 10.1042/bj2010569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jovanovi’c T, Ascenso C, Hazlett KRO, Sikkink R, Krebs C, Litwiller LM, Benson LM, Moura I, Moura JJG, Radolf JD, Huynh BH, Naylor S, Rusnak FM. J Biol Chem. 2000;275:28439–28448. doi: 10.1074/jbc.M003314200. [DOI] [PubMed] [Google Scholar]
- 32.Solomon EI, Hedman B, Hodgson KO, Dey A, Szilagyi RK. Coord Chem Rev. 2005;249:97–129. [Google Scholar]
- 33.Dey A, Glaser T, Moura JJG, Holm RH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2004;126:16868–16878. doi: 10.1021/ja0466208. [DOI] [PubMed] [Google Scholar]
- 34.Solomon EI, Gorelsky SI, Dey A. J Comp Chem. 2006;27:1414–1428. doi: 10.1002/jcc.20451. [DOI] [PubMed] [Google Scholar]
- 35.Neese F, Hedman B, Hodgson KO, Solomon EI. Inorg Chem. 1999;38:4854–4860. doi: 10.1021/ic990461p. [DOI] [PubMed] [Google Scholar]
- 36.Dey A, Okamura TA, Ueyama N, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2005;127:12046–12053. doi: 10.1021/ja0519031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hedman B, Frank P, Gheller SF, Roe AL, Newton WE, Hodgson KO. J Am Chem Soc. 1988;110:3798–3805. [Google Scholar]
- 38.Shadle SE, Hedman B, Hodgson KO, Solomon EI. Inorg Chem. 1994;33:4235–4244. [Google Scholar]
- 39.Agarwal BK. X-ray Spectroscopy. Springer-Verlag; Berlin: 1979. p. 276. ff. [Google Scholar]
- 40.Tyson TA, Roe AL, Frank P, Hodgson KO, Hedman B. Phys Rev B. 1989;39A:6305–6315. doi: 10.1103/physrevb.39.6305. [DOI] [PubMed] [Google Scholar]
- 41.Shadle SE, Penner-Hahn JE, Schugar HJ, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1993;115:767–776. [Google Scholar]
- 42.Frisch MJ, et al. Gaussian 03, Revision C 02. Gaussian, Inc; Wallingford CT: 2004. (Full Reference in supplementary information) [Google Scholar]
- 43.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 44.Schmidt M. Eur J Biochem. 1999;262:117–127. doi: 10.1046/j.1432-1327.1999.00359.x. [DOI] [PubMed] [Google Scholar]
- 45.Mulliken RSJ. Chem Phys. 1955;23:1833–1840. [Google Scholar]
- 46.Tenderholt AL. PyMOlyze. Stanford University; Stanford, CA USA: Version 1.1. [Google Scholar]
- 47.Miertus S, Scrocco E, Tomasi J. Chem Phys. 1981;55:117–129. [Google Scholar]
- 48.Gogonea V, Merz MK., Jr J Phys Chem A. 1999;103:5171–5188. [Google Scholar]
- 49.Note that the higher energy feature is hard to estimate as it strongly overlaps with the much strong S1S→C-Sσ* transition.
- 50.Van Vleck JH. Phys Rev. 1934;45:405. [Google Scholar]
- 51.Laporte O. Phys Rev. 1942;61:302–304. [Google Scholar]
- 52.The energy splitting between these triplet and broken-symmetry singlet dn+1 states was evaluated from DFT calculations to be 0.45 eV. From spin projection the T/S splitting is twice this value i.e. 0.9 eV.
- 53.Note that there is a weak shoulder in the data in Fig. 2 (red) at 2471.5 eV (~1 eV above the e feature) that is also visible in the 2nd derivative of the spectra (dashed red). However due to its overlap with the intense rising edge feature it could not be quantified.
- 54.A minimum in 2nd derivative indicates a maximum in Absorption.
- 55.Note that in Ref. 17 a high spin ground state was obtained using BP86 and a double zeta basis set. Using Gaussian 03 and ADF we found the low-spin ground state to be more stable using the BP86 functional irrespective of the quality of the basis set used.
- 56.Loew GH, Harris DL. Chem Rev. 2000;100:407–419. doi: 10.1021/cr980389x. [DOI] [PubMed] [Google Scholar]
- 57.Note that no calculations was performed on the H-bonded and α carbon frozen models using BP86 functional as the H-bonding stabilizes the HS state by only 2 Kcal/mole which will be insufficient to change the 5 Kcal/mole stable LS GS to a HS GS.
- 58.Szilagyi RK, Metz M, Solomon EI. J Chem Phys A. 2002;106:2994. [Google Scholar]
- 59.A fully optimized structure, i.e. no constraints, in B3LYP predicts that the low-spin ground state is 5 Kcal/mol more stable than the high-spin ground state.
- 60.Optimizations were attempted with only two constrains on the peptide fragment but they caused less than 0.01Å difference in geometry and less than 0.5 Kcal/mole difference in the relative energies of the spin states.
- 61.Pseudo-σ interaction implies that donor S3p orbital is tilted off the Fe-S bond vector.
- 62.Interestingly, the two trans-histidines, having short Fe-NHis bond lengths form strong Fe-N σ bonds and define the unique (z) axis while the dx2-y2 orbital forms the σ bond with two other histidines and the axial ligands. This description is in good agreement with the results from Johnson et. al. using EPR and VTVH MCD.10
- 63.Note that a single point calculation on the optimized H-bonded structure where the N-H’s were substituted by –O- i.e. amides were made esters, the Fe-S covalency was similar to the non H-bonded model. This indicates that the reduction of Fe-S covalency is due to H-bonding.
- 64.There is a large error bar (±3%) associated with this estimate due to the overlap of this higher energy feature with the rising edge transition.
- 65.Kennepohl P, Solomon EI. Inorg Chem. 2003;42:679–688. doi: 10.1021/ic020330f. [DOI] [PubMed] [Google Scholar]
- 66.Only the dihedral angles between the histidine rings were constrained which, reportedly, had no effect on the relative energies of the spin states of the structures examined.
- 67.Yang T, McNaughton RL, Clay MD, Jenney FE, Jr, Krishnan K, Kurtz DM, Jr, Adams MWW, Johnson MK, Hoffman BM. J Am Chem Soc. doi: 10.1021/ja064656p. (ASAP article) [DOI] [PubMed] [Google Scholar]
- 68.Noyes RM. J Am Chem Soc. 1962;84:513–522. [Google Scholar]
- 69.This is consistent with a recent report of an end-on bound Fe-OO intermediate trapped crystallographically in the E114A mutant. Katona G, Carpentier P, Nivière V, Amara P, Adam V, Ohana J, Tsanov N, Bourgeois1 D. Science. 2007;316:449. doi: 10.1126/science.1138885.
- 70.Lehnert N, Ho YNR, Que L, Jr, Solomon EI. J Am Chem soc. 2001;138:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 71.Derat E, Shaik S. J Phys Chem B. 2006;110:10526–10533. doi: 10.1021/jp055412e. [DOI] [PubMed] [Google Scholar]
- 72.Mathe C, Nivière V, Mattioli TA. J Am Chem Soc. 2005;127:16436–16441. doi: 10.1021/ja053808y. [DOI] [PubMed] [Google Scholar]
- 73.Note that another 2H sensitive decay pathway of the intermediate I has been proposed by Kurtz et. al which has a much slower rate (50 s−1). The possible candidate for this pathway could be protonation of the proximal Oxygen from a nearby ionizable residue or a H2O molecule.14
- 74.The S π and Fe d-π CT energy gap is calculated to be blue shifted by 1500 cm−1 consistent with this proposal.
- 75.Jovanovic T, Ascenso C, Hazlett KRO, Sikkink R, Krebs C, Litwiller R, Benson LM, Moura I, Moura JJG, Radolf JD, et al. J Biol Chem. 2000;275:28439–28448. doi: 10.1074/jbc.M003314200. [DOI] [PubMed] [Google Scholar]
- 76.Cooper JB, McIntyre K, Badasso MO, Wood SP, Zhang Y, Garbe TR, Young D. J Mol Biol. 1995;246:531–544. doi: 10.1006/jmbi.1994.0105. [DOI] [PubMed] [Google Scholar]
- 77.Jackson TA, Brunold TC. Acc Chem Res. 2004;37:461–470. doi: 10.1021/ar030272h. [DOI] [PubMed] [Google Scholar]
- 78.Han Wen-Ge, Lovell T, Noodleman L. Inorg Chem. 2002;41:205–218. doi: 10.1021/ic010355z. [DOI] [PubMed] [Google Scholar]
- 79.Lah MS, Dixon MM, Pattridge KA, Stallings WC, Fee JA, Ludwig ML. Biochem. 1995;34:1646–1660. doi: 10.1021/bi00005a021. [DOI] [PubMed] [Google Scholar]
- 80.Tierney DL, Fee JA, Ludwig ML, Penner-Hahn JE. Biochem. 1995;34:1661–1668. doi: 10.1021/bi00005a022. [DOI] [PubMed] [Google Scholar]
- 81.Solomon EI. Inorg Chem. 2006;45:8012–8025. doi: 10.1021/ic060450d. [DOI] [PubMed] [Google Scholar]
- 82.Martinis SA, Atkins WM, Stayton PS, Sligar SG. J Am Chem Soc. 1989;111:9253–9254. [Google Scholar]
- 83.In addition to this barrier due to a spin state change a low-spin FeIII-OOH species has a very covalent Fe-O bond which is calculated to make protonation uphill by 35 Kcal/mol.
- 84.The μ1-O2−-FeII species has higher charge (−0.53) on the distal Oxygen and a filled occupied π*v orbital at −3.9 eV having 40% distal O2p co-efficient relative to the μ2-O22−-FeIII which has a net total charge of (−0.37) and an orbital at −4.2 eV with 36% O2p character.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All optimized geometries, MO diagram of the H-bonded CN− bound form and the reaction scheme without H-bonding is available free of charge via the web at http://pucs.acs.org.