

Abstract
Tetrathiatriarylmethyl (TAM) radicals are commonly used as oximetry probes for electron paramagnetic resonance imaging (EPRI) applications. In this study, the electronic properties and the thermodynamic preferences for O2 addition to various TAM-type triarylmethyl (trityl) radicals were theoretically investigated. The radicals' stability in the presence of O2 and biological milieu were also experimentally assessed using EPR spectroscopy. Results show that H substitution on the aromatic ring affects the trityl radical's stability (tricarboxylate salt 1-CO2Na > triester 1-CO2Et > diester 2-CO2Et > monoester 3-CO2Et) and may lead to substitution reactions in cellular systems. We propose that this degradation process involves an arylperoxyl radical which can further decompose to alcohol or quinone products. This study demonstrates how computational chemistry can be used as a tool to rationalize radical stability in the redox environment of biological systems and aid in the future design of more biostable trityl radicals.
I. Introduction
Evaluating the concentration of O2 as well as O2-derived reactive species, including that of superoxide radical anion (O2·-) in in vivo and in vitro systems, is of utmost importance in the study of numerous pathophysiological processes.1-4 Over the past decade, major efforts have been made towards the development of electron paramagnetic resonance imaging (EPRI)5-7 of biological samples to provide improved image resolution and quality, and this method has evolved to become an important tool for imaging free radicals in organs4,8,9 and the entire body of small animals,10-12 tumors and normal tissues.13-17
The development of paramagnetic materials, such as nitroxides,18 triarylmethyl (trityl) radicals,19,20 and recently, spin-labeled dendrimers,21 as probes for EPRI applications is critical for the precise determination of O2 concentration and detection of superoxide radical anion (O2·-) in biological samples, including cells and tissues. 22-25 Trityl radicals have been the popular choice for EPRI applications due to their stability at physiological pH and long relaxation times which consequently give rise to narrow EPR linewidths. Moreover, trityl radicals offer some advantages over nitroxides, i.e., high analytical resolution at μM concentrations, stability in cells and tissues, and the flexibility to independently determine the concentrations of superoxide radical anion (O2·-) and O2 by EPR signal loss and line broadening, respectively.25
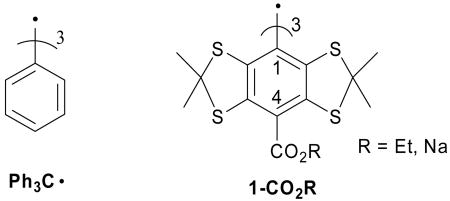
Trityl radicals have a long history in radical chemistry, ever since the initial report by Gomberg in 1900 of triphenylmethyl radical (Ph3C•).26 Tetrathiatriarylmethyl (TAM) radicals 1-CO2R belong to a family of trityl radicals which exhibit a very narrow linewidth (<100 mG),27 even in the presence of biological matrix, rendering this probe ideal for in vitro and in vivo EPR imaging applications.23 The perdeuterated analogue of 1-CO2Na has also found applications in magnetic resonance imaging (MRI)28 and Overhauser-enhanced magnetic resonance imaging (OMRI).29
Reddy et al. reported an elegant synthesis of 1-CO2Et and the corresponding salt, 1-CO2Na, and these authors also evaluated the sensitivities of these species to O2.27 However, the underlying mechanism of decay for 1-CO2Na in the presence of O2 was not reported. In a solid lattice, the reaction of the triphenylmethyl (Ph3C•) radical with O2 results in the formation of an equilibrium product, triphenylmethylperoxyl radical, Ph3COO• (eq. 1).30,31
| (1) |
In dichloromethane solution, a second order reaction was observed with Ph3C• and O2, finally yielding benzophenone (Ph2C=O) as a major product.32
In this paper, we explored the electronic and thermodynamic properties of TAM-type radicals with varying degrees of substitution (2-CO2Et and 3-CO2Et) and evaluated how these properties can affect radical stability in the presence of O2 and components of the biological milieu. We complement these experimental efforts with computational chemistry so as to understand and rationalize the radicals' stability.
II. Results and Discussion
II. A. EPR Spectroscopic Characterization
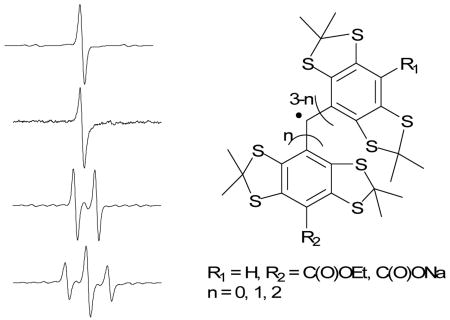
Tri- (1-CO2Et), di- (2-CO2Et) and mono- (3-CO2Et) ethoxycarbonyl derivatives of tetrathiatriarylmethyl (TAM) radical as well as its tri-sodium carboxylate salt (1-CO2Na) derivative were synthesized and characterized by minor modifications of the method reported by Reddy et al.27Figure 1 shows the X-band EPR spectrum for each of the trityl radical species with g values that range from 2.0041 to 2.0043, close to that reported for 1-CO2Et and 1-CO2Na of g ∼ 2.003.27 The singlet peaks in Figures 1a and 1b are consistent with a tri-substituted trityl radical, while the doublet and triplet spectral features for 2-CO2Et and 3-CO2Et, respectively, indicate the presence of a hydrogen hyperfine splitting due to the unsubstituted aromatic ring. The observed hyperfine splitting constant for both 2-CO2Et and 3-CO2Et trityl radicals of aH,para= 2.30 G in DMSO is close to our predicted isotropic hyperfine splitting of 2.11 or 2.22 G at the B3LYP/6-311+G**//B3LYP/6-31G* level of theory in the gas phase, and also similar to that reported experimentally for Ph3C· of aH,para = 2.745 G in toluene solution.33
Figure 1.

CW X-band EPR spectra of various trityl radicals in DMSO under anaerobic condition. (a) 1-CO2Et; (b) 1-CO2Na (in H2O); (c) 2-CO2Et; (d) 3-CO2Et. See Experimental Section for the EPR parameters.
Anaerobic peak-to-peak widths in DMSO for 1-CO2Et, 2-CO2Et and 3-CO2Et range from ΔBpp = 380 to 470 mG and are broader compared to the ΔBpp = 111 mG observed for 1-CO2Na in aqueous solution using a modulation amplitude of 0.01 G. Reddy et al.27 previously reported linewidths for 1-CO2Et and 1-CO2Na of ∼270 mG (in pyridine) and 84 mG (in water), respectively, at modulation amplitudes of 0.025-0.086 G. It should be noted, however, that the slight broadening observed in this study may be due to the presence of trace amounts of O2 since the capillary tubes were only sealed with inert clay after bubbling with Ar. The effect of O2 on the spectral profile of various trityl radicals was investigated as well. Results show that there is a significant linewidth broadening by about ΔΔBpp = 0.8-0.9 G for all of the ester-derivatived trityl radicals and about ΔΔBpp = 0.2 G broadening for 1-CO2Na when O2 was bubbled through DMSO (or water for 1-CO2Na) solutions. This difference in the ΔΔBpp between the ester and carboxylated-trityl radicals could be due to the higher solubility of O2 in DMSO compared to that in water.34 Purging the O2-saturated trityl radical solution with argon gave linewidths similar to that observed originally in the absence of O2, thereby demonstrating that the O2 with trityl radical interaction is reversible.
II. B. Computational Analysis of Structures, Properties and Decomposition Pathways
The measurement of O2 in biological systems requires long and continuous monitoring. Hence an investigation of the stability of trityl radicals in the presence of O2 is critical since O2 reaction with trityl radicals can lead to the degradation of the radical probe. To gain insight into the stability of these radicals in the presence of O2, the electronic as well as the thermodynamic properties of the trityl radical were theoretically investigated. Figure 2 shows the charges and spin (α-β) populations for various TAM-type compounds as well as for Ph3C• using the natural population analysis (NPA) partitioning scheme at the B3LYP/6-311+G**//B3LYP/6-31G* level. These data show that for TAM-type compounds, there is a 0.62 electron localization at the central carbon which is higher compared to the spin density (population) on the central carbon predicted for Ph3C• of 0.56e. However, the spin density of the para-C atoms in TAM-type compounds is in the range of 0.07-0.09e, but higher (0.12e) in Ph3C•.
Figure 2.

Spin populations (α−β) and atomic charges (in parentheses) of various trityl radicals at the B3LYP/6-311+G**//B3LYP/6-31G* level in the gas phase using the natural population analysis partitioning scheme.
A closer look at the molecular structures of 1-CO2Na and Ph3C• showed a significant difference in their respective aromatic ring conformations, i.e., trityl radical 1-CO2Na has a substantial out-of-plane ring twisting compared to that of Ph3C• radical.35 This difference in conformations may have a considerable effect on the electron density distribution and thereby could affect spin relaxation that can result in EPR spectral line broadening.
The preference for O2 addition to the trityl radical was investigated thermodynamically as to whether the O2 addition occurs at the central or para-carbon atoms (Scheme 1). Table 1 shows the thermodynamic values for O2 addition to various trityl radicals to form the peroxyl adduct (steps A and B), followed by subsequent H-atom abstraction (steps C and D) from various H-atom donors to form the hydroperoxyl adduct at the B3LYP/6-311+G**//B3LYP/6-31G* level of theory. Results show that, in general, O2 addition to TAM-type radicals is endothermic (steps A and B in Table 1), and the formation of the peroxyl adduct at the aromatic ring C is preferred by 2.5 to 6.5 kcal/mol compared to peroxyl adduct formation at the central carbon atom for all of the partially substituted TAM radicals. However, peroxyl adduct formation at the central carbon atom is preferred by 1.4 to 2.5 kcal/mol for the fully substituted trityl radicals such as 1-CO2Et, 1-CO2H and 1-CO2Na for the gas-phase calculations. The formation of the peroxyl adduct at the central carbon in Ph3C• is exothermic by -4.0 kcal/mol, and is preferred by 17.7 kcal/mol relative to O2 addition to the aromatic ring's carbon atom. Examination of the charge and spin populations for Ph3C• and 4-CO2Et shows only a small difference in their respective charge (∼0.05e) and spin population (∼ 0.06e) at the central C atom. The spin population on the para C of Ph3C•, however, is higher by only 0.03e compared to 4-CO2Et with no difference in their respective atomic charges. Based on the electronic properties predicted for Ph3C• and 4-CO2Et, the preference of O2 addition to the central C of Ph3C• appears to be due mainly to a steric effect. Although it has been previously36,37 shown that O2 adds preferentially to the carbon centers in delocalized radicals bearing the highest spin density, this behavior does not apply completely for trityl radicals for which steric effects are also important. The effect of solvation on the O2 addition to 1-CO2H and 1-CO2Na was investigated by employing the polarizable continuum model (PCM) for water as compared to the gas-phase energetics. The reactions for O2 addition to the aromatic ring shows no significant difference, while O2 addition reactions to the central carbon atom is preferred by 8∼10 kcal/mol in water compared to the gas-phase calculations. We have noted similar solvation effects for peroxyl radicals earlier.38
Scheme 1. Molecular Oxygen Reaction with Trityl Radicals by Addition to the Center Carbon Atom of the Trityl Radical (A) or by Addition to the Aromatic Ring of the Trityl Radical (B).

Table 1. Predicted Reaction Energies ΔE0 (kcal/mol) for the Formation of Various Trityl Radical-OOH Adducts at the B3LYP/6-311+G**//B3LYP/6-31G* Level in the Gas Phase.
| Reaction Energiesb ΔE0 (in kcal/mol) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Step A | Step B | Step C | Step D | Step A + C | Step B + D | ||||||||
| H2O | DMSO | MeSH | H2O | DMSO | MeSH | H2O | DMSO | MeSH | H2O | DMSO | MeSH | |||
| Ph3C• | -4.0 | 13.7 | 35.5 | 20.7 | 2.1 | 36.7 | 21.9 | 3.3 | 31.5 | 16.7 | -1.9 | 50.4 | 35.6 | 17.0 |
| 1-CO2H | 24.2 | 26.7 | 36.6 | 21.9 | 3.2 | 34.0 | 19.3 | 0.7 | 60.8 | 46.1 | 27.4 | 60.7 | 46.0 | 27.4 |
| 1-CO2Hc | 15.4 | 25.3 | 37.2 | 20.9 | 4.1 | 34.2 | 17.9 | 1.1 | 52.6 | 36.3 | 19.5 | 59.5 | 43.2 | 26.4 |
| 1-CO2Na | 22.8 | 24.2 | 36.8 | 22.0 | 3.4 | 32.2 | 17.5 | -1.2 | 59.6 | 44.8 | 26.2 | 56.4 | 41.7 | 23.0 |
| 1-CO2Nac | 14.3 | 22.8 | 37.0 | 20.7 | 3.9 | 34.8 | 18.5 | 1.7 | 51.3 | 35.0 | 18.2 | 57.6 | 41.3 | 24.5 |
| 1-CO2Et | 24.0 | 25.9 | 38.4 | 23.7 | 5.1 | 34.3 | 19.5 | 0.9 | 62.4 | 47.7 | 29.1 | 60.2 | 45.4 | 26.8 |
| 2-CO2Et | 22.5 | 17.7 | 38.5 | 23.8 | 5.1 | 35.0 | 20.3 | 1.6 | 61.0 | 46.3 | 27.6 | 52.7 | 38.0 | 19.3 |
| 3-CO2Et | 23.9 | 17.4 | 35.9 | 21.2 | 2.5 | 35.2 | 20.5 | 1.8 | 59.8 | 45.1 | 26.4 | 52.6 | 37.9 | 19.2 |
| 4-CO2Et | 19.7 | 17.2 | 37.3 | 22.5 | 3.9 | 35.2 | 20.4 | 1.8 | 57.0 | 42.2 | 23.6 | 52.4 | 37.6 | 19.0 |
Table 1 also shows that the energetics for H-atom abstraction by the peroxyl radical adducts from H2O, DMSO or MeSH to form the aryl hydroperoxyl adduct (steps C and D). Although H-atom abstraction is an endothermic process, abstraction is more preferred with DMSO as an H-atom donor compared to that with H2O, and most preferred with MeSH. The overall energetics (steps A + C and B + D) for the formation of hydroperoxyl adducts in the various solvents is highly endothermic. For all of the trityl radicals, the formation of the aromatic hydroperoxyl adduct (step B + D) is more preferred than the formation of the central carbon hydroperoxyl adduct (A + C) in the gas phase. The partially substituted trityl radicals are more favored to form aryl hydroperoxyl adducts than the fully substituted trityl radicals. This indicates that the stability of these radicals appears to be correlated with the number of H's on the aryl ring. However, based on the computational results, the 1-CO2Na trityl salt is more preferred to form aryl hydroperoxyl adducts than the fully ester-substituted trityl radicals 1-CO2Et. This is not consistent with the stability study results noted below. Our calculations, of course, are thermodynamic results and can not be used directly to represent kinetic results. Because of the computational expense, searching for the transition state (TS) of the reactions was not practical. Also, transition states for O2 addition to carbon-center radicals are often difficult to locate and the corresponding wave functions usually suffer from significant spin contamination, thereby rendering the results to be suspect.38b Based on Hammond's postulate, the TS should be late on the potential energy surface, and should resemble the product, peroxyl radical adduct, or hydroperoxyl adduct. We evaluated the reactions by the energy difference between product and starting material, not the activation barrier which would actually control the reaction process. A possible explanation of the inconsistency between computational and experimental results is that for the ester-substituted trityl radicals, their transition states have a similar correlation with the products and the thermodynamic energies are consistent with their kinetic values, while for the trityl salt, the transition state may have a different correlation with its product. These thermodynamic results are not exclusive, and other pathways for decomposition may be possible.
II. C. Experimental Stability of Trityl Radicals in Solution: EPR, MS and Product Studies
The stability in solution of various trityl radicals was investigated. Results show that each argon-purged solution of the esterifed trityls (1-CO2Et, 2-CO2Et and 3-CO2Et) gave a half-life of ∼4 days, while the carboxylate salt, 1-CO2Na did not decay for at least 17 days. This slow decomposition of the esterified trityls could be accounted for by the slow diffusion of O2 into the solution. However, in the presence of air, the half-life of 3-CO2Et was significantly shorter (∼1 day), while the half-lives of 2-CO2Et, 1-CO2Et and 1-CO2Na were the same as in the absence of O2. Product analysis of each of the trityl radicals' solutions using electrospray ionization (ESI) and matrix-assisted laser desorption ionization-time of flight (MALDI-ToF) mass spectrometry, before and after exposure to O2, was carried out (Figure 3). These analyses reveal that the same spectral pattern was observed for trityl solutions with ESI before and after exposure to O2 (Table 2). It should be noted, however, that ESI is an atmospheric pressure method, is an oxidizing environment and may lead to the formation of peroxyl adducts. In order to differentiate whether the observed mass spectra were due to O2 addition in the gas or liquid phase, trityl radical solutions were bubbled with O-18 labeled O2 (18O2) and the MS were acquired. Unfortunately, the peak for the 18O-labelled product was too small to be observed and could be due to the reversibility of O-16 O2 addition to these trityl radicals.
Figure 3.

Product ESI mass spectra of (a) 3-CO2Et and (b) 1-CO2Et; and MALDI-ToF mass fingerprint of (c) 1-CO2Na upon exposure to O2.
Table 2. ESI and MALDI-ToF Mass Fingerprints of Various Trityl Radicals in the Presence of O2.
| Entry |
Mobs (Mexact) |
M+Naobs (M+Naexact) |
M+Na+O-Hobs (M+Na+O-Hexact) |
M+Na+Oobs (M+Na+Oexact) |
M+Na+OHobs (M+Na+OHexact) |
M+Na+OOHobs (M+Na+OOHexact) |
|---|---|---|---|---|---|---|
| 1-CO2Et | 1083.036
(1083.036) |
1106.026
(1106.023) |
not observed
(1121.010) |
1121.998
(1122.018) |
1123.054
(1123.026) |
1139.066
(1139.021) |
| 2-CO2Et | 1011.030
(1011.011) |
1034.000
(1034.001) |
not observed
(1048.988) |
1049.988
(1049.996) |
1050.999
(1051.004) |
1067.047
(1067.000) |
| 3-CO2Et | 938.993
(938.990) |
961.981
(961.980) |
976.946
(976.967) |
977.970
(977.975) |
978.937
(978.983) |
994.976
(994.978) |
1-CO2Na: 999.110 (·C(ArCO2H)3), 1021.115 ((ArCO2Na)·C(ArCO2H)2), 1038.117 ((ArCO2Na)·C(ArCO2H)2+OH).
MALDI-ToF spectra for the carboxylate salt radical 1-CO2Na show only a very small peak corresponding to the OH adduct (Figure 3c), while OOH and OH adducts were produced from 1-CO2Et, 2-CO2Et and 3-CO2Et (Figure 3a-b and Table 2). This result is consistent with the EPR observation that in the presence of O2, 1-CO2Na exhibited the greatest stability compared to the three ester derivatives. The stability of these radicals, 1-CO2Na > 1-CO2Et > 2-CO2Et > 3-CO2Et, appears to be correlated with the number of H's on the aryl ring, as we predicted by the energetic calculations in the gas phase. Scheme 2 suggests an explanation consistent with this correlation, and shows the MS fragmentation pattern and the possible origin of the OH adduct as well as the quinone analogue. The scheme shows that the OH adduct could originate from the O-O homolytic cleavage of the peroxyl moeity. The formation of an (M+17) ion is evident in analysis of all of the ester trityl radicals 1-CO2Et, 2-CO2Et and 3-CO2Et, while a quinone-like product (M+15) was observed only for 3-CO2Et. While 2-CO2Et has one aromtic ring with a H, a species with a mass of (M+15) was not observed and is probably due to the smaller number of unsubstituted aromatic rings in 2-CO2Et compared to that in 3-CO2Et.
Scheme 2. Proposed mechanism for decomposition of an Unsubstituted Trityl Radical. (a) radical, (b) charged molecule pathways.

We then performed a product analysis for the decomposed 2-CO2Et and 3-CO2Et samples, and for which both samples possessed a lack of EPR signals. The products were separated by preparative HPLC, and the major products were analyzed by UV-vis, NMR, and high-resolution MS. Based on the MS results for both purified trityl reaction mixtures, quinone-like products (M+15) and trityl cation or anion (M) species are the major products. Even though these two products can be separated completely by HPLC because of their different polarity, trace quinone-like compound could always be detected from the M residue for both trityl compounds (Figure 4). Based on the MS pattern for the quinone-like compound (M+15) in the M residue, trace (M+16) and (M+17) ions can also be seen from the M residue for both decomposed samples. Furthermore, the relative intensities of (M+16) and (M+17) ions to (M+15) are higher in the M residue of 2-CO2Et than in that of 3-CO2Et. This also supports our previous assumption that the more unsubstituted aromatic rings, as in 3-CO2Et vs 2-CO2Et, will form more quinone-like (M+15) products. The MS results also indicate that the proposed radical pathway mechanism (Scheme 2a) may be a route for the decomposition of the trityl radical. There may also be a redox pathway in which trityl radicals lose their EPR signal by being oxidized or reduced to the cation or anion, respectively (Scheme 2b). (However, we should note that all attempts to obtain negative ion ESI-MS data were unsuccessful, and the MS results are most consistent with the formation of cationic species.) The formed trityl cation (or anion) could then form other products by reactions with nucleophiles (or electrophiles) and/or O2. UV-vis spectra of the quinone-like products have λmax peaks which are shifted to the red to 515 nm compared with their corresponding radicals (493 nm for 2-CO2Et and 481 nm for 3-CO2Et). This red shift is possibly due to the enhanced conjugation between the aromatic rings for the quinone-like products. The trityl cation or anion products, however, show blue shifts of their UV-vis spectra to 486 nm for 2-CO2Et and 473 nm for 3-CO2Et, respectively. NMR analysis was less conclusive as only 1H NMR could be obtained (see supporting information).
Figure 4.
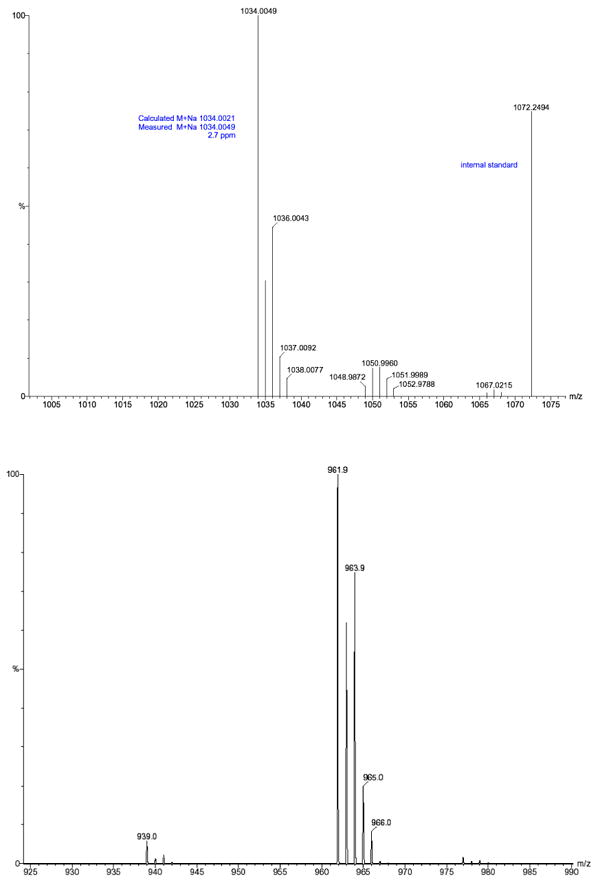
Product ESI mass spectra of M residues (trityl cation or anion) of (a) 2-CO2Et and (b) 3-CO2Et decomposition products.
II. D. Stability of Trityl Radicals in Intracellular Studies
So far, our studies show that the 3-CO2Et trityl radical is the least stable of all of the trityl radicals investigated in the presence of O2 alone. To test if these theoretical and experimental studies can be used to assess biostability of the trityls, their intracelluar stability in rat H9C2 cardiomyocytes was investigated. The ability of these compounds to penetrate into cells as well as their overall biostability were assessed for the four trityl radicals by incubating the compounds in the presence of rat H9C2 cardiomyocytes at various time intervals and subsequent washing of the cells to remove extracellular trityl radicals. After washing, the cells became colored (i.e., green or orange which are the original colors of the trityl radicals) indicating that there is absorption of these compounds inside the cell. EPR data reveal that among all of the trityl radicals used in the study, only the tri-substituted radicals, i.e., 1-CO2Na and 1-CO2Et, exhibited intracellular stability (Figure 5). In spite of the hydrophobic nature of the cell membrane, high permeability to cells was observed for the more polar 1-CO2Na and this may be due to the nature of the carrier solvent used, i.e., 1 μL DMSO per mL medium. The 2-CO2Et and 3-CO2Et trityl radicals, which have at least one H on the aryl-rings, showed only negligible EPR peak intensity after 1 hour of incubation with cells and suggests that these trityl radicals are susceptible to biodegradation in cellular systems.
Figure 5.

EPR signal intensity of 1-CO2Na (○) and 1-CO2Et (●) in rat H9C2 cardiomyocytes after 1, 2, 3, 4 and 5 hours of incubation and washing using phenol-red-free DMEM medium. (The 2-CO2Et and 3-CO2Et trityl radicals did not show an EPR signal after 1 hr of incubation and washing.)
II. E. Computational and CV Studies of the Oxidation/Reduction of the Trityl Radicals
In order to gain insights into the nature of the degradation process, the susceptibility of the trityl radicals to oxidation or reduction was investigated by calculating their respective electron affinities (EA) and ionization potentials (IP) as shown in Table 3. The partially substituted TAM-radicals, such as 2-CO2Et, 3-CO2Et and 4-CO2Et, gave lower EA and IP values compared to those of the fully substituted TAM-radicals (with the exception of the carboxylate salt, 1-CO2Na) indicating that the partially substituted TAM-radicals are harder to reduce and easier to oxidize compared to the fully substituted ones. The IP for the simplest trityl radical Ph3C• is intermediate, while the EA is lowest compared to the TAM-type radicals, indicating that the fully unsubstituted trityl radicals are the most difficult to be reduced. We therefore suggest that the loss of EPR signal in the decomposition of the trityl radicals leads to two processes: (1) formation of the trityl cation, which subsequently reacts with nucleophiles, and (2) O2-addition to the radical center, which subsequently forms the corresponding quinone product.
Table 3. Predicted Electron Affinities, Ionization Potentials, BDE's and Energies of Protonation of Various Trityl Radicals at the B3LYP/6-311+G**//B3LYP/6-31G* Level.
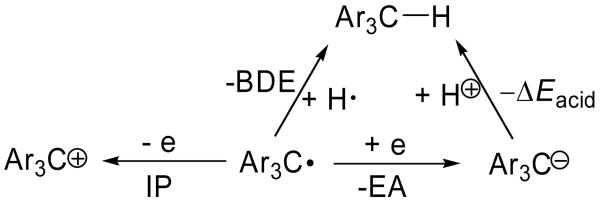 | ||||
|---|---|---|---|---|
| Ar3C•Radical | Electron Affinity (EA) kcal/mol (eV) | Ionization Potential (IP) kcal/mol (eV) | Ccentral-H Bond Dissociation Energy BDE (0K) kcal/mol | ΔEacid (0K) kcal/mol |
| Ph3C• | 32.7 (1.42) | 137.8 (5.98) | 80.4 | 362.8 |
| 1-CO2Na | 38.8 (1.68) | 118.1 (5.12) | 77.1 | 353.4 |
| 1-CO2Na (aq)a | 79.4 (3.44) | 109.8 (4.76) | 80.2 | 313.4 |
| 1-CO2Et | 60.7 (2.63) | 139.0 (6.03) | 77.2 | 331.7 |
| 2-CO2Et | 57.6 (2.50) | 137.2 (5.95) | 77.8 | 335.3 |
| 3-CO2Et | 54.4 (2.36) | 136.2 (5.91) | 77.9 | 338.6 |
| 4-CO2Et | 50.3 (2.18) | 134.8 (5.85) | 78.1 | 342.9 |
At the B3LYP/6-311+G**//B3LYP/6-31G* level using PCM.
Cyclic voltammetry (CV) was performed to obtain the redox potentials for the carboxylate salt 1-CO2Na and triester 1-CO2Et (Table 4). The experimental results showed that the carboxylate salt is easier to oxidize and to reduce than the triester. This is consistent with the computational results using the PCM calculations for the carboxylate salt and with the gas-phase calculations for the triester. Because the trityl radical salt (1-CO2Na) is more stable than the triester (1-CO2Et) in organic and biological solutions and 1-CO2Na is more easily oxidized and reduced, the decomposition of the trityl radical is more likely to be a radical process (Scheme 2a). It is possible that the redox reaction proceeds via an equilibrium; recall that the oxidation/reduction of 1-CO2Na is reversible, but is not reversible for 1-CO2Et (Table 4). Also, from the CV data (Table 4), the carboxylate salt 1-CO2Na is easier to oxidize than the 1-CO2Et triester,. Therefore, we conjecture that the rate-determining step for decomposition involves the corresponding trityl radical for the 1-CO2Na species, proceeding via peroxyl radical formation, but the rate-determining step for the ester radicals, such as 1-CO2Et, may involve the radical, or irreversibly formed cation. Such a scenario would be consistent with the stability of the carboxylate salt 1-CO2Na being greater than the corresponding esters (1-CO2Et) as well as the greater propensity to oxidize and to reduce the irreversibly formed cation. Such a scenario would be consistent with the stability of the carboxylate salt 1-CO2Na. However, which pathway, Scheme 2a (radical) or 2b (ionic), is the main cause of the decomposition of the trityl radicals is not fully clarified.
Table 4. Redox Potentials E1/2 of 1-CO2Na and 1-CO2Et Determined by Cyclic Voltammetrya relative to Ag / 4M AgCl.
| Compound | Solvent |
E1/2 (Ox1) (V)
(ΔEpc [mV]) |
E1/2 (Ox2) (V)
(ΔEp [mV]) |
E1/2 (Red1) (V)
(ΔEp [mV]) |
|---|---|---|---|---|
| TEMPO | aqueous | 0.52 (90) | ||
| TEMPO | CH3CN | 0.66 (85) | ||
| 1-CO2Na | aqueous | 0.45 (60) | 0.83 (50) | -0.65 (60) |
| 1-CO2Na | CH3CNb | 0.59 (60) | 0.97 (50) | -0.51 (60) |
| 1-CO2Et | CH3CN | 1.42d | 0.94e | -1.52 (400) |
Scan rate = 100 mV/s.
Using TEMPO to calibrate aqueous solution to acetonitrile.
ΔEp = |Epa – Epc|.
Anodic peak only.
Cathodic peak only.
II. F. Stability of Trityl Radicals: Bond Dissociation Energies
If the trityl radicals were decomposing by radical processes, it is important to understand the stability of the carbon-centered radical in this homologous series. A simple hypothesis would be that the more stable trityl radical (Ar3C•) would be derived from the parent aryl methane (Ar3C-H) derivative that had the lowest C-H bond dissociation energy (BDE). To assess the relative stability of various TAM radicals, the Ccentral-H BDE was calculated for the compounds under study (Table 3). The calculations reveal that there is less than 1.5 kcal/mol difference in the BDE's for generating these diverse radicals, and indicates that the unpaired electron on all of the TAM radicals is stabilized, as supported by the spin density (population) results shown in Figure 2. Ph3C-H, however, gave the highest BDE value of 80.4 kcal/mol compared to the TAM-type radicals of 77.2-78.1 kcal/mol, thereby indicating that substitution of the aromatic ring has a stabilizing effect on the radical formation and disfavors protonation (ΔEacid) of their respective carbanion analogues (Table 3). The partially substituted TAM-radicals, such as 2-CO2Et, 3-CO2Et and 4-CO2Et, gave higher BDE and ΔEacid values compared to those of the fully substituted TAM-radicals (with the exception of the carboxylate salt, 1-CO2Na) indicating that the H on the aromatic ring does not stabilize the trityl radical center and may contribute to the instability of the trityl radical.
IV. Conclusions
Over the years, the use of trityl radicals as probes for electron paramagnetic resonance imaging (EPRI) applications has become increasingly important. An understanding of the degradation pathways of trityl radicals is critical in the design of probes with improved stability. Electronic properties as well as the thermodynamics of O2 addition to various TAM-type radicals were theoretically investigated. Results show that the presence of hydrogen as a substituent on the aromatic ring affects the stability of the trityl radical by making the intermediate to be more susceptible to O2 addition at the aryl carbon bearing the H. Addition of O2 to the aromatic ring is more favored in TAM-type trityl radicals, while addition to the central carbon atom is more preferred in Ph3C• because of a steric effect. Mass spectrometric analyses reveal the formation of a quinone-type product in partially substituted trityl radicals in the presence of O2. The formation of the quinone intermediate after reaction with O2, as observed for 3-CO2Et, could be a potential precursor to the formation of gluthathione or Michael adducts with other nucleophiles that are present in cells as demonstrated by previous studies39-43 which showed the addition of certain thiols, such as glutathione, to quinones. Redox chemistry is also evident and suggests the formation of the trityl cation. Cell permeability studies show that fully substituted trityl radicals are biostable compared to the partially substituted ones. Predicted electron affinities and ionization potentials indicate that for TAM-type compounds, the less that the trityl radical is substituted, the easier it is to be oxidized. This study demonstrates how computational chemistry can be used as a tool to assess radical stability in complex systems and aid in the future design of more biostable trityl adducts.
Experimental Section
Although the preparations of 1-CO2Na and 1-CO2Et have been described in the literature,27 slight modifications in the reaction conditions were utilized.
Tris(8-ethoxycarbonyl-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′] bis[1,3]dithiol-4-yl)methanol (8), bis(8-ethoxycarbonyl)tris- (2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl) methanol (9), 8-ethoxycarbonyl-tris(2,2,6,6-tetramethylbenzo [1,2-d;4,5-d′]bis[1,3]dithiol-4-yl) methanol (10)
To a stirred solution of TMEDA (250 μL, 1.66 mmol) in benzene (1 mL) was added 2.5 M n-BuLi in pentane (0.67 mL, 1.67 mmol) dropwise at 0 °C. After being stirred for 30 min, a solution of 7 (193 mg, 0.2 mmol) in benzene (1 mL) was added to the above solution dropwise at 0 °C. After being stirred at 35-45 °C for 45 min, the resulting solution was added to a solution of diethyl carbonate (1.04 mL, 8.62 mmol) in benzene (1 mL) that was maintained at 0 °C. After the mixture was stirred at 45 °C for 2 h, saturated aqueous KH2PO4 was added. The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The red residue was crystallized from MeCN (4 mL) to give 90 mg of a crude orange solid. Flash chromatography on silica gel, using diethyl ether/hexanes (1:20) and then gradually increasing to ethyl acetate/hexanes (1:5) as eluant, provided: 45 mg (25%) of 8 as orange crystals that contained a small amount of inseparable impurities: mp > 280 °C (gradually turned black, dec); 1H NMR (400 MHz, CDCl3) δ 1.45 (t, 9H), 1.65 (s, 18H), 1.73 (s, 9H), 1.76 (s, 9H), 4.43 (m, 6H), 6.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.3, 28.6, 29.2, 31.9, 33.8, 60.85, 60.93, 62.3, 84.3, 121.3, 134.0, 139.3, 140.3, 141.4, 141.8, 166.2; IR (CHCl3) 3335, 2970, 1699, 1239, 1217, 1018, 728 cm-1; MS (ESI, (M+Na)+, m/z): 1123.03005 (observed), 1123.025363 (exact).
25 mg (15%) of 9 as orange crystals that contained a small amount of inseparable impurities: mp > 265 °C (gradually turned black, dec); 1H NMR (400 MHz, CDCl3) δ 1.45 (td, 6H), 1.63-1.82 (m, 36H), 4.42 (m, 4H), 6.68 (s, 1H), 7.18 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.2, 14.3, 27.5, 27.7, 29.3, 29.6, 30.1, 30.8, 31.6, 33.0, 34.0, 34.6, 34.9, 60.71, 60.74, 61.2(d), 61.8, 62.3, 63.2, 63.8, 84.1, 118.5, 121.24, 121.26, 130.6, 134.6, 134.7, 137.0, 137.7, 138.0, 139.1, 139.2, 139.6, 140.2, 140.7, 141.4, 141.7, 141.86, 141.89, 166.22, 166.24; MS (ESI, (M+Na)+, m/z): 1051.00782 (observed), 1051.004233 (exact).
15 mg (9%) of 10 as orange crystals that contained a small amount of inseparable impurities: mp > 255 °C (gradually turned black, dec); 1H NMR (400 MHz, CDCl3) δ 1.45 (t, 3H), 1.62-1.82 (m, 36H), 4.43 (m, 2H), 6.55 (s, 1H), 7.17 (t, 2H); 13C NMR (100 MHz, CDCl3) δ 14.3, 27.3, 27.9, 28.3, 29.1, 29.7, 31.6, 33.0, 33.3, 33.6, 34.3, 34.6, 35.0, 60.7, 62.34, 62.39, 63.46, 63.49, 63.54, 63.63, 83.9, 118.3, 118.4, 121.3, 130.7, 131.7, 135.6, 136.9, 137.2, 137.5, 137.6, 137.9, 139.04, 139.06, 139.20, 139.28, 140.8, 141.8, 142.0, 166.3; MS (ESI, (M+Na)+, m/z): 978.98481 (observed), 978.983103 (exact).
Tris(8-ethoxycarbonyl-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′] bis[1,3]dithiol-4-yl)methyl ester (1-CO2Et), bis(8-ethoxycarbonyl)tris (2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3] dithiol-4-yl)- methyl ester (2-CO2Et), 8-ethoxycarbonyl-tris(2,2,6,6-tetramethyl benzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl) methyl ester (3-CO2Et), Tris- (8-carboxyl-2,2,6,6-tetramethyl benzo-[1,2-d;4,5-d′]bis[1,3] dithiol -4-yl) methyl Sodium Salt (1-CO2Na)
To a stirred solution of 8 (11 mg, 0.01 mmol) in CH2Cl2 (2 mL) was added BF3·Et2O (10 μL, 0.08 mmol) dropwise at room temperature. After being stirred for 1 h, the resulting dark green-blue solution was treated with a solution of SnCl2 (3.2 mg, 0.017 mmol) in THF (0.4 mL). After 10 min, saturated aqueous KH2PO4 was added. The organic layer was separated, dried over Na2SO4, and concentrated in vacuo to give 10 mg of the crude trityl radical 1-CO2Et as an orange-brown solid: IR 2960, 2924, 2855, 1704, 1490, 1452, 1367, 1234, 1110, 1044 cm-1; UV 249, 411, 495 nm; MS (ESI, (M+Na)+, m/z): :1106.02081 (observed), 1106.022623 (exact).
To a stirred solution of 9 (10 mg, 0.01 mmol) in CH2Cl2 (2 mL) was added BF3·Et2O (10 μL, 0.08 mmol) dropwise at room temperature. After being stirred for 1 h, the resulting dark green-blue solution was treated with a solution of SnCl2 (3.2 mg, 0.017 mmol) in THF (0.4 mL). After 10 min, saturated aqueous KH2PO4 was added. The organic layer was separated, dried over Na2SO4, and concentrated in vacuo to give 10 mg of the crude trityl radical 2-CO2Et as an orange-brown solid: IR 2958, 2923, 2855, 1702, 1488, 1453, 1366, 1237, 1148, 1109, 1043 cm-1; UV 247, 405, 493 nm; MS (ESI, (M+Na)+, m/z): 1034.00046 (observed), 1034.001493 (exact).
To a stirred solution of 10 (9.5 mg, 0.01 mmol) in CH2Cl2 (2 mL) was added BF3·Et2O (10 μL, 0.08 mmol) dropwise at room temperature. After being stirred for 1 h, the resulting dark green-blue solution was treated with a solution of SnCl2 (3.2 mg, 0.017 mmol) in THF (0.4 mL). After 10 min, saturated aqueous KH2PO4 was added. The organic layer was separated, dried over Na2SO4, and concentrated in vacuo to give 9 mg of the crude trityl radical 3-CO2Et as an orange-brown solid: IR 2957, 2923, 2853, 1703, 1605, 1452, 1365, 1239, 1149, 1109, 1020 cm-1; UV 239, 449, 481 nm; MS (ESI, (M+Na)+, m/z): 961.98135 (observed), 961.980363 (exact).
The crude solid 1-CO2Et was dissolved in dioxane (0.2 mL), and 1M aqueous KOH (0.1 mL) was added. The resultant dark orange-brown solution was heated at 50 °C for 2 h. After being cooled to room temperature, the reaction mixture was diluted with water, and washed twice with ether. The aqueous layer was acidified with 1 M HCl, and the resulting orange brown precipitate was extracted with ether. The ether layer was separated, dried over Na2SO4, and concentrated in vacuo. The residue was dissolved in 0.1 M aqueous NaOH (0.3 mL), and concentrated in vacuo to give 9 mg of trityl radical 1-CO2Na as a dark green-yellow solid: IR ∼3400 (broad), 2923, 1585, 1425, 1260, 1150, 879 cm-1. UV 277, 469 nm;
Computational Methods
Density functional theory44,45 was applied in this study to determine the optimized geometry of each species.46-49 All calculations were performed using Gaussian 0350 at the Ohio Supercomputer Center. Optimized geometries were obtained at the B3LYP/6-31G* level. Electron spin densities (populations) were obtained from a natural population analysis (NPA) approach using single-point energies evaluated at the B3LYP/6-311+G** level with 6d functions.51 The effect of solvation on the gas-phase calculations was also investigated using the polarized continuum model (PCM).52-56 Because the vibrational frequency calculation for each trityl radical could not be finished in more than 14 days of cpu time, we employed the bottom-of-the-well energies to compare the respective radicals.
EPR Measurements
EPR measurements were carried out on an X band spectrometer with HS resonator at room temperature. General instrument settings are as follows: microwave power, 3 mW; modulation amplitude, 0.03 G; receiver gain 3.5 × 103; modulation frequency, 6 kHz; scan time, 40 s; time constant, 82 ms. Total volume of all solutions used for EPR measurement was 50 μL and was loaded into 50 μL micropipettes that were sealed with clay.
Mass Spectrometric Analysis
ESI/MS
Electrospray ionization (ESI) mass spectrometric analyses were performed for 1-CO2Et, 2-CO2Et and 3-CO2Et. The trityl solution (∼1mM) in tetrahydrofuran (THF) was initially purged with Ar or O2 gas for 4 min before mixing with THF/CH3OH solvent that was previously saturated with NaCl. This combination provided the necessary cationization for charging in the electrospray process for ion generation. Mass analysis was then performed in positive ion detection mode.
MALDI-TOF/MS
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) was used for 1-CO2Na with the mass spectrometer operated in linear, positive ion mode and with a N2 laser for laser desorption. Samples were prepared in 0.1% TFA at an approximate concentration of 50 pmol/μl. 2,5-Dihydroxybenzoic acid was prepared as a saturated solutions in 50% ACN/0.1% TFA (in water). Allotments of 1 mL of matrix and 1 mL of ∼1 mM aqueous solution of 1-CO2Na (purged with Ar or O2 gas for 4 min) were thoroughly mixed together; 0.5 mL of this was spotted on the target plate and allowed to dry prior to MS analysis.
Cell Studies
Rat cardiac H9C2 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin in 150 cm2 tissue culture flasks at 37°C in a humidified atmosphere of 5% CO2. The cells were fed every 2-3 days, and subcultured once they reached 80-90% confluence. For determination of penetration of the trityl radicals into cells, H9C2 cells were incubated with the trityl compounds introduced by 10 μL (1 mM) in 10:1 DMSO to water solution for various time points. Cells were then collected, and washed twice with PBS prior to EPR measurement.
Cyclic Voltammetry Measurements
Cyclic voltammetry was performed on a potentiostat and computer-controlled electroanalytical system. Electrochemical measurements were carried out in a 5 ml cell equipped with a gold working electrode (1.6 mm diameter), a platinum-wire auxiliary electrode, and a Ag/AgCl reference electrode. The gold electrode was cleaned before each run by rubbing (circular motion for approximately 10s) on a polishing pad with crystal solution and then washed with water-acetone. Solutions were degassed by bubbling with the argon gas. Background current (solvent with supporting electrolyte) corrections were done for all measurements. Half-wave potentials were calculated according to the relation E1/2 = (Epa + Epc)/2.
Acetonitrile and phosphate buffered saline were used without further purification. Tetrabutylammonium perchlorate (TBAP) and 2,2,6,6-tetrmethylpiperidine-1-oxyl (TEMPO) were used as purchase.
All measurements were performed in dry acetonitrile with 0.1 M TBAP as supporting electrolyte for triester trityl radical 1-CO2Et, or in phosphate aqueous solution for carboxylate salt trityl radical 1-CO2Na. The concentration of the investigated compounds was about 1 mM. Voltage was cycled between -2.0 and 0.0 V beginning at 0.0 V, between 0.0 V and 2.0 V beginning at 0.0 V, and -2.0 V and 2.0 V beginning at 0.0 V.
Supplementary Material
Geometries, energies, enthalpies, and free energies for all compounds and their corresponding adducts, 1H-NMR, EPR and MS spectra are available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH grants HL38324, HL63744 and HL65608. CMH acknowledges support from the NSF-funded Environmental Molecular Science Institute (CHE-0089147). The Ohio Supercomputer Center (OSC) is acknowledged for generous computational support of this research.
Contributor Information
Frederick A. Villamena, Email: frederick.villamena@osumc.edu.
Christopher M. Hadad, Email: hadad.1@osu.edu.
Jay L. Zweier, Email: jay.zweier@osumc.edu.
References
- 1.Bilenko MV. Ischemia and Reperfusion of Various Organs: Injury, Mechanisms, Methods of Prevention and Treatment. Nova Science Pub Inc.; Huntington, New York: 2001. [Google Scholar]
- 2.Clerch LB, Massaro DJ, editors. Oxygen, Gene Expression and Cellular Function. Vol. 105 Marcel Dekker, Inc; New York: 1997. [Google Scholar]
- 3.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford Univeristy Press; Oxford: 1999. [Google Scholar]
- 4.Zweier JL, Flaherty JT, Weisfeldt ML. Proc Natl Acad Sci U S A. 1987;84:1404. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berliner LJ. Applications of EPR imaging to materials, agriculture and medicine In Magnetic Resonance Microscopy: Methods and Applications in Material Science, Agriculture and Biomedicine. Weinheim; VCH: 1992. [Google Scholar]
- 6.Berliner LJ. In vivo EPR (ESR): Theory and Applications. Kluwer Academic Plenum; New York: 1993. [Google Scholar]
- 7.Eaton GR, Eaton SS, Ohno K. EPR Imaging and in vivo EPR. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 8.Zweier JL, Kuppusamy P. Proc Natl Acad Sci USA. 1988;85:5703. doi: 10.1073/pnas.85.15.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yordanov AT, Yamada K, Krishna MC, Russo A, Yoo J, English S, Mitchell JB, Brechbiel MW. J Med Chem. 2002;45:2283. doi: 10.1021/jm0105169. [DOI] [PubMed] [Google Scholar]
- 10.He G, Deng Y, Li H, Kuppusamy P, Zweier JL. Magn Reson Med. 2002;47:571. doi: 10.1002/mrm.10077. [DOI] [PubMed] [Google Scholar]
- 11.He G, Samouilov A, Kuppusamy P, Zweier JL. Molecular and Cellular Biochemistry. 2002;234:359. [PubMed] [Google Scholar]
- 12.Afeworki M, van Dam GM, Devasahayam N, Murugesan R, Cook J, Coffin D, Larsen JH, Mitchell JB, Subramanian S, Krishna MC. Magn Reson Med. 2000;43:375. doi: 10.1002/(sici)1522-2594(200003)43:3<375::aid-mrm9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell JB, Russo A, Kuppusamy P, Krishna MC. Ann N Y Acad Sci. 2000;899:28. doi: 10.1111/j.1749-6632.2000.tb06174.x. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell JB, Yamada K, Devasahayam N, Cook JA, Subramanian S, Krishna MC. J Nutr. 2004;134:3210S. doi: 10.1093/jn/134.11.3210S. [DOI] [PubMed] [Google Scholar]
- 15.Yamada KI, Kuppusamy P, English S, Yoo J, Irie A, Subramanian S, Mitchell JB, Krishna MC. Acta Radiol. 2002;43:433. doi: 10.1080/j.1600-0455.2002.430418.x. [DOI] [PubMed] [Google Scholar]
- 16.Subramanian S, Yamada K, Irie A, Murugesan R, Cook JA, Devasahayam N, Van Dam GM, Mitchell JB, Krishna MC. Magn Reson Med. 2002;47:1001. doi: 10.1002/mrm.10133. [DOI] [PubMed] [Google Scholar]
- 17.Kuppusamy P, Afeworki M, Shankar RA, Coffin D, Krishna MC, Hahn SM, Mitchell JB, Zweier JL. Cancer Res. 1998;58:1562. [PubMed] [Google Scholar]
- 18.Matsumoto K, Krishna MC, Mitchell JB. J Pharmacol Exp Ther. 2004;310:1076. doi: 10.1124/jpet.104.066647. [DOI] [PubMed] [Google Scholar]
- 19.Bowmana MK, Mailer C, Halpern HJ. J Magn Reson. 2005;172:254. doi: 10.1016/j.jmr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Matsumoto K, English S, Yoo J, Yamada K, Devasahayam N, Cook JA, Mitchell JB, Subramanian S, Krishna MC. Magn Reson Med. 2004;52:885. doi: 10.1002/mrm.20222. [DOI] [PubMed] [Google Scholar]
- 21.Yordanov AT, Yamada Ki K, Krishna MC, Mitchell JB, Woller E, Cloninger M, Brechbiel MW. Angew Chem Int Ed Engl. 2001;40:2690. doi: 10.1002/1521-3773(20010716)40:14<2690::aid-anie2690>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 22.Ilangovan G, Manivannan A, Li H, Yanagi H, Zweier JL, Kuppusamy P. Free Radic Biol Med. 2002;32:139. doi: 10.1016/s0891-5849(01)00784-5. [DOI] [PubMed] [Google Scholar]
- 23.Krishna MC, English S, Yamada K, Yoo J, Murugesan R, Devasahayam N, Cook JA, Golman K, Ardenkjaer-Larsen JH, Subramanian S, Mitchell JB. Proc Natl Acad Sci U S A. 2002;99:2216. doi: 10.1073/pnas.042671399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kutala VK, Parinandi NL, Zweier JL, Kuppusamy P. Arch Biochem Biophys. 2004;424:81. doi: 10.1016/j.abb.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Rizzi C, Samouilov A, Kumar Kutala V, Parinandi NL, Zweier JL, Kuppusamy P. Free Radic Biol Med. 2003;35:1608. doi: 10.1016/j.freeradbiomed.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 26.Gomberg M. J Am Chem Soc. 1900;22:757. [Google Scholar]
- 27.Reddy TJ, Iwama T, Halpern HJ, Rawal VH. J Org Chem. 2002;67:4635. doi: 10.1021/jo011068f. [DOI] [PubMed] [Google Scholar]
- 28.Williams BB, al Hallaq H, Chandramouli GV, Barth ED, Rivers JN, Lewis M, Galtsev VE, Karczmar GS, Halpern HJ. Magn Reson Med. 2002;47:634. doi: 10.1002/mrm.10089. [DOI] [PubMed] [Google Scholar]
- 29.Ardenkjaer-Larsen JH, Laursen I, Leunbach I, Ehnholm G, Wistrand LG, Petersson JS, Golman K. J Magn Reson. 1998;133:1. doi: 10.1006/jmre.1998.1438. [DOI] [PubMed] [Google Scholar]
- 30.Ayers CL, Janzen EG, Johnston FJ. J Am Chem Soc. 1966;88:2610. [Google Scholar]
- 31.Janzen EG, Johnston FJ, Ayers CL. J Am Chem Soc. 1967;89:1176. [Google Scholar]
- 32.Wang H, Parker VD. Acta Chim Scand. 1997;51:865. [Google Scholar]
- 33.Trapp C, Kulkarni SV. J Phys Chem. 1984;88:2703. [Google Scholar]
- 34.Che Y, Tokuda K, Ohsaka T. Bulletin of the Chemical Society of Japan. 1998;71:651. [Google Scholar]
- 35.Bowman MK, Mailer C, Halpern HJ. J Magn Reson. 2005;172:254. doi: 10.1016/j.jmr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 36.Pratt DA, Mills JH, Porter NA. J Am Chem Soc. 2003;125:5801. doi: 10.1021/ja034182j. [DOI] [PubMed] [Google Scholar]
- 37.Merle JK, Hadad CM. J Phys Chem A. 2004;108:8419. [Google Scholar]
- 38.(a) Liu J, Hadad CM, Platz MS. Org Lett. 2005;7:549. doi: 10.1021/ol047782b. [DOI] [PubMed] [Google Scholar]; (b) Fadden MJ, Barckholtz C, Hadad CM. J Phys Chem A. 2000;104:3004. [Google Scholar]
- 39.Seung S, Lee J, Lee M, Park J, Chung J. Chemico-Biological Interactions. 1998;113:133. doi: 10.1016/s0009-2797(98)00024-6. [DOI] [PubMed] [Google Scholar]
- 40.Karczewski JM, Peters JGP, Noordhoek J. Biochemical Pharmacology. 1999;57:27. doi: 10.1016/s0006-2952(98)00288-3. [DOI] [PubMed] [Google Scholar]
- 41.Briggs MK, Desavis E, Mazzer PA, Sunoj RB, Hatcher SA, Hadad CM, Hatcher PG. Chem Res Toxicol. 2003;16:1484. doi: 10.1021/tx0341512. [DOI] [PubMed] [Google Scholar]
- 42.Sachdeva B, Thomas B, Wang X, Ma J, Jones KH, Hatcher PG, Cornwell DG. Chem Res Toxicol. 2005;18:1018. doi: 10.1021/tx0496441. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Thomas B, Sachdeva R, Arterburn L, Frye L, Hatcher PG, Cornwell DG, Ma J. PNAS. 2006;103:3604. doi: 10.1073/pnas.0510962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Labanowski JW, Andzelm J. Density Functional Methods in Chemistry. Springer; New York: 1991. [Google Scholar]
- 45.Parr RG, Yang W. Density Functional Theory in Atoms and Molecules. Oxford University Press; New York: 1989. [Google Scholar]
- 46.Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 47.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 48.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 49.Hehre WJ, Radom L, Schleyer PV, Pople JA. Ab Initio Molecular orbital Theory. John Wiley & Sons; New York: 1986. [Google Scholar]
- 50.Frisch MJ, et al. Gaussian 03; Revision B.04. Gaussian, Inc.; Pittsburgh PA: 2003. [Google Scholar]
- 51.Reed AE, Weinhold FA, Curtiss LA. Chem Rev. 1998;98:899. [Google Scholar]
- 52.Tomasi J, Persico M. Chem Rev. 1994;94:2027. [Google Scholar]
- 53.Cossi M, Barone V, Cammi R, Tomasi J. Chem Phys Lett. 1996;255:327. [Google Scholar]
- 54.Barone V, Cossi M, Tomasi J. J Chem Phys. 1997;107:3210. [Google Scholar]
- 55.Barone V, Cossi M, Tomasi J. J Comput Chem. 1998;19:404. [Google Scholar]
- 56.Cossi M, Barone V. J Chem Phys. 1998;109:6246. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Geometries, energies, enthalpies, and free energies for all compounds and their corresponding adducts, 1H-NMR, EPR and MS spectra are available free of charge via the Internet at http://pubs.acs.org.