

Abstract
Objective
It has been suggested that polymorphisms in IL1 are correlated with severe and/or erosive rheumatoid arthritis (RA), but the implicated alleles have differed among studies. The aim of this study was to perform a broad and well-powered search for association between allelic polymorphism in IL1A and IL1B and the susceptibility to or severity of RA.
Methods
Key coding and regulatory regions in IL1A and IL1B were sequenced in 24 patients with RA, revealing 4 novel single-nucleotide polymorphisms (SNPs) in IL1B. These and a comprehensive set of 24 SNPs tagging most of the underlying genetic diversity were genotyped in 3 independent RA case-control sample sets and 1 longitudinal RA cohort, totaling 3,561 patients and 3,062 control subjects.
Results
No fully significant associations were observed. Analysis of the discovery case-control sample sets indicated a potential association of IL1B promoter region SNPs with susceptibility to RA (for RA3/A, odds ratio [OR] 1.27, P = 0.0021) or with the incidence of radiographic erosions (for RA4/C, OR 1.56, P = 0.036), but these findings were not replicated in independent case-control samples. No association with rheumatoid factor, anti-cyclic citrullinated peptide, or the Disease Activity Score in 28 joints was found. None of the associations previously observed in other studies were replicated here.
Conclusion
In spite of a broad and highly powered study, we observed no robust, reproducible association between IL1A/B variants and the susceptibility to or severity of RA in white individuals of European descent. Our results provide evidence that, in the majority of cases, polymorphism in IL1A and IL1B is not a major contributor to genetic susceptibility to RA.
Interleukin-1 (IL-1) is a key mediator of inflammation, with pleiotropic effects on several cells and signaling pathways. The activity defined as IL-1 reflects the function of 2 molecules, IL-1α and IL-1β. IL1A encodes IL-1α, which is cell-bound, and IL1B encodes IL-1β, a secreted cytokine. IL-1 is a critical mediator in several systemic autoinflammatory syndromes and in juvenile rheumatoid arthritis, as evidenced by a dramatic effect of anti-IL-1 therapy in those diseases (1–3). IL-1 also plays a pathogenic role in inflammation and tissue destruction in rheumatoid arthritis (RA) (4, 5). IL-1 blockade ameliorates arthritis in multiple mouse models and has been shown in clinical trials to improve human RA (6).
Although the existence of genetic factors that modulate susceptibility to RA is well established, only a few such genes have thus far been identified and confirmed (7). Other than that for the “shared epitope” alleles at the HLA locus (8), the most compelling evidence of an association exists for PTPN22 (9). Recently, a genome-wide association study not only confirmed the association between RA susceptibility and the HLA and PTPN22 loci but also uncovered 9 other loci (at a significance level of P = 10−5 to P = 10−7) that are potentially associated with RA (10). In addition, single-nucleotide polymorphisms (SNPs) in the regions of STAT4 (11,12), TRAF1/C5 (13, 14), and TNFAIP3 (15, 16) have recently been shown to be associated with RA susceptibility, but the functional consequence of these polymorphisms has not yet been elucidated.
The central role that IL-1 plays in inflammation suggests that allelic polymorphism in IL1 might have an impact on susceptibility to RA. Indeed, in mice, allelic polymorphism in Il1b influences the severity of arthritis in the K/BxN model (17). In humans, the situation is less clear. A genome-wide scan of a family cohort showed an excess of allele sharing for the IL1 gene cluster (18). A subsequent linkage study of the IL1 locus showed no linkage with susceptibility to RA, although subgroup analysis demonstrated linkage of IL1 in families with erosive RA if siblings did not share the HLA-DRB1 allele (19). Similarly, case-control association studies revealed no association of IL1 variants and susceptibility to RA but showed an association with severity or radiographic erosions or progression (19–24). The number of patients with RA who were screened in these studies was relatively small (<300 patients in all studies); therefore, these analyses were underpowered to show weak associations. In addition, these studies addressed a rather limited number of polymorphisms, focusing on some subset of only 2 polymorphisms in IL1B (−511 and +3954) and 2 polymorphisms in IL1A (−889 and +4845. These polymorphisms in IL1A are in tight linkage disequilibrium [LD] and thus provide the same information). International SNP databases currently list 88 SNPs in the IL1B gene alone; clearly, much of the existing variation has not yet been analyzed. In addition, it is possible that disease-specific variants found only in patients with RA exist and have not been catalogued in general SNP databases.
Given the importance of IL-1 as a mediator of inflammation, suggestive results from prior genetics studies in humans, and the clear evidence of an arthritis association in mouse models, we thought that a deeper and broader analysis of genetic variation at the IL1 loci was warranted. Therefore, we performed a comprehensive and well-powered study to determine the relevance of polymorphisms in IL1A and IL1B to susceptibility to RA or to its destructive consequences.
PATIENTS AND METHODS
Clinical samples
Clinical samples were obtained from 5 sources, as follows:
Centre d’Etude du Polymorphisme Humain (CEPH) samples from the International HapMap project (90 individuals from Utah with ancestry from northern and western Europe) (25).
The Brigham Rheumatoid Arthritis Sequential Study (BRASS). A prospective longitudinal study of patients with RA diagnosed by a rheumatologist (26). Clinical measures, including anti-cyclic citrullinated peptide (anti-CCP) antibodies, rheumatoid factor (RF), C-reactive protein, and the Disease Activity Score in 28 joints (DAS28) (27), were obtained at the baseline visit. Hand radiographs were evaluated by a radiologist for the presence of erosions. Only the 774 patients who identified their race as “white” were selected for analysis, in order to avoid population admixture effects.
North American Rheumatoid Arthritis Consortium (NARAC). The cases included 1,314 individuals from 619 multiplex families (primarily affected sibling pairs); at least 1 sibling had documented erosions on hand radiographs, and at least 1 sibling experienced disease onset between the ages of 18 years and 60 years (28). All of the NARAC patients satisfied the 1987 American College of Rheumatology (ACR; formerly, the American Rheumatism Association) criteria for the classification of RA (29). The control subjects were 1,103 individuals from the New York Cancer Project (NYCP), a population-based prospective study of healthy individuals. Approximately 2 control subjects were matched to a single randomly chosen affected sibling on the basis of sex, age (birth decade), and ethnicity (grandparental country/region of origin). (The number of cases does not equal the number of control subjects, because some case families contributed more or fewer than 2 siblings, and because 2 control subjects were not available for every case.) Only patients and control subjects who identified their ancestry as “white” were selected for this study. Complete data for PTPN22 for this case-control sample set have been published previously (9).
The Spanish case-control sample set from the University Clinical Hospital of Santiago de Compostela. The study group comprised 525 patients satisfying the ACR criteria for the classification of RA (29) and 504 control subjects ages 55 years or older who were undergoing elective nonorthopedic surgery (30). The patients and control subjects were white individuals of Spanish origin, all of whose known ancestors were of the same origin.
The Wichita Rheumatic Disease Data Bank (WRDDB)/National Inception Cohort of Rheumatoid Arthritis Patients (NICRAP)/Study of New Onset Rheumatoid Arthritis (SONORA) case-control sample set. Patients included 948 anti-CCP antibody-positive individuals in whom RA was diagnosed by a rheumatologist. These patients were selected from 3 independent registries, as follows: the WRDDB (in which the mean disease duration was 10 years) (31), the NICRAP (in which patients were enrolled within 6 months of the clinical diagnosis) (32), and SONORA (in which patients were enrolled within 3–12 months of the clinical diagnosis) (33). Control subjects included 1,455 unique individuals from the NYCP. Only patients and control subjects who identified their race as “white” were selected for this study.
Sequencing
The VISTA genome browser (34) was used to identify regions of homology with the rat and mouse genomes. Exons and noncoding regions showing homology with the rodent genomes were amplified by polymerase chain reaction (PCR) and then treated with exonuclease I and alkaline phosphatase. Bidirectional sequencing was performed. Sequencher software (Gene Codes Corp., Ann Arbor, MI) was used to align and compare chromatographs. Secondary peaks that were ≥30% of the maximum peak height were visually inspected for validity as heterozygotes.
Genotyping
Genotyping was performed primarily by primer extension of multiplex products, with detection by mass spectroscopy (35), using the Sequenom platform (36) and/or by allele-specific fluorogenic PCR (37). For quality control, PTPN22 was genotyped by both methods in the BRASS registry (99.9% concordance). For fluorogenic PCR, the concordance of duplicates was 100%. All SNPs included in the analysis had a P value greater than 0.001 for Hardy-Weinberg distribution and a genotyping efficiency >70% (all but 4 of the SNPs genotyped in NARAC had a genotyping efficiency >95%). Individuals were excluded if >50% of the genotypes were missing. In NARAC, 28 individuals were excluded, with 96% of the remaining individuals genotyped at 80% of the markers and 74% genotyped at 90% of the markers. In the BRASS registry, 31 individuals were excluded, with 98% of the remaining individuals genotyped at 90% of the markers. Genotyping efficiency was 99.97% in the Spanish samples and 99.96% in the WRDDB/NICRAP/SONORA samples.
Statistical analysis
Single-marker analysis
P values were calculated for binary variables using 2-by-2 contingency tables of allele counts and chi-square testing. For continuous variables, P values were determined using linear regression (Plink; online at http://pngu.mgh.harvard.edu/purcell/plink/ibdibs.shtml). Because >20 SNPs were tested in the NARAC and BRASS sample sets, we applied a conservative Bonferroni correction for multiple sampling, with a significance threshold of P = 10−3.
LD analysis
LD was investigated using Haploview (38). Blocks of high LD were defined according to the method described by Gabriel et al (36).
Haplotype analysis
Phased haplotypes were reconstructed using 2 different statistical frameworks: expectation-maximization using Haploview (38) and Whap (39), and Bayesian using Phase (40, 41). No significant differences were observed in the haplotypes derived using the different methods. For Whap, 500–1,000 repeats were performed for each analysis. Phase version 2.1.1 was run according to the authors’ recommendations. For each set of SNPs tested as a haplotype, linear regression and a likelihood ratio test of overall association were performed to determine an omnibus P value. For omnibus P values ≤0.05, a haplotype-specific chi-square test was performed for each haplotype.
RESULTS
Sequencing of IL1A and IL1B in patients with RA
To understand the full genetic diversity within IL1 in the context of RA, we sequenced IL1A and IL1B in a set of RA patients enrolled in the BRASS registry. Sequencing DNA from 24 patients (i.e., 48 chromosomes) provided 95% power to detect alleles with ≥5% frequency in the population (42). We performed bidirectional sequencing on all exons and promoter and noncoding regions that demonstrated a high degree of sequence similarity to the rat and mouse homologs (from the VISTA browser (34)) (Figure 1). The sequence data revealed 24 previously discovered SNPs, including 9 in IL1B and 15 in IL1A, and 4 novel SNPs in the noncoding regions of IL1B (submitted to the National Center for Biotechnology Information dbSNP database; accession nos. ss76859910, ss76859911, ss76859912, ss76859913). Hereafter, these SNPs will be referred to as RA1, RA2, RA3, and RA4, respectively. RA3 was subsequently reported as an SNP named “5164” (43). RA1, RA2, RA3, and RA4 were identified in 1 of 48, 1 of 48, 10 of 48, and 2 of 48 sequenced chromosomes, respectively. All 4 novel SNPs were then verified by genotyping using allele-specific fluorogenic PCR in DNA obtained from the BRASS registry and other sample sets (see below). No novel SNPs were identified in IL1A.
Figure 1.

A. Schematic diagram of the IL1A and IL1B loci. B. Linkage disequilibrium map of the SNPs genotyped in IL1A and IL1B in NARAC, as displayed by the Haploview software package. Haplotype blocks (black lines) are drawn according to confidence intervals (35). D′ values are shown in the boxes.
Discovery case-control samples for analyzing association with RA susceptibility
Querying an association between IL1 and RA by genotyping every known SNP in IL1A and IL1B would clearly be impractical. Instead, we followed a modified “tagging SNPs” strategy (44), which used a set of SNPs that captured the underlying diversity of the region based on the LD between SNPs, while also testing the novel and database-derived SNP alleles that had been observed by resequencing in patients with RA.
In order to comprehensively determine the LD structure in IL1A and IL1B, we genotyped a collection of SNPs in CEPH samples from the International HapMap project (25). This SNP set contained 15 of the 28 SNPs identified by resequencing that were selected for multiplex genotyping reaction compatibility. Information for an additional 5 of the SNPs identified from IL1A and IL1B was already available in HapMap at that time. We also genotyped 23 additional database SNPs chosen from the 20-kb region around each gene to ensure complete coverage of the genetic diversity in these regions.
By collating the genotype data with information available from the HapMap project, we determined LD across the region (36, 45). To select the final panel of SNPs to be genotyped in the case-control analysis, we then used Tagger software (46) to choose SNPs that adequately represented all common haplotypes. We preferentially included SNPs of interest by constraining Tagger to choose the 4 novel SNPs in IL1B and the 24 database-derived SNPs that had been identified by sequencing in patients with RA, including 4 SNPs that had previously been associated with aggressive and/or erosive arthritis (IL1B −511 [rs16944], IL1B +3954 [rs1143634], IL1A −889 [rs1800587], and IL1A +4845 [rs17561]) (19–21, 23, 24). We then eliminated some SNPs that had perfect or near-perfect proxies in the set.
This process ultimately led to identification of a set of 28 SNPs, including a minimally redundant subset of those selected by constraining the tagging algorithm, as well as 11 additional SNPs chosen by Tagger to provide complete coverage of the major haplotypes. Based on the LD structure in the CEPH samples, this set captured 61 of 66 haplotypes in a 20-kb region surrounding each gene, with an r2 value between the tagging SNPs and untested alleles of >0.8 and a mean r2 value of 0.984.
The 28 SNPs were genotyped in 1,314 RA patients from NARAC, which was used as our “discovery” sample set, because its RA patients have severe disease, with ~95% having evidence of joint erosions on hand radiographs. A total of 1,103 control samples were selected from the NYCP, as described previously (9). In a dominant model with 10% allele frequency, we had 90% power to detect a relative risk of 1.5 (47). The data were analyzed (by chi-square test) for correlation between the individual SNPs and susceptibility to RA (Table 1). Five SNPs in IL1A and 2 SNPs in IL1B showed differences between patients and control subjects, although none reached a Bonferroni-corrected threshold for significance (P = 0.001). The 5 SNPs in IL1A are in significant LD, with r2 values of 0.98–0.99 in the NARAC case-control data set. The most significant association was observed for RA3, with an odds ratio (OR) of 1.27 (95% confidence interval [95% CI] 1.09–1.48 [P = 0.0021]). Of the other SNPs initially identified by sequencing of RA samples, RA1 was shown to be monomorphic in all patients and control subjects, and the rare RA2 variant was observed in only 1 control subject. Because the NARAC collection includes affected siblings, it is possible that the OR could be inflated by using a simple case-control model. To examine this possibility, we performed the same analysis using only 1 sibling from each family (n = 619). The ORs for RA3 and RA4 were 1.37 (95% CI 1.13–1.67 [P = 0.0015]) and 1.49 (95% CI 1.05–2.11 [P = 0.0242]), respectively, suggesting that inclusion of siblings did not exaggerate the OR.
Table 1.
28 SNPs from IL1A and IL1B were genotyped in 1314 NARAC RA cases and 1103 controls. The allele that is more frequent in cases is shown in bold capital letters and the allele frequency and odds ratio are given for this allele. Genotype counts: (homozygotes of the allele more frequent in cases, heterozygotes, homozygotes of the alternate allele). P-values are from chi-squared analysis performed on the allele counts for single markers in RA patients versus controls.
Allelic Polymorphism in IL1 in Cases vs. Controls: NARAC
| Gene | SNP | SNP Info. | Alleles | Cases Freq (Gen. Cts) | Controls Freq (Gen. Cts) | OR | 95% CI | P value | |
|---|---|---|---|---|---|---|---|---|---|
| IL1A | rs6712572 | intron (CKAP2L) | T/g | 0.51 (332, 652, 296) | 0.50 (282, 536, 276) | 1.04 | 0.93 | 1.17 | 5.0E-01 |
| rs1304037 | 3′ UTR (IL1A) | A/g | 0.73 (686, 506, 92) | 0.70 (546, 444, 105) | 1.16 | 1.02 | 1.32 | 2.1E-02 | |
| rs3783550 | intron (IL1A) | C/a | 0.31 (133, 530, 609) | 0.30 (107, 416, 538) | 1.08 | 0.95 | 1.22 | 2.6E-01 | |
| rs17561 | missense (IL1A) | G/t | 0.73 (686, 513, 87) | 0.70 (545, 443, 104) | 1.17 | 1.03 | 1.33 | 1.5E-02 | |
| rs2856841 | intron (IL1A) | T/c | 0.73 (673, 502, 91) | 0.70 (537, 435, 107) | 1.17 | 1.03 | 1.32 | 1.8E-02 | |
| rs3783531 | missense (IL1A) | A/g | 0.00 (0, 9, 1282) | 0.00 (0, 7, 1089) | 1.09 | 0.41 | 2.94 | 8.6E-01 | |
| rs1609682 | intron (IL1A) | C/a | 0.31 (142, 516, 621) | 0.29 (110, 418, 559) | 1.10 | 0.97 | 1.24 | 1.4E-01 | |
| rs2856837 | intron (IL1A) | C/t | 0.73 (686, 507, 90) | 0.70 (560, 442, 104) | 1.15 | 1.02 | 1.31 | 2.8E-02 | |
| rs1800587 | 5′ UTR (IL1A) | C/t | 0.73 (687, 507, 89) | 0.70 (546, 445, 105) | 1.17 | 1.03 | 1.33 | 1.5E-02 | |
| rs6746923 | 5′ non-gene region | A/g | 0.42 (233, 615, 440) | 0.40 (176, 535, 383) | 1.06 | 0.94 | 1.19 | 3.4E-01 | |
| rs17597976 | 5′ non-gene region | A/g | 0.13 (29, 283, 979) | 0.12 (15, 242, 842) | 1.08 | 0.91 | 1.28 | 4.0E-01 | |
| rs11687624 | 5′ non-gene region | C/t | 0.44 (249, 643, 392) | 0.42 (192, 532, 372) | 1.10 | 0.98 | 1.24 | 9.0E-02 | |
| IL1B | rs4849125 | 3′ non-gene region | A/g | 0.70 (616, 539, 101) | 0.69 (507, 444, 108) | 1.08 | 0.95 | 1.23 | 2.3E-01 |
| rs1143642 | intron (IL1B) | C/t | 0.93 (1106, 171, 4) | 0.92 (922, 163, 10) | 1.22 | 0.98 | 1.51 | 7.5E-02 | |
| rs1143634 | synonomous (IL1B) | G/a | 0.78 (761, 416, 63) | 0.78 (663, 378, 55) | 1.02 | 0.89 | 1.17 | 7.6E-01 | |
| rs1143633 | intron (IL1B) | T/c | 0.37 (159, 580, 485) | 0.37 (154, 493, 445) | 1.00 | 0.89 | 1.13 | 9.4E-01 | |
| RA1 | intron (IL1B) | C/t | 1.00 (1300, 0, 0) | 1.00 (1099, 0, 0) | NA | NA | NA | NA | |
| rs1143627 | 5′ non-gene region | A/g | 0.66 (544, 544, 149) | 0.65 (465, 493, 135) | 1.04 | 0.92 | 1.17 | 5.5E-01 | |
| rs16944 | 5′ non-gene region | G/a | 0.66 (573, 548, 156) | 0.65 (466, 501, 134) | 1.06 | 0.94 | 1.20 | 3.5E-01 | |
| RA2 | 5′ non-gene region | C/t | 1.00 (1236, 0, 0) | 1.00 (1095, 1, 0) | NA | NA | NA | 2.9E-01 | |
| RA3 | 5′ non-gene region | A/g | 0.84 (837, 302, 41) | 0.80 (704,341, 45) | 1.27 | 1.09 | 1.48 | 2.1E-03 | |
| rs13013349 | 5′ non-gene region | G/a | 0.66 (537, 547, 153) | 0.65 (467, 493, 135) | 1.02 | 0.90 | 1.15 | 7.5E-01 | |
| rs13032029 | 5′ non-gene region | T/c | 0.47 (198, 451, 250) | 0.45 (190, 429, 276) | 1.09 | 0.95 | 1.24 | 2.1E-01 | |
| RA4 | 5′ non-gene region | C/g | 0.94 (829, 100, 2) | 0.92 (778, 123, 6) | 1.35 | 1.03 | 1.75 | 2.7E-02 | |
| rs4447608 | 5′ non-gene region | T/c | 0.48 (203, 469, 250) | 0.46 (200, 432, 270) | 1.06 | 0.93 | 1.21 | 3.6E-01 | |
| rs6735739 | 5′ non-gene region | C/t | 0.66 (568, 576, 156) | 0.66 (475, 495, 130) | 1.01 | 0.90 | 1.14 | 8.6E-01 | |
| rs6745746 | 5′ non-gene region | A/g | 0.46 (228, 637,332) | 0.45 (222, 534, 329) | 1.03 | 0.91 | 1.16 | 6.4E-01 | |
| rs12053091 | 5′ non-gene region | C/t | 0.27 (77, 346, 499) | 0.26 (69, 327, 504) | 1.06 | 0.92 | 1.23 | 4.1E-01 | |
Recent studies have demonstrated that the high-risk alleles at HLA and PTPN22 are more significantly associated with susceptibility to RA in the subset of patients who are positive for antibodies against CCP (48). We therefore investigated the degree of association of IL1 polymorphisms with RA in subgroups of patients defined by anti-CCP antibody status. No increased association was observed when we examined anti-CCP antibody-positive patients versus control subjects or RF-positive patients versus control subjects, or after stratification for HLA susceptibility alleles (HLA-DRB1*0101, *0401, *0404, *0405, *0408, or *1001) or sex (data not shown). However, when the patients and control subjects were stratified according to their PTPN22 genotype, the association signal for RA3 increased in the subset (912 patients and 933 control subjects) that was homozygous for the low-risk PTPN22 allele (OR 1.43, 95% CI 1.19–1.71 [P = 0.0000897], with allele frequencies for RA3/A of 0.85 in patients and 0.80 in control subjects). Conversely, the OR for RA4 and, to a lesser extent, rs1143642, was enhanced in the 382 patients and 158 control subjects with at least 1 copy of the high-risk allele at PTPN22 (for RA4, OR 2.48, 95% CI 1.37–4.48 [P = 0.002], with allele frequencies for RA4/C of 0.959 in patients and 0.905 in control subjects; for rs1143642, OR 1.68, 95% CI 1.03–2.72 [P = 0.0344], with allele frequencies of 0.941 in patients and 0.905 in control subjects).
Case-control replication of association with RA susceptibility
We attempted to replicate the association in 2 additional independent case-control sample sets by testing RA3 (which has the strongest suggestive association with susceptibility to RA) and RA4 (a novel and possibly RA-specific SNP variant). The first replication set included 948 anti-CCP antibody-positive individuals combined from 3 independent registries (WRDDB/NICRAP/SONORA) (31–33) and 1,455 unique control subjects from the NYCP. The second replication set comprised 525 patients and 504 control subjects from the University Clinical Hospital of Santiago de Compostela, Spain (30).
There was no population-specific variation in allele frequencies for RA3 and RA4 when comparing the NARAC and the replication samples. However, in contrast to the findings in NARAC, the frequency of the RA3/A disease-associated allele in the replication samples was actually lower in the patients with RA compared with control subjects, particularly in the Spanish patients (Table 2). For RA4, the higher frequency of the C allele in patients with RA was reproduced in the 2 replication sample sets, but these differences did not reach statistical significance (P = 0.09 and P = 0.3), regardless of PTPN22 stratification (data not shown).
Table 2.
RA3 and RA4 were genotyped in two independent replication case-control sample sets. First, they were genotyped in 948 CCP-positive cases from the WRDDB/NICRAP/SONORA cohort and 1455 controls from the NYCP. Next, they were genotyped in 525 RA cases and 504 controls from Northern Spain. Frequencies and odds ratios are shown for the allele that is capitalized and in bold. Genotype counts: (homozygotes of the allele capitalized and in bold, heterozygotes, homozygotes of the alternate allele). Chi-squared analysis of allele counts was performed for cases versus controls.
Allelic Polymorphism in RA3 and RA4 in Cases vs. Controls: Replication Sets
| SNP | Allele | Cases Freq (Gen. Cts.) | Controls Freq (Gen. Cts) | OR | 95% CI | P value | |
|---|---|---|---|---|---|---|---|
| WRDDB/NICRAP/SONORA | |||||||
| RA3 | A/g | 0.79 (589, 322, 37) | 0.80 (940, 452, 62) | 0.94 | 0.81 | 1.08 | 3.9E-01 |
| RA4 | C/g | 0.94 (844, 104, 0) | 0.93 (1261, 194, 0) | 1.23 | 0.96 | 1.57 | 9.7E-02 |
| SPANISH COHORT | |||||||
| RA3 | A/g | 0.77 (310, 185, 26) | 0.79 (318, 166, 20) | 0.88 | 0.71 | 1.08 | 2.2E-01 |
| RA4 | C/g | 0.92 (432, 73, 4) | 0.91 (415, 83, 5) | 1.18 | 0.86 | 1.61 | 3.0E-01 |
IL1 polymorphism and disease characteristics
Our second question was whether certain alleles of IL1 are associated with more severe or erosive disease, as suggested by prior studies involving smaller numbers of patients (19–24). We examined the association between the SNP alleles at IL1A and IL1B and the presence of erosions on hand radiographs in patients enrolled in the BRASS registry. For genotyping, we again chose a set of “tag” SNPs in IL1A and IL1B, based on LD structure in CEPH, with priority given to SNPs discovered by sequencing in RA samples. (The procedure used to identify SNPs was the same as that used for the NARAC set, but, for technical reasons, the panel of SNPs was slightly different.) We again included the 4 SNPs previously associated with aggressive or erosive arthritis (19–21, 23, 24). When analyzed in the CEPH samples, the 23 genotyped SNPs together captured 61 of 66 haplotypes in a 20-kb region surrounding each gene, with an r2 value between the tagging and untested alleles of >0.8 and a mean r2 value of 0.987.
In the BRASS cohort, 403 patients were found to have erosions, whereas 309 patients did not have erosions (80% power to detect a relative risk of 1.6). There was no association between erosion status and the previously reported IL1B −511, IL1B +3954, IL1A +4845, and IL1A −889 variants. The strongest association, which did not meet a corrected significance threshold of P = 0.001, was observed for the C allele at RA4 (OR 1.56, 95% CI 1.03–2.58 [P = 0.036]) (Table 3). In order to confirm this finding, we compared cases of erosive disease (n = 142) and cases of nonerosive disease (n = 272) in the cohort of Spanish patients for whom data on erosion status were available. In that cohort, RA4/C showed a frequency of 0.927 in the patients with erosive disease and a frequency of 0.942 in patients with nonerosive disease, for an OR of 0.78; these findings are in the opposite direction from those in the BRASS cohort.
Table 3.
24 SNPs from IL1A and IL1B were genotyped in 712 RA patients. A chi-squared analysis was performed comparing IL1 allele counts in the 403 of these patients that had erosions on plain radiographs of the hands versus the 309 that did not. Frequencies and odds ratios are shown for the allele that is capitalized and in bold. Genotype counts: (homozygotes of the allele capitalized and in bold, heterozygotes, homozygotes of the alternate allele). Chi-squared analysis of allele counts was performed for cases versus controls.
Allelic Polymorphism in IL1 in Patients With vs. Without erosions: BRASS Cohort
| Gene | SNP | SNP Info | Alleles | Eros pos. Freq (Gen. Cts.) | Eros neg. Freq (Gen. Cts) | OR | 95% CI | P value | |
|---|---|---|---|---|---|---|---|---|---|
| IL1A | rs6716572 | intron (CKAP2L) | T/g | 0.52 (108, 209, 93) | 0.51 (72, 154, 71) | 1.06 | 0.86 | 1.32 | 5.8E-01 |
| rs3783550 | intron (IL1A) | C/a | 0.28 (34, 160, 203) | 0.28 (24, 115, 153) | 1.01 | 0.79 | 1.25 | 9.6E-01 | |
| rs17561 | missense (IL1A) | G/t | 0.74 (223, 157, 30) | 0.70 (145, 117, 34) | 1.23 | 0.97 | 1.60 | 9.0E-02 | |
| rs3783531 | missense (IL1A) | A/g | 0.00 (0, 3, 406) | 0.00 (0, 3, 293) | 0.70 | 0.10 | 2.83 | 7.2E-01 | |
| rs1894399 | intron (IL1A) | G/a | 0.74 (220, 155, 26) | 0.71 (145, 116, 29) | 1.18 | 0.93 | 1.55 | 1.8E-01 | |
| rs1800587 | 5′ UTR (IL1A) | C/t | 0.74 (222, 158, 30) | 0.70 (146, 118, 34) | 1.21 | 0.95 | 1.57 | 1.2E-01 | |
| rs6746923 | 5′ non-gene region | A/g | 0.45 (80, 200, 127) | 0.40 (50, 136, 110) | 1.20 | 0.96 | 1.49 | 1.0E-01 | |
| rs17597976 | 5′ non-gene region | A/g | 0.14 (4, 104, 294) | 0.13 (7, 61, 226) | 1.12 | 0.81 | 1.46 | 4.9E-01 | |
| IL1B | rs4849125 | 3′ non-gene region | A/g | 0.72 (213, 155, 37) | 0.68 (137, 119, 37) | 1.22 | 0.96 | 1.57 | 1.0E-01 |
| rs7596684 | 3′ non-gene region | T/c | 0.82 (271, 117, 17) | 0.79 (180, 97, 16) | 1.19 | 0.91 | 1.61 | 2.1E-01 | |
| rs1143634 | synonomous (IL1B) | G/a | 0.79 (252, 129, 19) | 0.78 (167, 106, 10) | 1.09 | 0.84 | 1.45 | 5.4E-01 | |
| rs1143633 | intron (IL1B) | T/c | 0.38 (58, 191, 150) | 0.36 (34, 135, 114) | 1.12 | 0.89 | 1.38 | 3.3E-01 | |
| RA1 | intron (IL1B) | C/t | 1.00 (399, 0, 0) | 1.00 (281, 1, 0) | NA | NA | NA | 2.3E-01 | |
| rs1143627 | 5′ non-gene region | A/g | 0.67 (181, 168, 44) | 0.66 (120, 129, 31) | 1.07 | 0.85 | 1.37 | 5.6E-01 | |
| rs16944 | 5′ non-gene region | G/a | 0.67 (177, 177, 44) | 0.66 (118, 134, 30) | 1.05 | 0.84 | 1.34 | 6.7E-01 | |
| RA3 | 5′ non-gene region | A/g | 0.80 (254, 125, 17) | 0.82 (189, 84, 8) | 0.86 | 0.65 | 1.17 | 2.9E-01 | |
| rs13013349 | 5′ non-gene region | G/a | 0.67 (180, 176, 44) | 0.66 (119, 136, 28) | 1.04 | 0.83 | 1.33 | 7.2E-01 | |
| rs13032029 | 5′ non-gene region | T/c | 0.48 (93, 196, 107) | 0.46 (56, 142, 80) | 1.11 | 0.89 | 1.37 | 3.6E-01 | |
| RA4 | 5′ non-gene region | C/g | 0.94 (357, 36, 5) | 0.91 (232, 49, 0) | 1.56 | 1.03 | 2.58 | 3.6E-02 | |
| rs4447608 | 5′ non-gene region | T/c | 0.49 (94, 198, 106) | 0.46 (55, 149, 76) | 1.09 | 0.88 | 1.36 | 4.2E-01 | |
| rs6735739 | 5′ non-gene region | C/t | 0.67 (182, 172, 44) | 0.66 (118, 138, 26) | 1.05 | 0.83 | 1.34 | 6.9E-01 | |
| rs6745746 | 5′ non-gene region | A/g | 0.47 (82, 208, 105) | 0.45 (46, 160, 76) | 1.10 | 0.89 | 1.37 | 3.8E-01 | |
| rs12053091 | 5′ non-gene region | C/t | 0.25 (23, 148, 215) | 0.23 (17, 89, 161) | 1.12 | 0.87 | 1.41 | 3.9E-01 | |
Haplotype analysis
Because the analysis described above showed no reproducible association between polymorphism at single markers and the susceptibility to or severity of RA, we reconstructed phased haplotypes in the study subjects and searched for association between these haplotypes and disease incidence or characteristics.
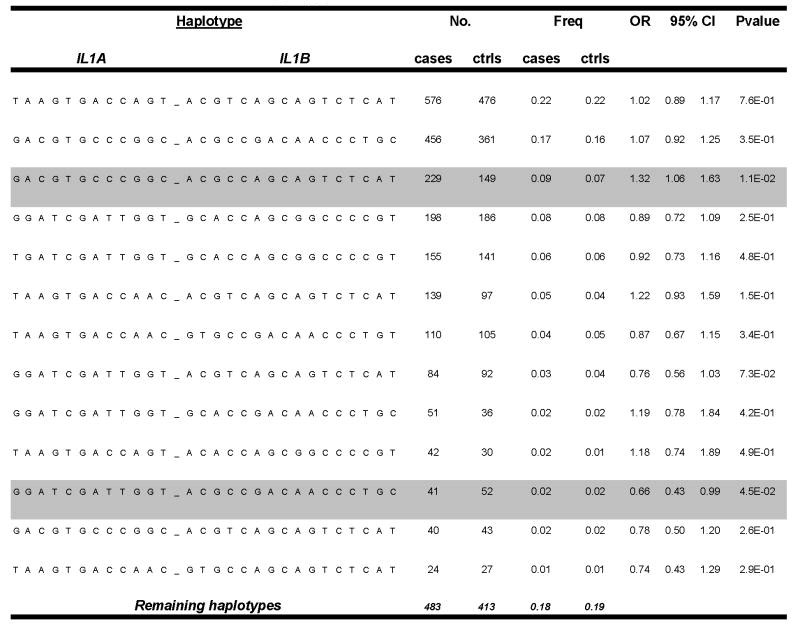
For NARAC patients and control subjects, we determined haplotypes defined by all of the SNPs, the SNPs around IL1A, and the SNPs around IL1B. For the extended IL1A haplotype, regression analysis provided an omnibus P value of 0.007, with 1 haplotype (GGATCGATTGGT) occurring in 17% of patients and 20% of control subjects (OR 0.82, 95% CI 0.70–0.94 [P = 0.006]) (Table 4). If only a single sibling from each family of cases was included, the magnitude of the OR for that haplotype was unchanged (OR 0.81, 95% CI 0.68–0.98 [P = 0.026]), suggesting that the potential association was not attributable to merely the inclusion of related individuals. In contrast to IL1A, the regression analysis for the extended IL1B haplotype and the extended IL1A-IL1B haplotype revealed P values of 0.295 and 0.233, respectively, suggesting no statistically significant skewing in the haplotype distribution. The haplotype-specific results (by chi-square test) for the extended IL1A-IL1B haplotype are shown in Table 5.
Table 4.
All of the SNPs within the 20kb region of IL1A were used to construct haplotypes for the NARAC cases and controls. Haplotypes of a frequency > 0.005 are shown. SNPs are ordered as in Table 1. OR = odds ratio and 95% CI = 95% confidence interval of the OR.
IL1A Haplotype Association with NARAC Cases vs. Controls
| Haplotype | No. | Freq. | OR | 95% CI | P value | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL1A | cases | ctrls | cases | ctrls | |||||||||||||||
| G | A | C | G | T | G | C | C | C | G | G | C | 776 | 607 | 0.30 | 0.28 | 1.12 | 0.99 | 1.27 | 8.3E-02 |
| T | A | A | G | T | G | A | C | C | A | G | T | 722 | 596 | 0.28 | 0.27 | 1.04 | 0.91 | 1.18 | 5.9E-01 |
| G | G | A | T | C | G | A | T | T | G | G | T | 443 | 443 | 0.17 | 0.20 | 0.82 | 0.70 | 0.94 | 6.0E-03 |
| T | A | A | G | T | G | A | C | C | A | A | C | 319 | 261 | 0.12 | 0.12 | 1.04 | 0.87 | 1.24 | 6.6E-01 |
| T | G | A | T | C | G | A | T | T | G | G | T | 229 | 203 | 0.09 | 0.09 | 0.95 | 0.78 | 1.16 | 6.2E-01 |
| T | A | C | G | T | G | C | C | C | G | G | C | 14 | 26 | 0.01 | 0.01 | 0.45 | 0.24 | 0.87 | 1.5E-02 |
| G | A | A | G | T | G | A | C | C | A | G | T | 10 | 20 | 0.00 | 0.01 | 0.42 | 0.20 | 0.90 | 2.2E-02 |
| Remaining Haplotypes | 83 | 42 | 0.03 | 0.02 | |||||||||||||||
Table 5.
All of the SNPs genotyped from IL1A and IL1B were used to construct haplotypes for the NARAC cases and controls. Haplotypes with a frequency of > 0.01 are shown. SNPs are ordered as in Table 1. OR = odds ratio and 95% CI = 95% confidence interval of the OR.
Extended IL1A-IL1B Haplotype Association with NARAC Cases vs. Controls
|
In addition, we analyzed SNP haplotypes defined as blocks determined by D′ confidence intervals (35) (Figure 1B), none of which provided an omnibus P value <0.05 in regression analysis (data not shown).
We similarly tested phased haplotypes reconstructed in the BRASS cohort for an association with erosion and other clinical variables. The omnibus P value for the regression was >0.05 for association with erosion status, anti-CCP antibodies, RF, and the DAS28 (data not shown).
DISCUSSION
Previous studies suggested an association between polymorphisms at IL1 and severe, erosive RA. We sought to perform a comprehensive, more definitive study adequately powered to determine whether IL1A or IL1B contributes to the risk or severity of RA. IL1A and IL1B were sequenced in patients with RA to fully assess the potential disease-relevant genetic diversity, and tagging SNPs that captured the majority of this diversity were chosen for further assessment. In the single-marker analysis, involving more than 3,500 patients and 3,000 control subjects in 4 independent sample sets, we observed no association between IL1 and RA susceptibility/severity that proved reproducible and met rigorous criteria for significance after correction for multiple comparisons.
The SNP demonstrating the strongest association with susceptibility to RA in the primary NARAC case-control set was RA3. Unfortunately, this association of RA3/A with susceptibility to RA was not validated in the replication samples. In fact, the allele distribution between patients and control subjects was in the opposite direction in the other 2 populations, suggesting that the lack of replication was not attributable to insufficient power only. The most likely explanation for the discrepancy is that the initial finding was a false positive and not representative of a true association. One alternative explanation is that the patients in the replication sets differed significantly from the discovery patients, and the association with disease susceptibility is only relevant in a subset of patients that was enriched in the NARAC cohort. Indeed, the patients in NARAC do represent a subset of RA patients with severe and aggressive disease, and, therefore, it is possible that the association with RA3 may be relevant only for severe erosive RA. If this were true, we would have expected to observe in the BRASS cohort an association of RA3/A with erosion status, which was not the case. If anything, the alternate RA3/G allele was associated with erosion. Theoretically, one could invoke the possibility that ethnic diversity is contributing to the difference between NARAC and the replication sample sets, but the allele frequencies for RA3 were similar in all groups.
Our analysis of the NARAC case-control samples was designed to have 90% power to detect a relative risk of 1.5 in a dominant model of an allele with 10% frequency (47). Therefore, our study was somewhat underpowered to detect weak associations with RA4 and other rare alleles. In fact, RA4/C showed a 1–2% greater frequency in patients than in control subjects in all 3 sample sets, and this difference was enhanced to 2–5% when stratified for the high-risk PTPN22 allele (Tables 1 and 2, and data not shown). Based on the P values (by chi-square test) and a conservative Bonferroni correction for the number of comparisons, this difference is not statistically significant.
Interestingly, the same allele was potentially associated with erosive RA versus nonerosive RA in the BRASS cohort, with RA4/C being 3% more frequent in patients with erosive disease (uncorrected P = 0.03). This potential association with erosion, however, was not replicated in the Spanish cohort. One possible issue concerning the use of erosion status as a marker of disease severity is that a radiograph is obtained at one point in time, and that erosive disease may ultimately develop in some of the patients who are erosion-negative or in those patients who would have had erosions in the absence of aggressive therapy. Therefore, the readout for each patient is influenced by the length of therapy, the severity of disease, and the aggressiveness of treatment.
Previous studies had shown association of rs16944, rs1143634, rs1800587, and rs17561 with erosive arthritis or radiographic progression, but the associated SNP and sometimes the associated allele differed among those studies (19–21, 23, 24). The disparities between these prior studies may suggest that the original findings were false-positive results reflective of the small number of patients studied. Indeed, our study in the BRASS cohort showed no statistically significant evidence of association with erosive disease status for any of these SNPs.
Although the single-marker analysis did not reveal a significant association between RA and IL1A, the haplotype analysis did uncover an IL1A haplotype, which was present in 17% of patients and 20% of control subjects, with a potential association with disease susceptibility. This finding could suggest that a certain set of SNP alleles may alter IL-1 function in an additive or synergistic manner or may indicate that a relevant allele is incompletely tagged with any of our single markers but is represented by one of the complex haplotypes. Because we did not genotype the entire set of IL1A and IL1B SNPs in the replication sample sets, we could not validate the haplotype association. At this point, we cannot exclude the possibility that certain haplotypes at IL1A may contribute to RA susceptibility. If this is the case, however, the magnitude of the effect is likely to be small.
We did not examine the relevance of polymorphisms in IL1RN, the gene that encodes the competitive antagonist of IL-1□ and IL-1□, IL-1 receptor antagonist (IL-1Ra). In fact, similar to the case with IL1A and IL1B, small studies have suggested an association with polymorphism in IL1RN and RA susceptibility and/or severity (49, 50), but results have been contradictory (20, 21), and therefore replication is needed. Given the importance of IL-1 in inflammatory arthritis, it may be interesting to perform a well-powered candidate gene study to examine the contribution of polymorphisms in other members of the IL-1 pathway, including the genes for the IL-1 receptors, IL-Ra, and components of the inflammasome.
In summary, based on our comprehensive study of variation at the IL1A and IL1B loci and genotyping in a large number of patients with RA and control subjects, we observed no evidence that common variants in IL1 contribute significantly to the risk or severity of RA in the majority of patients.
Acknowledgments
Scholarship, Arthritis National Research Foundation
K08 AI072044-01A1, National Institute of Allergy and Infectious Diseases, NIH
T32 AR 007530-21, National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH
R01 AR046580-08, National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH
Abbreviations
- RA
rheumatoid arthritis
- CCP
cyclic citrullinated peptides
- SNP
single-nucleotide polymorphism
- PCR
polymerase chain reaction
- SE
shared epitope
- DAS
disease activity score
- RF
rheumatoid factor
- LD
linkage disequilibrium
- r2
correlation of alleles at two different sites (measure of LD)
- OR
odds ratio
Footnotes
AUTHOR CONTRIBUTIONS
Dr. Mathis had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study design. Johnsen, Shadick, Weinblatt, Benoist, Mathis.
Acquisition of data. Johnsen, Plenge, Campbell, Dieguez-Gonzalez, Gomez-Reino, Shadick, Weinblatt, Gonzalez, Gregersen.
Analysis and interpretation of data. Johnsen, Plenge, Butty, Dieguez-Gonzalez, Gomez-Reino, Weinblatt, Gonzalez, Benoist, Mathis.
Manuscript preparation. Johnsen, Plenge, Butty, Weinblatt, Benoist, Mathis.
Statistical analysis. Johnsen, Plenge, Butty.
Provision of samples. Shadick.
References
- 1.Dinarello CA. The many worlds of reducing interleukin-1 [editorial] Arthritis Rheum. 2005;52:1960–7. doi: 10.1002/art.21107. [DOI] [PubMed] [Google Scholar]
- 2.Dinarello CA. Blocking IL-1 in systemic inflammation [review] J Exp Med. 2005;201:1355–9. doi: 10.1084/jem.20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52:1794–803. doi: 10.1002/art.21061. [DOI] [PubMed] [Google Scholar]
- 4.Goldring SR. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology (Oxford) 2003;42(Suppl 2):ii11–6. doi: 10.1093/rheumatology/keg327. [DOI] [PubMed] [Google Scholar]
- 5.Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford) 2003;42(Suppl 2):ii3–10. doi: 10.1093/rheumatology/keg326. [DOI] [PubMed] [Google Scholar]
- 6.Bresnihan B, Cobby M. Clinical and radiological effects of anakinra in patients with rheumatoid arthritis. Rheumatology (Oxford) 2003;42(Suppl 2):ii22–8. doi: 10.1093/rheumatology/keg329. [DOI] [PubMed] [Google Scholar]
- 7.Gregersen PK. Teasing apart the complex genetics of human autoimmunity: lessons from rheumatoid arthritis [review] Clin Immunol. 2003;107:1–9. doi: 10.1016/s1521-6616(02)00045-1. [DOI] [PubMed] [Google Scholar]
- 8.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 9.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–7. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee HS, Remmers EF, Le JM, Kastner DL, Bae SC, Gregersen PK. Association of STAT4 with rheumatoid arthritis in the Korean population. Mol Med. 2007;13:455–60. doi: 10.2119/2007-00072.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis: a genomewide study. N Engl J Med. 2007;357:1199–209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, et al. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis [published erratum appears in PLoS Med 2007;4:e358] PLoS Med. 2007;4:e278. doi: 10.1371/journal.pmed.0040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–82. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–3. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohmura K, Johnsen A, Ortiz-Lopez A, Desany P, Roy M, Besse W, et al. Variation in IL-1 gene expression is a major determinant of genetic differences in arthritis aggressivity in mice. Proc Natl Acad Sci U S A. 2005;102:12489–94. doi: 10.1073/pnas.0504325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrera P, Faure S, Prud’homme JF, Balsa A, Migliorini P, Chimenti D, et al. for the European Consortium on Rheumatoid Arthritis Families (ECRAF) European genetic study on rheumatoid arthritis: is there a linkage of the interleukin-1 (IL-1), IL-10 or IL-4 genes to RA? Clin Exp Rheumatol. 2001;19:709–14. [PubMed] [Google Scholar]
- 19.Cox A, Camp NJ, Cannings C, Di Giovine FS, Dale M, Worthington J, et al. Combined sib-TDT and TDT provide evidence for linkage of the interleukin-1 gene cluster to erosive rheumatoid arthritis. Hum Mol Genet. 1999;8:1707–13. doi: 10.1093/hmg/8.9.1707. [DOI] [PubMed] [Google Scholar]
- 20.Buchs N, Di Giovine FS, Silvestri T, Vannier E, Duff GW, Miossec P. IL-1B and IL-1Ra gene polymorphisms and disease severity in rheumatoid arthritis: interaction with their plasma levels. Genes Immun. 2001;2:222–8. doi: 10.1038/sj.gene.6363766. [DOI] [PubMed] [Google Scholar]
- 21.Cantagrel A, Navaux F, Loubet-Lescoulie P, Nourhashemi F, Enault G, Abbal M, et al. Interleukin-1, interleukin-1 receptor antagonist, interleukin-4, and interleukin-10 gene polymorphisms: relationship to occurrence and severity of rheumatoid arthritis. Arthritis Rheum. 1999;42:1093–100. doi: 10.1002/1529-0131(199906)42:6<1093::AID-ANR5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 22.Cvetkovic JT, Wallberg-Jonsson S, Stegmayr B, Rantapaa-Dahlqvist S, Lefvert AK. Susceptibility for and clinical manifestations of rheumatoid arthritis are associated with polymorphisms of the TNF-, IL-1, and IL-1Ra genes. J Rheumatol. 2002;29:212–9. [PubMed] [Google Scholar]
- 23.Genevay S, Di Giovine FS, Perneger TV, Silvestri T, Stingelin S, Duff G, et al. Association of interleukin-4 and interleukin-1B gene variants with Larsen score progression in rheumatoid arthritis. Arthritis Rheum. 2002;47:303–9. doi: 10.1002/art.10394. [DOI] [PubMed] [Google Scholar]
- 24.Jouvenne P, Chaudhary A, Buchs N, Giovine FS, Duff GW, Miossec P. Possible genetic association between interleukin-1 gene polymorphism and the severity of chronic polyarthritis. Eur Cytokine Netw. 1999;10:33–6. [PubMed] [Google Scholar]
- 25.The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 26.Shadick NA, Heller JE, Weinblatt ME, Maher NE, Cui J, Ginsburg GS, et al. Opposing effects of the D70 mutation and the shared epitope in HLA-DR4 on disease activity and certain disease phenotypes in rheumatoid arthritis. Ann Rheum Dis. 2007;66:1497–502. doi: 10.1136/ard.2006.067603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prevoo ML, van ’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–8. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- 28.Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Etzel C, et al. for the North American Rheumatoid Arthritis Consortium. Screening the genome for rheumatoid arthritis susceptibility genes: a replication study and combined analysis of 512 multicase families. Arthritis Rheum. 2003;48:906–16. doi: 10.1002/art.10989. [DOI] [PubMed] [Google Scholar]
- 29.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Lopez J, Perez-Pampin E, Gomez-Reino JJ, Gonzalez A. Regulatory polymorphisms in extracellular matrix protease genes and susceptibility to rheumatoid arthritis: a case-control study. Arthritis Res Ther. 2006;8:R1. doi: 10.1186/ar1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfe F, Michaud K, Gefeller O, Choi HK. Predicting mortality in patients with rheumatoid arthritis. Arthritis Rheum. 2003;48:1530–42. doi: 10.1002/art.11024. [DOI] [PubMed] [Google Scholar]
- 32.Fries JF, Wolfe F, Apple R, Erlich H, Bugawan T, Holmes T, et al. HLA-DRB1 genotype associations in 793 white patients from a rheumatoid arthritis inception cohort: frequency, severity, and treatment bias. Arthritis Rheum. 2002;46:2320–9. doi: 10.1002/art.10485. [DOI] [PubMed] [Google Scholar]
- 33.Bombardier C, Deaton RL, Gregerson P, Massarotti E, Formica C, Weisman MH. Pattern of DMARD use in a North American cohort of patients with early rheumatoid arthritis (RA) (SONORA) [abstract] Arthritis Rheum. 2002;46(Suppl 9):S344. [Google Scholar]
- 34.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(Web Server issue):W273–9. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Koster H. Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci U S A. 1999;96:10016–20. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 37.Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity. 2005;22:385–96. doi: 10.1016/j.immuni.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 39.Purcell S, Daly MJ, Sham PC. WHAP: haplotype-based association analysis. Bioinformatics. 2007;23:255–6. doi: 10.1093/bioinformatics/btl580. [DOI] [PubMed] [Google Scholar]
- 40.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–9. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kruglyak L, Nickerson DA. Variation is the spice of life. Nat Genet. 2001;27:234–6. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 43.Chen H, Wilkins LM, Aziz N, Cannings C, Wyllie DH, Bingle C, et al. Single nucleotide polymorphisms in the human interleukin-1B gene affect transcription according to haplotype context. Hum Mol Genet. 2006;15:519–29. doi: 10.1093/hmg/ddi469. [DOI] [PubMed] [Google Scholar]
- 44.De Bakker PI, Burtt NP, Graham RR, Guiducci C, Yelensky R, Drake JA, et al. Transferability of tag SNPs in genetic association studies in multiple populations. Nat Genet. 2006;38:1298–303. doi: 10.1038/ng1899. [DOI] [PubMed] [Google Scholar]
- 45.Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–32. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- 46.De Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 47.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–50. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 48.Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005;77:1044–60. doi: 10.1086/498651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carreira PE, Gonzalez-Crespo MR, Ciruelo E, Pablos JL, Santiago B, Gomez-Camara A, et al. Polymorphism of the interleukin-1 receptor antagonist gene: a factor in susceptibility to rheumatoid arthritis in a Spanish population. Arthritis Rheum. 2005;52:3015–9. doi: 10.1002/art.21287. [DOI] [PubMed] [Google Scholar]
- 50.Kapetanovic MC, Lindqvist E, Sturfelt G, Saxne T, Truedsson L, Eberhardt K. The impact of IL-1Ra and MBL gene polymorphisms on joint damage after 5 and 10 years in patients with early rheumatoid arthritis. J Rheumatol. 2007;34:894–5. [PubMed] [Google Scholar]