

Abstract
Isotactic polyethylenimines with (S)-benzyl side chains were synthesized from 4-(S)-4-benzyl-2-oxazolines. When α-ketoacids were subjected to transamination in the presence of this polymer, and a pyridoxamine coenzyme modified with hydrophobic chains, enantioselectivity towards the natural isomer (L>D) were observed, followed by racemization of the amino acid products. However, the racemization did not occur when the coenzyme was covalently attached to the polymer.
Natural enzymes catalyze reactions with regio- and stereospecificity. Chemists, inspired by the natural enzymes, try to mimic them in order to develop synthetic systems that can achieve high selectivity in chemical transformations.1 Achievement of high chiral induction in the amino acid products from transaminase mimics has attracted a number of research groups worldwide.
Besides our work in this field, Schultz used catalytic antibodies,2 Imperiali used polypeptide-pyridoxamine chimeras,3 Destefano used an adipocyte lipid binding protein,4 Jorgensen used metal-chiral ligand systems,5 and Nicholls used a molecularly imprinted polymer6 as transaminase mimics. Our group has achieved a high degree of chiral induction in transamination reactions with pyridoxamine systems having rigidly attached chiral side chains7 and, more recently, enzyme mimics consisting of pyridoxamines covalently linked to reduced peptide have induced moderate to high chirality.8
Transaminase enzymes perform a series of general acid and general base catalyzed reactions in chiral environments to afford chiral amino acids.9 Polyethylenimines (PEIs) titrate over a range of pH 3 to 13, having both effective amino group general bases and ammonium group general acids near neutrality, and thus can act as efficient transaminase mimics. Recently, we have described transaminase mimics consisting of pyridoxamines covalently linked to branched PEIs, carrying long-chain alkyl chains. They converted α-ketoacids to amino acids with up to a 240,000-fold rate enhancement relative to the reaction with simple pyridoxamine at the same total pyridoxamine concentration.10-12 We have also used non-covalent polymer-pyridoxamine systems as enzyme mimics, in which pyridoxamines carrying hydrophobic sidechains bound—along with the substrates—into hydrophobic regions of the PEIs.13 The latter showed even better rate enhancement, as much as 725,000-fold with the hydrophobic indolepyruvic acid that forms tryptophan.14 In all these cases the polymer, being achiral, formed racemic mixtures of the product amino acids.
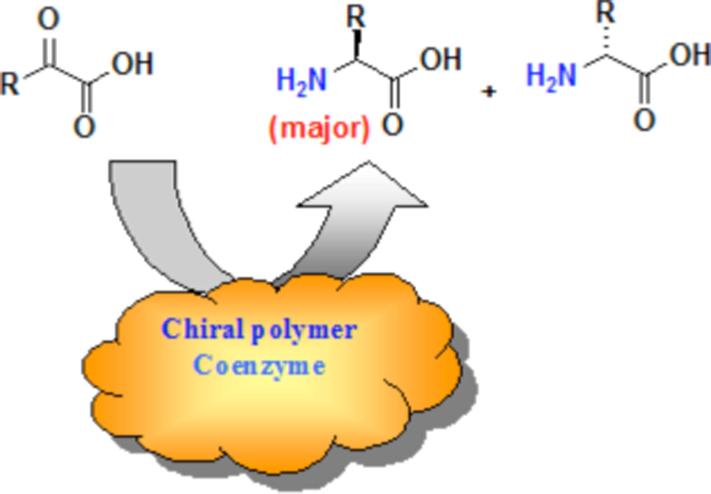
Incorporating the features of hydrophobicity in a chiral environment, and general acid general base catalyst, we have briefly described a transaminase mimic with non-covalently bound pyridoxamine-chiral polyethylenimine systems (Scheme 1).15
Scheme 1. Transamination with 1.
Conditions: Reaction conditions: 5.0 × 10−4 mol/L pyridoxamine 4, 10 g/L PEI, 5 × 10−4 EDTA, 5 × 10−3 mol/L 3-methyl-2-oxopyruvic acid, t = 20−25 °C. pH values reported here are as read with a glass-electrode calibrated against aqueous buffers.
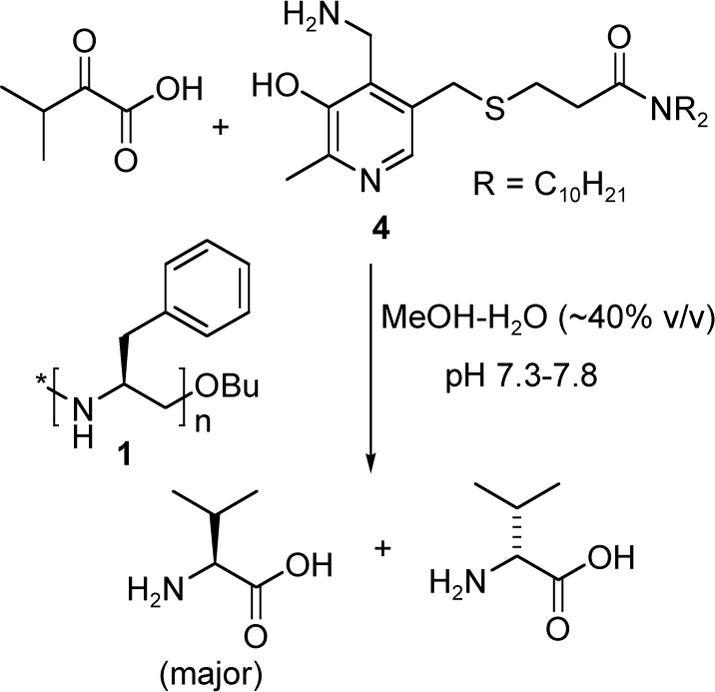
The chiral PEI 1 with (S)-benzyl side chains was synthesized from 4-(S)-4-benzyl-2-oxazoline 3 via a cationic ring opening polymerization using methyl tosylate as the initiator and 1-butanol as the terminator, and subsequent deformylation of the N-formamide groups of poly(N-formyl-1-benzylethylenimine) 2 with a base (Scheme 2), following Saegusa's method.16,17 The methyl group of the butoxy group served as the NMR probe for the determination of degree of polymerization. Complete absence of the formyl groups in the hydrolyzed product 1 was confirmed by IR spectroscopy, where the strong formyl C$#xFF1D;O stretching at 1668 cm−1 of 2 disappered, and also by 1H NMR spectroscopy, where the broad formyl proton peak around δ 7.7 of the precursor was absent.
Scheme 2.
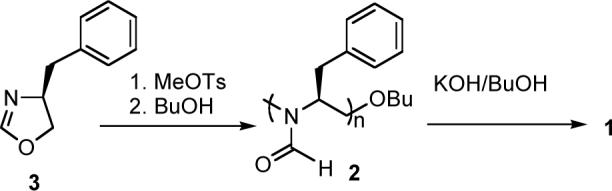
Synthesis of 1.
The average degree of polymerization (DP) with 2 mol% initiator (compared to the oxazoline) was ca. 50, with 3 mol% was ca. 30, and with 6 mol% was ca. 13. The polydispersity of the chiral PEI's determined by GPC were 1.01, 1.07 and 1.20 for the 50-mer, 30-mer and the 13-mer respectively. The low polydispersity values are in accordance with the highly living nature of the polymerization, where the DP values are very close to the oxazoline/initiator ratio.18 Prior to our work, Goodman et al. had synthesized optically active polyoxazolines (including the 50 mer 2) following Saegusa's method, and from circular dichroism studies had shown that polyoxazolines obtained via this kind of cationic ring opening polymerization are isotactic in nature.19
The (S)-benzyl side chains of the polymers 1 can form a chiral hydrophobic pocket in which the non-polar alkyl chains of the pyridoxamine coenzyme 413 can bind, and thus the transamination reaction can take place in a chiral environment, as in the natural system, to give chiral induction in the amino acid products. Indeed, high enantioselectivity was achieved when the 50-mer in ca. 40% aqueous methanol (v/v%) at pH 7.3−7.8 produced L/D valine with a ratio of 83/17 (ca. 5−7% conversion), but unfortunately the amino acid racemized at higher conversion. The ratio of L/D valine with the 30 and 13-mers were slightly less (78/22 and 71/29, respectively). Enantioselectivity at the initial stage of transamination, followed by racemization of the amino acid products, was also reported by Bernauer with Cu(II) complexes of C2-symmetric ligands and Schiff bases of ketoacids and pyridoxamine.20
Interestingly, racemization was never observed with pyridoxamines that were covalently attached to other catalytic units, including amines.7,8 We believe that the Schiff base product between a pyridoxal and the amino acid can move into water and racemize before hydrolysis. Thus in order to overcome the problem of racemization, we have now explored pyridoxamine systems that are covalently attached to a chiral isotactic linear polyethylenimine carrying (S)-benzyl side chains. When the cofactor is covalently attached to the polymer, it is in a chiral environment where racemization is slow.
PEI 1 was acylated with the N-hydroxysuccinimide ester of 3,3’-dithiopropionic acid (Scheme 3). The disulfide bonds were reduced with sodium borohydride to produce the thiolate groups in situ, which were then alkylated using 5-bromomethyl analog of pyridoxamine to yield the chiral PEI-pyridoxamine system 5. The polymer was precipitated from a solution of sodium carbonate to remove any unbound 3-thiopropionic acid and pyridoxamine impurities, centrifuged, washed with water, and lyophilized. From the 1H NMR integration, it was found that ca. 9 % of the polymer nitrogens carried the pyridoxamine units. From the intensity of the UV-vis spectrum of 5 at 318 nm, comparing it with the absorption of a standard solution of pyridoxamine, the amount of pyridoxamine attached to the polymer was estimated to be ca. 10% of the maximum possible value, in reasonable agreement with the NMR results.
Scheme 3.
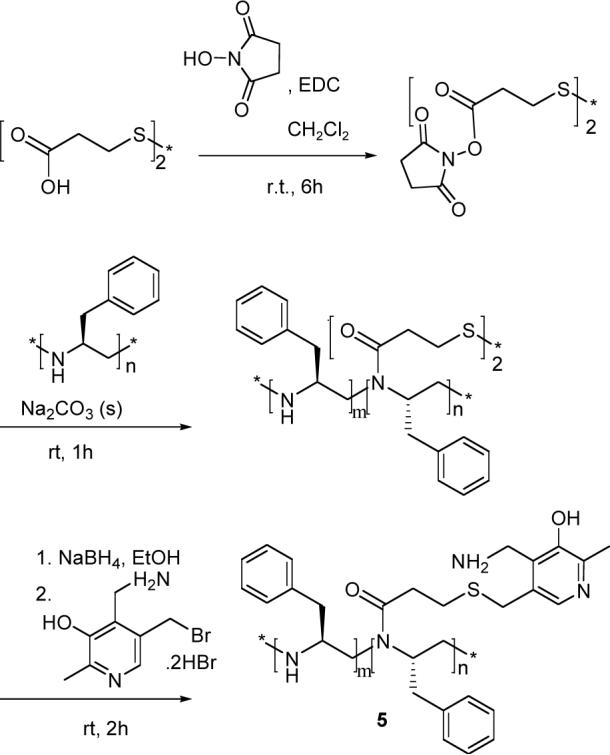
Synthesis of 5.
When the sodium salt of 3-methyl-2-oxobutanoic acid was subjected to transamination with the chiral PEI-pyridoxamine system 5 in aqueous-methanol (35−40 % v/v) at pH 5.5 in presence of EDTA, and the product, after derivatization with o-phthalaldehyde and N-acetylcysteine, was analyzed by reverse phase HPLC,21 an enantiomeric ratio of 58/42 ± 3 % of valine (L>D) was observed. In this case the reaction time or the conversion did not affect the relative amounts of the two enantiomers to any significant extent, which indicated that the problem of racemization that was observed in the non-covalent system was eliminated with this covalently attached system. Under the same conditions, when phenylpyruvic acid was subjected to transamination, the ratio, of L/D phenylalanine was found to be 69/31± 2 %, an increase that can be attributed to better hydrophobic binding of this ketoacid to the polymer-pyridoxamine system. 4-Hydroxyphenylpyruvic acid, which is less hydrophobic than phenylpyruvic acid, gave a ratio of 63/37 ± 2 % of the L/D tyrosine. Thus, with this system the highest enantioselectivity so far was achieved with phenylalanine, reflecting its stronger interaction with the hydrophobic polymer.
We examined whether a shorter spacer, instead of a thiopropionyl spacer, could bring the pyridoxamine reaction center closer to the chiral units, and increase the stereoselectivity. Thus we synthesized the chiral PEI-pyridoxamine system 6 via a substitution reaction of the Boc protected pyridoxamine dimesyl ester 7 with the chiral PEI, and subsequent deprotection of the mesyl group with sodium ethoxide in ethanol, followed by the deprotection of the Boc group with trifluoroacetic acid (Scheme 4). The resulting pyridoxamine-PEI 6 was purified by the method descibed previously. The 1H NMR spectrum of 6 indicated that ca. 8% of the polymer nitrogens were attached to the pyridoxamine (UV-vis measurement indicated 9%).
Scheme 4.
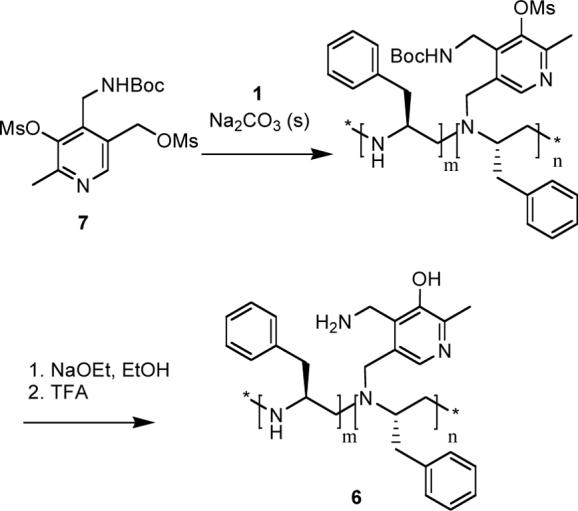
Synthesis of 6.
When phenylpyruvic acid was transaminated in the presence of this system under conditions identical to those described above, the ratio of L/D phenylalanine was 71/29 ± 3 %, essentially the same as obtained with the PEI-pyridoxamine system 5. Changing the length of the tether did not enhance the chiral induction significantly, and both of these systems are less effective in inducing chirality than the non-covalently bound pyridoxamine (4)-chiral PEI (1) system. However, the benefit of using the covalent systems 5 and 6 was that the problem of racemization encountered in the non-covalent system was solved here.
We envisaged that one reason for the lower values of chiral induction in these cases (with 5 and 6) could be that the pyridoxamine coenzyme 4 was able to bind to the most hydrophobic part of the polymer, whereas with the covalently bound pyridoxamine-polymer systems 5 and 6, the mobility of the pyridoxamine moiety is lost and it may link to the most accessible part of the polymer, not necessarily the most catalytically effective part. We thus decided to block the more accessible part of the chiral PEI by acylating it, so that the pyridoxamines could be attached to the interior part of the polymer.
PEI 1 was partially N-acylated using 0.2 equivalents 4-t-butylbenzoylchloride to give polymer 8 (Scheme 5). The NMR spectrum indicated that about 15% of the polymer nitrogens were acylated. Polymer 8 was then alkylated with 5-deoxymethyl-5-bromomethylpyridoxamine in dichloromethane-DMF in the presence of sodium carbonate to give 9. When 0.25 equivalent (with respect to polymer 8) of 5-deoxymethyl-5-bromomethylpyridoxamine was used, it was found that only ca. 8% of the polymer nitrogens of 9 were attached to the pyridoxamines. Removal of the 4-t-butylbenzoyl groups of the polymer 9 with potassium hydroxide in ethanol gave 10.
Scheme 5.
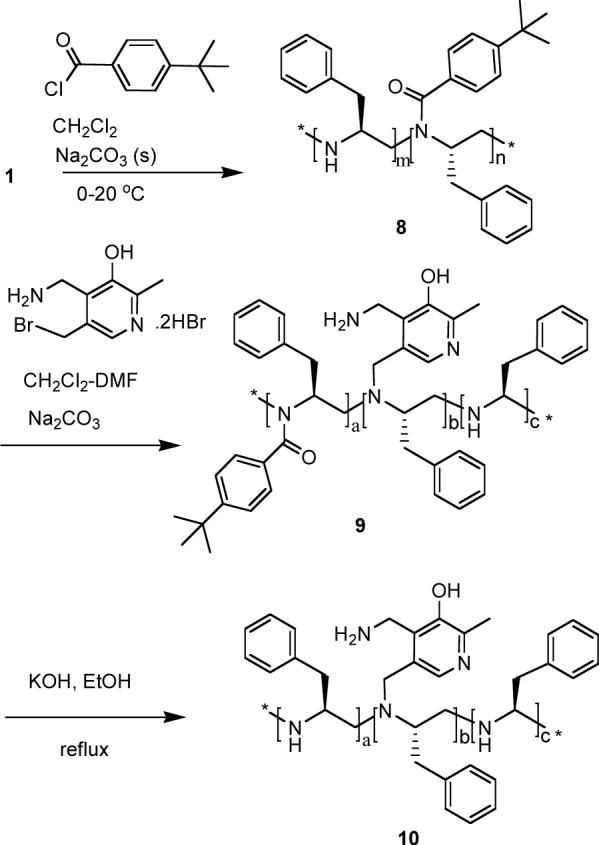
Synthesis of 10.
When phenylpyruvic acid was subjected to transamination with polymer 10, the ratio of L/D phenylalanine was found to increase up to 76/24 ± 3 %, (highest 79/21).
The efficacy of PEI chains in transamination with 5, 6, and 10 is established by the rate accelaration relative to pyridoxamine without any PEI under identical conditions. Polymer 5, 6, and 10 showed rate enhancement of the pseudo first order reactions with factors of 1369, 1178 and 1021 respectively at pH 5.5. The agreement between different runs was within 15%.
Thus, in this work we have demonstrated enantioselectivity in transamination reaction with a new isotactic PEI in a less than fully aqueous environment. The reversibly bound systems were somewhat more effective than the covalently linked version, but the reversible systems showed product racemization that was not seen in the covalent cases. Regeneration experiments to afford turnover, as we have described with our nonchiral systems,13,14 are currently under progress.
Supplementary Material
Acknowledgement
We thank Ms. Meghann White for her help with the measurement of the polydispersity. We thank NSF and NIH for financial support of this work.
Footnotes
Supporting Information Available. Experimental procedures and characterization data for the polymers. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Breslow R. In: Artificial Enzymes. Breslow R, editor. Wiley-VCH; Weinhelm, Germany: 2005. [Google Scholar]
- 2.Pham T;,R, Schultz PG, Sugasawara R, Schultz PG. J. Am. Chem. Soc. 1991;113:6670–6672. [Google Scholar]
- 3.Imperiali B, Roy RS. J. Am. Chem. Soc. 1994;116:12083–12084. [Google Scholar]
- 4.Kuang H, Brown ML, Davies RR, Young EC, Distefano MD. J. Am. Chem. Soc. 1996;118:10702–10706. [Google Scholar]
- 5.Bachman S, Knudsen K, Jorgensen KA. Org. Biomol. Chem. 2004;2:2044–2049. doi: 10.1039/b404381b. [DOI] [PubMed] [Google Scholar]
- 6.Svenson J, Zheng N, Nicholls IA. J. Am. Chem. Soc. 2004;126:8554–8560. doi: 10.1021/ja039622l. 2004. [DOI] [PubMed] [Google Scholar]
- 7.Zimmerman SC, Breslow R. J. Am. Chem. Soc. 1984;106:1490–1491. [Google Scholar]
- 8.Zhou WJ, Yerkes N, Chruma JJ, Liu L, Breslow R. Bioorg. Med. Chem. Lett. 2005;15:1351–1355. doi: 10.1016/j.bmcl.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 9.a Bruice TC, Benkovic S. Bioorganic Mechanisms. Vol. 2. W. A. Benjamin; New York: 1966. Chapter 8. [Google Scholar]; b Walsh C. Enzymatic Reaction Mechanisms. W. H. Freeman; San Fransisco: 1979. Chapter 24. [Google Scholar]
- 10.Liu L, Rozenman M, Breslow R. J. Am. Chem. Soc. 2002;124:12660–12661. doi: 10.1021/ja028151k. [DOI] [PubMed] [Google Scholar]
- 11.Zhou WJ, Liu L, Breslow R. Helv. Chim. Acta. 2003;86:3560–3567. [Google Scholar]
- 12.Liu L, Breslow R. J. Am. Chem. Soc. 2002;124:4978–4979. doi: 10.1021/ja025895p. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Zhou WJ, Chruma J, Breslow R. J. Am. Chem. Soc. 2004;126:8136–8137. doi: 10.1021/ja048671a. [DOI] [PubMed] [Google Scholar]
- 14.Chruma JJ, Liu L, Zhou W, Breslow R. Bioorg. Med. Chem. 2005;13:5873–5883. doi: 10.1016/j.bmc.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Breslow R, Bandyopadhyay S, Levine M, Zhou W. ChembioChem. 2006;7:1491–1496. doi: 10.1002/cbic.200600120. [DOI] [PubMed] [Google Scholar]
- 16.Saegusa T, Fujii H, Ikeda H. Polym. J. 1972;3:35–39. [Google Scholar]
- 17.Saegusa T, Fujii H, Ikeda H. Macromolecules. 1972;5:108. [Google Scholar]
- 18.Kobayashi S, Iijima S, Igarashi T, Saegusa T. Macromolecules. 1987;20:1729–1734. [Google Scholar]
- 19.Oh YS, Yamazaki T, Goodman M. Macromolecules. 1992;25:6322–6331. [Google Scholar]
- 20.Chuard T, Gretillat F, Bernauer K. Chimia. 1993;47:215–217. [Google Scholar]
- 21.Liu L, Rozenman M, Breslow R. Bioorg. Med. Chem. 2002;10:3973–3979. doi: 10.1016/s0968-0896(02)00334-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.