

Abstract
Growing evidence suggests that changes in the ion buffering capacity of glial cells can give rise to neuropathic pain. In the CNS, potassium ion (K+) buffering is dependent on the glia-specific inward rectifying K+ channel Kir4.1. We recently reported that the satellite glial cells that surround primary sensory neurons located in sensory ganglia of the peripheral nervous system also express Kir4.1, whereas the neurons do not. In the present study, we show that, in the rat trigeminal ganglion, the location of the primary sensory neurons for face sensation, specific silencing of Kir4.1 using RNA interference leads to spontaneous and evoked facial pain-like behavior in freely moving rats. We also show that Kir4.1 in the trigeminal ganglion is reduced after chronic constriction injury of the infraorbital nerve. These findings suggests that neuropathic pain can result from a change in expression of a single K+ channel in peripheral glial cells, raising the possibility of targeting Kir4.1 to treat pain in general and particularly neuropathic pain that occurs in the absence of nerve injury.
Keywords: microglia, chronic constriction injury, primary sensory neurons, infraorbital nerve, trigeminal system, orofacial pain
Introduction
Peripheral neuropathic pain is a condition resulting from a primary lesion or dysfunction of the peripheral nervous system. One characteristic of neuropathic pain is its resistance to common analgesics and is currently the focus of intense research aimed at developing alleviating treatments. A contributing factor to the generation of neuropathic pain is a change in the excitability of primary sensory neurons, causing them to become spontaneously active or discharge at a much lower threshold than normal (Fried et al., 2001; Tsuzuki et al., 2003; Cherkas et al., 2004; Robinson et al., 2004). Such hyperexcitability gives rise to paresthesia (prickling or tingling sensation on the skin), shooting pains and allodynia (painful response to nonpainful stimuli) in the cutaneous territory served by the injured nerve. A major determinant of neuronal excitability is the extracellular concentration of potassium ions (K+) (Haydon, 2001), which is primarily regulated by ion channels expressed by satellite glial cells (SGCs) (Hanani, 2005) that surround primary sensory neurons. SGCs demonstrate marked K+ permeability (Hosli et al., 1978), and trigeminal SGCs show mainly inward K+ conductance (Cherkas et al., 2004). Conventional models of neuronal ion balance predict that a disruption of the ability of SGCs to maintain normal levels of extracellular K+ would be associated with altered excitability of primary sensory neurons (Laming et al., 2000; Walz, 2000) and would thus lead to abnormal sensory perception. In support of this model, hyperkalemia, direct K+ injection, or inhibition of K+ uptake all lead to neuronal hyperexcitability (Traynelis and Dingledine, 1988; Janigro et al., 1997; Kiernan et al., 2002).
In the brain, the high K+ buffering capacity of astrocytes (Takumi et al., 1995; Poopalasundaram et al., 2000) is conferred by the glia-specific Kir4.1 channel subunit, acting as homomeric channels or as heteromeric channels by coassembly with the Kir5.1 channel subunit (Butt and Kalsi, 2006). Because Kir5.1 subunits do not form functional homomeric channels (Pessia et al., 1996), the Kir4.1 subtype remains the major component for inward rectifying K+ conductance in CNS glia. In addition, we recently found that Kir4.1 is also expressed by trigeminal SGCs (Vit et al., 2006). In the present study, we report that the expression of Kir4.1 is reduced in the ganglion of animals that had been subjected to a standard peripheral nerve injury. In view of these findings, we examined the role of Kir4.1 in establishing nociceptive thresholds in the trigeminal ganglia of rats without nerve injury. We found that silencing Kir4.1 in the trigeminal ganglion using RNA interference (RNAi) resulted in a reversible change in nociceptive threshold and nociceptive-related behavior, showing that neuropathic pain can result from a change in expression of a single K+ channel in SGCs in the absence of nerve injury.
Materials and Methods
Animals
Adult male Sprague Dawley rats (270–330 g; Charles River Laboratories, Wilmington, MA) were housed on a 12 h light/dark cycle and given food and water ad libitum. Procedures for the maintenance and use of the rats conformed to the regulations of the University of California, San Francisco Committee on Animal Research and were performed in accordance with the guidelines of the National Institutes of Health regulations on animal use and care (publication 85-23, revised 1996). Primates were obtained from ITR Laboratories Canada (Montreal, Quebec, Canada) and were housed, and were experiments performed according to procedures approved by the local AstraZeneca Animal Care Committee and in accordance with The Care and Use of Experimental Animals of Canadian Council on Animal Care.
Cannula implant for injection in the maxillary division of the trigeminal ganglion
Rats were anesthetized with ketamine–xylazine and placed in a stereotaxic apparatus. The skull was exposed, and a burr hole was drilled above the location of the maxillary division (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) of the left trigeminal ganglion at 6.5 mm anterior to interaural zero and 2.3 mm lateral to the midline. A guide cannula pedestal (Plastics One, Roanoke, VA) was fixed to the skull over the burr hole using four stainless steel screws and dental acrylic cement. The guide cannula extended into the burr hole,1 mm below the pedestal but did not touch the surface of the cortex. At least 7 d were allowed for recovery from surgery before injection into the trigeminal ganglion.
Chronic constriction injury of the infraorbital nerve
A 2 cm skin incision was made along the upper edge of the orbit to expose the skull and nasal bone. Gentle retraction of the orbital content exposed ∼5 mm of the infraorbital nerve (ION), and two ligatures (5.0 chromic gut) were loosely tied around the exposed nerve. The incision was closed using 6.0 silk.
Synthesis of dsRNA and preparation for injection
To reduce Kir4.1 expression, we used naked long double-stranded RNA (dsRNA) sequences that have been shown to produce robust, specific and sustained gene silencing in vivo (Bhargava et al., 2004; la Fleur et al., 2005) from day 2 to day 10 after administration, after which expression reverts to baseline (Bhargava et al., 2004; la Fleur et al., 2005). A BLAST (basic local alignment search tool) search of the nonredundant rat database identified a 602 bp sequence that is not homologous to any other sequences and therefore specifically targets only Kir4.1.
cDNA of genes of interest were produced by reverse transcription of 1 μg of total RNA followed by a 30-cycle PCR using gene-specific primers. These cDNAs were then cloned into a pTOPO vector (Invitrogen, Carlsbad, CA). The specific forward and reverse primer sequences used to make Kir4.1 dsRNA corresponded to nucleotides 18–35 and 603–620, respectively (GenBank accession number NM_053804). Rat β-globin sequences were used as nonspecific dsRNA control as described previously (Bhargava et al., 2004). Sense and antisense RNA were synthesized from cDNA inserts by using MegaScript RNA kit (Ambion, Austin, TX) according to the specification of the manufacturer. Naked long dsRNA for β-globin was used for control injections because it does not affect gene expression in nervous tissue (Bhargava et al., 2004; Vit et al., 2006).
Before the injection in the trigeminal ganglion, 15 μg of dsRNA of either Kir4.1 or globin dsRNA were mixed with lipofectamine (Invitrogen) in a final volume of 5 μl and let stand at room temperature. After a 30 min incubation, the red fluorescent marker 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (Invitrogen) was added to the mixture at a final concentration of 10 μm.
Injection into the trigeminal ganglion
Injection was performed with the rats under isoflurane anesthesia. A 33-gauge beveled stainless steel cannula (Plastics One) was inserted through the guide cannula to 9.5 mm below the cortical surface. The injection cannula was connected to a 25 μl Hamilton syringe attached to a microinjection pump set to deliver 2 μl over a 1 min period.
Behavioral testing
For all experiments, a treatment-blind observer conducted behavioral testing between 10:00 A.M. and 4:00 P.M. On each testing day, rats were brought into the behavior room at least 30 min before the test session to habituate them to the environment.
Spontaneous eye closure.
The rats were placed individually in a Plexiglas testing chamber (10 × 15 × 19 cm) in a quiet, darkened room. Mirrors were placed behind the side and back walls of the chamber so the head and eyes of the rat were always visible to the observer. Rats were habituated to the testing chamber for 3 d before the first session. The number of eye closures and the side (left, right, or bilateral) were recorded for three periods of 2 min at 5 min intervals. Eye closures were not counted during periods of face grooming. The sessions were also videotaped (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The results are expressed as the mean number of eye closures per minute.
Mechanical (von Frey hair) testing.
Three von Frey hairs of increasing stiffness, 2, 10, and 50 g, corresponding to log units 4.31, 5.07, and 5.88, respectively, were used. Five or six consecutive applications were performed at 5 s intervals on different areas of the vibrissal pad and in the perioral and perinasal territory. The observer scored the behavioral response of the rats based on the method of Vos et al. (1994) as follows: 0, no detection; 1, detection and exploration of the von Frey hair (sniffing, licking); 2, head withdrawal and/or single grabbing movement; 3, attack (includes biting) and/or escape (includes burrowing) and/or multiple grabbing movements; and 4, active asymmetrical grooming (at least three face wash strokes) directed toward the stimulated facial area. For each hair, the highest score was recorded and the results are presented as the average of the scores obtained from the three different hairs. The average value is presented because the analysis of scores from each hair separately gave the same results when compared between groups.
Operant drinking box (operant conflict paradigm: lick test).
Rats were tested for avoidance of an innocuous stimulus when attempting to obtain a reward of sugar-sweetened water (20% sucrose) delivered from a drinking tube. Rats were acclimated to sweetened water in their home cage and drank in the test chamber without being water deprived (Davis, 1973). The testing apparatus (Med Associates, St. Albans, VT) consisted of a chamber (20 × 20 × 20 cm) with a 5 × 5 × 3 cm alcove in one wall with a stainless steel drinking spout located at the rear. A conductance-measuring device (Lickometer; Ugo Basile, Comerio, Italy) attached to the drinking spout counted the number of licks when the rat drank from the spout. Four brushes (1 cm deep) were positioned on the left side of the drinking alcove (ipsilateral to the injection in the trigeminal ganglion), so that the rats had to maintain contact with the brushes to drink. The brushes were positioned to stimulate the rat's vibrissal pad as well as the perinasal and perioral area.
The rat was placed in the test chamber for a minimum of 5 min or after 10 attempts to drink and then returned to the home cage. The number of successful and unsuccessful licking attempts and the time required to achieve 10 attempts were recorded. A licking episode (successful attempt) was when the rat had a minimum of six licks before withdrawing its head from the drinking alcove and could be one continuous series of licks or several periods of licking without removing the head from the drinking area. An unsuccessful attempt was when the rat licked five or less times before removing its head from the opening. The limit of five licks was based on preliminary testing showing that less than five licks was part of exploratory behavior, whereas drinking episodes were always more than five licks. The number of licks was corrected for the weight of the rat. The results are presented as the number of licks per licking episode and per kilogram weight of rat, the number of unsuccessful attempts, and the time required to achieve 10 attempts.
Testing schedule and statistics.
Three weeks (day −21) before the dsRNA injection, the rats were habituated to 20% sucrose in water in their home cage. Rats were then tested every other day in the lick-test chamber for five sessions consisting of two trials on days −18 and −16 and a single trial on days −14, −12, and −10. Habituation to the environment and procedure for the blink test and the orofacial von Frey test was performed during the last three sessions (days −14, −12, and −10). Baseline values of naive rats for all tests were collected on day −8. Cannulas were implanted on day −7. The lick test was done on day −3, and baseline values of implanted rats were recorded again on day −1 for licking behavior, blink, and orofacial von Frey hair testing. On day 0, rats were randomly separated into two groups. One group received an injection of globin dsRNA, and the other was injected with Kir4.1 dsRNA into the maxillary division of the left trigeminal ganglion. Licking behavior, blink, and orofacial von Frey hair testing was then performed on days 1, 3, 5, 7, 10, and 13 after injection of the dsRNA. For tissue processing and analysis, four animals in each group were killed on days 5 and 14.
Statistics.
All results are expressed as mean ± SEM. All the recorded data were computed and analyzed using SigmaStat software (Systat Software, San Jose, CA). Differences between the two dsRNA groups (globin and Kir4.1) and changes over time were analyzed with a mixed repeated-measure (RM) ANOVA, followed by Bonferroni's post hoc comparisons between groups. Significant changes after dsRNA injection were established by comparison with baseline values (naive animals) using Student's t test for paired data. For eye closure and von Frey tests, in each group, differences between ipsilateral and contralateral sides to the injection were tested over time with a two-way RM ANOVA, followed by multiple comparisons between both sides using the paired t test. The magnitude of the effect on dsRNA-injected rats was determined on day 5 (day of maximum effect) by comparing with a 10 d chronic constriction injury (CCI) (day of maximum effect) of the ION (and sham operation). These different days were chosen to compare the effects when they were at a maximum in the two experiments to give a more meaningful comparison. The differences between dsRNA–day 5 or CCI of the ION–day 10 and their respective baseline values were calculated. A one-way ANOVA was performed to assess the difference among the four groups. The Bonferroni's post hoc test was used for multiple comparisons between groups. Results were considered statistically significant at p < 0.05.
Tissue processing
Histology.
Rats were deeply anesthetized intraperitoneally with 100 mg/kg pentobarbital and perfused transcardially with paraformaldehyde fixative. The brains and trigeminal ganglia were postfixed in 10% Formalin for 5 h and 30 min, respectively, and then placed in 30% sucrose in PBS, pH 7.4, for 48 h. Brainstems were marked ventrally on the right side, and 50 μm transverse sections of the entire brainstem were cut on a freezing microtome. Left and right ganglia from each animal were embedded together in tissue freezing medium and cut longitudinally at 10 μm on a cryostat.
Male monkeys were preanesthetized with ketamine and killed with sodium pentobarbital, and dorsal root ganglia were rapidly dissected, snap frozen at −40°C in isopentane for 20 s, and stored at −80°C until sectioning. All frozen tissues were transversely sectioned at 14 μm on a cryostat and mounted on slides for immunocytochemistry.
For electron microscopy, rats were perfused with 2% glutaraldehyde/2% paraformaldehyde and postfixed for 5 h. The ganglia were then cut at 50 μm on a vibratome, and the slices were osmicated (1% in phosphate buffer for 1 h), dehydrated, and embedded in Epon. Thin sections were cut, stained with lead citrate and uranyl acetate, and viewed in the electron microscope.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining.
Apoptosis was analyzed using the In Situ Cell Death Detection kit (Roche Diagnostics, Indianapolis, IN) according to the specifications of the manufacturer.
Immunohistochemistry.
Floating (brainstem, 50 μm) or on-slide sections (trigeminal ganglia, 10 μm) were processed in the same manner but with different primary antibodies. Sections were blocked in 5% normal goat serum (NGS), 0.3% Triton X-100 (Sigma, St Louis, MO) in PBS for 1 h and then incubated in the primary antiserum: OX-42 (1:2000; BD Biosciences, Sparks, MD), substance-P (SubP) (1:8000; Eugene Tech International), Kir4.1 (1:4000 with amplification; Alomone Labs, Jerusalem, Israel), Kir5.1 (1:2000 with amplification; Santa Cruz Biotechnology, Santa Cruz, CA), Kir 2.1 (1:100; Alomone Labs), Kir2.3 (1:1000; Alomone Labs), glial-specific glutamate transporter (GLAST) (1:16,000; Millipore Bioscience Research Reagents, Temecula, CA), rat endothelial cell antibody-1 (RECA-1) (1:5000 with amplification; Serotec, Raleigh, NC), isolectin B4 (IB4) (1:1000; Sigma), neurofilament 160 (NF160) (1:200; Sigma), neurokinin 1 (NK1) (1:150,000 with amplification; from Dr. Steve Vigna at University of Washington, Seattle, WA), or activating transcription factor 3 (ATF3) (1:5000; Santa Cruz Biotechnology) in 5% NGS, 0.3% Triton in PBS for 24 h. For fluorescence analysis, a fluorescein (FITC)-tagged secondary antibody (Vector Laboratories, Burlingame, CA), diluted 1:400 in 0.3% Triton/PBS was used for 1 h. Sections were then washed, mounted on slides, and coverslipped with Vectashield (Vector Laboratories). For light microscopy, a biotinylated secondary was used then the sections were washed and incubated for 1 h in a 1:400 ABC Elite (PK 6100; Vector Laboratories) solution diluted in 0.3% Triton/PBS. To visualize the antibody–antigen complex, we used the nickel–diaminobenzidine protocol. Alternatively, after ABC Elite incubation, amplification was performed by placing sections in biotinylated tyramide for 5 min, followed by incubation for 1 h in a 1:400 FITC–streptavidin solution (Vector Laboratories) for immunofluorescence. All antisera were tested for specificity by preabsorption of the antibody with the appropriate peptide. Also, omitting the primary antibody resulted in absence of labeling in all immunocytochemistry.
Quantification of immunolabeling and statistics.
Densitometry for assessment of immunostaining levels was performed in a treatment-blinded manner using a modified procedure described previously (Goff et al., 1998). A digital image of immunolabeled sections was captured, and the threshold of the image was set to a uniform value. The number of black pixels was counted using the Scion (Frederick, MD) Image program in a standardized square. Immunostaining was analyzed on three sections for each trigeminal ganglion or trigeminal brainstem subnucleus (caudalis, interpolaris/oralis, and main sensory) per animal, the counts were averaged for each nucleus, and the densitometry data are presented as the mean number of pixels per square micrometer. The densitometry was not used to measure the absolute value of immunostained marker but to determine relative differences between groups. To avoid variability in experimental procedures that could affect the densitometric analysis, immunohistochemistry for all rats to be compared with each other was done on the same days. To quantify the reduction of Kir4.1 protein, we counted the number of immunopositive pixels in standard-sized bins across the trigeminal ganglion (maxillary and mandibular divisions) from digital images. To account for staining variation, we expressed the lowest values from the maxillary division (i.e., the injection site) as a percentage of the highest value obtained from the mandibular division, which was outside the injection site.
Protein extraction, Western blot, and quantification.
Protein extracts were obtained from trigeminal ganglia as described previously (Vit et al., 2006). The amount of protein was determined using the Bio-Rad (Hercules, CA) protein assay. Protein samples (20 μg) were then resolved by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membrane. Equal loading and transferring of proteins was monitored by Ponceau-S staining of the membrane. Immunoblots were performed for ATF3 (1:1000 dilution) and Kir4.1 (1:400). Films were scanned and band intensity was quantified using NIH Image J software. The number of black pixels was counted in a standardized square positioned over each band.
Macrophage inflammation assay.
To evaluate the potential inflammatory capacity of our dsRNA, we tested RAW-ELAM macrophages (Hume et al., 2001), which harbor a luciferase reporter construct driven by nuclear factor-κB (NF-κB) activation (most or perhaps all innate immune pattern recognition receptor activation induces expression of downstream targets via NF-κB activation). Cells were seeded in 96 well plates. Negative controls were untreated cells, and positive controls were cells treated with 100, 250, or 500 ng/ml lipopolysaccharide (LPS) (a potent activator of NF-κB). Experimental conditions included treatment with Kir 4.1 and globin dsRNA in concentrations similar to those injected into the trigeminal ganglia (50 mg/ml). Cells were lysed after 6 h treatment and then read by a luminometer that microinjects substrate and converts luciferase luminosity into luciferin concentration, which in turn is directly proportional to NF-κB activation.
Results
SGC-confined Kir4.1 expression decreases in the trigeminal ganglion after CCI of the ION
To first establish that in the trigeminal ganglion Kir4.1 was confined to SGCs (Fig. 1) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), we used double-label immunofluorescence for Kir4.1 and GLAST (Danbolt, 2001) (Fig. 1E) and antibodies against Kir4.1 and NF160 [a protein unique to neurons (Oblinger, 1987)] (Fig. 1F). Every SGC surrounding all sizes of neuron were Kir4.1 immunopositive in both the rat trigeminal ganglion (Fig. 1D,E1,E3) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) and dorsal root ganglia (Fig. 1G) as well as in the primate dorsal root ganglion (Fig. 1H). Neither blood vessel endothelial cells (Fig. 1I) nor Schwann cells (Fig. 1J) demonstrated positive staining for Kir4.1 by immunohistochemistry.
Figure 1.
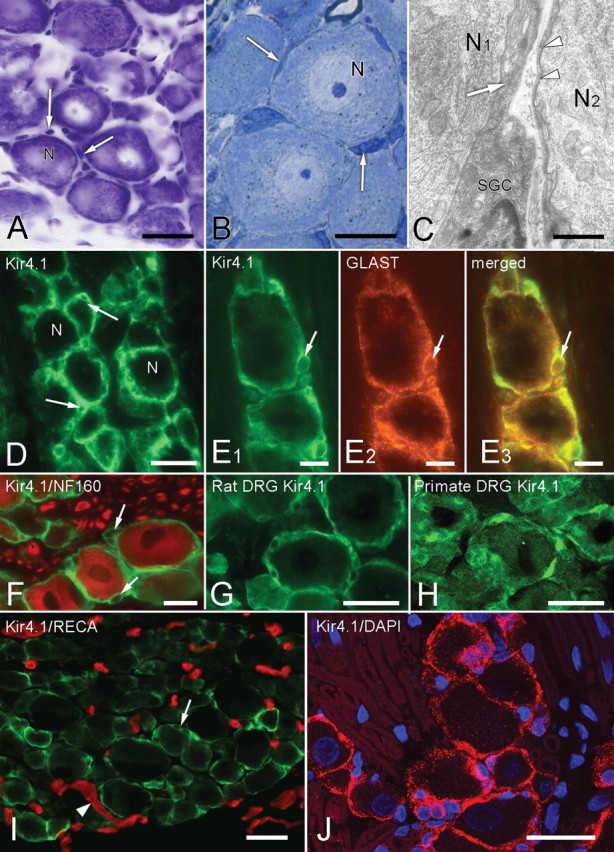
Morphology and immunohistochemistry of SGCs in sensory ganglia. A, Nissl-stained section showing trigeminal ganglion neurons (N) and the nuclei of surrounding SGCs (arrows). B, Semithin Epon section, 0.1 μm, showing closely packed neurons with surrounding SGCs (arrows). C, Electron micrograph demonstrating that the darkly stained, often attenuated SGC cytoplasm (arrow and arrowheads) closely surrounds the primary sensory neurons (N1, N2). The separation between the neuronal and SGC membranes is ∼20 nm. D, SGCs are immunopositive for Kir4.1. The unstained nuclei of SGCs (arrows) give a characteristic signet-ring appearance. N indicates the cell bodies of sensory neurons. E1–E3, Double immunolabeling with Kir4.1 (green, E1) and GLAST (red, E2) show extensive overlap in the merged image (yellow, E3), confirming that Kir4.1 is confined to SGCs. The arrow indicates the same SGC nucleus in each image. F, Immunolabels for Kir4.1 (green) and the neuronal marker NF160 (red) do not overlap. Arrows indicate unstained SGC nuclei. G, H, SGCs in rat (G) and primate (H) dorsal root ganglion are also Kir4.1 immunopositive. I, Section of control trigeminal ganglion immunostained for Kir4.1 (green, arrow) and blood vessels (RECA-1 antibody, red, arrowheads). There is no overlap of the two labels. J, Control trigeminal ganglion immunostained for Kir4.1 (red) and nuclei (DAPI, blue). The only Kir4.1-immunopositive structures are the SGCs surrounding unstained neurons. Nuclei (arrows) in the surrounding unstained regions belong to Schwann cells, and the outline of unstained myelin sheaths (arrowheads) is visible. Scale bars: A, D, G, H, I, J, 30 μm; B, E, F, 15 μm; C, 5 μm.
We next determined whether expression of Kir4.1 in the trigeminal ganglion was changed after a nerve injury that also caused a change in nociceptive threshold. To do this, we performed a CCI of the ION, a standard model of orofacial neuropathic pain (Vos et al., 1994). The ION carries sensation for the vibrissal pad. Ten days after the CCI, the trigeminal ganglia were harvested, and Western blot analysis showed that Kir4.1 expression was reduced by ∼40% (Fig. 2) (pixel intensity: ipsilateral, 9.5 ± 1.3; contralateral, 16.0 ± 1.7; p < 0.01; n = 6). Western blot analysis of ATF3 (a marker of nerve injury) was performed as a marker of nerve injury. ATF3 expression was dramatically increased ipsilateral to the CCI of the ION (Fig. 2) (pixel intensity: ipsilateral, 32.2 ± 3.5; contralateral, 6.5 ± 0.6; p < 0.001; n = 6).
Figure 2.

Changes in Kir4.1 expression after CCI of the ION. Ten days after CCI of the ION, there is a significant decrease in the expression of Kir4.1 in the trigeminal ganglion ipsilateral to the nerve injury and an increase in expression of the cell injury marker ATF3.
Silencing of Kir4.1 induces behavioral changes characteristic of neuropathic pain-like behavior
Having established that the Kir4.1 was confined to SGCs in the ganglion and that Kir4.1 expression was decreased after nerve injury, we wanted to determine whether decreased expression of Kir4.1 in the absence of injury was sufficient to produce reduced nociceptive thresholds. We therefore injected dsRNA against Kir4.1 into the maxillary division of the trigeminal ganglia to temporarily reduce the expression of the protein and then performed behavioral testing during the period of suppression. A fluorescent dye (DiI) was coinjected to help localize the site of the injection (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). DiI injected alone at the concentration used here had no toxic effects on cells or behavioral effects (data not shown). To test for evoked nociceptive responses, we used the orofacial von Frey hair test (Vos et al., 1994) and mechanically stimulated the vibrissal pad of the rat's snout with 2, 10, and 50 g hairs. The results are presented as the average of the scores obtained from the three von Frey hairs. After Kir4.1 dsRNA injection, the rats showed a significant increase in aversive responses to all von Frey hairs on the ipsilateral side but not to stimulation to the side contralateral to the injection (RM ANOVA; ipsilateral, F = 5.9, p < 0.001; contralateral, F = 1.3, p = 0.283) (Fig. 3A1). The elevated mechanical sensitivity was observed from day 1 to day 10 after injection, after which the sensory responses returned to normal. In control animals treated with β-globin dsRNA, the sensory response score remained unchanged (Fig. 3A1). We also confirmed that the surgery and cannula implantation used for dsRNA injection had no effect on sensory responses (before and after implantation; RM ANOVA, ipsilateral, F = 0.7, p = 0.409; contralateral, F = 0.3, p = 0.617) (Fig. 3A1).
Figure 3.
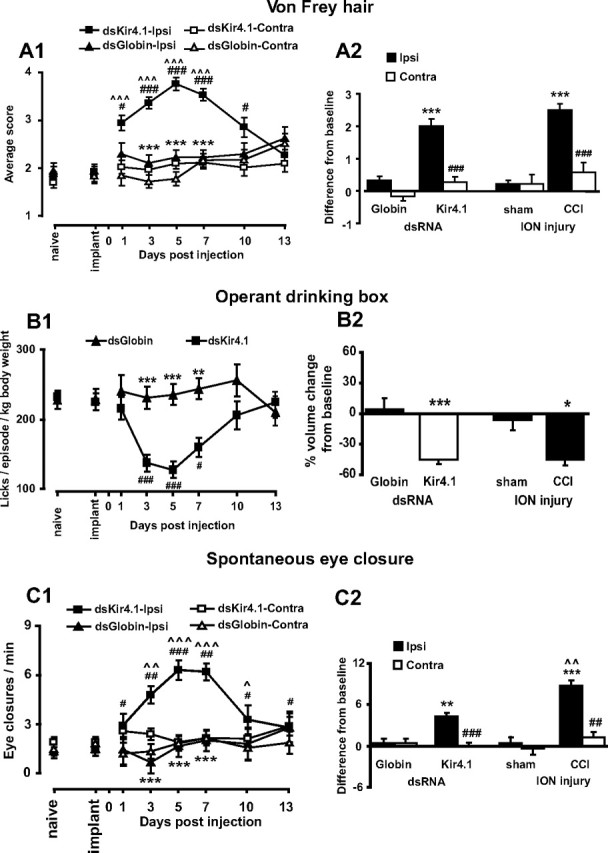
Silencing Kir4.1 in the trigeminal ganglion leads to pain-like behavior. A1, Rats show increased sensitivity to von Frey hair testing beginning 1 d after Kir4.1 dsRNA injection. This sensitivity reaches a maximum at day 5. A2, When compared with CCI rats, there is no difference between groups. Note that, in all graphs, the values are taken at day 10 after the CCI when nociceptive scores were at the maximum value. B1, Kir4.1 dsRNA reduces the number of licks per episode of drinking. B2, The reduction in drinking behavior is similar to the CCI group. C1, After Kir4.1 dsRNA injection, the number of eye closures on the injected side increases to a maximum on day 5 and then declines thereafter. C2, There is an increase from baseline for both Kir4.1 dsRNA-injected and CCI rats, but, in the CCI group, the eye closure rate is greater than in the Kir4.1 dsRNA group. A1, B1, C1, #p < 0.05, ##p < 0.01, ###p < 0.001 compared with preinjection of dsRNA (implant); **p < 0.01, ***p < 0.001 compared with Kir4.1 dsRNA; ^p < 0.05, ^^ p < 0.01, ^^^p < 0.001 compared with contralateral side (n = 10 per group for Kir4.1 and globin for all comparisons). A2, C2, ##p < 0.01, ###p < 0.001 compared with ipsilateral side. A2, B2, C2, *p < 0.05, **p < 0.01; C2, ***p < 0.001 compared with appropriate control; ^^p < 0.01 compared with Kir4.1 dsRNA (n = 10 per group for Kir4.1 and globin groups and n = 4 per group for CCI and sham groups).
We then wanted to compare the behavioral effects of silencing Kir4.1 with a standard model of neuropathic pain and so chose the CCI of the ION (Vos et al., 1994). The von Frey scores after silencing Kir4.1 or CCI were similar for both ipsilateral and contralateral sides (ANOVA, F = 3.0, p = 0.108), whereas scores in control rats treated with β-globin dsRNA were similar to sham-injured rats (Fig. 3A2). We also found that systemic morphine (4 mg/kg) had no effect on the von Frey scores of Kir4.1 dsRNA rats but resulted in a small decrease in von Frey scores of the CCI rats (Kir4.1 dsRNA: before morphine, 3.5 ± 0.2; morphine, 3.6 ± 0.2; p > 0.05; n = 10; CCI: before morphine, 4.0 ± 0.2; morphine, 3.5 ± 0.2; p < 0.01; n = 4). The morphine-induced reduction of von Frey scores has been reported (for review, see Zhao et al., 2004) and appears to be influenced by the pain model as well as the route of morphine administration. We interpret the difference in response to morphine between dsKir4.1 dsRNA injection and CCI as an additional example of the heterogeneity in causative factors for reduced nociceptive thresholds.
We next used an operant conflict paradigm (Neubert et al., 2005) in which the rat presses the side of its snout ipsilateral to the injection against a stiff-bristled brush to gain access to sugar-sweetened water. The brush is innocuous to normal rats and does not affect their drinking (data not shown). Rats with silenced Kir4.1 drank less than β-globin injected rats (Fig. 3B1) and maintained contact with the brush for less time than control rats. All other measured parameters (volume per lick, number of drinking attempts, and time to make 10 attempts) were not significantly different in rats treated with dsRNA against Kir4.1 and control rats (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). When the brush was removed, the Kir4.1 dsRNA-treated rats drank normally. Finally, the amount of water consumed was similar between Kir4.1 dsRNA-treated rats and rats with a CCI of the ION (ANOVA, F = 0.6, p = 0.463) (Fig. 3B2), showing that silencing Kir4.1 resulted in a reduction of nociceptive threshold similar to that resulting from CCI.
Within 3 d of injecting Kir4.1 dsRNA, rats were closing the eye ipsilateral to the injection more frequently (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). This spontaneous behavior is similar to the grooming response seen after CCI of the ION (Vos et al., 1994) and is also characteristic of the unilateral wincing seen in patients with trigeminal neuralgia when they experience shooting pains. The number of ipsilateral eye closures reached a maximum on days 5–7 after injection and then decreased to a value close to baseline by day 10. The increase in spontaneous eye closures was not related to the presence of the injection guide cannula because all treatment groups showed the same number of eye closures before the injection of dsRNA (RM ANOVA, ipsilateral, F = 0.8, p = 0.388; contralateral, F = 0.2, p = 0.639; n = 10). On the side ipsilateral to the injection, the increase in spontaneous eye closures was specific to Kir4.1 dsRNA when compared with the control β-globin dsRNA (RM ANOVA, ipsilateral, F = 4.5, p < 0.001; contralateral, F = 0.7, p = 0.661; n = 10).
We also compared eye closures between rats 5 d after Kir4.1 dsRNA injection and rats 10 d after a CCI of the ION (when nociceptive scores were at the maximum value for both groups). There was a significant increase in eye closures in the nerve-injured (CCI of the ION) animals and in Kir4.1 dsRNA-injected rats (ANOVA, F = 10.2, p = 0.008; n = 10) (Fig. 3C2), and this behavior was identical between groups. When we administered morphine (4 mg/kg, i.p.) to the Kir4.1 dsRNA-treated (day 5) and CCI (day 10) rats, the eye closures returned to baseline values in both groups (Kir4.1 dsRNA before morphine: vehicle, 6.2 ± 1.0; morphine, 3.25 ± 0.5; p < 0.05; n = 10; CCI before morphine, 10.8 ± 1.1; morphine, 1.7 ± 0.4; p < 0.05; n = 4), indicating that this nociceptive behavior was different from allodynia.
Kir4.1 expression is reduced in SGCs of the trigeminal ganglion after Kir4.1 dsRNA injection
At day 5 after dsRNA administration, there was a significant decrease in Kir4.1 expression as shown by immunocytochemistry and Western blotting. The decrease was approximately fourfold in the maxillary division of the trigeminal ganglion in Kir4.1 dsRNA-injected rats (9.2 ± 4.3%) when compared with β-globin dsRNA-treated (53.7 ± 6.3%) or naive (60.2 ± 2.1%) rats (ANOVA, F = 33.4, p < 0.001, n = 10) (Fig. 4A1–A3). Note that, when measuring the immunolabel, the reference values from β-globin dsRNA-treated and naive rats are <100% because the sample bins include nonlabeled fiber bundles. SGCs surrounding all sizes of neuron were equally affected. The reduction in Kir4.1 expression was confirmed by Western blot analysis (Fig. 5).
Figure 4.

Kir4.1 dsRNA injection into the trigeminal ganglion. A1, Injection of Kir4.1 dsRNA into the trigeminal ganglion inhibits the expression of Kir4.1 as shown by reduced SGC immunolabeling (green). A2, A3, Pseudocolor representation of the squares depicted in A1 shows that Kir4.1 inhibition is greatest close (A2) to the injection site indicated by the red fluorescent dye DiI (A1, arrow) when compared with a distant area (A3). B, C, Serial sections from a Kir4.1 dsRNA injection site showing that, after the disappearance of Kir4.1 immunoreactivity (B, green), SGCs are still present as indicated by GLAST labeling (C, green). The asterisk shows the same blood vessel on each section (B, C). D, Kir5.1 immunolabeling (green) is still present in SGCs after Kir4.1 dsRNA injection (arrow shows DiI injection site). E, Immunostaining for SK3 (green) from the region of the trigeminal ganglion injected with Kir4.1 dsRNA indicated by the presence of DiI (red, arrow). There is no reduction of the SGC-specific SK3. F, Kir2.3 immunostaining (green) from the region of the trigeminal ganglion injected with Kir4.1 dsRNA indicated by the presence of DiI (red). There is no reduction of the Kir2.3 immunostaining. G, Section from a Kir4.1 dsRNA injection site (arrows indicate DiI, showing we are in the injection site) on day 14 showing the return of Kir4.1 expression (green). H, Kir4.1 dsRNA injection site showing only a single ATF3-immunopositive neuron (arrow). Inset, Higher magnification of the ATF3-immunolabeled neuron. The asterisk indicates the location of the center of DiI injection. I, Large numbers of ATF3-positive neurons (arrows) are present after CCI of the ION. Scale bars: A, H, 300 μm; B, C, 90 μm; D, I, 30 μm; E, 60 μm; G, I, 180 μm.
Figure 5.

Changes in Kir4.1 expression 5 d after Kir4.1 dsRNA or globin dsRNA was injected into the trigeminal ganglion. There is a significant decrease in the expression of Kir4.1 in the Kir41. dsRNA-injected ganglia and no change in the globin dsRNA-injected ganglia. I, Ipsilateral ganglion; C, contralateral ganglion.
Kir4.1 immunolabeling returned to normal by day 14 (53.9 ± 6.6%, Kir4.1–14 d, n = 6 vs Kir4.1–5 d, n = 9, p < 0.001; Kir4.1–14 d vs control animals, n = 9, p = 0.340) (Fig. 4G). To rule out the possibility that the reduction in Kir4.1 immunoreactivity was attributable to death of SGCs, adjacent sections were Nissl stained (Fig. 6A,B) or immunostained for GLAST (Fig. 4C) and showed that SGCs were still present and surrounding neurons at the injection site. To confirm the specificity of the Kir4.1 knockdown, we immunostained for four K+ channels, the Kir 2.1, Kir 2.3, and Kir5.1 subunits and the small conductance calcium-activated K+ channel SK3. Kir5.1 and SK3 expression was present in SGCs and unchanged in both dsRNA treatment groups (Fig. 4D,E). Because Kir4.1 and Kir5.1 are known to heterodimerize (Butt and Kalsi, 2006), this finding suggests that the expression of one heteromer does not affect expression of the other heteromer. Kir2.3 was expressed in the cell bodies of sensory neurons as shown by double immunolabeling with GLAST (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). The expression of Kir2.3 was unchanged after injection of Kir4.1 dsRNA (Fig. 4F). Kir2.1 was not found in neurons or SGCs in the trigeminal ganglion.
Figure 6.

Effect of Kir4.1 dsRNA injection in the trigeminal ganglion. A, B, Nissl-stained sections from Kir4.1 dsRNA-treated (A) and globin dsRNA-treated (B) ganglia. There is no difference in the number of neurons ipsilateral to the injection between Kir4.1 dsRNA (20 ± 1 neurons/100,000 μm2) and globin dsRNA (19 ± 1 neurons/100,000 μm2) groups (ANOVA, F = 2.1, p = 0.180, n = 3). C–H, Neurons (arrows) are immunolabeled for substance-P (C, D, red fluorescence), IB4 (E, F, green fluorescence), or CGRP (G, H, blue fluorescence) in Kir4.1 dsRNA-treated (C, E, G) and globin dsRNA-treated (D, F, H) ganglia. There is no difference in the percentage of substance-P- or IB4-positive neurons ipsilateral to the injection between Kir4.1 dsRNA (substance-P, 12 ± 1%; IB4, 32 ± 1%; CGRP, 34 ± 1%) and globin dsRNA (substance-P, 14 ± 1%; IB4, 33 ± 1%; CGRP, 32 ± 1%) groups (ANOVA; substance-P, F = 2.3, p = 0.204; IB4, F = 0.3, p = 0.815; CGRP, F = 1.1, p = 0.566; n = 3). Scale bars: A–H, 60 μm.
Nociception and inflammatory markers in the trigeminal ganglion were unchanged
After silencing Kir4.1, there was no increase in markers associated with other models of neuropathic pain (Munglani et al., 1996), including SubP, calcitonin gene-related peptide (CGRP), or IB4 in the trigeminal ganglia (Fig. 7C–H). SubP and its receptor NK1 were also unchanged in the trigeminal spinal nucleus, in which primary afferents from the trigeminal ganglion synapse (Fig. 7D,E). ATF3, an indicator of cell stress or nerve injury, was found in very few cells in the dsRNA-injected ganglia (ATF3-positive neurons: globin dsRNA, 15 ± 3; Kir4.1 dsRNA, 13 ± 5). This contrasted with the dramatic increase in ATF3-positive neurons observed after CCI of the ION (ATF3-positive neurons: CCI, 152 ± 13, F = 109, p < 0.001) (Fig. 4H,I). There was no difference in terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining (a marker of apoptotic cell death) or Nissl staining (Fig. 7A,B) between dsRNA-treated and control rats. Using hematoxylin and eosin staining, as well as ED1 immunostaining, we observed a limited number of inflammatory cells along the injection tracks (a normal finding), but no difference was found between rats injected with water, DiI alone, or either of the dsRNA (data not shown).
Figure 7.
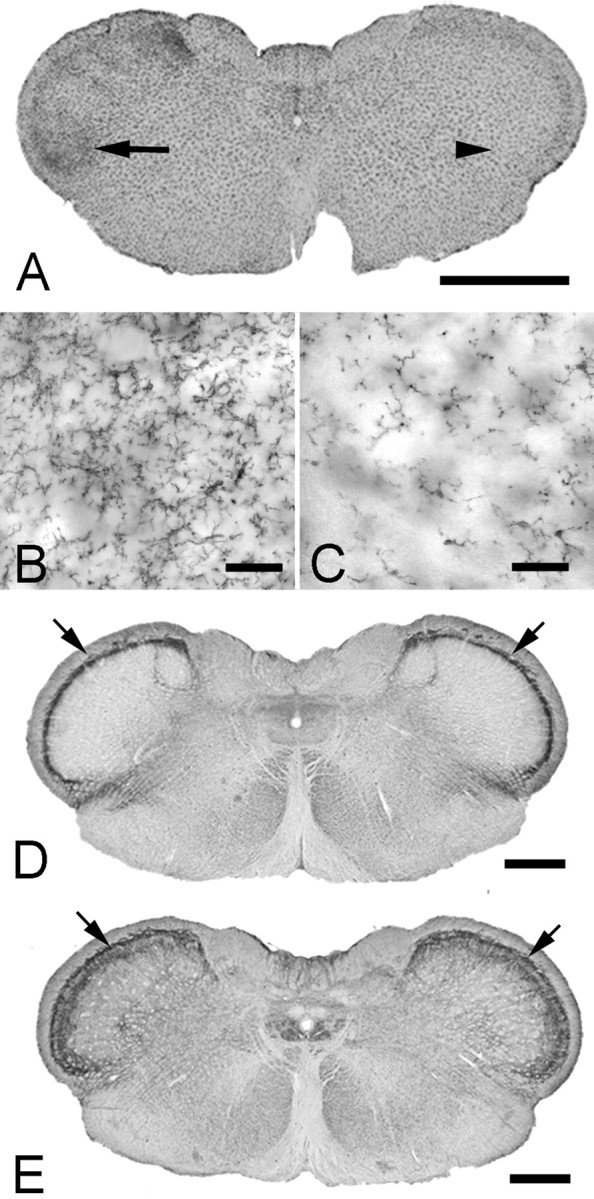
Effect of Kir4.1 dsRNA injection in the spinal trigeminal nucleus. A, Kir4.1 dsRNA injection into the trigeminal ganglion results in microglia activation (OX-42 immunostaining) in the brainstem. The arrow and arrowhead indicated the regions shown at high magnification in B and C, respectively. D, E, There is no change in substance-P or NK1 receptor staining in the trigeminal nucleus caudalis after Kir4.1 suppression in the trigeminal ganglion. Sections of the brainstem from a rat with Kir4.1 dsRNA injected into the left trigeminal ganglion immunostained for substance-P (D) or NK1 (E). Densitometry quantification of the immunostaining (arrows) shows no difference in the ipsilateral/contralateral pixel ratio between Kir4.1 dsRNA (substance-P, 1.2 ± 0.1; NK1, 1.0 ± 0.1) and globin dsRNA (substance-P, 0.9 ± 0.1; NK1, 1.0 ± 0.0) groups (ANOVA; substance-P, F = 2.4, p = 0.315; NK1, F = 0.5, p = 0.531). Scale bars: A, 2 mm; B, C, 50 μm; D, E, 1 mm.
To confirm the morphological finding that there was limited macrophage activation, we used a culture assay to quantify the effects of Kir4.1 and globin dsRNA on macrophage activation (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). Both Kir4.1 and globin dsRNA caused a small increase (∼12% for Kir4.1) in macrophage activation compared untreated cultures but far less than the activation resulting from LPS, which resulted in a more than threefold increase. However, there was no significant difference in macrophage activation between Kir4.1 and globin dsRNA treatment despite large difference in effect of these two constructs on behavior and Kir4.1 protein expression.
Injection of Kir4.1 dsRNA in the trigeminal ganglion activated brainstem microglia
Mechanical damage or chemical stimulation of a peripheral nerve can activate glial cells in the CNS and can thus contribute to the generation and maintenance of pain-like behavior (Wieseler-Frank et al., 2004; Obata et al., 2006). We therefore examined the brainstem trigeminal nuclear complex. Five days after Kir4.1 dsRNA, we found that microglia were activated (Fig. 7A,B) in all subnuclei of the ipsilateral trigeminal complex as shown by an increase in OX-42 immunostaining, whereas globin dsRNA-injected rats showed no change in microglial activation (Fig. 7C) (pixel intensity, ratio of ipsilateral/contralateral: globin dsRNA, 1.09; Kir4.1 dsRNA, 2.34; ANOVA, F = 5.6, p = 0.018, n = 6). Because SGCs have no physical connection with the brainstem, this finding reinforces the idea that the central glial activation results from the altered activity of primary sensory neurons (Banati, 2002) and does not need to involve physical damage to a nerve. Fourteen days after Kir4.1 dsRNA injection, microglial immunostaining was back to normal.
Discussion
Our results show that the reduction of the Kir4.1 channel in SGCs is sufficient to produce behavioral changes in the rat that are characteristic of neuropathic pain-like behavior. This conclusion is based on the concurrent reduction of both nociceptive threshold and Kir4.1 expression after Kir4.1 dsRNA injection and the subsequent return to normal nociceptive coincident with the return of Kir4.1 expression. The absence of detectable changes in expression of other genes in the trigeminal ganglion and lack of behavioral effect of injection of globin dsRNA are additional evidence that the behavioral changes are specific to the targeted gene. Concerns have been raised over immune activation by RNAi. For example, Hornung et al. (2005) reported that some small interfering RNA sequences are detected by the innate immune pattern recognition receptor TLR7 (Toll-like receptor 7) and can potently stimulate interferon-α production (Hornung et al., 2005). Other reports indicate that both single-stranded or double-stranded RNA (either sense or antisense) can trigger immune-mediated expression of proinflammatory cytokines, but the effect appears to be sequence dependent (Sioud and Iversen, 2005). Our macrophage assay showed a minimal amount of activation by either Kir4.1 dsRNA or globin dsRNA, and there was not a significant difference between the two dsRNA. If an immune response was a component of the behavioral and morphological response, we would expect that these two dsRNAs should produce dissimilar immune activation. Our observation that Kir4.1 expression decreases after CCI of the ION is additional evidence that the reduction of this protein contributes to increased nociceptive responses.
It is also significant that the changes in SGCs rather than neuronal expression of Kir4.1 result in lowered nociceptive threshold. As suggested by others (Hibino et al., 1999; Hanani, 2005), we confirmed that Kir4.1 is expressed only by SGCs in peripheral ganglia, which mirrors the situation in the CNS, in which Kir4.1 is located in astrocytes rather than neurons (Higashi et al., 2001). It has been proposed that SGCs in sensory ganglia play a role in nerve injury-mediated pain (Hanani, 2005; Miyagi et al., 2006; Bhangoo et al., 2007). Our findings add to the previous reports that, in a number of pathologies, a change in glial function is determinant in the initiation and maintenance of behavior characteristic of increased nociception (Hansson, 2006; Kim et al., 2007; Sun et al., 2007).
A recent advance related to nociceptive sensation concerns the role played by specific ion channels in the initiation (Caterina et al., 1997) and transmission of nociceptive stimuli (Cox et al., 2006; Waxman, 2006; Zimmermann et al., 2007). The absence (Cox et al., 2006) or mutation (Fertleman et al., 2007; Sheets et al., 2007) of a single neuronal sodium channel profoundly alters pain sensation. Similar effects have been suggested for alterations of potassium channels. For example, mice lacking the TREK-1 (TWIK-related K+ channels) have a lowered nociceptive threshold (Alloui et al., 2006). It has also been proposed that deactivation of the Kir3.1 channel subunit resulting from peripheral nerve injury and inflammation might underlie hyperalgesia (Ippolito et al., 2005), suggesting that potassium channel openers may be used to treat neuropathic pain (Wua and Dworetzky, 2005; Kobayashi and Ikeda, 2006). As yet, potassium channelopathies leading to altered pain sensation have not been described in humans.
We postulate that the behavioral changes reported here are a direct consequence of the decrease in the homodimeric (Kir4.1/4.1) and/or the heterodimeric (Kir4.1/5.1) forms of the K+ channel (Butt and Kalsi, 2006), resulting in hyperexcitability of primary sensory neurons. In turn, the increased excitability of primary sensory neurons has been associated with neuropathic pain-like behavior (Fried et al., 2001; Tsuzuki et al., 2003; Cherkas et al., 2004; Robinson et al., 2004). During RNAi, the K+ buffering capacity of SGCs (Haydon, 2001; Butt and Kalsi, 2006) would significantly decrease, with a consequent increase in extracellular K+. It should also be noted that, although other K+ channels are present on both SGCs (Vit et al., 2006) and CNS glial cells (Horio, 2001), the best evidence indicate they cannot compensate for the silencing of Kir4.1 (Kucheryavykh et al., 2007).
Conventional models of neuronal ion balance predict that, if normal levels of extracellular K+ are not maintained, the excitability of primary sensory neurons would increase, and this could lead to abnormal sensory perception (Laming et al., 2000; Walz, 2000). An increase in cortical neurons excitability has been described in mice with reduced Kir currents and lower expression of Kir2.1 and Kir6.1 channel subunits (Jansen et al., 2005). It has also been shown that extracellular K+ increases in Kir4.1−/− mice (Neusch et al., 2006; Djukic et al., 2007), and, although an increase in neuronal excitability was not reported, in one case there was an enhancement of synaptic potentiation. Inward rectifying K+ channels are not confined to astrocytes or SGCs; Kir2.1 and Kir2.3 channels are also present on Schwann cells at the nodes of Ranvier in peripheral nerves (Mi et al., 1996). In this latter case, the authors propose that an increase in extracellular K+ reduces axonal excitability so the purpose of the Kir channels is to remove excess K+ to maintain axonal excitability, although their study does not address this issue directly. In a study in which this issue was studied experimentally (Kocsis et al., 1983), the excitability of axons was found to be dependent on the external K+ concentration with increased excitability at low concentrations and decreased excitability at higher concentrations. Others also suggest that whether neuronal excitability is increased or decreased depends on the amount of K+ increase (Laming et al., 2000; Walz, 2000; Djukic et al., 2007) rather than an increase per se. The heterogeneity of K+ channel types, their very specific and confined cellular location, and the importance of level the extracellular K+ concentration in determining excitability precludes making definitive statements about conditions within the ganglia without experimental evidence, but overall the evidence supports the notion that an increase in extracellular K+ concentration resulting from Kir4.1 suppression is sufficient to alter neuronal excitability.
Glial cells are essential for the recycling of the excitatory neurotransmitter glutamate (Anderson and Swanson, 2000; Danbolt, 2001; Suchak et al., 2003), and we have shown recently that the glutamate transporter GLAST is highly expressed by SGCs in the trigeminal ganglion (Vit et al., 2006). Silencing Kir4.1 would result in a decreased (less negative) resting potential of SGCs, which would in turn lead to a reduction in the activity of glutamate transporters (Laming et al., 2000; Djukic et al., 2007; Kucheryavykh et al., 2007) with a consequent increase in extracellular levels of glutamate (Kucheryavykh et al., 2007). Thus, the decrease in nociceptive threshold observed after silencing Kir4.1 could be principally attributable to an increase of extracellular K+, an increase of extracellular glutamate, or a combination of the two.
We also found an increase in microglial activation in the brainstem trigeminal nuclei after silencing Kir4.1. Clearly, there is no physical connection between the SGCs and CNS glial cells, so the activation of central glia must have resulted from changes in ganglion neuronal activity or release of unknown glial activation factors by central terminals of these neurons. Again, we found no obvious phenotypic changes in trigeminal ganglion neurons after Kir4.1 silencing, indicating that damage or overt changes in ganglion neurons are not necessary to initiate central glial activation. The activation of central glia is believed to both increase and prolong the effects of peripherally induced nociception (Watkins et al., 2007). The manner in which this occurs remains to be clarified.
There has been a long-standing debate over the neurogenic or ideogenic origin of chronic pain that occurs without a lesion (Hodgkiss, 2000). Our experiments support growing evidence that conditions as severe and complex as neuropathic pain can result from changes to a single ion channel (Waxman, 2006) and may, at least in part, be responsible for neuropathic pain in patients that have no indication of nerve injury (Martinez-Lavin, 2006). Our conclusion is that the reduction of Kir4.1 expression in SGC is sufficient to reduce nociceptive thresholds, but we do not know whether it is a necessary condition for all types of neuropathic pain. The normal diversity of genetic makeup could result in individual differences in channel expression, resulting in a subpopulation with different sensitivities to nociceptive stimuli and susceptibilities to develop chronic pain in the absence of obvious cause (Gjerstad, 2007; Stamer and Stuber, 2007). From a treatment perspective, peripherally acting K+ channel modulators may thus be uniquely suited to treat such “gliopathic” pain.
Footnotes
This work was supported by the National Institutes of Health. We thank Cris Cua, Prema S. Idumalla, and Julia Hoy for their invaluable technical assistance and Christopher Sundberg for the macrophage assay. We also thank Pamela Derish, Terence Doherty, and Melinda Gordon for their comments on this manuscript and Dana Im for editorial assistance. We are also indebted to A. Morinville (AstraZeneca, Montréal, Quebec, Canada) for providing the Kir4.1 immunocytochemistry on the primate trigeminal ganglia.
References
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, Borsotto M, Romey G, Heurteaux C, Reeh P, Eschalier A, Lazdunski M. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006;25:2368–2376. doi: 10.1038/sj.emboj.7601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Banati RB. Brain plasticity and microglia: is transsynaptic glial activation in the thalamus after limb denervation linked to cortical plasticity and central sensitisation? J Physiol (Paris) 2002;96:289–299. doi: 10.1016/s0928-4257(02)00018-9. [DOI] [PubMed] [Google Scholar]
- Bhangoo SK, Ren D, Miller RJ, Chan DM, Ripsch MS, Weiss C, McGinnis C, White FA. CXCR4 chemokine receptor signaling mediates pain hypersensitivity in association with antiretroviral toxic neuropathy. Brain Behav Immun. 2007;21:581–591. doi: 10.1016/j.bbi.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava A, Dallman MF, Pearce D, Choi S. Long double-stranded RNA-mediated RNA interference as a tool to achieve site-specific silencing of hypothalamic neuropeptides. Brain Res Brain Res Protoc. 2004;13:115–125. doi: 10.1016/j.brainresprot.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cherkas PS, Huang TY, Pannicke T, Tal M, Reichenbach A, Hanani M. The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain. 2004;110:290–298. doi: 10.1016/j.pain.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Davis JD. The effectiveness of some sugars in stimulating licking behavior in the rat. Physiol Behav. 1973;11:39–45. doi: 10.1016/0031-9384(73)90120-0. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, Griesemer DA, Goutieres F, Kirkpatrick M, Malmros IN, Pollitzer M, Rossiter M, Roulet-Perez E, Schubert R, Smith VV, Testard H, Wong V, Stephenson JB. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome) Neurology. 2007;69:586–595. [Google Scholar]
- Fried K, Bongenhielm U, Boissonade FM, Robinson PP. Nerve injury-induced pain in the trigeminal system. The Neuroscientist. 2001;7:155–165. doi: 10.1177/107385840100700210. [DOI] [PubMed] [Google Scholar]
- Gjerstad J. Genetic susceptibility and development of chronic non-malignant back pain. Rev Neurosci. 2007;18:83–91. doi: 10.1515/revneuro.2007.18.1.83. [DOI] [PubMed] [Google Scholar]
- Goff JR, Burkey AR, Goff DJ, Jasmin L. Reorganization of the spinal dorsal horn in models of chronic pain: correlation with behaviour. Neuroscience. 1998;82:559–574. doi: 10.1016/s0306-4522(97)00298-4. [DOI] [PubMed] [Google Scholar]
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48:457–476. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Hansson E. Could chronic pain and spread of pain sensation be induced and maintained by glial activation? Acta Physiol (Oxf) 2006;187:321–327. doi: 10.1111/j.1748-1716.2006.01568.x. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Hibino H, Horio Y, Fujita A, Inanobe A, Doi K, Gotow T, Uchiyama Y, Kubo T, Kurachi Y. Expression of an inwardly rectifying K(+) channel, Kir4.1, in satellite cells of rat cochlear ganglia. Am J Physiol. 1999;277:C638–C644. doi: 10.1152/ajpcell.1999.277.4.C638. [DOI] [PubMed] [Google Scholar]
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- Hodgkiss A. From lesion to metaphor: chronic pain in Br, French and German medical writings, 1800–1914. Clio Med. 2000;58:i–iii. 1–218. [PubMed] [Google Scholar]
- Horio Y. Potassium channels of glial cells: distribution and function. Jpn J Pharmacol. 2001;87:1–6. doi: 10.1254/jjp.87.1. [DOI] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Hosli L, Andres PF, Hosli E. Neuron-glia interactions: indirect effect of GABA on cultured glial cells. Exp Brain Res. 1978;33:425–434. doi: 10.1007/BF00235564. [DOI] [PubMed] [Google Scholar]
- Hume DA, Underhill DM, Sweet MJ, Ozinsky AO, Liew FY, Aderem A Macrophages exposed continuously to lipopolysaccharide and other agonists that act via toll-like receptors exhibit a sustained and additive activation state. BMC Immunol. 2001;2:11. doi: 10.1186/1471-2172-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DL, Xu M, Bruchas MR, Wickman K, Chavkin C. Tyrosine phosphorylation of K(ir)3.1 in spinal cord is induced by acute inflammation, chronic neuropathic pain, and behavioral stress. J Biol Chem. 2005;280:41683–41693. doi: 10.1074/jbc.M507069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janigro D, Gasparini S, D'Ambrosio R, McKhann G, 2nd, DiFrancesco D. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci. 1997;17:2813–2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia. 2005;46:1871–1880. doi: 10.1111/j.1528-1167.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Walters RJ, Andersen KV, Taube D, Murray NM, Bostock H. Nerve excitability changes in chronic renal failure indicate membrane depolarization due to hyperkalaemia. Brain. 2002;125:1366–1378. doi: 10.1093/brain/awf123. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, Choi SY, Park K, Kim JS, Akira S, Na HS, Oh SB, Lee SJ. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282:14975–14983. doi: 10.1074/jbc.M607277200. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ikeda K. G protein-activated inwardly rectifying potassium channels as potential therapeutic targets. Curr Pharm Des. 2006;12:4513–4523. doi: 10.2174/138161206779010468. [DOI] [PubMed] [Google Scholar]
- Kocsis JD, Malenka RC, Waxman SG. Effects of extracellular potassium concentration on the excitability of the parallel fibres of the rat cerebellum. J Physiol (Lond) 1983;334:225–244. doi: 10.1113/jphysiol.1983.sp014491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–281. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- la Fleur SE, Wick EC, Idumalla PS, Grady EF, Bhargava A. Role of peripheral corticotropin-releasing factor and urocortin II in intestinal inflammation and motility in terminal ileum. Proc Natl Acad Sci USA. 2005;102:7647–7652. doi: 10.1073/pnas.0408531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laming PR, Kimelberg H, Robinson S, Salm A, Hawrylak N, Muller C, Roots B, Ng K. Neuronal-glial interactions and behaviour. Neurosci Biobehav Rev. 2000;24:295–340. doi: 10.1016/s0149-7634(99)00080-9. [DOI] [PubMed] [Google Scholar]
- Martinez-Lavin M. Fibromyalgia is a neuropathic pain syndrome. J Rheumatol. 2006;33:827–828. [PubMed] [Google Scholar]
- Mi H, Deerinck TJ, Jones M, Ellisman MH, Schwarz TL. Inwardly rectifying K+ channels that may participate in K+ buffering are localized in microvilli of Schwann cells. J Neurosci. 1996;16:2421–2429. doi: 10.1523/JNEUROSCI.16-08-02421.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi M, Ohtori S, Ishikawa T, Aoki Y, Ozawa T, Doya H, Saito T, Moriya H, Takahashi K. Up-regulation of TNFalpha in DRG satellite cells following lumbar facet joint injury in rats. Eur Spine J. 2006;15:953–958. doi: 10.1007/s00586-005-1031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munglani R, Harrison SM, Smith GD, Bountra C, Birch PJ, Elliot PJ, Hunt SP. Neuropeptide changes persist in spinal cord despite resolving hyperalgesia in a rat model of mononeuropathy. Brain Res. 1996;743:102–108. doi: 10.1016/s0006-8993(96)01026-8. [DOI] [PubMed] [Google Scholar]
- Neubert JK, Widmer CG, Malphurs W, Rossi HL, Vierck CJ, Jr, Caudle RM. Use of a novel thermal operant behavioral assay for characterization of orofacial pain sensitivity. Pain. 2005;116:386–395. doi: 10.1016/j.pain.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, Hulsmann S. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–1852. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain. 2006;7:816–822. doi: 10.1016/j.jpain.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Oblinger MM. Characterization of posttranslational processing of the mammalian high-molecular-weight neurofilament protein in vivo. J Neurosci. 1987;7:2510–2521. [PMC free article] [PubMed] [Google Scholar]
- Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 1996;15:2980–2987. [PMC free article] [PubMed] [Google Scholar]
- Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, Gallo V, Wilkin GP. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–372. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Robinson PP, Boissonade FM, Loescher AR, Smith KG, Yates JM, Elcock C, Bird EV, Davies SL, Smith PL, Vora AR. Peripheral mechanisms for the initiation of pain following trigeminal nerve injury. J Orofac Pain. 2004;18:287–292. [PubMed] [Google Scholar]
- Sheets PL, Jackson JO, 2nd, Waxman SG, Dib-Hajj SD, Cummins TR. A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol (Lond) 2007;581:1019–1031. doi: 10.1113/jphysiol.2006.127027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sioud M, Iversen PO. Ribozymes, DNAzymes and small interfering RNAs as therapeutics. Curr Drug Targets. 2005;6:647–653. doi: 10.2174/1389450054863653. [DOI] [PubMed] [Google Scholar]
- Stamer UM, Stuber F. Genetic factors in pain and its treatment. Curr Opin Anaesthesiol. 2007;20:478–484. doi: 10.1097/ACO.0b013e3282ef6b2c. [DOI] [PubMed] [Google Scholar]
- Suchak SK, Baloyianni NV, Perkinton MS, Williams RJ, Meldrum BS, Rattray M. The “glial” glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings. J Neurochem. 2003;84:522–532. doi: 10.1046/j.1471-4159.2003.01553.x. [DOI] [PubMed] [Google Scholar]
- Sun S, Cao H, Han M, Li TT, Pan HL, Zhao ZQ, Zhang YQ. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain. 2007;129:64–75. doi: 10.1016/j.pain.2006.09.035. [DOI] [PubMed] [Google Scholar]
- Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T, Kiyama H, Sohmiya K, Nakanishi S, Kurach Y. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995;270:16339–16346. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Tsuzuki K, Fukuoka T, Sakagami M, Noguchi K. Increase of preprotachykinin mRNA in the uninjured mandibular neurons after rat infraorbital nerve transection. Neurosci Lett. 2003;345:57–60. doi: 10.1016/s0304-3940(03)00487-7. [DOI] [PubMed] [Google Scholar]
- Vit JP, Jasmin L, Bhargava A, Ohara PT. Satellite glial cells in the trigeminal ganglion as a determinant of orofacial neuropathic pain. Neuron Glia Biol. 2006;2:247–257. doi: 10.1017/s1740925x07000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos BP, Strassman AM, Maciewicz RJ. Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat's infraorbital nerve. J Neurosci. 1994;14:2708–2723. doi: 10.1523/JNEUROSCI.14-05-02708.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–146. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Neurobiology: a channel sets the gain on pain. Nature. 2006;444:831–832. doi: 10.1038/444831a. [DOI] [PubMed] [Google Scholar]
- Wieseler-Frank J, Maier SF, Watkins LR. Glial activation and pathological pain. Neurochem Int. 2004;45:389–395. doi: 10.1016/j.neuint.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Wua YJ, Dworetzky SI. Recent developments on KCNQ potassium channel openers. Curr Med Chem. 2005;12:453–460. doi: 10.2174/0929867053363045. [DOI] [PubMed] [Google Scholar]
- Zhao C, Tall JM, Meyer RA, Raja SN. Antiallodynic effects of systemic and intrathecal morphine in the spared nerve injury model of neuropathic pain in rats. Anesthesiology. 2004;100:905–911. doi: 10.1097/00000542-200404000-00021. [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]