

Abstract
(6R)-5,6,7,8-tetrahydrobiopterin (BH4) is a key cofactor involved in the electron transfer to P450 heme of nitric oxide synthase. We calculated the electronic structure of the neutral, cation and anion forms of BH4 in gas phase, solution (both dielectric and explicit water), and in the protein environment using the density functional theory method (B3LYP/6-31+G(d,p)). Subsequently, we derived the ionization potential (IP) and electron affinity (EA) of the cofactor in these chemical environments. We found that the electronic structure of BH4 is susceptible to the presence of an external electric field, and that conformational changes in the structure of BH4 alone do not affect its electronic structure significantly. In gas phase, water, and protein environment the neutral BH4 is the most stable species, while, in dielectric, the anion becomes the most stable species. The IP of BH4 in the protein environment is about half of that in gas phase and its EA isabout five times smaller than in gas phase. Our results indicate that changes in the external electric field created by moving charged amino acid residues around BH4 may lead to configurations that have the BH4 ion as stable as, or more stable than the neutral form, thus facilitating the electron transfer.
Keywords: density functional theory, ionization potential, electron affinity, enzyme cofactor, tetrahydrobiopterin, nitric oxide synthase, dielectric, solution
Introduction
(6R)-5,6,7,8-Tetrahydrobiopterin (BH4) is a cofactor for several enzymes and can generate or scavenge reactive oxygen species1. Molecules that are related to biopterin include pterins, lumazines, alloxazines, folates, and riboflavins. The common structural feature of this group of molecules is a core of two or three fused heterocyclic six-member rings. Pterins are very reactive, they form chelates (five-membered rings) with metals through the O4 and N5 atoms (see Scheme 1 for atom labeling). In BH4 the pyrazine ring is hydrogenated, thus adding four more hydrogen atoms, two of which are bound to the two nitrogen atoms of this ring. The pKa of N5 is 5.62. In solution, BH4 can react with O23, hydrogen peroxide2,4, and peroxynitrite5. Glutathione, ascorbic acid, and dihydropteridine reductase (DHPR) reduce the quininoid form of BH4 6,7 and dihydrofolate reductase (DHFR) reduces the dihydro form (BH2)8. It has been suggested that the auto-oxidation proceeds through a radical species, which involves the N5 atom2. The substitution of the hydrogen atom bound to N5 with methyl9, or the replacement of N5 with a methylene bridge10, in BH4, decrease the susceptibility to auto-oxidation. Electronic structure calculations on BH4 in gas phase show that C4a has maximum electron density, and that the adjacent N5 enhances the reactivity of C4a1. The reduced susceptibility to oxidation of N5-alkylated BH4 was postulated to originate from the capability of the 5-methyl substituent to block the access of O2 to the C4a atom10.
Scheme 1.
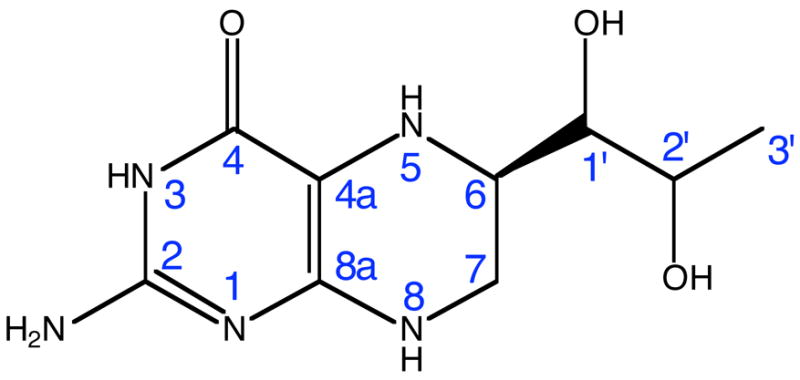
Tetrahydrobiopterin has redox functions in aromatic amino acid hydroxylase (AAH)11–13 and nitric oxide synthase (NOS)14,15. In the latter it binds close to P450 heme of NOS and is implicated in the timely transfer of one electron to heme in the first step of arginine oxidation to N-hydroxyarginine by NOS. This paper presents such electronic properties of BH4 such as the ionization potential (IP) and electron affinity (EA) calculated in gas phase, solution, and protein environment, casting light on the possible scenarios of electron transfer from BH4 to P450 heme of NOS.
Theoretical Calculations
Quantum mechanical calculations
The optimized geometry and vibrational frequencies of neutral, cation and anion forms of BH4have been calculated by quantum mechanics using the density functional theory method (B3LYP Hamiltonian: Becke3 exchange16 and Lee-Yang-Parr correlation17 functionals) with the 6-31+G(d,p) basis set. Single point calculations have also been performed with Møller-Plesset second order perturbation theory18–23 with the 6-31+G(d,p) basis set and B3LYP hybrid method using the 6-311+G(d,p) and 6-311++G(d,p) basis sets. The electrostatic potential-derived (ESP) charges for atoms in neutral BH4 (used in deriving OPLS parameters24) were obtained from the wave function of the optimized-geometry using the Merz-Kollman25 method as implemented in the Gaussian98 program26. The IP and EA of BH4 in gas phase, dielectric27, explicit water (TIP4P28) and protein environment (NOS) were calculated at B3LYP/6-31+G(d,p) level. Graphical representations of the highest occupied molecular orbital (HOMO), singly occupied molecular orbital (SOMO), and lowest unoccupied molecular orbital (LUMO) data in CUBE files, in addition to spin densities, were created using the VMD program29 and the POV-Ray visualization program30.
Force field parametrization
In order to perform energy minimization of BH4 in water and protein environment (NOS) we have developed OPLS parameters24 for the following species: neutral BH4, P450 heme and [Zn(Cysteine)4] 2-complex species. The molecular mechanics optimized geometry and vibrational frequencies of neutral BH4, methylthiolate dioxy-iron (Fe2+) porphyrin complex and the tetramethylthiolate-zinc complex [Zn(MeS)4]2- were calculated using the OPLS force field24 as implemented in the molecular dynamics simulation program Gromacs31. The OPLS force field parameters used for atom types of BH4, P450 heme, and [Zn(Cysteine)4]2- complex have been derived in the following way: (a) OPLS atom types similar to those found in these species were first identified and assigned guess parameters; (b) the geometrical parameters (equilibrium bond lengths, bond angles, Ryckaert-Bellemans coefficients32, and improper dihedral angles) were adjusted using a genetic algorithm (GA) optimization program33 such that the root mean square deviation (RMSD) in the internal coordinates of the quantum and molecular mechanics calculated structures is minimum; (c) the OPLS force constants for bond stretching and angle bending were adjusted (using the GA) such that the RMSDs between the vibrational frequencies of BH4, methylthiolate dioxy-iron (Fe2+) porphyrin and the tetramethyltiolate-zinc complexes calculated by quantum mechanics and molecular mechanics methods were less than the RMSDs calculated for a few amino acids with OPLS parameters (defined in the OPLS force field implemented in the Gromacs program31).
Molecular mechanics calculations
The Protein Data Bank structure 1NSI34 was used to obtain an energy-minimized structure of the solvated iNOSoxy dimer. The structure was prepared as follows: (a) one oxygen molecule was bound to Fe2+ of heme of each monomer by modifying the PDB file (the first electron transfer reduces Fe3+ to Fe2+, which allows oxygen to bind to the iron ion, and the second electron transfer is facilitated by BH4); (b) hydrogen atoms were added and protonation states were assigned (corresponding to pH=7) using the Gromacs utility pdb2gmx. In this particular configuration of 1NSI, there are 3 charged amino acids within 0.5 nm of BH4 (chain A): Arg199A, Arg381A and Glu479B. It seems that among these charged amino acid residues only Arg381A interacts significantly with BH4 (Arg381A makes a H-bond (0.205 nm) with O4 of BH4, which is 0.242 nm in the crystal structure). Because the crystal structure (1NSI) indicates that the C-NH2 bonds of the guanidinium moiety of Arg381A are equal, we considered Arg381 (in both monomers) to be protonated (charge +1 e.u.). Following the same argument we also considered Arg199 to be protonated. Note that this residue together with Trp463 and BH4 stack on top of each other like a sandwich (with Trp463 in between) and it is reasonable to believe that the positive charge of Arg199, which is delocalized on the guanidinium moiety, involves this residue in the π-stacking interaction between Trp463 and BH4. The residue Glu479B, which is 0.466 nm (0.545 nm in the crystal structure) far from the hydroxy tail of BH4 (docked in chain A), does not interact with the cofactor and was considered to be deprotonated. There is no experimental evidence that these residues should have a different protonation state than the one assigned in this study. (c) The iNOSoxy structure was solvated in a box of 19529 water molecules (TIP4P); (d) eight Na+ ions were added at random positions in the solvent to neutralize the negative charge of the dimer. The potential energy of the solvated iNOSoxy was minimized using the OPLS force field24 implemented in Gromacs31.
Results and Discussion
The geometry and the electronic structure of neutral, cation and anion BH4 Geometry
Figure 1 shows the quantum mechanically optimized geometry of BH4. This figure also displays a comparison between the bond lengths of the neutral (black), cation (red) and anion (blue) forms of BH4 in the pyrimidine (aromatic) and tetrahydropyrazine rings. One interesting structural detail of neutral BH4 is the non-equivalence of the N5 and N8 nitrogen atoms (Scheme 1). The atom N5 is hybridized sp3 (C4A-N5 bond length is .1417 nm, C4A-N5-H bond angle is 108.9º and C4-C4A-N5-H dihedral angle is −25.3º), while atom N8 is hybridized sp2 (C8A-N8 bond length is .1365 nm, C8A-N8-H bond angle is 116.5º and N1-C8A-N8-H dihedral angle is −7.3º). This structural aspect is important because it was hypothesized that in the BH4 cation the spin density of the unpaired electron is mainly localized at N5.35 Table 1 shows that the geometry of neutral BH4 calculated at B3LYP/6-31+G(d,p) level is in good agreement with the structure of BH4 (without H atoms) bound to chain A of the iNOSoxy (PDB id: 1NSI) dimer. The root mean square deviation of the bond lengths given in Table 1 is 0.0026 nm and the RMSDs for bond and dihedral angles are 2.4º and 3.9º, respectively. Figure 1 shows one noticeable aspect of the difference in structure between the neutral, cation and anion forms of BH4.The alternation of bond lengths involving C4A, C8A, N5 atoms is reversed in the cation as compared with the neutral and anion BH4: C4A-N5 is .1417 in neutral and .1425 in anion versus .1340 nm in cation, while C4A-C8A is .1386 in neutral and .1387 in anion versus .1431 nm in cation. Table 2 shows a comparison of a subset of bond and dihedral angles (mostly defining the pyrimidine and tetrahydropyrazine rings). As in the case of bond lengths, a substantial change in bond angle (8.2º) is observed between the cation and neutral BH4 and involves the N5 atom (e.g. the C4A-N5-C6 angle is 113.8º (neutral), 115.0º (anion), and122.0º (cation)). Overall, the anion geometry is closer to the geometry of neutral BH4 than the cation geometry is. This may be one of the reasons that the electron affinity (EA) of BH4 in gas phase is less than 1 eV, while the ionization potential is around 6 eV (vide infra).
Figure 1.
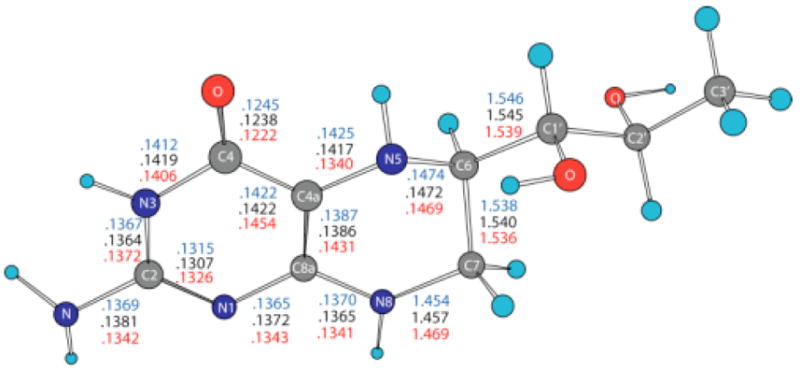
Bond lengths (nm) in BH4 neutral (black), cation (red) and anion (blue). The geometries have been optimized in gas phase using density functional theory (B3LYP/6-31+G(d)).
Table 1.
Comparison between the X-Ray Crystal Structure of Tetrahydrobiopterin bound to Nitric Oxide Synthase, and the Structure Calculated by Density Functional Theory and Molecular Mechanics (using the OPLS Force Field).
| Internal coordinate | Geometry | ||||
|---|---|---|---|---|---|
| QMa | Crystalb | DQM-Cc | M Md | DQM-MMe | |
| Bond lengthf | |||||
| N1-C2 | .1307 | .1346 | −.0039 | .1322 | −.0015 |
| N1-C8A | .1372 | .1371 | .0001 | .1389 | −.0017 |
| C2-N3 | .1364 | .1352 | .0012 | .1349 | .0015 |
| N3-C4 | .1419 | .1402 | .0017 | .1403 | .0016 |
| C4-C4A | .1422 | .1417 | .0005 | .1420 | .0002 |
| C4A-N5 | .1417 | .1448 | −.0031 | .1408 | .0009 |
| C8A-N8 | .1365 | .1333 | .0032 | .1382 | −.0017 |
| N5-H | .1019 | - | - | .1023 | −.0004 |
| N5-C6 | .1472 | .1514 | −.0042 | .1480 | −.0008 |
| C6-C7 | .1540 | .1509 | .0031 | .1556 | −.0016 |
| C7-N8 | .1457 | .1455 | .0002 | .1468 | −.0011 |
| N8-H | .1009 | - | - | .1013 | −.0004 |
| RMSDg | .0026 | .0012 | |||
| MAXh | .0042 | .0017 | |||
| Bond anglei | |||||
| C4-C4A-N5 | 119.4 | 117.6 | 1.8 | 118.5 | .9 |
| C4A-N5-C6 | 115.5 | 111.0 | 4.5 | 116.0 | −.5 |
| C4A-N5-H | 108.9 | - | - | 110.1 | −1.2 |
| C6-N5-H | 113.9 | - | - | 113.6 | .3 |
| N5-C6-C7 | 108.6 | 108.9 | −.3 | 107.7 | .9 |
| C6-C7-N8 | 110.5 | 111.7 | −1.2 | 109.9 | .6 |
| C8A-N8-C7 | 120.7 | 119.9 | .8 | 119.8 | .9 |
| C8A-N8-H | 116.5 | - | - | 117.2 | −.7 |
| C7-N8-H | 120.9 | - | - | 120.3 | .6 |
| RMSD | 2.4 | .9 | |||
| MAX | 4.5 | 1.2 | |||
| Dihedral anglei | |||||
| C4-C4A-N5-H | −25.3 | - | - | −22.4 | −2.9 |
| N1-C8A-N8-H | −7.3 | - | - | −9.7 | 2.4 |
| N5-C4A-C8A-N8 | −3.4 | −1.9 | 1.3 | −1.1 | 2.2 |
| C8A-C4A-N5-C6 | 24.9 | 29.0 | −4.1 | 23.1 | 1.8 |
| C4A-C8A-N8-C7 | 10.7 | 1.6 | 9.1 | 9.7 | 1.0 |
| C4A-C8A-N8-H | 175.1 | - | - | 171.4 | 3.7 |
| C4A-N5-C6-C7 | −49.0 | −53.8 | 4.8 | −48.8 | −.2 |
| C4A-N5-C6-H | 70.7 | - | - | 69.5 | 1.2 |
| N5-C6-C7-N8 | 53.6 | 53.8 | −.2 | 54.5 | −.9 |
| C6-C7-N8-C8A | −36.1 | −28.6 | −7.5 | −37.1 | 1.0 |
| C6-C7-N8-H | 160.0 | - | - | 161.9 | −1.9 |
| RMSD | 3.9 | 4.3
3.2j |
|||
| MAX | 9.1 | 10.2
6.4j |
|||
The structure of BH4 obtained by calculation using the density functional theory (B3LYP/6-31+G(d,p)) with the Gaussian98 program 26.
The structure of BH4 used in this comparison was extracted from the crystal structure of iNOSoxy dimer (1NSI)34.
Difference between the same internal coordinate measured in the quantum mechanically calculated (DFT) structure and the crystal structure of BH4.
The structure of BH4 calculated with the molecular mechanics program Gromacs 31 using the OPLS force field 41 derived by the GA method.
Difference between the same internal coordinate measured in the DFT structure and the molecular mechanics optimized structure of BH4.
in nm.
Root-mean square deviation for all bond lengths in BH4.
Maximum difference.
in degree.
Calculated without considering the dihedral angles that contain at least one hydrogen atom.
Table 2.
Bond and Dihedral Angles in the Optimized Gas Phase Geometriesa of the Neutral, Cation and Anion Forms of Tertrahydrobiopterin.
| Neutral | Cation | N-Cb | Anion | N-Ac | |
|---|---|---|---|---|---|
| Bond angle c | |||||
| N1-C8a-C4a | 123.7 | 122.6 | 1.1 | 124.2 | −0.5 |
| C4-C4a-C8a | 119.3 | 119.4 | 0.1 | 119.2 | 0.1 |
| C4a-C8a-N8 | 119.6 | 117.8 | 1.8 | 119.4 | 0.2 |
| N5-C4a-C8a | 120.8 | 121.3 | −0.5 | 121.6 | −0.8 |
| C4a-N5-C6 | 113.8 | 122.0 | −8.2 | 115.0 | −1.2 |
| N5-C6-C7 | 108.4 | 108.7 | −0.3 | 108.6 | −0.2 |
| N5-C6-C1′ | 106.6 | 109.2 | −2.6 | 106.4 | 0.2 |
| C6-C7-N8 | 110.8 | 110.4 | 0.4 | 110.4 | 0.4 |
| C7-N8-C8a | 122.3 | 121.8 | 0.5 | 121.0 | 1.3 |
| Dihedral angled | |||||
| C4-C4a-N5-C6 | −148.4 | −170.8 | 22.4 | −153.6 | 5.2 |
| C4a-N5-C6-C7 | −53.9 | −34.7 | −19.2 | −48.6 | −5.3 |
| C4a-C8a-N8-C7 | 0.3 | 12.9 | −12.6 | 10.8 | −10.5 |
| C4a-N5-C6-C1′ | −175.8 | −159.0 | −16.8 | −170.3 | −5.5 |
| N5-C6-C7-N8 | 50.6 | 47.0 | 3.6 | 53.5 | −2.9 |
| C6-C7-N8-C8a | −25.7 | −39.8 | 14.1 | −36.4 | 10.7 |
calculated at B3LYP/6-31+G(d,p) level of density functional theory
neutral – cation
neutral – anion
In degree (°).
Charges and spin density
Electrostatic potential-derived (ESP) charges have been calculated using the ChelpG36 and Merz-Kollman (MK)25 methods as implemented in the Gaussian98 program26. The ChelpG-derived charges (not shown) are slightly smaller than the MK-derived charges in the pyrimidine ring of BH4, but are larger in the tetrahydropyrazine ring of BH4. The partial charge of N 5 (ChelpG: - 0.4974 e.u.; MK: −0.4958 e.u.) is about half the partial charge of N8 (ChelpG: −0.8119 e.u.; MK: −0.8089 e.u.). Figure 2 shows MK charges for the neutral (black), cation (red), and anion (blue) forms of BH4. The C4A, N5 and N8 atoms are negatively charged (-.3213, -.4974 and -.8119 e.u., respectively) in neutral BH4. Removing one electron from BH4 makes these atoms less negative (-.1622, -.2462 and -.5854 e.u., respectively), but adding one electron causes a small change in the partial charges of these atoms (-.3645, -.4835 and -.8947 e.u., respectively). On the other hand, the partial charges on the C6 and C7 atoms (in neutral BH4: .1300 and .3712 e.u., respectively) affect the anion more (.5432 and .8039 e.u., respectively) than in the cation (.1395 and .1335 e.u., respectively). It should be noted that the charge of N3 atom is less negative in the anion (-.4850 e.u.) compared to the neutral and cation forms (-.7400 and -.7226 e.u., respectively). The semi-occupied molecular orbital (SOMO) of the anion is mostly localized on the side of the pyrimidine ring containing the N3 atom and NH2 group (vide infra), whereas the virtual SOMO is mostly localized near the hydroxy tail of BH4. Figure 3 gives the spin densities for the cation (red) and anion (blue). In the case of the cation, more than half of the spin density is localized on the C4A and N5 atoms, which supports an earlier suggestion that much of the spin density is localized on atom N5. The results for the anion are more difficult to rationalize since the spin densities oscillate from positive to negative values. However, a graphical representation of the spin density for the anion (vide infra) indicates that it is mostly localized on the side of the pyrimidine ring containing the N3 atom andthe NH2 group.
Figure 2.
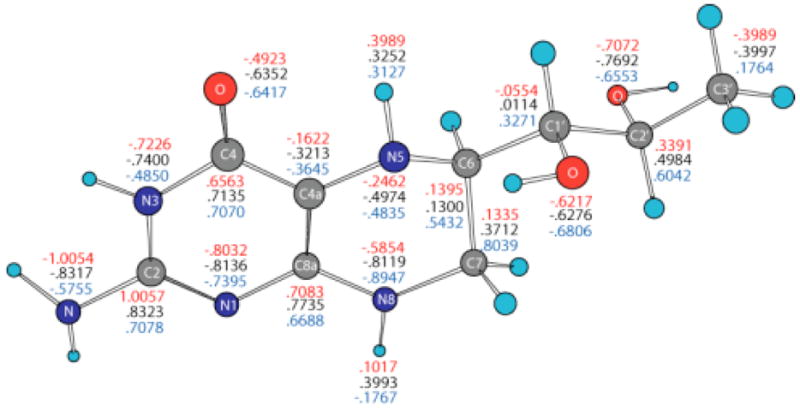
Merz-Kollman electrostatic potential-derived partial atomic charges for atoms in BH4 neutral (black), cation (red) and anion (blue) calculated by density functional theory (B3LYP/6-31+G(d)) in gas phase.
Figure 3.
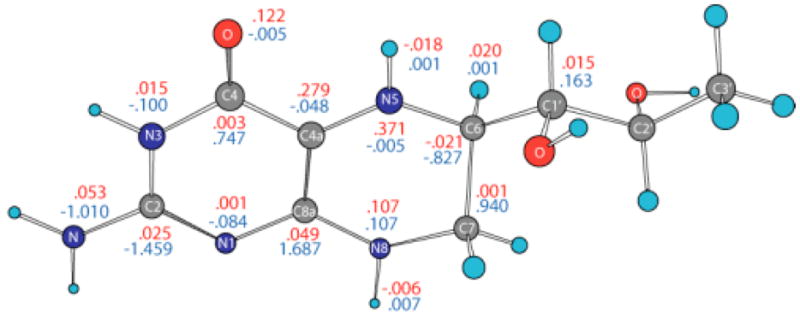
Spin densities for BH4 cation (red) and anion (blue) calculated by density functional theory (B3LYP/6-31+G(d)) in gas phase.
OPLS force field for BH4, P450 Heme, and Zn(Cys)4complex
The quality of the OPLS force field parameters (for BH4 atom types) was assessed by comparing the optimized geometry of BH4 calculated by quantum mechanics and molecular mechanics methods, and the geometry of BH4, extracted from the crystal structure of the oxygenase domain from inducible nitric oxide synthase (iNOSoxy, PDB id: 1NSI). The discussion here will be focused on the geometry of the pyrimidine and tetrahydropyrazine rings of BH4. Table 1 shows the comparison of the quantum mechanical, molecular mechanical and crystal structures using a subset of the internal coordinates of the atoms that compose these two rings. The difference between the quantum mechanical structure, on the one hand, and the crystal and molecular mechanical structures, on the other hand, are expressed as RMSDs for bond lengths, bond angles and dihedral angles, respectively. The root mean square deviation between the QM and MM structures (QM-MM) for bond lengths is 0.0012 nm. For bond angles the QM-MM is 0.9º, and for dihedral angles the QM-MM RMSD is 4.3º. The QM-MM RMSD decreases to 3.2º when the dihedral angles that include hydrogen atoms are excluded (the crystal structure has no hydrogen atoms). Table 1 shows that the MM optimized geometry is very close to the QM optimized and crystal structure geometries.
To produce realistic molecular dynamics (MD) simulations it is essential that the derived force field parameters correctly reproduce not only the equilibrium geometry, but also the dynamics of the molecule (i.e. the normal modes of vibration). The OPLS parameters that modulate the MM-calculated vibrational frequencies are the force constants for bond stretching, angle bending and dihedral angle torsion. By comparing the QM- and MM-derived vibrational frequencies we assess the quality of theseforce constants. The root mean square deviation of 90 normal modes of vibration for BH4 is 38 cm−1 withthe largest difference being 131 cm−1 for normal mode 75 (see table in the Supporting Information). Table 3 gives a comparison between the QM-MM RMSD of vibrational frequencies calculated for BH4, glycine (94 cm−1) and tyrosine. (65 cm−1). Table 3 shows that the force field derived in this work for BH4 is of better quality than the average OPLS parameters used for amino acids. In addition to the BH4 cofactor, iNOSoxy dimer contains a P450 heme cofactor and a Zn-cysteine bridge between the two monomers37. In order to perform an energy minimization of the iNOSoxy dimer in water we derived OPLS parameters for P450 heme cofactor33 and a [Zn(Cys)4]2- complex38. Table 3 and 4 list the RMSDs for geometrical parameters (Table 4) as well as for vibrational frequencies (Table 3) for these two moieties.
Table 3.
Root mean square deviation (RMSD) of Vibrational Frequencies for Tetrahydrobiopterin, Dioxy-Iron Porphirin Methylthiolate Complex, Tetramethylthiolate Zn Complex, Glycine and Tyrosine.
The vibrational modes were calculated with the OPLS force field obtained by the GA method and implemented in Gromacs program31.
Root mean square deviation in cm−
Dioxy-iron porphyrin methylthiolate complex.
Tetramethylthiolate Zn complex.
Table 4.
Root mean square deviation (RMSD) between the Quantum Mechanical and the Molecular Mechanical Optimized Structures of Tetrahydrobiopterin, Dioxy-Iron Porphirin Methylthiolate Complex, and Tetramethylthiolate Zn Complex.
| Compound | RMSD a | ||
|---|---|---|---|
| Bond length [nm] | Bond angle [º] | Dihedral angle [°] | |
| BH4 | 0.0013
0.0012 |
0.89
1.57 |
20.6
15.0 |
| O2-Fe-PP-SMe b | 0.0010
0.0010 |
0.98
0.98 |
2.15
2.15 |
| [Zn(MeS)4] 2- c | 0.0036
0.0036 |
4.62
4.62 |
23.20
23.20 |
First row: RMSD for parameterized bond lengths, bond angles and dihedral angles; second row: total RMSD.
Dioxy-iron porphyrin methylthiolate complex.
Tetramethylthiolate Zn complex.
The IP and EA of BH4 in gas phase, solution and protein environment
The IP and EA of BH4 in different chemical environments (e.g. vacuum, solution, protein environment) are probably the most relevant electronic properties for understanding the involvement of BH4 in the electron transfer in NOS because, unlike in other enzymes, the cofactor in NOS is oxidized/reduced without changing protonation states1. Calculations show that the wave function of BH4 is highly polarizable and an external electric field shifts the frontier molecular orbitals (i.e. HOMO, LUMO, and SOMO for the cation and anion) and thus changes the IP and EA of BH4. We calculated the IP and EA of BH4 in gas phase, dielectric27 (dielectric constant 80.0), explicit water (TIP4P28) and protein environment (iNOSoxy) using the density functional theory at B3LYP/6-31+G(d,p) level.
Gas phase
In gas phase the neutral BH4 is more stable than both its cation and anion. Because there are less geometrical differences between the anion and neutral BH4 than between the cation and neutral BH4 (vide supra), one expects that the difference in energy between the anion and neutral BH4 to be smaller than the difference between the cation and neutral BH4. Table 5 lists the IP and EA of BH4 calculated in different chemical environments. The IP increases with the use of more sophisticated basis sets, e.g. from 6.09 eV (6-31+G(d,p)) to 6.77 eV (6-311+G(d,p) and 6-311++G(d,p)). The electron affinity, however, does not show a definite trend; e.g. −0.39 eV (6-31+G(d,p)), −0.44 eV (6-311+G(d,p)), −0.11 eV (6-311++G(d,p)). MP2 calculations performed on BH4 using the 6-31+G(d,p) basis set yield 7.09 eV for the IP and −1.96 eV for EA of BH4. It is worthy of note that the IP and EA change only slightly with the change in the conformation of BH4 and that they are mostly susceptible to the action of an external electric field. Thus, when the calculation is performed on the geometry of BH4 in solution, or on the geometry of BH4 in protein environment without the external electric field due to point charges, the IP and EA change from 6.09 and −0.39 eV in gas phase, to 6.60 and −0.42 eV in water, to 6.37 and −0.43 eV for BH4 bound to chain A, and 6.47 and −0.44 eV for BH4 bound to chain B of iNOSoxy dimer. Figure 4 shows graphical representations of the HOMO and LUMO orbitals for the neutral BH4 and of HOMO, SOMO and LUMO orbitals of the BH4 cation and anion. The pictures were obtained by visualizing CUBE files (obtained with Gaussian9826) with the VMD29 and POV-Ray30 programs. Figure 4 shows that the HOMO of neutral BH4 is localized on the two rings, while the LUMO is mostly localized near the pyrimidine ring (close to the N3 atom and NH2 group). The SOMO (occupied) of the anion is similar to the LUMO of the neutral BH4 (localized mostly in the same region), though the virtual SOMO is mainly localized near the hydroxy tail of BH4. The localization of the occupied SOMO of the anion confirms that the unpaired electron is basically unbound in gas phase (the EA is negative). On the other hand, the occupied SOMO of the cation spreads over the entire molecule, whereas the virtual SOMO is mostly localized on the two rings of BH4 and is very similar to the HOMO of the neutral BH4. Another distinctive feature of these frontier orbitals is that the LUMO (for α and β electrons) of the cation are very different from the LUMO of the neutral species and SOMO of the anion (Figure 4). This observation is valid for all chemical environments of BH4 used in this study (vide infra).
Table 5.
Ionization Potential and Electron Affinitya of (6R)-5,6,7,8-Tetrahydrobiopterin in Gas Phase, Dielectric, Water and Protein Environment.
| Environment | Energyb |
IPc | EAc | ||
|---|---|---|---|---|---|
| Neutral | Cation | Anion | |||
| Gas phase | −851.509486 | −851.285676 | −851.495086 | 6.09 | −0.39 |
| −851.7029332d | −851.4540438 | −851.6868365 | 6.77 | −0.44 | |
| −851.7033758e | −851.4545306 | −851.6993969 | 6.77 | −0.11 | |
| −849.083620f | −848.825127 | −849.011603 | 7.09 | −1.96 | |
| −851.467803g | −851.225248 | −851.452508 | 6.60 | −0.42 | |
| −851.475396h | −851.241385 | −851.459583 | 6.37 | −0.43 | |
| −851.478942i | −851.241096 | −851.4626326 | 6.47 | −0.44 | |
|
| |||||
| Waterj | −1195.536599 | −1195.2724002 | −1195.523429 | 7.19 | −0.36 |
|
| |||||
| Dielectrick | −851.556900 | −851.379800 | −851.587815 | 4.82 | 0.84 |
|
| |||||
| Proteinl | −12441.231311 | −12441.072356 | −12441.139355 | 4.33 | −2.50 |
| −12441.221713 | −12441.096451 | −12441.099584 | 3.41 | −3.32 | |
Calculations performed by density functional theory at B3LYP/6-31+G(d,p) level unless otherwise specified. All calculations were performed with Gaussian98 program 26.
In Hartree.
In eV.
B3LYP/6-311+G(d,p) level.
B3LYP/6-311++G(d,p) level.
MP2/6-31+G(d,p) level.
Calculation on the optimized geometry of BH4 in solution without the external electric field generated by the surrounding water.
Calculation on the optimized geometry of BH4 in protein environment (chain A of iNOSoxy dimer) without the external electric field generated by the surrounding amino acids, water molecules and P450 heme.
Calculation on the optimized geometry of BH4 in protein environment (chain B of iNOSoxy dimer) without the external electric field generated by the surrounding amino acids, water molecules and P450 heme.
Calculation on the optimized solution geometry of BH4 with external electric field generated by point charges representing TIP4P water molecules. The cofactor’s molecular mechanics force field is described by OPLS parameters 24 obtained as described in the Methodology section. The optimization was performed on BH4 immersed in a box of 603 water (TIP4P) molecules.
Calculation on the gas phase optimized geometry of BH4 with the external electric field generated by a polarizable dielectric (dielectric constant = 80).
Calculation on the optimized geometry of BH4 in protein environment (iNOSoxy) with external electric field generated by the point charges representing surrounding amino acids, water molecules and P450 heme. The molecular mechanics force fields for BH4, P450 heme and [Zn(Cys)4]2− complex are described by OPLS parameters 24 obtained as described in the Methodology section. The optimization was performed on the iNOSoxy dimer immersed in a box of 19529 water molecules (TIP4P) and 8 counter ions (Na+). First row: data for BH4 bound at chain A of the iNoxy dimer; second raw: data for BH4 bound to chain B.
Figure 4.


HOMO and LUMO orbitals for BH4 neutral (N), HOMO SOMO, and LUMO orbitals for the BH4 cation (C) and anion (A) calculated by density functional theory (B3LYP/6-31+G(d)) in gas phase with Gaussian 98. The MO pictures have been obtained from CUBE Gaussian98 files and displayed with the VMD and POV-Ray programs.
Solution
The IP and EA of BH4 have been calculated both in dielectric and explicit water (TIP4P). The calculation in dielectric (dielectric constant 80.0) gives the effect of an average electric field on the IP and EA due to water polarization, and the calculation in explicit water introduces anisotropy in the electric field due to specific positions of water molecules and the presence of hydrogen bonds. The calculations on BH4 in dielectric and explicit water show a substantial effect from the external electric field on the IP and EA of BH4. Calculation in dielectric gives the average effect of water polarization on the electronic structure of BH4. Thus the IP decreases to 4.82 eV, while the EA becomes positive (0.84 eV), which makes the anion more stable than neutral BH4 in dielectric. This result suggests that one electron reduction of BH4 in water should be easy, while one electron oxidation may not39,40. The calculation in explicit water (Table 5) shows that the IP (7.19 eV) and EA (−0.36 eV) values are closer to the vacuum values rather than the dielectric values. Figure 5 shows that neutral BH4 makes nine H-bonds with surrounding water molecules (in this particular energy minimized configuration). Five of these H-bonds are made with the BH4 tail, while the remaining four H-bonds are made with the NH2 group, N1, O4, H5 and H8 (Scheme 1). Figures 6 and 7 show pictures of HOMO and LUMO orbitals for the neutral, and HOMO, SOMO, and LUMO for the cation and anion in dielectric (Figure 6) and explicit water (Figure 7). One distinctive feature of the LUMO of neutral BH4 calculated in either dielectric or water is that it resembles the LUMO of the gas phase cation rather than the LUMO of the gas phase neutral BH4. There is little difference between the HOMO of neutral BH4 and the SOMO of the cation (dielectric and water), and the same holds for LUMO. However, the HOMO of the cation (explicit water) show different polarization of α and β electrons: the α electron is localized mainly in the hydroxy tail of BH4, while the β electron is localized on parts of the two rings of BH4 containing N1 and N8 atoms. In contrast, the SOMO of the anion in dielectric are very similar to the SOMO of gas phase BH4 anion (i.e. localized near/on the pyrimidine ring), while the SOMO in water are mostly localized in the tail of BH4, probably due to specific interactions with water molecules. The α and β electrons in LUMO of the anion have different polarization in water and similar polarization in dielectric. This result demonstrates the limitation on using dielectric calculations as a substitute for explicit solvent environment.
Figure 5.
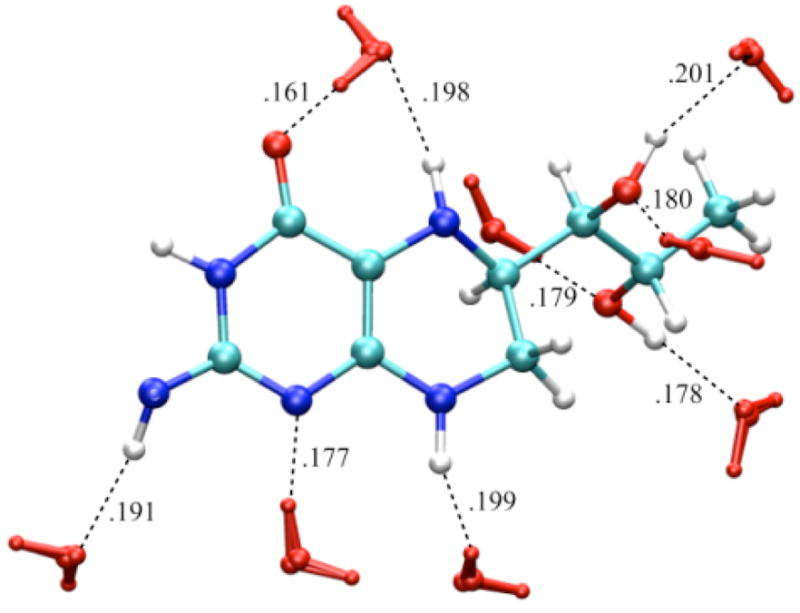
Water configuration (red) around neutral BH4 neutral: the water (TIP4P) box containing one molecule of BH4 (OPLS) was energy minimized using Gromacs program. There are nine H-bonds (black dashed lines) between BH4 and water within 0.2 nm.
Figure 6.


HOMO and LUMO orbitals for BH4 neutral (N); HOMO SOMO, and LUMO orbitals for the BH4 cation (C) and anion (A) calculated by density functional theory (B3LYP/6-31+G(d)) in dielectric (dielectric constant 80.0) with Gaussian 98. The MO pictures were obtained from CUBE Gaussian98 files and displayed with the VMD and POV-Ray programs.
Figure 7.


HOMO and LUMO orbitals for BH4 neutral (N), HOMO SOMO, and LUMO orbitals for the BH4 cation (C) and anion (A) calculated by density functional theory (B3LYP/6-31+G(d)) in water (TIP4P) with Gaussian 98. The MO pictures were obtained from CUBE Gaussian98 files and displayed with the VMD and POV-Ray programs.
Protein environment
We finally calculated the IP and EA of BH4 docked in the active site of chains A and B of iNOSoxy dimer. Figure 8 shows that in this particular configuration of the enzyme, there are slightly different chemical environments for BH4 docked to chain A (top pane) and chain B (bottom pane). One important feature of cofactor docking to NOS is that the carboxylic oxygen of one propionate group of heme makes bifurcated H-bonds (Figure 8) with H3 (0.192 nm in chain A and 0.188 nm in chain B) and one hydrogen atom of the NH2 group of BH4 (0.178 nm in chain A and 0.189 nm in chain B). Another distinctive feature is the sandwich arrangement (π-stacking) that BH4, Trp463, and Arg199 make together (vide supra). The distance between BH4 and Trp463 is 0.375 nm in chain A and 0.381 nm in chain B. The interactions of BH4 with heme, the π-stacking residues (Trp463 and Arg199), and Arg381 seem to be critical for the function of the cofactor. Tetrahydrobiopterin bound to chain A makes nine H-bonds (Figure 8, top pane): four H-bonds are made with water molecules, while the other five H-bonds are made with P450 hemeA, Ile462A, Trp463A and Ser118A. Tetrahydrobiopterin bound to chain B makes seven H-bonds (Figure 8, bottom pane); three H-bonds are made with water molecules and the remaining four H-bonds are made with P450 hemeB, Ile462B, Arg381B and Ser118B.
Figure 8.
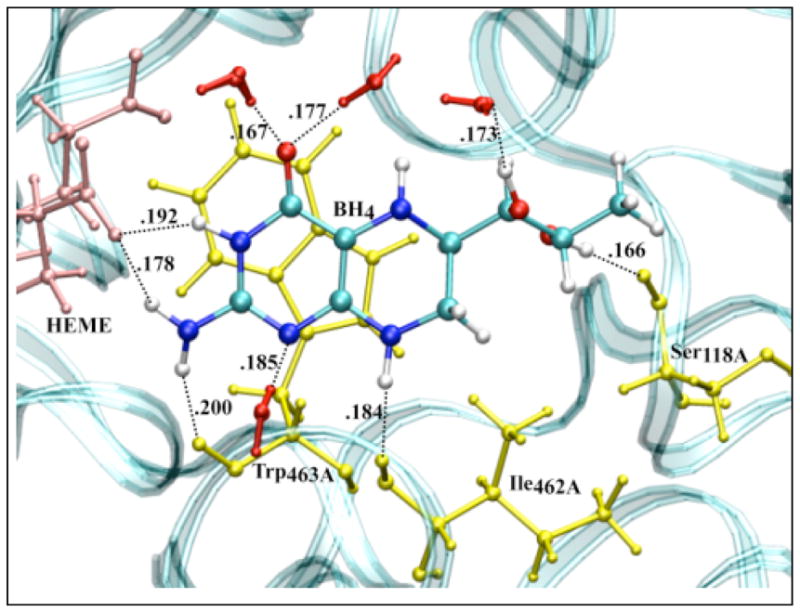
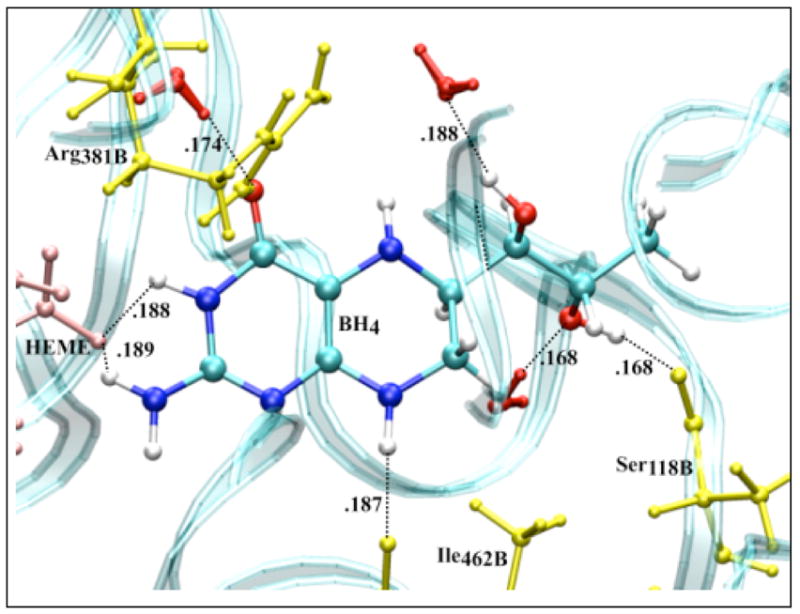
Amino acid residues (yellow), P450 heme (pink) and solvent (red) configuration around neutral BH4 bound to chain A (top) and chain B (bottom) of iNOSoxy dimer. The water (TIP4P) box containing a iNOSoxy dimer (OPLS) was energy minimized using Gromacs program. There are nine H-bonds (black dashed lines) between BH4 (chain A) and amino acid residues, heme and water within 0.2 nm, and seven H-bonds for BH4 bound to chain B.
Figure 9 shows pictures of the HOMO and LUMO orbitals of the neutral, and HOMO, SOMO, and LUMO orbitals of the cation and anion BH4 bound to chain A (left panes) and chain B (right panes). As in the case of BH4 solvated in water, the HOMO of the neutral BH4 and the SOMO of the cation are similar and spread over the two rings of BH4. In contrast, the occupied SOMO of the anion (slightly different for chains A and B, but similar with the LUMO of neutral BH4) are localized near the BH4 hydroxy tail, probably because of the presence of the heme propionate group in the vicinity of the pyrimidine ring of BH4. This should increase the electron repulsion if the unpaired electron is localized in this region; this interaction makes the anion even more unstable. On the other hand, the virtual SOMO (which are slightly different for chains A and B) are mainly localized on the pyrimidine ring. The α and β electrons in the LUMO of the anion have different polarization, as is the case of BH4 solvated in water.
Figure 9.




HOMO and LUMO orbitals for BH4 neutral (N), HOMO, SOMO and LUMO orbitals for the BH4 cation (C) and anion (A) from both iNOSoxy monomers (chains A and B), calculated by density functional theory (B3LYP/6-31+G(d)) in protein environment with Gaussian 98. The MO pictures have been obtained from CUBE Gaussian98 files and displayed with the VMD and POV-Ray programs.
Figure 10 shows the spin densities of the cation (left panes) and the anion (right panes) species of BH4 in gas phase, solvated in dielectric and explicit water, and bound to chain A and chain B of iNOSoxy dimer. While the cation’s spin densities are very similar in all environments, being mostly localized at the junction between the pyrimidine and tetrahydropyrazine rings (including atoms N5 and N8), the spin densities found for the anion are much different. For gas phase and dielectric the spin density is mostly localized on the side of the pyrimidine ring, whereas the spin density is localized in the tail for BH4 in explicit water and protein environment. Table 5 shows that the IP and EA of both BH4 cofactors (chains A and B) are about 2 eV (IP) and 2.5 eV (EA) lower, respectively, in protein environment than either in gas phase or explicit water. More importantly, there is a significant difference between the IP and EA of BH4 bound to chains A and B, even though their values in vacuum (calculated for the geometries in protein environment without the electric field of the point charges) differ little (Table 5). The ionization potential and electron affinity of BH4 bound to chain B are 0.92 eV and 0.82 eV lower, respectively, than those calculated for BH4 bound to chain A. Figure 11 (top pane) shows that charged amino acid residues of iNOSoxy dimer make a multipole structure around BH4 with a total charge (chain A BH4 magenta curve, chain B BH4 cyan curve) that varies dramatically as a function of the thickness of the layer. The multipole structure for a particular layer (hypothetical) which can have a net charge, demonstrates a fluctuation in the IP (chain A BH4 black curve, chain B BH4 red curve) and EA (chain A BH4 green curve, chain B BH4 blue curve) as depicted in the bottom pane of Figure 11. This feature suggests that protein dynamics can dramatically alter the configuration of this multipole structure and thus substantially change the IP and EA of BH4. Table 5 shows that IP of BH4 in protein environment is about half of its value in gas phase and its EA is about five times smaller, which indicates that ionization becomes more likely and reduction less likely, in protein environment than in gas phase. We argue that during protein movement the multipole structure made of layers of charged residues around BH4 can drop the IP further such that BH4 could donate an electron to heme via the propionate bridge. For the anion to mediate the electron transfer it needs to acquire an electron from the reductase part of NOS before transferring it to heme. If BH4 has to act as a molecular switch and deliver an electron to heme in a timely manner, it is less likely that the electron transfer would depend on another molecular event (i.e. the delivery of one electron from the reductase part of NOS to BH4), which should be much slower. It should be also noted that the negatively charged propionate localizes the SOMO of BH4 anion in its tail, that is, on the opposite side of the cofactor.
Figure 10.


Spin densities for the BH4 cation and anion calculated by density functional theory (B3LYP/6-31+G(d)) in gas phase, dielectric, water and protein environment. The pictures have been obtained from CUBE Gaussian98 files and displayed with the VMD and POV-Ray programs.
Figure 11.
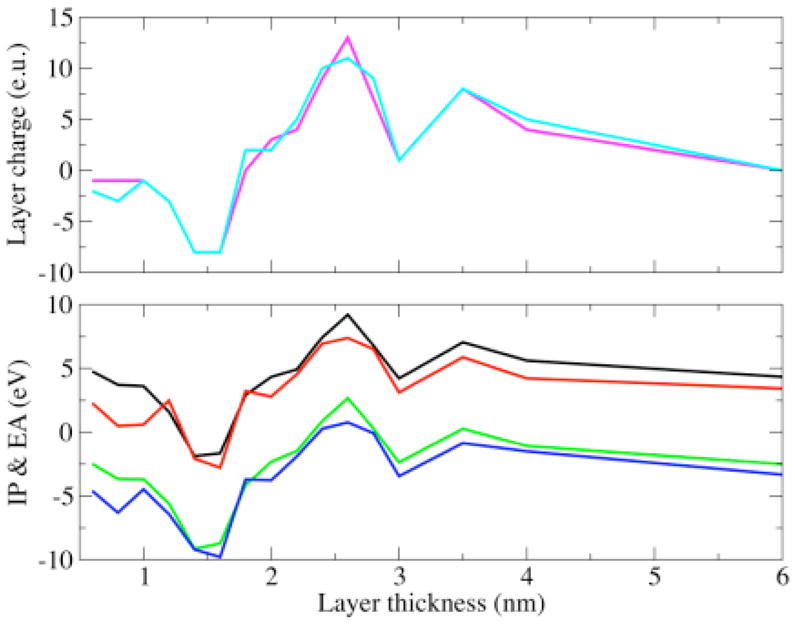
Bottom pane: hypothetical change in the IP and EA of BH4 due to the charge of a protein layer (of certain thickness) around BH4: IP of BH4 bound to chain A (black curve), IP of BH4 bound to chain B (red curve), EA of BH4 bound to chain A (green curve), EA of BH4 bound to chain B (blue curve), Top pane: Variation of the charge on the protein layer (of certain thickness) around BH4: total charge on the protein layer around BH4 bound to chain A (magenta curve), total charge on the protein layer around BH4 bound to chain B (cyan curve).
Conclusions
Our density functional theory calculations show that the electronic properties of BH4 such as IP and EA are not very sensitive to conformational changes rather they change dramatically in the presence of an external electric field. Calculations of the IP and EA show that, in gas phase, water, and in an energy-minimized protein environment, the neutral BH4 is the most stable species. The anion is more stable than the cation in all environments. An average polarization of water (provided in the form of a polarizable dielectric) makes the anion the most stable species in solution. The cation, however is still 4.8 eV less stable, making the one electron oxidation of BH4 in solution difficult. The specific docking of BH4 in iNOSoxy (i.e., in the vicinity of P450 heme with its pyrimidine ring next to a heme propionate group) may favor the cation as electron transfer mediator because the IP of BH4 is then about half of what it is in gas phase and the EA is about five times smaller. However, the susceptibility of the BH4 wave function to external electric field fluctuations generated by moving charged residues should allow protein dynamics to produce proper configurations in which the cation (or the anion) becomes nearly as stable as, or even more stable than the neutral BH4 and thus facilitating the electron transfer to heme. A paramagnetic species is observed during enzyme turnover, but there is no experimental procedure to distinguish whether an anion or a cation is involved in the electron transfer because both are radical species and produce the same electron spin resonance spectrum.
Supplementary Material
Table with calculated vibrational frequencies of tetrahydrobiopterin and Complete Reference 26. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the National Institutes of Health (grant number: 1R15GM070469-01), the Department of Energy (grant number: DE-FG02-03ER15462), the National Center for Supercomputer Applications (NCSA) at University of Illinois and the Ohio Supercomputer Center.
References
- 1.Wei CC, Crane BR, Stuehr DJ. Chem Rev. 2003;103:2365. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 2.Eberlein G, Bruice TC, Lazarus RA, Henrie R, Benkovic SJ. J Am Chem Soc. 1984;106:7916. [Google Scholar]
- 3.Davis MD, Kaufman S, Milstien S. Eur J Biochem. 1988;173:345. doi: 10.1111/j.1432-1033.1988.tb14004.x. [DOI] [PubMed] [Google Scholar]
- 4.Vasquez-Vivar J, Whitsett J, Martasek P, Hogg N, Kalyanaraman B. Free Radical Biol Med. 2001;31:975. doi: 10.1016/s0891-5849(01)00680-3. [DOI] [PubMed] [Google Scholar]
- 5.Milstien S, Katusic Z. Biochem Biophys Res Commun. 1999;263:681. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- 6.Komori Y, Hyun J, Chiang K, Fukuto JM. J Biochem (Tokyo) 1995;117:923. doi: 10.1093/oxfordjournals.jbchem.a124797. [DOI] [PubMed] [Google Scholar]
- 7.Toth M, Kukor Z, Valent S. Mol Hum Reprod. 2002;8:271. doi: 10.1093/molehr/8.3.271. [DOI] [PubMed] [Google Scholar]
- 8.Curtius HC, Heintel D, Ghisla S, Kuster T, Leimbacher W, Niederwieser A. Eur J Biochem. 1985;148:413. doi: 10.1111/j.1432-1033.1985.tb08855.x. [DOI] [PubMed] [Google Scholar]
- 9.Moad G, Luthy CL, Benkovic PA, Benkovic SJ. J Am Chem Soc. 1979;101:6068. [Google Scholar]
- 10.Lazarus RA, Wallick DE, Dietrich RF, Gottschall DW, Benkovic SJ, Gaffney BJ, Shiman R. Fed Proc. 1982;41:2605. [PubMed] [Google Scholar]
- 11.Fitzpatrick PF. Annu Rev Biochem. 1999;68:355. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 12.Flatmark T, Stevens RC. Chem Rev. 1999;99:2137. doi: 10.1021/cr980450y. [DOI] [PubMed] [Google Scholar]
- 13.Kappock TJ, Caradonna JP. Chem Rev. 1996;96:2659. doi: 10.1021/cr9402034. [DOI] [PubMed] [Google Scholar]
- 14.Gorren AC, Mayer B. Curr Drug Metab. 2002;3:133. doi: 10.2174/1389200024605154. [DOI] [PubMed] [Google Scholar]
- 15.Wei C, Wang Z, Meade A, McDonald J, Stuehr D. J Inorg Biochem. 2002;91:618. doi: 10.1016/s0162-0134(02)00432-4. [DOI] [PubMed] [Google Scholar]
- 16.Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 17.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 18.Moller C, Plesset MS. Phys Rev. 1934;46:618. [Google Scholar]
- 19.Head-Gordon M, Pople JA, Frisch MJ. Chem Phys Lett. 1988;153:503. [Google Scholar]
- 20.Frisch MJ, Head-Gordon M, Pople JA. Chem Phys Lett. 1990;166:275. [Google Scholar]
- 21.Frisch MJ, Head-Gordon M, Pople JA. Chem Phys Lett. 1990;166:281. [Google Scholar]
- 22.Head-Gordon M, Head-Gordon T. Chem Phys Lett. 1994;220:122. [Google Scholar]
- 23.Saebo S, Almlof J. Chem Phys Lett. 1989;154:83. [Google Scholar]
- 24.Jorgensen W, Tirado-Rives J Am Chem Soc. 1988;110:1657. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 25.Besler BH, Merz KMJ, Kollman PA. J Comput Chem. 1990;11:431. [Google Scholar]
- 26.Frisch MJ, et al. Gaussian 98. A.11. Gaussian Inc; Pittsburgh: 1998. [Google Scholar]
- 27.Barone V, Cossi M. J Phys Chem A. 1998;102:1995. [Google Scholar]
- 28.Jorgensen WL, Chandrasekhar J, Madura JD. J Chem Phys. 1983;79:926. [Google Scholar]
- 29.Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1966;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 30.Buck DK. Persitance of Vision Racetracer. 3.6. Persistence of Vision Pty. Ltd; Williamstown, Victoria, Australia: 2004. [Google Scholar]
- 31.Lindahl E, Hess B, van der Spoel D. J Mol Mod. 2001;7:306. [Google Scholar]
- 32.Ryckaert JP, Bellemans A. Far Disc Chem Soc. 1978;66:95. [Google Scholar]
- 33.Shy J, Gogonea V. OPLS Molecular Mechanics Force Field for P450 Heme. ACS Meeting-in-Miniature; 2005; Baldwin College, Cleveland, Ohio. [Google Scholar]
- 34.Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL. J Biol Chem. 1999;274:21276. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 35.Wei CC, Wang ZQ, Arvai AS, Hemann C, Hille R, Getzoff ED, Stuehr DJ. Biochemistry. 2003;42:1969. doi: 10.1021/bi026898h. [DOI] [PubMed] [Google Scholar]
- 36.Breneman CM, Wiberg KB. J Comp Chem. 1990;11:361. [Google Scholar]
- 37.Raman CS, Li H, Martasek P, Kral V, Masters BSS. Cell. 1998;95:939. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 38.Shy J, Gogonea V. OPLS Molecular Mechanics Force Field for [Zn(Cys)4]2- Complex [Google Scholar]
- 39.Gorren ACF, Kungl AJ, Schmidt K, Werner ER, Mayer B. Nitric Oxide. 2001;5:176. doi: 10.1006/niox.2001.0332. [DOI] [PubMed] [Google Scholar]
- 40.Boutros J, Bayachou M. Electrochem Comm. to be submitted. [Google Scholar]
- 41.Damm W, Frontera A, Tirado-Rieves J, Jorgensen W. J Comput Chem. 1997;18:1955. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table with calculated vibrational frequencies of tetrahydrobiopterin and Complete Reference 26. This material is available free of charge via the Internet at http://pubs.acs.org.