

The chamigrene subclass of sesquiterpenes, characterized by a spiro[5.5]undecane core, is an ever-growing family of natural products (Figure 1).1 Well over one hundred members have been isolated thus far, and many of these compounds exhibit a diverse array of biological activity.1c In particular, elatol (1),2 one of the most widely studied chamigrenes, displays antibiofouling activity,3a–b antibacterial activity (including human pathogenic bacteria),3c–e antifungal activity,3f and cytotoxicity against HeLa and Hep-2 human carcinoma cell lines.3g Despite the interesting bioactivity and compact structure of these molecules, no general strategy for their preparation has been developed, and, to the best of our knowledge, no total synthesis of elatol has been reported in the thirty-three years since its original isolation.4,5
Figure 1.
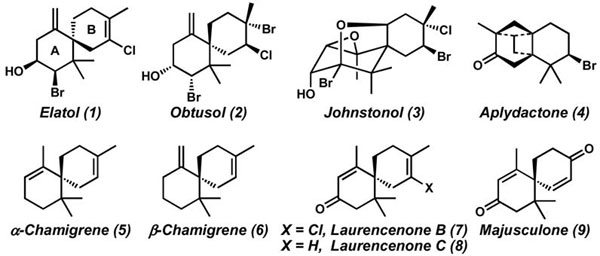
Examples of chamigrene natural products.
Structurally, elatol (1) consists of a densely functionalized A ring bearing three stereocenters, including an all-carbon quaternary stereocenter, which is vicinal to a second, non-stereogenic quaternary carbon. Within the B ring is also a fully substituted chlorinated olefin. We envisioned a strategy toward these challenging motifs based on methodological advances recently reported by our laboratories. Specifically, enantioselective decarboxylative allylation6 could generate the all-carbon quaternary stereocenter, while ring-closing metathesis (RCM)7 could be employed to concomitantly provide the tetrasubstituted olefin and the spirocyclic core of 1 (Scheme 1). Importantly, this approach serves as a general platform to access the chamigrene family.
Scheme 1.
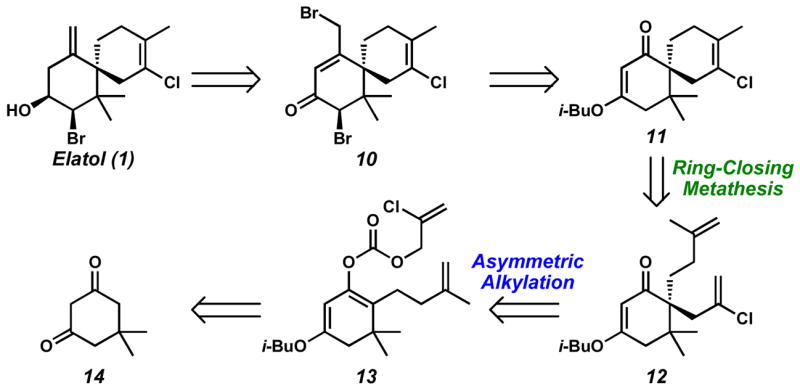
We envisioned 1 to ultimately arise from sequential reductive olefin transposition and diastereoselective reduction of α-bromoketone 10. In turn, compound 10 would be obtained from bromination of the enone resulting from 1,2-addition of a methyl anion to spirocycle 11. Intermediate 11 itself could be the product of RCM of α,ω-diene 12. Although generation of a fully substituted chlorinated olefin via RCM has not been previously reported,8 we anticipated that the improved reactivity of catalyst 227 (vide infra) might be sufficient for this transformation. Access to 12 would be possible via enantioselective decarboxylative allylation of an appropriately substituted vinylogous ester derivative (i.e., 13), employing the Pd(0) complex of a phosphinooxazoline (PHOX) ligand. This would constitute a previously unexplored substrate class with this catalyst system.9 Finally, enol carbonate 13 could be derived from commercially available dimedone (14).
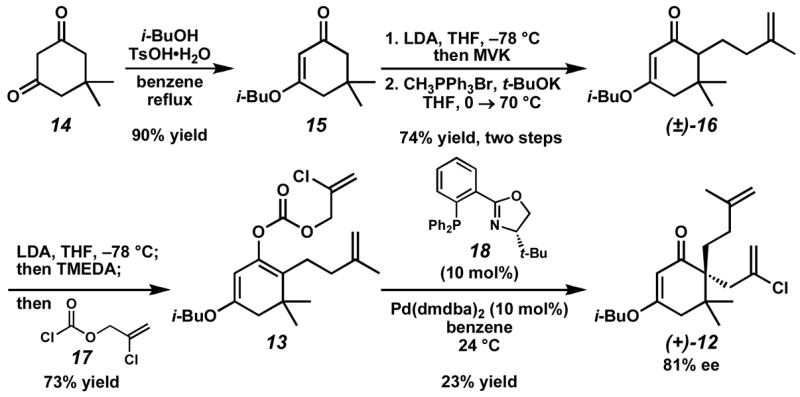
Our synthetic efforts began with the condensation of isobutyl alcohol and dimedone (14) to provide known vinylogous ester 15 (Scheme 2).10 Direct alkylation of vinylogous ester 15 with 4-iodo-2-methyl-1-butene was sluggish; however, a two-step procedure involving conjugate addition to methyl vinyl ketone (MVK) followed by Wittig methylenation afforded olefin (±)-16 in good yield. Selective enolization of vinylogous ester (±)-16 and trapping with chloroformate 17 allowed access to enol carbonate 13 in 73% yield. In our initial attempt, application of our standard reaction conditions11 for Pd-catalyzed asymmetric alkylation to enol carbonate 13 provided desired alkylation adduct (+)-12, but in low yield.12
Scheme 2.
a dmdba = bis(3,5-dimethoxybenzylidene)acetone.
We reasoned that the poor reactivity of enol carbonate 13 in the enantioselective allylation reaction could stem from one of three possibilities: (1) slow oxidative addition to the allyl carbonate moiety; (2) slow decarboxylation to reveal the active enolate intermediate; or (3) slow alkylation of the enolate intermediate to provide the desired product 12. In order to discern between these scenarios, we ran a set of control reactions outlined in Scheme 3. Exposure of enol carbonate 13 to conditions developed in our laboratories for enantioselective decarboxylative protonation13 led to rapid formation of olefin 16.14 Furthermore, removal of the 2-chloro substituent on the allyl fragment resulted in facile decarboxylative allylation of enol carbonate 19 to yield bis(olefin) 20. Based on these results, we concluded that slow alkylation, not slow oxidative addition or decarboxylation, was most likely the problematic step in this transformation.
Scheme 3.

a Conditions: (a) HCO2H, Pd(OAc)2 (10 mol%), 18 (12.5 mol%), MS 4Å, benzene, 40 °C, (b) Pd(dmdba)2 (10 mol%), 18 (13 mol%), benzene, 40 °C.
In order to enhance the reactivity of our π-allyl Pd(II) electrophile, we attempted to increase its electrophilicity by incorporating electron-withdrawing substituents into the PHOX ligand framework.15 Ultimately, asymmetric alkylation employing ligand 21 in benzene at 11 °C afforded the best balance between reactivity and selectivity, providing vinylogous ester 12 in 82% yield and 87% ee (Scheme 4). Gratifyingly, when α, ω-diene 12 was subjected to our standard RCM reaction conditions with catalyst 22, the desired fully substituted chloroalkene (+)-11 was produced in 97% yield.16 Addition of methyllithium in the presence of CeCl3 then provided (+)-laurencenone B ((+)-7)17 after acid-mediated elimination and hydrolysis.18 Enone (+)-7 was subsequently bis-halogenated with Br2 to generate d***** 10 in ≥8:1 dr.19 Finally, the crude α-bromoketone 10 was doubly reduced with DIBAL to afford elatol (1) (3.9:1 syn:anti,20 11:1 SN2′:SN2). Overall, enantioenriched (+)-laurencenone B ((+)-7) was prepared in seven steps and 34% yield from dimedone (14), while enantioenriched (+)-elatol (1) was prepared in nine steps and 11% yield.21
Scheme 4.

We have successfully developed a concise enantioselective route to the chamigrene natural product family, culminating in the first total syntheses of elatol (1) and (+)-laurencenone B ((+)-7), as well as the first preparation of a fully substituted chlorinated olefin via RCM. Moreover, we have demonstrated the ability of the key enantioselective alkylation reaction to access sterically-encumbered enantioenriched vinylogous esters. The application of these methods to the syntheses of other chamigrene natural products and a full exploration of both vinylogous esters in enantioselective decarboxylative alkylation and vinyl chlorides in RCM are the focus of ongoing studies.
Supplementary Material
Experimental details are available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank the NIH-NIGMS (R01 GM080269-01, postdoctoral fellowships to D.E.W. and I.C.S.), Abbott, Amgen, Bristol-Myers Squibb, Merck, and Caltech for generous funding; Materia, Inc. for their kind donation of catalyst 22 used in these studies; Professors Mercedes Cueto and Karen L. Erickson for their kind donation of natural samples of elatol (1); Brinton Seashore-Ludlow for experimental assistance; and Professor Peter B. Dervan and David M. Chenoweth for use of their HPLC.
References
- 1.(a) Erickson KL. Constituents of Laurencia. In: Scheuer PJ, editor. Marine Natural Products: Chemical and Biological Perspectives. V. Academic Press; New York: 1983. pp. 131–257. [Google Scholar]; (b) Dorta A, Díaz-Marrero AR, Cueto M, D’Croz L, Maté JL, Darias J. Tetrahedron Lett. 2004;45:7065–7068. [Google Scholar]
- 2.Originally isolated from the marine alga Laurencia elata: Sims JJ, Lin GHY, Wing RM. Tetrahedron Lett. 1974;15:3487–3490.
- 3.Granado I, Caballero P. Sci Mar. 1995;59(Supl 1):31–39.de Nys R, Leya T, Maximilien R, Afsar A, Nair PSR, Steinberg PD. Biofouling. 1996;10:213–224. doi: 10.1080/08927019609386281.Martín JD, Pérez C, Ravelo JL. J Am Chem Soc. 1986;108:7801–7811. doi: 10.1021/ja00284a052.Vairappan CS, Daitoh M, Suzuki M, Abe T, Masuda M. Phytochemistry. 2001;58:291–297. doi: 10.1016/s0031-9422(01)00243-6.Vairappan CS. Biomol Eng. 2003;20:255–259. doi: 10.1016/s1389-0344(03)00067-4.König GM, Wright AD. J Nat Prod. 1997;60:967–970. doi: 10.1021/np970181r.(g) HeLa: IC50 = 4.1 μM (lag phase), 1.3 μM (log phase); Hep-2: IC50 = 2.4 μM (lag phase), 2.0 μM (log phase); Dias T, Brito I, Moujir L, Paiz N, Darias J, Cueto M. J Nat Prod. 2005;68:1677–1679. doi: 10.1021/np050240y.
- 4.For the preparation of elatol (1) via the degradation of isoobtusol, see: González AG, Darias J, Díaz A, Fourneron JD, Martín JD, Pérez C. Tetrahedron Lett. 1976;17:3051–3054.González AG, Martín JD, Martín VS, Martínez-Ripoll M, Fayos J. Tetrahedron Lett. 1979;20:2717–2718.González AG, Martín JD, Martín VS, Norte M, Pérez R. Tetrahedron Lett. 1982;23:2395–2398.
- 5.For lead references on the total syntheses of other chamigrene natural products, see: Taber DF, Sikkander MI, Storck PH. J Org Chem. 2007;72:4098–4101. doi: 10.1021/jo070257g.Srikrishna A, Lakshmi BV, Mathews M. Tetrahedron Lett. 2006;47:2103–2106.Chen YJ, Wang CY, Lin WY. Tetrahedron. 1996;52:13181–13188.Hatsui T, Wang JJ, Takeshita H. Bull Chem Soc Jpn. 1994;67:2507–2513. Also see ref 3c.
- 6.(a) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 7.Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]
- 8.For the preparation of trisubstituted chloroalkenes via RCM, see: Chao W, Weinreb SM. Org Lett. 2003;5:2505–2507. doi: 10.1021/ol034775z.Chao W, Meketa ML, Weinreb SM. Synthesis. 2004:2058–2061.For the preparation of a fully substituted vinyl fluoride via RCM, see: Marhold M, Buer A, Hiemstra H, van Maarseveen JH, Haufe G. Tetrahedron Lett. 2004;45:57–60.
- 9.Trost has recently reported the use of vinylogous ester and thioester derivatives in enantioselective decarboxylative allylation using a chiral bis(phosphine)-Pd(0) complex: Trost BM, Bream RN, Xu J. Angew Chem, Int Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421.
- 10.House HO, Fischer WF., Jr J Org Chem. 1968;33:949–956. [Google Scholar]
- 11.Use of Pd2(dba)3 in lieu of Pd(dmdba)2 led to significantly less conversion.
- 12.For convenience, the enantioselective allylation reaction with 13 was optimized in the opposite enantiomeric series.
- 13.Mohr JT, Nishimata T, Behenna DC, Stoltz BM. J Am Chem Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]
- 14.A separate experiment revealed no reactivity in the absence of a Pd(0) catalyst.
- 15.For preliminary results on the rate acceleration of enantioselective decarboxylative allylation of enol carbonates with electron-deficient PHOX ligands, see: Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s.
- 16.(H2IMes)(PCy3)(Cl)2Ru=CHPh produced significant product for this transformation under similar reaction conditions (2.5 mol% catalyst, C6D6, 60 °C), but at a slower rate than catalyst 22: 85% conversion after 24 h as measured by 1H NMR.
- 17.(a) For the isolation of laurencenone B (7) from the marine alga Laurencia obtusa, see: Kennedy DJ, Selby IA, Thomson RH. Phytochemistry. 1988;27:1761–1766.Neither the absolute configuration nor the optical rotation was specified. (b) For the preparation of (+)-laurencenone B ((+)-7) via the degradation of elatol (1), see: Brennan MR, Erickson KL, Minott DA, Pascoe KO. Phytochemistry. 1987;26:1053–1057.
- 18.Discrepancies between the published 1H NMR data for the natural product (ref 17a) and that of the synthetic material exist. No 13C NMR data was available for comparison. 1H and 13C NMR data for semisynthetic (+)-laurencenone B ((+)-7) (ref 17b) matched that of our synthetic material. See the supporting information for a detailed comparison.
- 19.Purification of α-bromoketone 10 was hampered by its poor stability to silica gel and reverse phase HPLC.
-
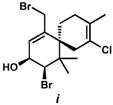
20.Determined by quenching the reaction at −78 °C in a separate run, resulting in the isolation a 3.9:1 mixture of alcohol diastereomers favoring i.
- 21.Synthetic (+)-elatol ((+)-1) was identical in all respects to a natural sample provided by Prof. Mercedes Cueto except for the magnitude of its optical rotation: [α]23D +92.09° (c 0.22, CHCl3, synthetic), [α]25D +109.78° (c 0.045, CHCl3, natural). Based on 87% ee, the expected [α]D value for the synthetic material would be +95.5°, which differed from the observed value by 3.6%.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details are available free of charge via the Internet at http://pubs.acs.org.