

Abstract
High throughput technologies, including array-based chromatin immunoprecipitation, have rapidly increased our knowledge of transcriptional maps—the identity and location of regulatory binding sites within genomes. Still, the full identification of sites, even in lower eukaryotes, remains largely incomplete. In this paper we develop a supervised learning approach to site identification using support vector machines (SVMs) to combine 26 different data types. A comparison with the standard approach to site identification using position specific scoring matrices (PSSMs) for a set of 104 Saccharomyces cerevisiae regulators indicates that our SVM-based target classification is more sensitive (73 vs. 20%) when specificity and positive predictive value are the same. We have applied our SVM classifier for each transcriptional regulator to all promoters in the yeast genome to obtain thousands of new targets, which are currently being analyzed and refined to limit the risk of classifier over-fitting. For the purpose of illustration we discuss several results, including biochemical pathway predictions for Gcn4 and Rap1. For both transcription factors SVM predictions match well with the known biology of control mechanisms, and possible new roles for these factors are suggested, such as a function for Rap1 in regulating fermentative growth. We also examine the promoter melting temperature curves for the targets of YJR060W, and show that targets of this TF have potentially unique physical properties which distinguish them from other genes. The SVM output automatically provides the means to rank dataset features to identify important biological elements. We use this property to rank classifying k-mers, thereby reconstructing known binding sites for several TFs, and to rank expression experiments, determining the conditions under which Fhl1, the factor responsible for expression of ribosomal protein genes, is active. We can see that targets of Fhl1 are differentially expressed in the chosen conditions as compared to the expression of average and negative set genes. SVM-based classifiers provide a robust framework for analysis of regulatory networks. Processing of classifier outputs can provide high quality predictions and biological insight into functions of particular transcription factors. Future work on this method will focus on increasing the accuracy and quality of predictions using feature reduction and clustering strategies. Since predictions have been made on only 104 TFs in yeast, new classifiers will be built for the remaining 100 factors which have available binding data.
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s11693-006-9003-3 and is accessible for authorized users.
Keywords: Transcription factor, SVM, Machine learning
Background
Understanding transcriptional regulation is one of the key challenges of the post-genomic era. Transcription factors control the expression of their target genes by binding specific sequences of bases, typically 10–15 nt in length, in a region upstream of transcription initiation. Sequences bound by a TF are not identical to each other and only represent a preferred pattern of nucleotides within a binding motif. The complete regulation of a gene will often depend on the co-operative or antagonistic effects of several transcription factors with potentially overlapping binding sites. Thus, the regulatory code for a gene is composed of a pattern of degenerate motifs concealed within the promoter.
Many methods for predicting additional target sites for a TF have been proposed. Founding work in TF binding site representation involved the use of position specific scoring matrices (PSSMs) (Stormo 2000; Workman and Stormo 2000; Schneider et al. 1986; Schneider and Stephens 1990), which contain the frequency of nucleotide bases at each position in a possible binding site, or motif. New predictions are sites which match the PSSM based on a score threshold (Stormo 2000). Supervised learning tools such as support vector machines (SVM) can be used to categorize new genes when given a set of genes known to be regulated by a certain factor and a set known not to be co-regulated. Unsupervised methods begin with less well-defined information, for example a set of genes from a microarray study which show similar expression over many experiments. Such genes could be hypothesized to be regulated by common factors and thus contain some set of common but unknown sequence patterns in their promoters. These patterns can then be discovered by statistical overrepresentation or by local search algorithms such as Gibbs sampling. Several unsupervised techniques for predicting binding sites have been reported (Conlon et al. 2003; Keles et al. 2004; Wang et al. 2002; Bussemaker et al. 2001; Birnbaum et al. 2001; Zhu et al. 2002; Pritsker et al. 2004; Elemento and Tavazoie 2005), and a comprehensive review of current motif-discovery methods is available (Tompa et al. 2005).
The approach reported here is a supervised pattern classification scheme designed to integrate a large number of heterogeneous data sources in order to more accurately predict the association of a transcription factor and its target. In particular, we explore the use of support vector machines, which are able to incorporate high-dimensional data sets (many features). SVM classifiers have previously been used for the prediction of protein homology (Jaakola et al. 1999), secondary structure (Hua and Sun 2001a), and sub-cellular localization (Hua and Sun 2001b). As sequence classifiers they have also been useful in predicting translation start sites (Zien et al. 2000), mRNA splice sites, and signal peptide cleavage sites (Wang et al. 2005). More broadly they show good performance in the identification of normal and cancerous tissue samples (Furey et al. 2000) as well as prediction of gene function (Pavlidis and Noble 2001).
Few groups have published work on supervised classification schemes for predicting new transcription factor targets. We briefly reviewed some of these previously (Holloway et al. 2006). One method includes linear discriminant analysis (LDA) to select from a set of potentially co-regulated genes those that are most likely to share common transcription factors (Simonis et al. 2004). Another approach uses Bayesian networks to learn the combinatorial relationships of TFs and targets that underlie specific gene expression experiments (Beer and Tavazoie 2004). Finally, in an approach similar to ours, SVMs have been applied to microarray data in order to predict TF–target associations (Qian et al. 2003).
Although some of these techniques work well, they either do not effectively incorporate the large amount of regulatory data available in ChIP–chip interactions or they base their classification on only one or two types of genomic data. Our approach easily combines 26 large genomic datasets, adaptively weighting each data source based on its ability to correctly classify a training set. The combination of heterogeneous data reduces false positive predictions while maintaining high accuracy. Genomic data combination using SVMs has been demonstrated before. Protein sequence similarity, protein–protein interactions, protein hydrophobicity, and gene expression data were successfully combined to predict the functional group of a set of proteins, and the combination of data was shown to significantly outperform individual methods (Lanckriet et al. 2004).
We provide accuracy measurements on our classifiers based on leave-one-out cross validation, and we benchmark our results against randomized datasets. Our full set of predictions for 104 TFs based on all combined methods can be downloaded from our website, http://www.cagt10.bu.edu/SSBPaper/MachineLearningTFSSB.htm.
SVMs: background
We consider 26 different datasets sequentially, train a classifier on each, and then construct a composite classifier which is a weighted combination of the 26. For each training set, we develop an allocation rule for every TF. Let N be the size of the training set for a particular TF (the collection of positive and negative examples, i.e., genes which do and do not bind it). Each gene has a set of attributes forming a vector that contributes to the distinction between positive and negative sets. As an example, an attribute vector for a gene could be an ordered list consisting of the number of times each possible 4-mer occurs in the upstream region. The collection of such vectors is the feature space, F. Each gene would then be characterized by a 256 component feature vector. The SVM generates a hyperplane of D = 255 dimensions in the feature space separating positives from negatives (d will henceforth be an index over the features of the dataset). We write a vector in F as xi = (xi1, xi2, xi3... xid), the components xid representing, for the example above, the count of the dth k-mer in the ith gene. Then the equation for a hyperplane has the form
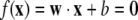 |
1 |
where x = (x1, x2, …, xd) and w≡ (w1, w2, …,wd). For D = 2, this is a straight line in variables x = (x1, x2) with slope − w1/w2 and intercept − b/w2.
Geometrically w is a vector perpendicular to the hyperplane H, the magnitude | wd| of its dth component weighting the corresponding dimension. The function f(x) is assumed normalized (through scaling of w) so that the closest (positive, negative) pair x+i and x−i have values f(x+) = 1 and f(x−) = − 1, respectively. Then the SVM problem is to find w and b such that the attribute vectors of all genes in the positive set are above the hyperplane H1 defined by
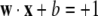 |
and all in the negative set are below hyperplane H2 defined by
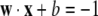 |
and that the margin (distance between H1 and H2) is maximal. Thus the goal is to find a separator that maximizes the margin, or distance between the positive and negative classes. This construction is essentially a choice of scaling for w, b, in particular requiring that the length |w| be minimal, since this maximizes the margin under the above normalization. Maximizing the margin is a convex optimization problem which is generally solved using standard Lagrangian methods (Sholkopf and Smola 2002). Typically, as in our case, perfect separation cannot be achieved. When error-free decisions are not possible the method can be readily generalized to allow any specified amount of misclassification, with a suitable penalty function.
An important aspect of the solution is that the data enter only in the form of a kernel matrix K, whose entries Kij are dot products of all pairs xi, xj of feature vectors. In the case that all components of the feature vector are truly independent, the Lagrangian is a linear function of the elements of the kernel, and the linear dot product is used with Kij = xi·xj. When the elements are correlated, the Lagrangian is written as a non-linear function of the inner products of the attribute vectors (see below). In particular, the non-linear dot products are defined for data points by Kij = K(xi, xj), where the given positive definite function K(x, y) is known as the kernel function. Such non-linear products are equivalent to assuming that an unspecified higher dimensional feature space F1 exists into which F is mapped and in which the separating hyperplane is linear. This yields a Lagrangian with matrix entries given by this alternative dot product. The implicit choice of F1 is made by changing the type of inner product used (see Table 1). For a more detailed development of SVMs, see the excellent reference texts (Sholkopf and Smola 2002, Tan et al. 2005). For a detailed two-dimensional example see Holloway et al. (2006).
Table 1.
Four common kernels tested
| Kernel | Parameters | Description |
|---|---|---|
| Linear | None | K (x, y) = x·y |
| Polynomial | Poly degree d | K (x, y) = (x·y + 1)d |
| Gaussian radial basis function (RBF) | σ | 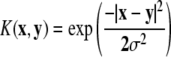 |
| Gaussian | σ | 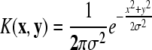 |
These are the four common kernel functions, the parameters which must be set by the user, and their mathematical description
Post-processing can be an essential task in pattern classification problems, particularly if one wishes to extract the highest quality predictions from a classifier. A naïve way to extract the most significant (positive) prediction from an SVM classifier is to select those data points which are most distant from the separator (distance given by w·xi + b for data point i). The interpretation is that those distant points are most unlike the negative set and contain the strongest positive character. A more informative method is to rank data by P(yi = 1|w·xi + b); i.e. by the posterior probability of a positive classification, given the distance of example xi from the hyperplane. Platt observed that these posterior probabilities could be well approximated by fitting the SVM output to the form of a sigmoid function (Platt 1999), and developed a procedure to generate the best-fit sigmoid to an SVM output for any dataset. The result is the posterior probability P(yi = 1|w· xi + b) for each data point in the training set (see Platt 1999) for further details). This probability places a confidence level on any new prediction made in the yeast genome and, most importantly, results in an ability to identify high-confidence predictions for future experiments.
Methods
We have tested a variety of sequence and non-sequence based classifiers for predicting the association of TFs and genes. All together 26 separate data sources (each yielding a feature map and kernel) are combined to build classifiers for each transcription factor. The 26 data sources comprise a family of sequence-based methods (e.g., k-mer counts, TF motif conservation in multiple species, etc), expression data sets, phylogenetic profiles, gene ontology (GO) functional profiles, and DNA structural information such as promoter melting temperature, DNA bending, and DNA accessibility predictions (see Table 2).
Table 2.
Abbreviations of datasets used to generate classifiers
| Abbreviation | Description | |
|---|---|---|
| 1 | MOT | Motif hits in S. cerevisiae |
| 2 | CON | Motif hits conservation 18 organisms |
| 3 | PHY | Phylogenetic profile |
| 4 | EXP | Expression correlation |
| 5 | GO | GO term profile |
| 6 | KMER | K-mers—4,5,6-mers |
| 7 | S1 | Split 6-mer 1 gap kkk_kkk |
| 8 | S2 | Split 6-mer 2 gaps kkk__kkk |
| 9 | S3 | Split 6-mer 3 gaps kkk___kkk |
| 10 | S4 | Split 6-mer 4 gaps kkk____kkk |
| 11 | S5 | Split 6-mer 5 gaps kkk_____kkk |
| 12 | S6 | Split 6-mer 6 gaps kkk______kkk |
| 13 | S7 | Split 6-mer 7 gaps kkk_______kkk |
| 14 | S8 | Split 6-mer 8 gaps kkk________kkk |
| 15 | M01 | 6-mer with 1 mismatch (count 0.1) |
| 16 | M05 | 6-mer with 1 mismatch (count 0.5) |
| 17 | ENT | Condition specific TF–target correlation |
| 18 | BIT | Nucleotide sparse binary encoding |
| 19 | CRV | Promoter curvature prediction |
| 20 | HC | Homolog conservation |
| 21 | HYD | Hydroxyl cleavage |
| 22 | KPo | Kmer median positions from start |
| 23 | KPr | Kmer Probabilities (− log pval) |
| 24 | MT | Promoter melting temperature − 20 bp window |
| 25 | DG | Promoter melting Delta G profile − 20 bp win |
| 26 | BND | Promoter bend prediction |
Abbreviations for each dataset and a short description are given
Our positive and negative training sets are taken from ChIP–chip experiments (Harbison et al. 2004; Lee et al. 2002), Transfac 6.0 Public (Matys et al. 2005), and a list curated by Young et al. from which we have excluded indirect evidence such as sequence analysis and expression correlation (Young Lab Web Data, http://www.staffa.wi.mit.edu/cgi-bin/young_public/navframe.cgi?s=17&f=evidence). Only ChIP–chip interactions of p-value ≤ 10−3 (i.e., a high confidence level) are considered positives (Harbison et al. 2004). The Transfac and curated list represent a manually annotated set which will later be used separately during SVM comparison to PSSM performance. For the purposes of SVM, however, all manually curated and high-throughput sets are grouped together, making a total of 9,104 positive interactions.
Negative sets pose a greater challenge since no defined negatives exist in the literature; however, since a particular TF will regulate only a small fraction of the genome, a random choice of negatives seems acceptable. In fact, test cases with a few TFs show good classification performance with random negatives (unpublished work). Nevertheless, a safer set of negatives would be those showing no binding by experiment under some set of conditions. Along those lines, we have chosen for each TF 175 genes with the highest p-values (generally > 0.8) under all conditions tested in genomic ChIP–chip analyses (Harbison et al. 2004; Lee et al. 2002). Clearly all experimental conditions have not been sampled and this does not guarantee that our choices are truly never bound by the TF, but this choice of negatives should maximize our chances of selecting genes not regulated by the TF of interest.
All promoter sequences have been collected from RSA tools (van Helden 2003), Ensembl (Birney et al. 2006), or the Broad Institute’s Fungal Genome Initiative (Galagan et al. 2003; Dean 2005). For yeast, promoters are defined as the 800 bp upstream of the coding sequence. The motif hit conservation dataset required promoter regions from 17 other genomes. Those genomes, their sources, and the length of the promoter regions are described in our previous report (Holloway et al. 2006). Sequences are masked using the dust algorithm and the RepeatMasker software (Tatusov and Lipman 2005; Smit et al. 2005) where appropriate, to exclude low complexity sequences and known repeat DNA from further analysis. PSSM scans (for datasets 1 and 2, below) are performed with the MotifScanner algorithm (Aerts et al. 2003). MotifScanner assumes a sequence model where regulatory elements are distributed within a noisy background sequence (Aerts et al. 2003). The algorithm requires input of a background sequence model, which in this case is a transition matrix of a third order Markov model generated from the masked upstream regions of each genome. MotifScanner only requires one parameter be set by the user, i.e. the threshold score for accepting a motif as a binding site. Several thresholds have been tested and the results we have used to create SVM kernels are all at a setting of 0.15, which has been found to be a reasonable middle ground, making approximately 560 predictions per TF. Settings beyond 0.2 produce too many false hits to be useful. The PSSMs themselves are obtained from Transfac 6.0 Public and from (Harbison et al. 2005), which are a mix of experimentally derived motifs and those generated by motif-discovery procedures.
Datasets using k-mers rather than PSSMs are generated using the fasta2matrix (Pavlidis et al. 2004) program which lists all possible k-mers and counts the occurrence of each within a set of promoters. Gapped k-mers are detected using custom scripts written as Matlab m-files. The expression data used include 1011 microarray experiments compiled by Ihmels and co-workers, which can be downloaded with permission from the authors (Ihmels et al. 2005).
Each data set is normalized so that each feature in the training set has mean of 0 and standard deviation of 1. Gene Ontology, phylogenetic profile, and TF–target correlation data are not normalized since their data are binary. Finally, since the ultimate goal is data integration the number of training examples for a given TF must be the same for every dataset used to make a classifier. When examples are missing in a dataset, as is the case with the GO and COG (phylogenetic profiles based on the Clusters of Orthologous Groups database) based classifiers, random values sampled from the rest of the training set are used to fill in the missing vectors.
All classifier construction and validation was performed in Matlab (The Mathworks: http://www.mathworks.com/) using the Spider machine learning library (Weston et al. 2005). Mapping of predicted binding targets to biological pathways was done using the Pathway Tools Omics Viewer at SGD (Christie et al. 2004). See our supplementary methods section for an expanded description of the analyses below.
Description of analysis
A separate classifier is developed for each TF based on each independent dataset. The four kernel functions in Table 1 (linear, rbf, Gaussian, and polynomial) are tested using leave one out cross validation, and the function with the highest F1 score (below) is chosen as best for that particular TF–dataset combination. A flow diagram of our method can be seen in Fig. 1. Let TP denote the count of true positives, FN false negatives, etc. The F1 statistic is a robust measure that represents a harmonic mean between sensitivity (S), and positive predictive value (PPV). It is defined by
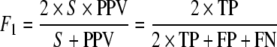 |
Fig. 1.

Flow diagram: synthesizing a single classifier for each TF from several data sets. A classifier is constructed for each individual TF for each genomic dataset, using every one of four possible kernel functions (26 datasets × 104 TFs × 4 kernel functions = 10816 kernels from which SVM classifiers are built). For each of these classifiers optimal parameters are chosen by cross-validation. For each dataset and each TF, the best performing of the four kernel functions is selected, reducing the number of classifiers to 2704 (26 datasets × 104TFs). Finally, the datasets are combined based on F1 score of their best performing kernel so that there is only one classifier per TF
If we choose the classifier with the best F1 statistic, each TF now has one classifier for each type of genomic data (26 classifiers total). For every classifier the C parameter (the trade-off between training error and margin) must be specified, and some kernel functions require a second parameter, e.g., the polynomial degree k for a polynomial kernel or a standard deviation σ (which controls the scaling of data in the feature space) for a Gaussian or radial basis function (RBF) kernel. The values for these parameters are chosen by a grid-selection procedure in which many values are tested over a specified range using 5-fold cross validation. The ROC score is used to choose the best values. As an example for an RBF kernel a range of C values from 2−5 to 200 is tested with a range of σ values from 2−15 to 23. The best combination of values is then chosen to make the final classifier.
The performance of any parameter-optimized classifier is determined using leave-one-out cross validation. Once the best kernel function K(x, y) (with optimized parameter values) has been chosen for a particular TF–dataset pair, the next step is to combine the datasets to create a composite classifier. To that end, the K(x, y) is used to create a kernel matrix for each of the 26 datasets. Before weighting and combining kernels for each data set, all kernel matrices are normalized according to
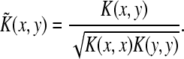 |
This normalization effectively adjusts all points to lie on a unit hypersphere in the feature space F, and the diagonal elements in every kernel matrix the will be 1. This assures that no single kernel has matrix values that are comparatively larger or smaller than other kernels, so all matrices initially have the same contribution to the combination.
Datasets can be combined by adding kernel matrices together; however, an unweighted linear combination ignores dataset dependent performance—in fact some datasets do not perform better than random for some TFs. To avoid this problem, we determine whether the number of true positives predicted using a particular dataset is significantly different (p ≤ 0.05) than what would be achieved by random guessing. We calculate the probability of observing more than g true positives given the training set size N, the total number of known positives L (i.e., TP + FN), and the number of positively classified examples, M (i.e., TP + FP)
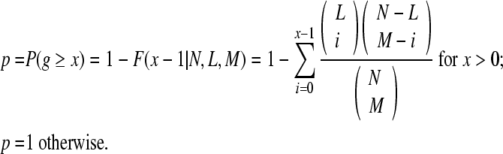 |
Here p is the probability of drawing x or more true positives at random. Datasets that do not meet the p-value cutoff are eliminated from the analysis for a particular TF.
Finally, the significant datasets (each represented by a kernel matrix Kij) must be weighted based on their performance. Using a scheme (described below) with weights equal to the F1 score of each classifier, the underlying 26 kernel matrices are scaled and added into a single unified kernel corresponding to the given transcription factor. Once the weighting is complete, an overall leave-one-out cross-validation is employed to estimate the error of the combined classifier. Although individual kernels were tuned on the entire set of examples for each dataset independently, the C parameter of the final, combined SVM was determined only on the training set during cross-validation. Nevertheless, to measure the danger of overfitting the most useful performance benchmark is perhaps the random data controls shown in Fig. 2. Also, the use of Platt’s posterior probabilities as a post-processing filter can help in choosing the truly relevant targets once the procedure is applied to the entire genome. As further validation we employed an alternative scheme for data combination on a few test cases. The feature vectors for several datasets were directly concatenated and recursive feature elimination (Guyon et al. 2002) was applied to select the most relevant features for classifier construction completely independent of test data. This is a more computationally intensive procedure requiring many datasets to be loaded into memory simultaneously and hundreds of SVMs to be fit iteratively in order to weight data features. The results for these tests appeared similar to the results obtained by the procedures outlined in this manuscript, and we will describe these results on a larger set of transcription factors in a future publication.
Fig. 2.

SVM performance. Performance of each dataset and combined datasets ordered by increasing F1 score. Cumulative results for all transcription factors were used to plot the sensitivity, positive-predictive-value, and the F1 statistic for each dataset and data combination. Dataset abbreviations are given in Table 3. The combined classifiers, labeled 26st (linear weighting), 26sq (square weighting), and 26t (tangent square weighting) on the far right, perform better than any dataset alone, with the squared tangent weighting giving the best result overall. Three random datasets also appear in the table, R (randomized k-mer counts), RH (randomized 10% selection of each dataset), and RN (normally distributed random numbers)
Three simple weighting schemes have been compared. In all cases the primary weight for a method is determined by computing its ratio with the best performing method. Our first weighting scheme is linear and simply multiplies the mth matrix Km = Kmij by its scaled F1 score αm and computes a sum, yielding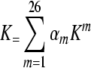 . A second scheme is non-linear and squares the weights of the first method before multiplying, yielding
. A second scheme is non-linear and squares the weights of the first method before multiplying, yielding
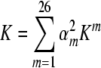 . This will not change the weight of the best performing method, which will be scaled to 1, but will decrease the relative weights of poorer methods. Our third scheme, which is the most non-linear, takes the squared tangent (an effective sigmoidal function) of the primary weight, yielding
. This will not change the weight of the best performing method, which will be scaled to 1, but will decrease the relative weights of poorer methods. Our third scheme, which is the most non-linear, takes the squared tangent (an effective sigmoidal function) of the primary weight, yielding
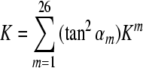 . This more steeply penalizes poorly performing methods while increasing relative weights of the best methods (e.g., instead of weight 1, the best method will have a weight of 2.43).
. This more steeply penalizes poorly performing methods while increasing relative weights of the best methods (e.g., instead of weight 1, the best method will have a weight of 2.43).
Genomic datasets
1 PSSM motif counts (MOT, Table 2 item 1)
Position-specific weight matrices (PSSM) for 104 transcription factors have been used to scan 800 bp promoters in S. cerevisiae for each gene in a training set, and the number of hits for each PSSM has been counted. These counts are the features (i.e., components) of 104 dimensional feature vectors. It is clear that a greater number of “hits” by a PSSM in the upstream region of a gene will imply a greater likelihood that the TF corresponding to the matrix will actually bind the gene. For each prediction there is a probability that it will be true, P(True|hit). If a certain upstream region of a gene has more than one hit, the probability that the TF binds to the gene will increase (Supplementary Figure 1). This method aims to better predict TF binding by taking into account the number and types of binding motifs in a promoter.
2 PSSM hit conservation (Table 2 item 2)
Conservation of a TF binding site is determined by counting hits of the TF probability matrix (PSSM) in orthologous upstream regions from several organisms. Orthology information was taken from the Homologene database (Wheeler et al. 2005) for all organisms except for sensu stricto and sensu lato yeasts, which was obtained from Washington University and the Whitehead Broad Institute (Cliften et al. 2003a, b; Kellis et al. 2003; Kellis 2003).
In this analysis, a hit by a PSSM in the upstream region of an ortholog is defined as a conserved motif. In this way, conservation of a potential binding site is being measured rather than the exact nucleotide string. This is because a PSSM may identify sequences that are different in nucleotide composition but still match the probability matrix. This is a loose conservation criterion that makes sense biologically, since natural selection will act to preserve a binding site, and not necessarily an exact nucleotide string.
The stronger the conservation of a potential binding site, the more likely the site is to be real (See Supplementary Figure 2). These data are assembled into a 104 dimensional feature vector for each gene in yeast. Each feature represents a transcription factor motif and the value of the attribute is the number of genomes in which the binding site is conserved.
3 Kmers, mismatch kmers, and gapped kmers (Table 2 and 6–16)
PWMs may fail to detect binding sites if the binding site collection used to generate them is incomplete (in the case of experimental data) or if the motif discovery procedure is inaccurate (as may occur in the case of computationally generated matrices). In this case, the distribution of all k-mers in a gene’s promoter may be used to predict whether it is bound or not-bound by a TF. K-mer counts in promoters have been used previously with SVMs to predict genes’ functions (Pavlidis and Noble 2001). Here, several strategies are used to generate a variety of datasets based on k-mer strings. First, one dataset of feature vectors is created by decomposing all yeast promoters into counts of all k-mers of length 4, 5, and 6. Similarly, 6-mers with variable length center gaps (of the form kkk − {x}n − kkk) are counted in each promoter to form sequence datasets allowing gaps of size 1–8 (Table 2, items 4–11). This allows detection of split motifs such as the binding site for Abf1, RTCRYNNNNNACGR. Finally, we construct two datasets with 6-mer counts allowing one mismatch in any 6-mer (Table 2 items 12–13). A mismatched base pair is counted with a value of 0.1 in the first dataset, and 0.5 in the second.
Given a set of true positives and true negatives for each TF, the SVM classifies genes based on their complete promoter content as represented by these k-mer distributions. As we point out in the “Discussion section”, k-mer counts are the single best performing method for distinguishing transcription factor targets.
It should be noted that our sequence based kernels are very similar to sequence kernels used in previous work. Specifically, our kernels are inspired by the spectrum kernel (Leslie et al. 2002), the (g,k)-gappy kernel (Leslie and Kuang 2003) and the mismatch kernel (Leslie et al. 2004) which have been proposed for sequence classification (see Supplementary Methods for a more complete description). Finally, the kernels used here take into account the reverse complements of each k-mer. This means, for instance, that the 3-mers “AAA”, and “TTT” are counted together as one unit since the presence of one necessitates the other on the opposite strand of DNA.
4 GO annotation (Table 2 item 5)
GO term annotation can be used to detect possible transcriptional targets. The targets of a transcription factor have often been shown to have similar function and a gene’s GO annotation can be used to measure its functional similarity to known targets (Allocco et al. 2004). For this method, all GO Biological Process terms in yeast become features for genes, such that every gene will have a binary vector, with a 1 for the terms which are annotated to it, and 0 otherwise. Parent terms of direct annotations also receive a 1. There are 2,155 possible terms for yeast, giving a vector of the same length. Since only about one-third of yeast genes are annotated with GO terms, a feature matrix generated with GO data is sparse, consisting mostly of zeros. Imputing zeros for genes unannotated in GO can potentially bias the result of the classifier (for instance, if many negatives are missing and hence are described using zero vectors it may be trivial to separate these from the positives). Instead, the binary vector is filled in with random data according to the background distribution of term annotation in the yeast genome. Despite using random data, the vectors are still sparse and the best 800 GO terms are selected using the Fisher score criterion during the classifier construction for each TF. The Fisher criterion gives high scores to features that have large differences in mean between the positive and negative classes in relation to variance. This feature selection is performed in the SPIDER data mining package (Bishop 1995).
5 Phylogenetic profiles (Table 2 item 3)
Co-evolution of a transcription factor’s targets may indicate regulation. A phylogenetic profile of a gene is simply the pattern of occurrence of its orthologs across a set of genomes. Genes with similar patterns have been shown to participate in the same physical complexes or have similar biochemical roles within the cell (Wu et al. 2003). It has also been postulated that transcription factors and their targets co-evolve (Gasch et al. 2004). Therefore it seems reasonable that a group of commonly regulated genes could share a similar pattern of inheritance. Phylogenetic profiles here were parsed from the COG database, which contains orthology information between S. cerevisiae and 65 other microbial genomes. Each gene in the positive and negative set is represented by a 65 component binary vector, a component being 1 if the gene’s ortholog is present in the corresponding genome, and zero otherwise. As with the GO data, gene attribute vectors are binary, containing 65 elements, one for each genome in COG. Also, since many genes have not been annotated to COG groups, it is necessary to generate random vectors for missing genes as described for the GO example above.
6 TF–target expression correlation as a method to predict regulation
Analysis of transcription factor motif-matching outputs shows that false positive predictions are numerous even in cases of low sensitivity. Expression analysis provides a means to discover targets missed by sequence based methods. Several studies have shown that genes with similar expression patterns are likely to share similar regulation and, conversely, genes regulated by the same TF are more likely to be co-expressed (Allocco et al. 2004; Yu et al. 2003).
Two strategies are often useful for discovering transcription factor targets using expression data. Often genes are turned on and off as the expression levels of their controlling TFs are altered. Thus one method is to find targets of some TFs by finding TF/gene pairs that have correlated expression patterns (Zhu et al. 2002). A second approach involves identifying groups of co-expressed genes, and hypothesizing that this co-expression is due to co-regulation by the same TF(s) (Ihmels et al. 2002, 2004). In the two sub-sections below, we describe how each of these strategies can be used to construct data vectors for SVM learning.
6.1 TF–target correlations measured by profile entropy minimization (Table 2item 17)
The approach described in (Mellor and DeLisi 2004) addresses the problem of discovering condition specific regulation by searching for the conditions under which a regulator’s profile is maximally associated with a target’s profile, for example, when the TF and target have correlated expression. This essentially chooses the set of experiments where the TF most clearly and significantly controls the expression of a potential target. In this analysis correlations with a p-value of 10−10 are chosen in order to extract the most significant regulatory relationships and reduce false predictions. Significant relationships are coded as 1’s in gene’s feature vector, so that every gene is described by a binary list whose length is the number of TFs (104 in this case).
6.2 Target–target correlations (Table 3item 4)
Table 3.
High ranking k-mer alignment and comparison to known binding site

Weight vectors for each TF classifier are used to rank all k-mers. Known TF motifs appear in the middle column and high ranking k-mers are assembled in the right column showing correspondence with the known motif. Standard nucleotide abbreviations are used. Some less common abbreviations are W = {A or T}, R = Purine, Y = Pyrimidine, S = {C or G}, K = {T or G}, M = {C or A}, D = not C
For purposes of representing expression correlation between targets, we use normalized log2 ratios for each gene across 1,011 experiments (Bergman et al. 2003). Each gene’s expression profile is normalized to a mean of 0 and standard deviation of 1. This expression profile is then the vector of features used by the SVM to represent any example gene (each gene will have 1,011 features). In this case, the dot product between such gene vectors is analogous to a Pearson correlation and naturally fits into the SVM framework. Given many known targets of a transcription factor as positive cases, the SVM can identify a new target based on how closely its expression resembles that of the known examples.
7 Sparse binary encoding of promoters (Table 2 item 18)
Efforts to encode strings into kernel representations have progressed for many applications. The mismatch, gap, and k-mer kernels mentioned above have been used mainly for protein classification, translation initiation site detection, and mRNA splice site identification. Another straightforward sequence representation is the sparse bit encoding (Zien et al. 2000). In this simple scheme each nucleotide in a sequence is encoded by 4 bits, only one of which is set to 1. The nucleotide is identified as A, C, T, or G based on the position of the “1” in each such set. This leaves an 800·4 = 3200 dimensional vector to describe each example sequence, and the dot product of two vectors results simply in the number of nucleotides shared between the two sequences.
8 Promoter curvature and bend predictions (Table 2 items 19 and 26)
It is well known that sequence-dependent DNA bending can be an important aspect of protein–DNA interactions. Some prominent examples of proteins that induce DNA bending are the TATA-binding protein (TBP) (Masters et al. 2003), catabolite activating protein (CAP), and the yeast Mcm1 transcription factor (Acton et al. 1997). A specific sequence of nucleotides that is more prone to bending into the proper configuration would provide a ready-made site for transcription factor binding. The particular bend and curve properties of known target genes may help discriminate them from non-targets.
Using the “Banana” algorithm in the EMBOSS toolkit, bend and curvature predictions were made along the promoters of all yeast genes. These were used as two separate genomic methods from which to generate classifiers for all 104 TFs, one based on bend predictions and one based on curve. Specifically, bending refers to the tendency of adjacent base pairs to be non-parallel (twists and short bends of ∼3 bp), whereas curvature refers to the tendency of the double-helix axis to follow a non-linear path for a distance of several base pairs (broad loops and arcs, ∼9 bp window). Banana follows the method of Goodsell and Dickerson (1994) which is consistent with published experimental data (Satchwell et al. 1986). The output of the Banana algorithm becomes the feature values along a promoter for each example gene. For more details on the method see our Supplementary methods, reference (Goodsell and Dickerson 1994) or see the EMBOSS website (http://www.emboss.sourceforge.net/apps/banana.html).
9 Homolog conservation (Table 2 item 20)
This method is akin to the phylogenetic profiles taken from the COG database described above. Because COG uses a strict definition of orthology, namely bi-directional best hits within a group of at least three organisms, many genes are not allocated to any ortholog group. The method described here relaxes the definition of orthology to allow a profile to be constructed for any gene, while still discriminating between well-conserved sequences and weakly conserved sequences (Snitkin et al. personal communication). These phylogenetic profiles are constructed using BLASTP to compare yeast proteins to 180 prokaryotic genomes. The resulting best hit E-values are then discretized by placing them into one of six bins based on empirically determined E-value cut-offs. The bin numbers range from 0 (no significant hit) to 5 (very significant). Thus, a typical example gene will have 180 features, each corresponding to a different genome, with values ranging from 0 to 5 indicating the strength of the best BLASTP hit of that gene’s protein to another genome.
10 Hydroxyl cleavage—DNA accessibility (Table 2 item 21)
It is possible that strands of DNA sharing little sequence similarity may still share common structural motifs. Transcription factors may seek out these structural cues for binding, thereby identifying conserved structural motifs when no strong consensus sequence can be detected. Experiments show that hydroxyl (OH) radical cleavage is an effective probe for DNA structure, in that strand breaking mirrors the accessible surface areas of the sugar-phosphate backbone (Balasubramanian et al. 1998; Parker et al. 2005; Tullius and Greenbaum 2005). A database of DNA sequences and their hydroxyl cleavage patterns has been published (Parker et al. 2005). This database allows accurate prediction of backbone accessibility for any sequence by sequentially examining every 3-mer in a sequence and looking up its experimental cleavage intensity as measured by phosphor imaging of cleaved, radio-labeled DNA separated by electrophoresis (Balasubramanian et al. 1998).
Predictions of this sort are generated for all sequences in the yeast genome and the individual 3-mer cleavage intensities along each promoter serve as feature vectors for TF–target classification. This method could prove useful in identifying potential targets when k-mer counts and other sequence based methods fail.
11 Kmer median positions from start (Table 2 item 22)
A potential transcription factor binding site may be functional only when within a certain distance from other binding motifs or from the start site of transcription. When such positional constraints exist, they can be used to filter out sites which would otherwise become false positive predictions.
For each k-mer in a sequence, we record its median distance from the transcription start. This dataset will be useful in classifying targets for a transcription factor only if the factor shows positional bias in promoter binding.
12 K-mer likelihoods (Table 2 item 23)
Although k-mer counts may describe promoter composition, the abundance of non-informative sequences may hide the few k-mers which meaningfully contribute to class separation. Those k-mers which are statistically over-represented in a promoter can often be transcription factor binding sites, and this fact has been effectively used to identify biologically significant patterns (Cora et al. 2004; van Helden and Collado-Vides 1998; Haverty et al. 2004). For every possible k-mer 4, 5, and 6 long we calculate the probability that the k-mer has x occurrences in a gene’s promoter. The negative log of these probabilities are then the features used for SVM classification.
Background k-mer counts are obtained from RSA (van Helden 2003; van Helden and Collado-Vides 1998) tools. The prior probability (f) for a k-mer to be found in any position is calculated by dividing the total number of counts in the background sequence set by the total number of possible positions in the background set (here, the background set is the full set of 800 bp yeast promoters). Given this prior probability for a k-mer, the expected number of occurrences of the k-mer in any sequence can be calculated by
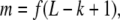 |
where L is the length of the sequence and k is the length of the k-mer.
The goal is then to calculate the probability of finding the observed number of counts by chance given the expected number for a promoter. This can be done simply by using the probability density function of the Poisson distribution with mean m. This method for calculating k-mer likelihoods is similar to the method described in (van Helden 2004). Thus, for each gene, a p-value will be calculated for each k-mer which represents the likelihood that the k-mer appears as many times as observed by chance. A feature vector for a gene is then the vector of probabilities describing all k-mers.
13 Promoter melting temperature profile and promoter Delta G profile (Table 2 items 24 and 25)
It is widely known that the initiation of transcription by polymerase involves melting of the DNA double helix. Several experiments have indicated that differences in melting temperature (Tm) of DNA can influence the rate of transcription by assisting or obstructing DNA melting by polymerase (Flickinger 2005), and there is evidence that torsional strain can play a role in duplex destabilization and opening (Benham 1992). Furthermore, it has been shown that sites thought to be susceptible to stress-induced duplex destabilization (SIDD) match well with gene regulatory regions (Benham 1996). It is therefore possible that transcription factors binding DNA may induce conformational adjustments in the promoter which slightly alter the stability of the helix. This change in stability may indirectly change the frequency or likelihood of transcription initiation. Indeed, recent models have shown correlation between sites of local promoter melting, regulatory sites, and initiation sites (Choi et al. 2004).
If certain transcription factors influence a target’s expression by altering promoter stability, its targets may contain a specific melting temperature or free-energy signature in their promoter regions. This signature could potentially distinguish targets from non-targets much as sequence motifs do. To include this information in a classifier the EMBOSS (Rice et al. 2000) toolbox is used to calculate the melting and free energy profiles of all yeast promoters using a sliding window of 20 bp. Thus, for every 20 bp increment along each upstream region, a Tm value and a Gibbs free energy (ΔG at 25°C) is calculated. For these calculations EMBOSS uses the nearest-neighbor thermodynamics from (Breslauer et al. 1986; Baldino 1989). The Tm profile and the free energy profile become separate feature vectors for each gene, thereby providing two additional datasets which can be used for classification.
PSSM comparison
Using the same positive and negative sets as for the SVM procedure, PSSMs are also used to make predictions across the yeast genome at various score thresholds to serve as a comparison to predictions made by SVM. The threshold used for PSSM scanning was adjusted for each TF such that the overall specificity is held constant at 0.95 to match the SVM results. Other choices of threshold do not appear to improve performance. Loosening the threshold begins to dramatically increase false positive predictions beyond a prior of 0.2. By making detection stricter, false predictions are reduced along with sensitivity.
Results and discussion
After data pre-processing, the analysis begins with the independent evaluation of each dataset on each TF. Several kernel functions are tested and any necessary parameters are optimized before a final classifier is constructed (see “Methods”). A schematic of our procedure is given in Fig. 1. Once parameter optimized classifiers are constructed for each TF–dataset pair, all of the datasets, represented by the optimized kernel matrices, are combined using a weighting scheme based on their F1 scores. The hyper geometric test is used to filter out datasets which do not perform better than random (accept p-value ≤ 0.05) for a particular TF. Accuracy estimates for the combined classifier are made using a final leave-one-out cross validation.
Three simple weighting schemes have been tried (see “Methods”), and the primary weight for a method is the ratio of its F1 score with that of the best performing method. The first scheme simply multiplies all kernel matrices by their scaled F1 scores and sums them. The second scheme squares the weights before multiplying. This has the effect of decreasing weights of poorly performing methods. Our third scheme uses the squared tangent of the primary weight. This will more severely penalize poor performers while boosting the weights of the best methods (e.g., instead of weight 1, the best method will have a weight of 2.43).
We have been able to accurately classify the known targets of many transcription factors in S. cerevisiae. Figure 2 shows the performance of classifiers generated on each individual dataset (see also Supplementary Table 1). The combination of datasets performs better than any individual type of data, but the best single method achieves a sensitivity of 71% and a positive predictive value of 0.82. The combined datasets are labeled STD for weighting based on simply the scaled F1 measure, SQU for weighting based on squared, scaled F1 measure, and TAN for weighting based on the tangent squared F1 measure, as described in “Methods”. Other abbreviations can be found in Table 2. Almost all methods perform much better than random. The exceptions are GO term annotation and phylogenetic profiles. For phylogenetic profiles this is not unexpected, since only 30% of the yeast genome has an established ortholog in the COG database. This absence of data means that many positive examples can no longer contribute to classification, leading to poor performance for most TFs. The situation is similar for GO term annotation, where many genes are poorly annotated or have no known function.
The performance statistics mentioned in Fig. 2 are a summary of those for all 104 combined classifiers. Since there are 9,104 known positives for all regulators, a sensitivity of 71% indicates that, considering all 104 classifiers, we recover 71% of the known data. This means that classifiers for some TFs have much higher sensitivities or PPVs while other classifiers perform no better than random.
The most powerful individual data set uses k-mer counts allowing 1-missmatch per k-mer. However, the combination of all of the methods shows increased sensitivity and precision over all individual methods. The squared-tangent weighting function performs the best overall, reaching a sensitivity of 73% and a positive predictive value of 0.89. Looking only at the top 20 TFs, we see a sensitivity and PPV of 88.2% and 0.9, respectively. Our results show that combining datasets increases sensitivity only incrementally over classifiers built on simple k-mer counts alone, and that it produces a small improvement in positive predictive value. Thus, combining methods results in the modest reduction of false positive classifications.
The use of the hypergeometric distribution to test the significance of a dataset for each TF allows us to assess how useful a particular data type is for target identification. Figure 3 plots the percentage of TFs for which each dataset has been found to be significant at p ≤ 0.05. Overall, sequence based methods (k-mer counts, mismatch and gapped k-mer counts, and k-mer likelihoods) show the best overall coverage, being significant for almost all transcription factors. Structural descriptions of the promoter region differ greatly in their usefulness, varying from DNA curve prediction, useful for ∼15% of TFs, to melting temperature profiles and free energy values, significant for over 60% of TFs tested.
Fig. 3.

Percentage of TFs for which each dataset is significant (p ≤ 0.05). Percentage of TFs is on the left axis and datasets are numbered along the bottom with a key given to the right of the diagram (see Table 3 for descriptions of method abbreviations)
In work with genomic datasets having large numbers of features (e.g., k-mer counts, expression measurements) there is always an inherent risk of over-fitting when the number of positives and negatives are relatively small. To give a more practical portrayal of our method and prevent an overly optimistic view of the results, it is illuminating to compare our results with those from classifiers obtained by training on random data. Thus three random datasets have been constructed as controls and their results displayed in Fig. 2. The first, abbreviated R is simply randomly shuffled k-mer count data. The second (RH) is created by shuffling a composite dataset composed of a random 10% selection of each individual dataset. The third (RN) is a normally distributed random set of numbers with mean of 0 and standard deviation of 1.
Although performance is much better than random it is doubtful from these results that predictions obtained by applying our classifiers to the entire genome would yield truly reliable targets without further processing. A simple classification of all potential targets with our 104 classifiers returns, on average, ∼800 new targets for each TF. The conditional probabilities given as output from Platt’s method (Platt 1999) allows the selection of possible targets at a desired probability threshold. For instance, one can select predictions for which the probability of being a positive is greater than 0.99. In some of the examples below, the top targets were selected in this fashion and compared to the full set of known positive genes.
Another method to reduce the risk of over-fitting, which we reserve for our future work, is application of sophisticated dimension reduction techniques to discover significant features in different datasets based on classifier performance. Feature selection and clustering will allow the most relevant features from different datasets to be retained while large portions of redundant and irrelevant information are discarded. In some cases this has been shown to increase classifier accuracy. In other cases, the reduction in the complexity of the problem is worthwhile since other learning algorithms, like k-nearest-neighbors or Bayes networks, which are difficult to train on large feature sets, could be compared efficiently on the smaller set of features. Although it is clear that combination of data slightly increases performance it is natural to ask whether such complexity of data is worthwhile when k-mer based data alone contributes a large portion of the classification accuracy. Dimension reduction techniques can help address this by potentially eliminating thousands of features. This will make it simpler to classify new sequences for which not all datasets are available since only the most relevant features need be present. In practice, it is likely that only a few data types will be needed to make useful predictions for most applications. K-mer counts, k-mer overrepresentation, and an improved measure of sequence conservation might comprise a baseline dataset for further refinement.
The dynamics of the individual classifiers can also be examined based on distributions of sensitivity and F1 score as compared to the random classifier. Figure 4a, c show the distribution of F1 score and sensitivity, respectively, for normal random data. Figure 4b, d show the same distributions but for actual data (26 method combination with tangent weights). The sensitivities and F1 scores for actual data have distributions heavily shifted to the right as opposed to those for random data. Although the majority of classifiers are comparatively good, several TFs have poor performance, something which warrants further inspection. There are four classifiers for which the F1 score and sensitivity are zero (YHL020C, YNL139C, YER068W, and YER161C). These factors have comparatively few known targets compared to others. On average these four TFs have 10 targets each (one of them has only three positives) in their training sets compared to an average of 88 targets for most regulators. This low number of positive examples is likely the cause of the poor performance. Figure 5 shows a plot of sensitivity vs. TF sorted by increasing number of positives for all classifiers. The general trend shows that classifiers having more positives give better performance.
Fig. 4.

Random vs. combined classifiers. (a) Distribution of F1 scores for normal random classifiers, (b) the same distribution on classifiers made from 26 dataset combinations for all TFs. (c) Sensitivity distribution for normal random classifiers and (d) the sensitivity distribution for the 26 dataset classifiers for all TFs
Fig. 5.

Sensitivity as a function of increasing positives. Classifiers for each TF were sorted according to increasing number of positives and the trend in their sensitivity is shown. Generally, classifiers with more positive examples perform better
Biological insights—promoter melting
Beyond categorizing genomic datasets as useful or not for classification purposes, the significance of a particular dataset has potential biological implications for a TF. To see if this could be explored based on our results, the factor YJR060W was chosen for further examination, since the promoter melting temperature profile is significant for this TF at p = 0.0037. Figure 6 shows a plot of the average promoter melting temperature curve (calculated using a 20 bp window and moving in steps of 1 bp) over all genes in yeast (solid blue), the average curve for genes in this TF’s negative set (dashed blue), the average in the TF’s positive set (dashed red), and the average in the most significant 33 predicted targets of the TF (solid red). The top 33 targets have Platt conditional probabilities P (positive | distance from separator) ≥ 0.99 and are obtained from the predictions made using the combination of all datasets, thus representing the best predictions we can make for this TF. This is equivalent to choosing predictions significant with a p-value of 0.01. These most significant targets contain 18 new predictions which are not part of the original positive set.
Fig. 6.

Melting temperature curves YJR060W. Using a 20 bp window for DNA melting temperature calculation, the temperature plots are presented for the average over all 5571 yeast genes (solid blue), positive targets for YJR060W (dashed red), negatives for YJR060W (dashed blue), and high confidence targets (solid red—P(true|distance to separator) ≥ 0.99) determined using Platt’s method for probability assignment to SVM output. Under the graph is an indicator displaying hits to the YJR060W consensus sequence in the top 33 targets. Consensus hits are distributed throughout the 800 bp upstream space
Clearly, the positive and negative groups for this TF contain average differences in promoter melting temperature. This difference is magnified when only the best targets are examined. The best 33 predictions have a very different melting signature from the negative set and the average yeast gene. A two-sample t-test was used to find the significance of this difference from the average curve. The purple over bar in Fig. 6 shows the window positions where the best targets have an average value which is significant at p ≤ 0.01. Almost all positions show a significant increase in melting temperature, with the exception of several positions proximal to the transcription start site. Considering that the transcription machinery must unwind the helix in this region, it is not unexpected that the melting temperature here would be smaller, as this would lower the activation energy needed to dissociate the strands.
As reviewed in “Methods”, there is ample support for the idea that melting temperature can influence transcription (Flickinger 2005), and that torsional strain can affect the stability of the DNA duplex (Benham 1992). Experiments have also shown that sites susceptible to this kind of destabilization correlate well with regulatory regions (Benham 1996). In light of the high melting temperature of promoter targets of YJR060W, it is possible that duplex destabilization plays a role in regulation by this TF. Indeed, experiments have shown that YJR060W functions largely in recruiting chromatin remodelling factors to proximal promoters (Kent et al. 2004). The exact mechanism for this recruitment is not fully understood, but it is required for transcription at some promoters and complementary to additional binding factors at others (Kent et al. 2004). In any case a possible hypothesis is that duplex stability is an important mechanism for regulation at these promoters and that YJR060W binding affects this stability either by conformational change induced by its binding or induced by the recruitment of chromatin remodelling factors. The conformational changes may alter the torsional strain on the DNA and thus affect the melting temperature prior to transcription.
Biological insights—binding site detection
Our results demonstrate that there is clearly a signal identifying ChIP–chip positives from other genes. Other groups have had less success confirming the validity of the ChIP–chip data, and this has led some to consider that as many as 50% (Simonis et al. 2004) to 60% (Gao et al. 2004) of the targets produced by ChIP–chip are false positives in the assay. The fact that the high throughput results are chosen to be significant with p ≤ 0.001 indicates that the transcription factors do in fact bind their targets. It is certainly possible that this binding does not always translate into changes in gene expression, that the changes are not large enough to be considered significant, or perhaps that the conditions under which binding would result in expression change were not tested. In any case, our classifier appears to pick up the information necessary to identify target genes.
To find this signal we have looked at the results of various individual datasets and extracted the attributes which contribute most to a transcription factor’s classifier. Support vector machines are often considered a “black box” method, since their results are not as readily interpretable as, for instance, the probability assessment of Bayesian classifiers. Nevertheless, the w vector described above can give an indication of which features in the data are important to the classification. Features whose components wi are large correspond to dimensions in feature space where positives and negatives are more widely separated. Thus by examining a single dataset, e.g. k-mer counts, it is possible to determine the k-mer(s) most responsible for differences between positives and negatives. To this end, w-vectors from the k-mer count dataset have been calculated for each linear TF classifier and examined to determine which k-mers had the largest weights. We compare these k-mers to known binding sites for each factor. Results for the best 10 TFs can be seen in Table 3, where the highest ranked k-mers are manually assembled to show their correspondence with known binding motifs. In most cases the k-mers with the highest weights match closely the reported binding site for the TF, showing that the algorithm is choosing meaningful features for classification. For example, the DNA binding protein Cep1 is known the bind the consensus TCACGTG and regulate cell cycle and stress response genes. The highest weighted k-mer in the classifier is CACGT, and the top 4 k-mers all overlap precisely with the known site (CACGT, CGTG, TCACG, TCACGT).
Biological insights—microarray expression
The ability to identify the primary conditions under which a transcription factor exerts control would be a critical component of any focused study of gene regulation. As we have seen, the w vector generated on a dataset indicates which of its components are most important for discriminating targets. In the case of gene expression classifiers, w elucidates which expression conditions are discriminatory. Intuitively, these are the conditions in which we would expect to see differential regulation of true targets. Given the predictions made using the combination of all methods, and the w obtained from the linear classifier built on expression data alone, we can see whether the predicted targets have differential regulation, and identify conditions where the TF is likely to act.
By the hypergeometric test, expression data is a significant predictor (p = 6.12e − 14) of targets for Fhl1, a forkhead-like TF known to be involved in rRNA processing and ribosomal protein gene expression. The w for this TF’s classifier from expression data has been calculated and sorted to determine the conditions having the highest weight. Figure 7 shows a plot of expression values over the top 25 conditions for the average yeast gene (solid blue), the average for genes in Fhl1’s negative set (dashed blue), the average in the positive set (dashed red), and the average in the most significant (P(true) ≥ 0.99) 48 targets of this TF (solid red).
Fig. 7.

Expression plot of Fhl1 targets over top 25 discriminative conditions. Average expression is plotted over all 5571 yeast genes (solid blue), over the negative set for Fhl1 (dashed blue), the positive targets (dashed red), and the most significant targets (solid red), P(true | distance from classifier) ≥ 0.99. The best targets have expression significantly different than the average or negative genes. The chosen expression conditions, ranked by w-vector from the expression based classifier, are shown under the graph with numbers indicating the position of the conditions in the graph. These conditions make sense since Fhl1 is regulated by the TOR signalling pathway, which is blocked by rapamycin. There is also some support in the literature for TOR having a role in meiosis and stress response
For 23 of the top 25 conditions the highly significant targets show expression which is different from both the average and the negative sets (t-test p-value ≤ 0.01). Most importantly, the best 10 ranked conditions contain six where yeast cells were treated with rapamycin and two involving meiosis/sporulation. This result is satisfying since rapamycin treatment specifically inhibits the Target of Rapamycin (TOR) signalling pathway, which is known to activate ribosomal protein expression as well as regulate several other pathways in yeast. Inhibition of TOR directly prevents Fhl1 from binding at promoter sites, thereby down-regulating expression of ribosomal protein genes (Martin et al. 2004), explaining why Fhl1 targets show differential expression in these experiments. Furthermore, although Fhl1 has not been directly implicated in meiosis, TOR pathway kinases are required for meiosis (Zheng and Schreiber 1997), indirectly suggesting that Flh1 might be involved. This is a reasonable suggestion since Fhl1 has been shown to alter its activity in response to factors (mainly Sfp1 which is also under TOR control) controlling progression to Start in the yeast cell cycle. Thus the most highly ranked experiments seem to correlate well with the real biological roles of the TF, indicating that the SVM can correctly rank important experimental conditions. Our method can identify differential regulation as an important predictor of target genes (hypergeometric test) and use the SVM-based classifier to make testable hypothesis about which conditions show biological effects of transcription factor activity.
Biological insights—PSSM comparison
We have found that support vector classification performs better than a simple weight matrix scan, and the combination of 26 methods outperforms any one method by itself. In some sense, a direct comparison with these PSSMs is not entirely fair since a majority of the weight matrices used here were created by motif discovery procedures rather than directed experimentation (such as DNA footprinting). Also, carefully constructed variants of PSSMs, which may take into account motif conservation in multiple species or interdependence of bases, can offer state of the art motif detection. Unfortunately, sufficient data is not always available to build such detailed models. The purpose of our comparison is simply to highlight the improved performance of classification methods relative to the commonly available binding site models. Figure 8 shows the result of a comparison between simple PSSM scanning using the MotifScanner algorithm and predictions by SVM on combined data. The leftmost grouping is a result from scans using PSSMs for all 104 TFs against the positive and negative sets on which the SVMs were trained. The MotifScanner score threshold was chosen individually for each TF so that the specificity on the training set was held constant at 0.95. This makes comparison to the SVM classifiers more straightforward as overall specificity for the SVMs is 0.95. The grouping on the right restates the performance of the SVMs with 26 combined datasets on the full set of positives. The SVM classifiers outperform PSSMs in the number of detected positives. It is clear that loosening the thresholds for the PSSMs would allow for better coverage but degrade performance by increasing the number of false positive predictions. Support vector machine classifiers offer a good balance between sensitivity and false prediction.
Fig. 8.

SVM vs. PSSM scan. Left: PSSMs for 104 TFs scanned against positive and negative sets. Overall specificity is held constant to 0.95 to match that of the SVM results. Right: Overall results for SVM classifiers trained on weighted combination of 18 datasets
Biological insights—pathway control
Finally, we have applied the combined classifier for each TF to all promoters in the yeast genome in order to expand the known binding repertoire of each factor. On average, each classifier produced approximately 884 new targets. Although it is unlikely that this set is free of false positives, examining the data in the context of biochemical pathways can shed light on significant predictions, which can quickly elucidate new sites which are good candidates for further study.
Gcn4 is a transcription factor in yeast known to control genes in the amino acid biosynthetic pathway (Hinnebusch 1992), and SVM predictions match well with the known biology of Gcn4 control mechanisms. The final classifier for this TF has an F1 score of 0.89, sensitivity of 0.86, and PPV of 0.92. This TF is a master regulator which has known targets in at least 12 amino acid biosynthetic pathways and has been shown by gene expression to induce at least 1/10th of the yeast genome (Hinnebusch and Natarajan 2002). Figure 9 highlights some known targets of Gcn4 in methionine/threonine biosynthesis in the aspartate family pathway. Branch-points from this pathway can ultimately lead to the amino acids methionine, threonine, lysine, and isoleucine. This group is of particular interest to humans since they are essential and not synthesized in the human metabolism. Gcn4 is known to regulate the genes Hom3, Thr1 and Thr4 leading to threonine, lysine, and isoleucine. However, predictions by SVM indicate it also directly targets committed steps of methionine biosynthesis by binding Met2, Met17, and Met6, which are interesting targets for further study.
Fig. 9.

GCN4 and amino acid biosynthesis. Predictions by SVM match well with the known biology of Gcn4 control mechanisms. Pathway map generated taken from the Pathway Tool Omics Viewer at SGD (Christie et al. 2004)
Previously Gcn4 was known to indirectly influence synthesis of methionine by activating Met4, a transcription factor specific to methionine biosynthesis and sulphur metabolism (Mountain et al. 1993). It is feasible that regulation of these enzymes by both Gcn4 and target Met4 represents a transcriptional feed-forward loop. Such loops have been described before and can be advantageous to an organism by exhibiting sign-sensitive delay, since it may be useful to have a quick response when shifting to an OFF state and a slow response when turning back ON (Mangan et al. 2003).
The Rap1 DNA binding factor is a widely known regulator in the cell cycle, acting as a repressor or activator depending on its context. Rap1 is also a key element in the structure of yeast telomeres, where it plays a role in telomere silencing (Pina et al. 2003). In a seemingly contradictory role, Rap1 has also been shown to regulate several glycolytic enzymes, as shown in Fig. 10. The specificity of this glycolytic regulation is dependent on a second factor, Gcr2, which binds to the Rap1/Gcr1 complex but does not contact DNA directly (Deminoff and Santangelo 2001). New predictions by SVM in the pathways of sugar metabolism show good correspondence with expectations for Rap1 (Fig. 10). Most interestingly, the new predictions include both isoforms of the enzyme phosphofructokinase. This step, where fructose-6-phosphase is converted into fructose-1,6-bisphosphate, is the crucial step in sugar breakdown where most metabolic flux through the pathway is controlled (Zubay 1996).
Fig. 10.

Rap1 and glycolytic/TCA cycle reaction. Glycolysis leading to acetate and ethanol are shown. The gray box on the left contains a pathway overview of glycolysis, fermentation and the TCA cycle, where red connections are known and yellow are predicted. Rap1 can be seen to regulate key control points in glycolysis and the TCA cycle. Pathway map generated taken from the Pathway Tool Omics Viewer at SGD (Christie et al. 2004)
Also of significance is the prediction that Rap1 regulates malate dehydrogenase in the TCA cycle. Malate dehydrogenase is unique in the TCA cycle in that it has a very small equilibrium constant, meaning that the forward reaction from malate to oxaloacetate is highly unfavorable. This is generally overcome during aerobic growth since the subsequent reaction is extremely favorable (large free energy release). However, in the absence of oxygen the cell still requires certain intermediates which can now not be made in the normal way. Running the malate dehydrogenase reaction in reverse, a favorable direction, can provide a way to synthesize these intermediates (Zubay 1996). Rap1 is already known to regulate the conversion of acetaldehyde to ethanol via alcohol dehydrogenase, and the possible complementary control of malate dehydrogenase suggests a possible role for Rap1 in regulation of fermentative growth.
Conclusions
We have seen that support vector machines can accurately classify transcription factor binding sites using a wide range of genomic data types. Combining various information sources can reduce false positives and incrementally increase sensitivity, while post-processing of the data to assign posterior probabilities allows the selection of high confidence targets. Although the maximal margin of SVMs is resistant to over-fitting, it can be further abrogated by selecting the best features for classifier construction. Feature selection and clustering techniques can be used in future work to refine predictions and more efficiently compare the SVM to other learning machines (KNN, Bayes, and Neural Network) which do not easily handle high dimensional or correlated data.
Based on k-mer data, SVMs appear to be isolating appropriate features for classification where many known transcription factor binding sites overlap with highest ranked k-mers. Examination of melting temperature classifiers for YJR060W demonstrates the unique biological features of targets for that TF. Similarly, expression-based classifiers for Fhl1 show the conditions under which Fhl1 acts on its targets, pointing the way to testable hypotheses supported by data in the literature. Finally, targets of Gcn4 and Rap1, when put into the context of biological pathways, correspond well to published experiments and show the effectiveness of integrated classifiers for building system-wide gene regulatory networks. Future work will then involve development of methods to discover biologically significant features in different datasets based on classifier performance and intelligent dimension-reduction techniques to reduce noise and improve accuracy.
Electronic supplementary material
Acknowledgements
We acknowledge Steve Parker and Tom Tullius for providing the DNA hydroxyl cleavage predictions from their database, and Adam Gustafson and Evan Snitkin for the Homolog Conservation (method 8) profiles for yeast genes.
Footnotes
Electronic Supplementary Material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s11693-006-9003-3 and is accessible for authorized users.
Authors' contributions
DH coded the required software in Matlab and Perl, conceived of many of the design implementations, and wrote this article. All authors made contributions to this manuscript and developed the experimental design. CD initially conceived and motivated this work. All authors read and approved the final manuscript.
Contributor Information
Dustin T. Holloway, Email: dth128@bu.edu
Mark Kon, Email: mkon@bu.edu.
Charles DeLisi, Email: delisi@bu.edu.
References
- Acton T, Zhong H, Vershon A (1997) DNA-binding specificity of Mcm1: operator mutations that alter DNA-bending and transcriptional activities by a MADS box protein. Mol Cell Biol 17:1881–1889 [DOI] [PMC free article] [PubMed]
- Aerts S, Thijs G, Coessens B, Staes M, Moreau Y, De Moor B (2003) Toucan: deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res 31:1753–1764 [DOI] [PMC free article] [PubMed]
- Allocco D, Kohane I, Butte A (2004) Quantifying the relationship between co-expression, co-regulation, and gene function. BMC Bioinformatics 5:18 [DOI] [PMC free article] [PubMed]
- Balasubramanian B, Pogozelski WK, Tullius TD (1998) DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. PNAS 95:9738–9743 [DOI] [PMC free article] [PubMed]
- Baldino F (1989) High-resolution in situ hybridization histochemistry. Meth Enzymol 168:761–777 [DOI] [PubMed]
- Beer MA, Tavazoie S (2004) Predicting gene expression from sequence. Cell 117:185–198 [DOI] [PubMed]
- Benham CJ (1992) Energetics of the strand separation transition in superhelical DNA. J Mol Biol 225:835–847 [DOI] [PubMed]
- Benham CJ (1996) Duplex destabilization in superhelical DNA is predicted to occur at specific transcriptional regulatory regions. J Mol Biol 255:425–434 [DOI] [PubMed]
- Bergman S, Ihmels J, Barkai N (2003) Iterative signature algorithm for the analysis of large-scale gene expression data. Phys Rev 67:031902 [DOI] [PubMed]
- Birnbaum K, Benfey PN, Shasha DE (2001) cis Element/transcription factor analysis (cis/TF): a method for discovering transcription factor/cis element relationships. Genome Res 11:1567–1573 [DOI] [PMC free article] [PubMed]
- Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, Cutts T et al (2006) Ensembl 2006. Nucleic Acids Res 34:D556–D561 [DOI] [PMC free article] [PubMed]
- Bishop C (1995) Neural networks for pattern recognition. Oxford University Press
- Breslauer KJ, Frank R, Blocker H, Marky LA (1986) Predicting DNA duplex stability from the base sequence. PNAS 83:3746–3750 [DOI] [PMC free article] [PubMed]
- Bussemaker H, Li H, Siggia E (2001) Regulatory element detection using correlation with expression. Nat Genet 27:167–171 [DOI] [PubMed]
- Choi CH, Kalosakas G, Rasmussen KO, Hiromura M, Bishop AR, Usheva A (2004) DNA dynamically directs its own transcription initiation. Nucleic Acids Res 32:1584–1590 [DOI] [PMC free article] [PubMed]
- Christie KR, Weng S, Balakrishnan R, Costanzo MC, Dolinski K, Dwight SS, Engel SR, Feierbach B, Fisk DG, Hirschman JE et al (2004) Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res 32:D311–D314 [DOI] [PMC free article] [PubMed]
- Cliften PF et al [http://www.genetics.wustl.edu/saccharomycesgenomes/]. 2003a
- Cliften PF, Johnston M et al (2003b) Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301:71–76 [DOI] [PubMed]
- Conlon EM, Liu XS, Lieb JD, Liu JS (2003) Integrating regulatory motif discovery and genome-wide expression analysis. PNAS 100:3339–3344 [DOI] [PMC free article] [PubMed]
- Cora D, Di Cunto F, Provero P, Silengo L, Caselle M (2004) Computational identification of transcription factor binding sites by functional analysis of sets of genes sharing overrepresented upstream motifs. BMC Bioinformatics 5:57 [DOI] [PMC free article] [PubMed]
- Dean R (2005) Fungal Genomics Laboratory at North Carolina State University and the Broad Institute: Magnaporthe Sequencing Project: [ http://www.fungalgenomics.ncsu.edu, http://www.broad.mit.edu ]
- Deminoff SJ, Santangelo GM (2001) Rap1p requires Gcr1p and Gcr2p homodimers to activate ribosomal protein and glycolytic genes, respectively. Genetics 158:133–143 [DOI] [PMC free article] [PubMed]
- Elemento S, Tavazoie S (2005) Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol 6:R18 [DOI] [PMC free article] [PubMed]
- Emboss Website: [ http://www.emboss.sourceforge.net/apps/banana.html]
- Flickinger RA (2005) Transcriptional frequency and cell determination. J Theor Biol 232:151–156 [DOI] [PubMed]
- Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D (2000) Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 16:906–914 [DOI] [PubMed]
- Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma L-J, Smirnov S, Purcell S et al (2003) The genome sequence of the filamentous fungus Neurospora crassa. Nature 422:859–868 [DOI] [PubMed]
- Gao F, Foat B, Bussemaker H (2004) Defining transcriptional networks through integrative modelling of mRNA expression and transcription factor binding data. BMC Bioinformatics 5:31 [DOI] [PMC free article] [PubMed]
- Gasch A, Moses A, Chiang D, Fraser H, Berardini M, Eisen M (2004) Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLOS Biol 2:2202–2219 [DOI] [PMC free article] [PubMed]
- Goodsell D, Dickerson R (1994) Bending and curvature calculations in B-DNA. Nucleic Acids Res 22:5497–5503 [DOI] [PMC free article] [PubMed]
- Guyon I, Weston J, Barnhill S, Vapnik V (2002) Gene selection for cancer classification using support vector machines. Mach Learn 46:389–422 [DOI]
- Harbison C, Fraenkel E, Young R (2005) Web site: [http://www.jura.wi.mit.edu/fraenkel/download/release_v24/final_set/Final_InTableS2_v24.motifs]
- Harbison C, Fraenkel E, Young R et al (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431:99–104 [DOI] [PMC free article] [PubMed]
- Haverty P, Hansen U, Weng Z (2004) Computational inference of transcriptional regulatory networks from expression profiling and transcription factor binding site identification. Nucleic Acids Res 32:179–188 [DOI] [PMC free article] [PubMed]
- van Helden J (2003) Regulatory sequence analysis tools. Nucleic Acids Res 31:3593–3596 [DOI] [PMC free article] [PubMed]
- van Helden J (2004) Metrics for comparing regulatory sequences on the basis of pattern counts. Bioinformatics 20:399–406 [DOI] [PubMed]
- van Helden J, Collado-Vides J (1998) Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J Mol Biol 281:827–842 [DOI] [PubMed]
- Hinnebusch A (1992) General and pathway-specific regulatory mechanisms controlling the synthesis of amino acid biosynthetic enzymes in saccharomyces cerevisiae. In: Broach JR, Jones EW, Pringle JR (eds) The molecular and cellular biology of the yeast Saccharomyces: gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 319–414
- Hinnebusch AG, Natarajan K (2002) Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell 1:22–32 [DOI] [PMC free article] [PubMed]
- Holloway D, Kon M, DeLisi C (2006) Machine learning methods for transcription data integration. IBM J Res Develop Syst Biol 50: (in press)
- Hua S, Sun Z (2001a) A novel method of protein secondary structure prediction with high segment overlap measure:support vector machine approach. J Mol Biol 308:397–407 [DOI] [PubMed]
- Hua S, Sun Z (2001b) Support vector machine approach for protein subcellular localization prediction. Bioinformatics 18:721–728 [DOI] [PubMed]
- Ihmels J, Barkai N et al (2002) Revealing modular organization in the yeast transcriptional network. Nat Genet 31:370–377 [DOI] [PubMed]
- Ihmels J, Bergman S, Barkai N (2005) Barkai Lab: [http://www.barkai-serv.weizmann.ac.il/GroupPage/]
- Ihmels J, Bergman S, Barkai N (2004) Defining transcription modules using large-scale gene expression data. Bioinformatics 20:1993–2003 [DOI] [PubMed]
- Jaakola T, Diekhans M, Haussler D (1999) Using the Fisher kernel method to detect remote protein homologies. In: Proc Int Conf INtell Syst Mol Biol, AAAI Press, pp 149–158 [PubMed]
- Keles S, van der Laan MJ, Vulpe C (2004) Regulatory motif finding by logic regression. Bioinformatics 20:2799–2811 [DOI] [PubMed]
- Kellis M Website: [ http://www.broad.mit.edu/annotation/fungi/comp_yeasts/], 2003
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES (2003) Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423:241–254 [DOI] [PubMed]
- Kent NA, Eibert SM, Mellor J (2004) Cbf1p is required for chromatin remodelling at promoter-proximal CACGTG motifs in yeast. J Biol Chem 279:27116–27123 [DOI] [PubMed]
- Lanckriet G, Cristianini N, Jordan M, Noble WS (2004) A statistical framework for genomic data fusion. Bioinformatics 20:2626–2635 [DOI] [PubMed]
- Lee IT et al (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298:799–804 [DOI] [PubMed]
- Leslie C, Kuang R (2003) Fast kernels for inexact string matching. In: 16th Annual Conference on Computational Learning Theory and 7th Kernel Workshop Proceedings, pp 114–128
- Leslie C, Eskin E, Noble WS (2002) The Spectrum Kernel: a string kernel for SVM protein classification. In: Proceedings of the Pacific Symposium on Biocomputing, pp 564–575 [PubMed]
- Leslie CS, Eskin E, Cohen A, Weston J, Noble WS (2004) Mismatch string kernels for discriminative protein classification. Bioinformatics 20:467–476 [DOI] [PubMed]
- Mangan S, Zaslaver A, Alon U (2003) The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks. J Mol Biol 334:197–204 [DOI] [PubMed]
- Martin DE, Soulard A, Hall MN (2004) TOR regulates ribosomal protein gene expression via PKA and the forkhead transcription factor FHL1. Cell 119:969–979 [DOI] [PubMed]
- Masters KM, Parkhurst KM, Daugherty MA, Parkhurst LJ (2003) Native human TATA-binding protein simultaneously binds and bends promoter DNA without a slow isomerization step or TFIIB requirement. J Biol Chem 278:31685–31690 [DOI] [PubMed]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K et al (2005) TRANSFAC(R) and its module TRANSCompel(R): transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34:D108–D110 [DOI] [PMC free article] [PubMed]
- Mellor J, Wu J, DeLisi C (2004) Constructing networks with correlation maximization methods. Genome Informatics 15:149–159 [PubMed]
- Mountain H, Bytrom A, Korch C (1993) The general amino acid control regulates MET4, which encodes a methionine-pathway-specific transcriptional activator of Saccharomyces cerevisiae. Mol Microbiol 9:221–223 [DOI] [PubMed]
- Parker S, Greenbaum J, Benson G, Tullius TD (2005) Structure-based DNA sequence alignment. In: poster: 5th International Workshop in Bioinformatics and Systems Biology
- Pavlidis P, Noble WS (2001) Gene functional classification from heterogeneous data. In: RECOMB Conference Proceedings, pp 249–255
- Pavlidis P, Wapinski I, Noble WS (2004) Support vector machine classification on the web. Bioinformatics 20:586–587 [DOI] [PubMed]
- Pina B, Fernandez-Larrea J, Garcia-Reyero N, Idrissi F (2003) The different (sur)faces of Rap1p. Mol Genet Genomics 268:791–798 [DOI] [PubMed]
- Platt JC (1999) Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. In: Smola A, Bartlett P, Scholkopf D, Schuurmans D (eds) Advances in large margin classifiers. MIT Press, Cambridge, pp 61–74
- Pritsker M, Liu Y-C, Beer MA, Tavazoie S (2004) Whole-genome discovery of transcription factor binding sites by network-level conservation. Genome Res 14:99–108 [DOI] [PMC free article] [PubMed]
- Qian J, Lin J, Luscombe NM, Yu H, Gerstein M (2003) Prediction of regulatory networks: genome-wide identification of transcription factor targets from gene expression data. Bioinformatics 19:1917–1926 [DOI] [PubMed]
- Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277 [DOI] [PubMed]
- Satchwell S, Drew H, Travers A (1986) Sequence periodicities in chicken nucleosome core DNA. J Mol Biol 191:659–675 [DOI] [PubMed]
- Schneider T, Stephens R (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18:6097–6100 [DOI] [PMC free article] [PubMed]
- Schneider TD, Stormo GD, Gold L (1986) A Ehrenfeucht: information content of binding sites on nucleotide sequences. J Mol Biol 188:415–431 [DOI] [PubMed]
- Sholkopf B, Smola AJ (2002) Learning with Kernels. MIT Press, Cambridge
- Simonis N, Wodak SJ, Cohen GN, van Helden J (2004) Combining pattern discovery and discriminant analysis to predict gene co-regulation. Bioinformatics 20:2370–2379 [DOI] [PubMed]
- Smit A, Hubley R, Green P (2005) Repeatmasker Open 3.0:[http://www.repeatmasker.org]
- Stormo GD (2000) DNA binding sites: representation and discovery. Bioinformatics 16:16–23 [DOI] [PubMed]
- Tan P-N, Steinbach M, Kumar V (2005) Introduction to data mining. Pearson Education
- Tatusov RL, Lipman DJ (2005) dust. NCBI Toolkit: [http://www.ncbi.nlm.nih.gov/]
- The Mathworks: [ http://www.mathworks.com/]
- Tompa M et al (2005) Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 23:137–144 [DOI] [PubMed]
- Tullius TD, Greenbaum JA (2005) Mapping nucleic acid structure by hydroxyl radical cleavage. Curr Opin Chem Biol 9:127–134 [DOI] [PubMed]
- Wang W, Cherry JM, Botstein D, Li H (2002) A systematic approach to reconstructing transcription networks in Saccharomyces scerevisiae. PNAS 99:16893–16898 [DOI] [PMC free article] [PubMed]
- Wang M, Yang J, Chou K-C (2005) Using string kernel to predict signal peptide cleavage site based on subsite coupling model. Amino Acids 28:395–402 [DOI] [PubMed]
- Weston J, Elisseeff A, Bakir G, Sinz F et al (2005) SPIDER: object oriented machine learning library version 6: [http://www.kyb.tuebingen.mpg.de/bs/people/spider/]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W et al (2005) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 33:D39–D45 [DOI] [PMC free article] [PubMed]
- Workman CT, Stormo GD (2000) ANN-Spec: a method for discovering transcription factor binding sites with improved specificity. In: Pac Symp Biocomput, pp 467–478 [DOI] [PubMed]
- Wu J, Kasif S, DeLisi C (2003) Identification of functional links between genes using phylogenetic profiles. Bioinformatics 19:1–7 [DOI] [PubMed]
- Young Lab Web Data: [http://www.staffa.wi.mit.edu/cgi-bin/young_public/navframe.cgi?s=17&f=evidence]
- Yu H, Luscombe N, Qian J, Gerstein M (2003) Genomic analysis of gene expression relationships in transcriptional regulatory networks. Trends Genet 19:422–427 [DOI] [PubMed]
- Zheng X-F, Schreiber SL (1997) Target of rapamycin proteins and their kinase activities are required for meiosis. PNAS 94:3070–3075 [DOI] [PMC free article] [PubMed]
- Zhu Z, Pilpel Y, Church G (2002) Computational identification of transcription factor binding sites via a transcription-factor-centric-clustering (TFCC) algorithm. J Mol Biol 318:71–81 [DOI] [PubMed]
- Zien A, Ratsch G, Mika S, Scholkopf B, Lengauer T, Muller K-R (2000) Engineering support vector machine kernels that recognize translation initiation sites. Bioinformatics 16:799–807 [DOI] [PubMed]
- Zubay G (1996) Biochemistry, 4th edn. Columbia University, WCB Publishers, pp 297–335
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.