

Abstract
Trypanosoma brucei, the agent of human sleeping sickness and ruminant nagana, is the most genetically tractable representative of the domain Excavata. It is evolutionarily very distant from humans, with a last common ancestor over 1 billion years ago. Frataxin, a highly conserved small protein involved in iron-sulfur cluster synthesis, is present in both organisms, and its deficiency is responsible for Friedreich's ataxia in humans. We have found that T. brucei growth-inhibition phenotype caused by down-regulated frataxin is rescued by means of human frataxin. The rescue is fully dependent on the human frataxin being imported into the trypanosome mitochondrion. Processing of the imported protein by mitochondrial processing peptidase can be blocked by mutations in the signal peptide, as in human cells. Although in human cells frataxin must be processed to execute its function, the same protein in the T. brucei mitochondrion is functional even in the absence of processing. Our results illuminate remarkable conservation of the mechanisms of mitochondrial protein import and processing.
Keywords: mitochondria protein processing, kinetoplastida, evolutionary conservation, function rescue, import
Trypanosomes and related kinetoplastid flagellates are responsible for several serious diseases of humans and animals. Trypanosoma brucei, the agent of human sleeping sickness, has become a widely used model organism, because it is among the few protists amenable to all main approaches of reverse genetics. Hardly any other eukaryote contains as many oddities as T. brucei. Its mitochondrion is an illustrious example of a departure from solutions generally adopted by the mitochondria of most other eukaryotic cells (1). These unusual features and the enormous evolutionary distance from mammals (2) would seem to disqualify trypanosomes for functional analysis of human genes. Yet it seems worth trying, because the unicellular background may be particularly useful for discerning the ancestral functions of proteins that have been proposed to participate in multiple different capacities and have been found in different cellular compartments in multicellular organisms. Moreover, because currently favored model organisms fall in only 2 of the 5 major domains of life, namely Plantae and Unikonts (2), there is a need to establish model organisms representing the other domains. Trypanosomes belong to the arguably most ancient domain, Excavata. We therefore decided to test the potential of trypanosomes for functional analysis of human genes by selecting human frataxin.
Frataxin is a conserved multifunctional protein that plays a role in iron storage, iron-sulfur (Fe-S) cluster and heme synthesis, repair of oxidatively damaged Fe-S clusters in aconitase, and control of reactive oxygen species (ROS) and has been proposed as a component of the respiratory pathway (3–5). Frataxin has been implicated further in complex phenotypes such as longevity and antioxidant defense. Its dysfunction causes Friedreich's ataxia, a common wasting disorder in humans (6, 7). The bacterial homologue of frataxin, CyaY, is evolutionarily rather divergent from it; moreover, CyaY is not essential, because prokaryotes operate several functionally distinct Fe-S cluster assembly pathways (8, 9). Accordingly, the yeast model would seem to be more suitable than prokaryotes for the investigation of frataxin function (10–12). Indeed, our knowledge about this versatile protein comes from yeast and to some extent also from other model eukaryotes (13–15). Recently, however, key insights into the functions of frataxin have come from studies that used conditional knockout mice (16) and from tissue cultures obtained from patients afflicted with Friedreich's ataxia (17, 18). These studies are extremely important but face the difficulty of determining whether the complex phenotypes seen in frataxin mutants or in tissues deficient for frataxin are primary or whether they arise as a consequence of secondary defects caused by the disruption of Fe-S cluster assembly.
We have sought to ascertain which mitochondrial import signal will be sufficient and efficient for recruiting human frataxin (frataxin) into the trypanosome mitochondrion. We demonstrate that frataxin must be confined to the mitochondrion to compensate for the lack of T. brucei frataxin (Tb-frataxin) down-regulated by RNAi. Moreover, our observation that frataxin is processed in an equally complex manner in T. brucei as in human cells, although this processing does not seem to be essential for the functions assayed, has interesting evolutionary implications. We suggest that our results justify using T. brucei for functional analysis of human genes, unorthodox as this may seem.
Results
Preparation of Constructs.
In human cells, frataxin is synthesized as a precursor that is, upon import into the mitochondrion, proteolytically matured with at least 2 sequential cleavages by mitochondrial processing peptidase (MPP) (10), although an alternative processing has been postulated recently (18) (Fig. 1A). To investigate the effect of different cellular localizations of frataxin on promoting cell survival, we took advantage of a T. brucei cell line in which Tb-frataxin can be eliminated inducibly by RNAi (14). For this purpose, we have prepared 3 constructs containing the frataxin gene that differ in their 5′ regions, allowing constitutive expression of the inserted gene upon stable integration in the trypanosome genome (Fig. 1B). The full-length construct (frataxin1–210) includes the 55-aa frataxin mitochondrial import signal. In another construct (frataxinTb-56–210) the frataxin gene (56–210 aa) is preceded by the 14-aa mitochondrial import signal for T. brucei dihydrolipoamide dehydrogenase. Finally, a third construct (frataxin56–210) contains frataxin without any import signal, leaving only mature form 1 (m1) (Fig. 1B). All 3 constructs were introduced into T. brucei Tb-frataxin RNAi knockdown cells. Although Tb-frataxin and frataxin share only 33% aa sequence similarity, both seem to have a conserved 3-dimensional structure composed of 6 β sheets and 2 α helices [supporting information (SI) Fig. S1].
Fig. 1.
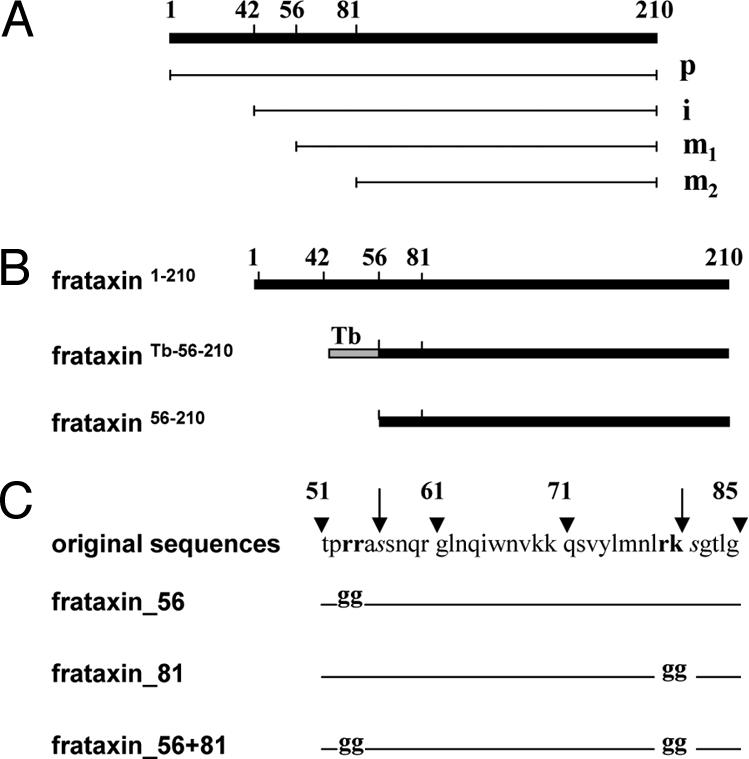
Schematic representation of human frataxin processing and constructs. (A) Frataxin precursor (1–210) (p), its intermediate processing product (42–210) (i), mature form 1 (56–210) (m1), and mature form 2 (81–210) (m2). (B) Three different constructs expressing frataxin in the pABPURO vector with or without mitochondrial import sequence: full-length human frataxin (frataxin1–210), frataxin preceded by the 14-aa mitochondrial import sequences of the T. brucei dihydrolipoamide dehydrogenase (frataxinTb-56–210), and frataxin without import signal (frataxin56–210). (C) Three constructs expressing different site-specific mutants of full-length of frataxin in the pABPURO vector. The arginines 53, 54, and 79 and lysine 80 were replaced with glycines, producing constructs frataxin_56, frataxin_81, and frataxin_56 + 81. Arrowheads and arrows indicate aa positions and MPP cleavage sites, respectively.
Survival of T. brucei Is Promoted by Frataxin1–210 but Not by Frataxin56–210.
Upon RNAi induction, Tb-frataxin is virtually eliminated within 2 days, and cell growth is completely inhibited by day 7 (14). However, the picture is dramatically different in other cell lines. RNAi-induced cells expressing frataxin1–210 or frataxinTb-56–210 retain almost the same growth as their noninduced counterparts (Fig. 2 A and B). However, the presence of frataxin56–210 did not rescue at all of the growth of cells lacking Tb-frataxin (Fig. 2C).
Fig. 2.

Effect of constructs on parasite growth. Numbers of noninduced cells (gray squares) and RNAi-induced cells (black triangles). (A) Growth of the noninduced and RNAi-induced Tb-frataxin knockdown transfected with constitutively expressed frataxin1–210. (B) Growth of the noninduced and RNAi-induced Tb-frataxin knockdown transfected with frataxinTb-56–210. (C) Growth of the noninduced and RNAi-induced Tb-frataxin knockdown transfected with frataxin56–210. Cell densities were measured daily to day 12 using a Beckman Coulter Z2 cell counter. The arrow represents the sampling site for experiments.
Western blot analysis with α-Tb-frataxin antibodies confirms elimination of the protein in all cell lines by day 2 upon RNAi induction (Fig. 3). Antibodies against human frataxin (α-frataxin) reveal strong constitutive translation of the protein; however, a strikingly different processing pattern appeared in individual cell lines. Surprisingly, frataxin was processed in T. brucei exactly as in human cells (Fig. 3A), producing a large abundance of the fully processed mature form 2 (m2). In human cells, cleavage of the precursor after Gly-41 is followed by another MPP-mediated processing after Ala-55 or Lys-80, releasing either 155- or 130-aa m1 or m2, respectively (18). The processing pattern of frataxin1–210 in the transfected flagellates is virtually identical with that described in human cells. We thus propose that the very weak uppermost band is the preprocessed protein (p), the prominent upper band is frataxin without the import signal (i), the weak lower band represents m1, and, finally, the fully processed and abundant mature protein m2 is the lowest strong band. The distinctive feature of this pattern, compared with human cells, is the joint presence, although in different amounts, of m1 and m2 (Fig. 3A).
Fig. 3.
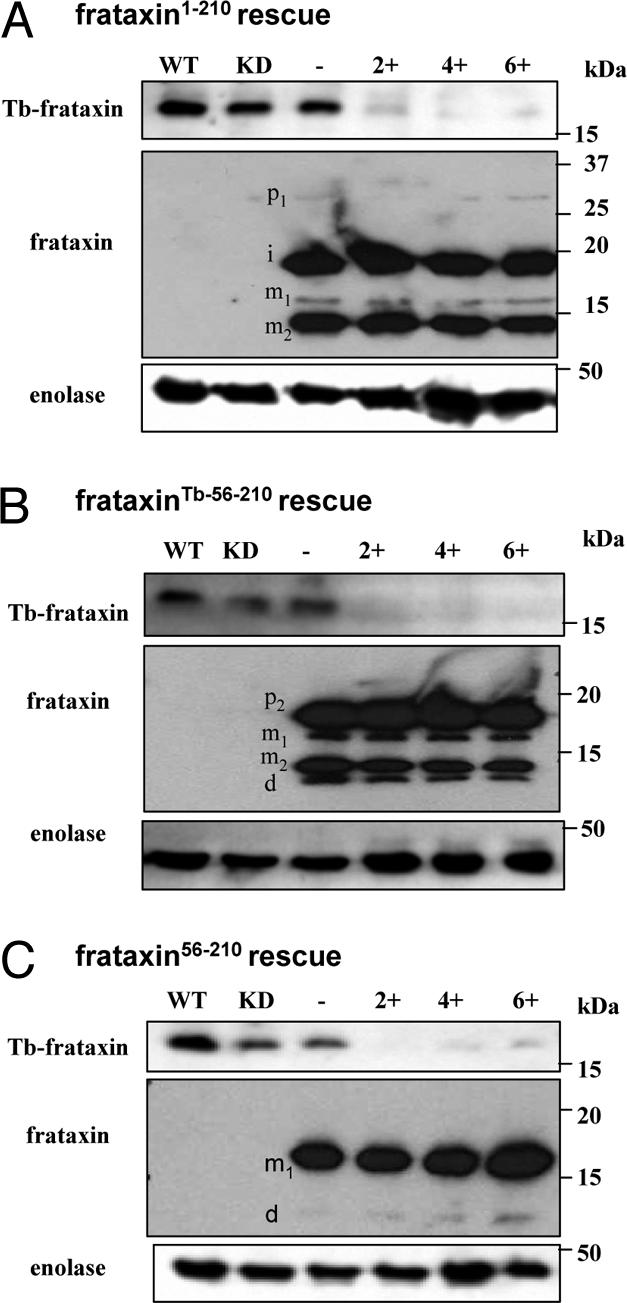
Western blot analysis of frataxin in rescued cell lines. Analysis of frataxin in noninduced and induced cell lines. (A) Cells transfected with frataxin1–210, (B) frataxinTb-56–210, and (C) frataxin56–210. Upon RNAi induction, Tb-frataxin was completely eliminated on day 2 postinduction (Upper); different forms of frataxin were stably and constitutively expressed. 2.5 × 106 cells were analyzed by SDS/PAGE and immunoblotted with α-Tb-frataxin, α-frataxin, and α-enolase antibodies. Cytosolic enolase (Lower) was used as a loading control. −, noninduced cell lines; +, RNAi-induced cell lines (2, 4, and 6 days of tetracycline induction); d, degradation product; i, intermediate form; KD, noninduced Tb-frataxin RNAi knockdown; m1, mature form 1; m2, mature form 2; p1, preprocessed (precursor) form 1; p2, (precursor) form 2; WT, wild-type T. brucei procyclic cells.
A somewhat different but also complex processing pattern appeared in the cells bearing frataxinTb-56–210 (Fig. 3B). Because of the length of the T. brucei mitochondrial import signal used, the preprocessed form (p2) is shorter. Because of its inefficient removal, the preprocessed protein is the most abundant form. Both mature forms m1 and m2 are present, although, as in the frataxin1–210 cells, the m2 form is much more abundant (Fig. 3B). We consider the weak, fastest migrating band to be a degradation product (d). The simplest pattern, composed of the highly abundant unprocessed cytosolic m1 and its very weak putative degradation product (d), is found in the frataxin56–210 cells (Fig. 3C).
Frataxin1–210 Is Efficiently Targeted and Processed in the T. brucei Mitochondrion.
To explain the dramatic differences in processing among the tested frataxin constructs in procyclic T. brucei, we subjected all 3 cell lines to fluorescence microscopy. Using α-frataxin antibody we show that in the frataxin1–210 cells all frataxin is distributed abundantly and evenly throughout the reticulated mitochondrion, as confirmed by its colocalization with the mitochondrial RNA-binding protein 2 (MRP2) and DAPI-stained kinetoplast DNA (Fig. 4A). Colocalization with MRP2 was less prominent in the frataxinTb-56–210 cells, which appeared to contain frataxin both in the organelle and the cytoplasm, the nucleus being the only cellular compartment clearly devoid of frataxin (Fig. 4A). Finally, in the frataxin56–210 cells, the target protein was abundantly present in the cytoplasm and did not colocalize with MRP2 (Fig. 4A).
Fig. 4.

Mitochondrial localization of frataxin in rescued cell lines. (A) Immunolocalization of frataxin. Cells stably expressing frataxin1–210, frataxinTb-56–210, or frataxin56–210 were fixed and treated with α-frataxin mouse monoclonal antibody and α-MRP2 rabbit polyclonal antibody, followed by donkey α-mouse antibody (green) and donkey α-rabbit antibody (red). Finally, the cells were stained with DAPI. The merged images show complete (frataxin1–210), partial (frataxinTb-56–210), and lack of co-localization (frataxin56–210) of frataxin with the mitochondrial protein MRP2. Merge 1 is given for colocalization of frataxin with MRP2. Merge 2 is given for colocalization of frataxin and DAPI. (B) Immunoblot analysis of frataxin expression in whole-cell lysates and extracted cytosolic and mitochondrial fractions of T. brucei frataxin RNAi knockdown transfected with constitutively expressed frataxin1–210, frataxinTb-56–210, or frataxin56–210. MRP2 and enolase proteins were used as mitochondrial and cytosolic controls, respectively. The labeling of p2, i, m1 and m2 as in Figs. 1 and 3.
These observations were corroborated by cell fractionation. Subcellular fractions were analyzed with antibodies against MRP2 (mitochondrial marker) and enolase (cytosolic marker), as controls. This approach confirmed that the processing of frataxin is conserved between the protist and human cells showed that the 55-aa import signal of frataxin is sufficient for its efficient import into the T. brucei mitochondrion (Fig. 4B). In contrast, the trypanosome import signal proved rather inefficient in targeting the frataxin into the organelle. As expected, in the absence of any import signal, frataxin56–210 was confined to the cytosol (Fig. 4B). The lengthy fractionation procedure probably is the cause of the appearance of an abundant degradation band (d) that is less pronounced in total cell lysates (Fig. 3).
Extramitochondrial Frataxin56–210 Does Not Restore Activities of Cytosolic and Mitochondrial Fe-S Proteins.
The rescue of the growth phenotype (Fig. 2) strongly indicates that the full-size frataxin1–210 is able to reverse the deficiency of Tb-frataxin almost completely. We now have dissected the phenotypes of the rescued cells. A marker Fe-S protein, aconitase, is present in the cytosol and mitochondrion of trypanosomes, and its activity depends on proper biogenesis of Fe-S clusters (19). Because in cells with down-regulated Tb-frataxin, cytosolic and mitochondrial activities of aconitase drop to ≈ 20% and 40% of the wild-type level, respectively (Fig. 5A), the subcellular fractions were tested for the ability of frataxin to restore aconitase activity in both compartments. Indeed, in the RNAi-induced frataxin1–210 cells, the activity of cytosolic and mitochondrial aconitase is restored to ≈70% and 90% levels, respectively (Fig. 5B). Such a powerful rescue of activity, however, did not occur in the frataxinTb-56–210 cells, where, respectively, <30% and 60% of the aconitase activity was present 5 days after RNAi induction (Fig. 5C). Finally, the activity was not restored in any compartment of the cells containing only cytosolic frataxin (frataxin56–210) (Fig. 5D). Immunoblot analysis of mitochondrial and cytosolic markers unequivocally confirmed the purity of both cellular fractions (Fig. 5).
Fig. 5.

Rescue effects on aconitase activity. The percentage of specific aconitase activity in total cell lysate, cytosol, and mitochondria in wild-type cells (striped bars), noninduced cells (white bars), and induced cells (gray bars). The mean and the SD values of 3 independent inductions are shown. (A) RNAi knockdown cell line for Tb-frataxin. (B–D) Cells stably expressing frataxin1–210 (B), frataxinTb-56–210 (C), or frataxin56–210 (D). The purity of cellular fractions obtained from noninduced (−) and induced (+) cells of these described cell lines was controlled with α-enolase (cytosolic marker) and α-MRP2 antibodies (mitochondrial marker).
Assaying the activity of another Fe-S cluster containing protein with dual cellular localization, fumarase, reveals a pattern virtually identical with that described for aconitase. In the absence of Tb-frataxin, fumarase activity drops to <20% of the wild-type level, and the activity stays at this level when frataxin is restricted to the cytosol (frataxin56–210). However, exclusive mitochondrial localization of frataxin1–210 leads to an almost full rescue of fumarase activity, whereas only a moderate rescue is observed in the frataxinTb-56–210 cells (Fig. S2A). Finally, the activity of another mitochondrially located Fe-S cluster enzyme, succinate dehydrogenase, confirmed that the mitochondrial localization of frataxin is a prerequisite for the Fe-S cluster assembly pathway in T. brucei (Fig. S2B). As anticipated, the activity of threonine dehydrogenase, an enzyme lacking Fe-S clusters, remains at the wild-type level in all studied cells (Fig. S2C).
Mitochondrial Frataxin Is Needed for Mitochondrial Maintenance.
Because respiratory complexes are needed to uphold mitochondrial membrane potential, the disruption of the assembly of Fe-S clusters abundant in these protein complexes is expected to have direct consequences on the inner membrane potential. As in other eukaryotes, the uptake of tetramethylrodamine ethyl ester (TMRE) by the T. brucei mitochondrion can be quantified by flow cytometry. A significant decrease of membrane potential in cells interfered against Tb-frataxin (Fig. S3) can be rescued not only by highly efficient import of frataxin1–210 into the mitochondrion, but surprisingly, also by frataxinTb-56–210 that is recruited to the organelle with a low efficiency. Once again, frataxin56–210, confined to the cytosol, failed to elicit any effect on mitochondrial membrane potential (Fig. S3).
The down-regulation of frataxin triggers the accumulation of ROS in trypanosomes (14) (Fig. S3) and in other eukaryotes (7). In agreement with the other assays, the full-length frataxin1–210 lowers the amount of ROS to the noninduced level, whereas in its cytosolic localization frataxin exerted no effect on the very high ROS levels in the RNAi cells (Fig. S3). It is worth noting that the inefficient import of frataxin into the T. brucei mitochondrion (frataxinTb-56–210) is, in contrast to its effect on membrane potential, insufficient for rescuing the ROS phenotype (Fig. S3).
Processing of Frataxin in T. brucei Is Not Necessary for its Function.
Based on the results recently obtained with site-specific mutants of frataxin in cells of patients suffering from Friedreich's ataxia (18), 3 mutant constructs (frataxin_56, frataxin_81, and frataxin_56+81) were generated by the replacement of critical amino acids (Fig. 1C) at the cleavage sites. Upon their stable transfection into the Tb-frataxin RNAi cells and inducible down-regulation of Tb-frataxin (data not shown) the in vivo maturation of mutant frataxins was followed using specific antibodies. When cleavage at the MPP processing site 56 is abrogated (frataxin_56), the resulting pattern is identical to the pattern found in cells expressing frataxin1–210, differing only in the m1 form, which cannot be detected even in overexposed Western blots (Fig. 6). Mutation of the downstream MPP processing site 81 (frataxin_81) leads to prominent changes in the pattern, namely the overaccumulation of the otherwise very rare m1 form and the disappearance of the m2 form. The most dramatic effect is associated with the Tb-frataxin RNAi cells expressing the frataxin_56+81, in which both mature forms are missing, with the consequent overaccumulation of preprocessed (p) and intermediate (i) proteins (Fig. 6).
Fig. 6.
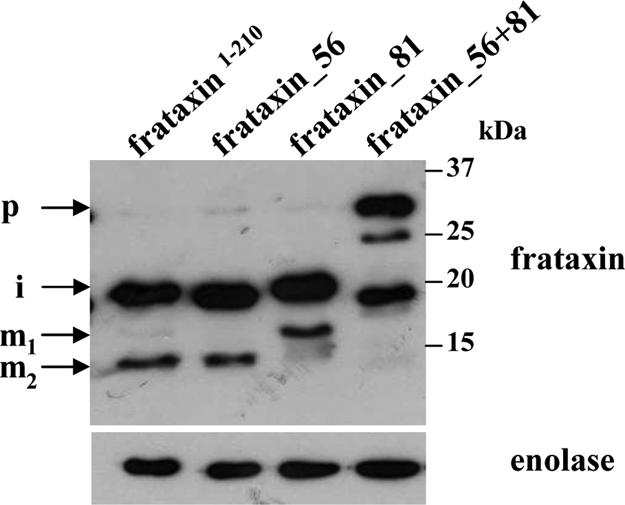
Processing of mutated frataxin in T. brucei. Western analysis of total lysates of the T. brucei Tb-frataxin RNAi knockdown cells stably expressing frataxin1–210 (frataxin1–210), frataxin1–210 with mutated processing site 56 (frataxin_56), frataxin1–210 with mutated processing site 81 (frataxin_81), and frataxin1–210 with mutated processing sites 56 and 81 (frataxin_56+81). The labeling of p, i, m1 and m2 as in Figs. 1 and 3. α-Enolase antibodies were used as a loading control.
The effect of the misprocessed frataxin on the survival of noninduced and induced T. brucei RNAi knockdowns was assessed by measurement of the activities of aconitase and fumarase (Fig. 7). Despite blocking 1 or both processing sites, the aconitase and fumarase activities were restored to the same extent as observed in cells containing frataxin1–210. This result was particularly unexpected for the cell line containing frataxin_56+81 (Fig. 7). Threonine dehydrogenase activities were the same in all cell lines, regardless of RNAi induction (data not shown).
Fig. 7.

Rescue effects by unprocessed frataxin on the activities of fumarase and aconitase. The percentage of specific aconitase (A) and fumarase (B) activities in total cell lysate in wild-type cells (striped bars), noninduced cells (white bars), and induced cells (gray bars). The mean and the SD values of 3 independent inductions are shown. Measurements were performed in the lysate of noninduced and induced Tb-frataxin RNAi knockdown cells and in the lysate from these cells stably expressing either frataxin1–210, frataxin with mutated processing site 56, frataxin with mutated processing site 81, and frataxin with both processing sites mutated. The T. brucei Tb-frataxin RNAi knockdown cells transfected with an empty pABPURO vector were used as a control.
Discussion
Along with diplomonads and trichomonads, kinetoplastids are organisms with relatively short organellar targeting sequences (20–22). Despite a rather distant evolutionary relationship, their targeting signals are interchangeable: the mitosomal signal of Giardia can target proteins into the hydrogenosome of Trichomonas, and vice versa (23), whereas the hydrogenosomal import sequence of Trichomonas is able to import a protein into the T. brucei mitochondrion (14). Finally, the mitochondrial targeting sequence of Trypanosoma cruzi is sufficient for the import of a protein into the mitosome of Entamoeba histolytica (24). The shared simplicity of the presequences, along with the apparent absence of several components of the protein import machinery in these early-diverged protist lineages, has been interpreted recently as an ancestral condition (23–28).
Our results indicate otherwise. Despite its length, the targeting signal of frataxin very efficiently mediates import into the T. brucei mitochondrion. The efficacy is by far superior to 1 of the experimentally tested trypanosome mitochondrial import signals (21). Such an efficient import mediated by a long signal is unexpected because in the available kinetoplastid genomes, potential homologues of the translocase in the outer mitochondrial membraneseem to be missing altogether, and only a single inner membrane translocase (TIM) has been identified so far (27). This TIM protein is equally diverged from the TIM22 and TIM23 translocases and is essential in T. brucei (28, 29). In human and yeast cells, matrix proteins, such as frataxin, traverse the inner membrane exclusively via the TIM23 translocase (30). A successful and efficient import of frataxin into the matrix therefore suggests that the single known TIM protein is capable of translocating both matrix and membrane-spanning proteins or that the genome of T. brucei harbors highly diverged translocases that remain undetected.
There are at present 2 views concerning the distribution of frataxin in human cells. Several research groups postulate that, in addition to its prevailing mitochondrial localization, frataxin also is present in the cytosol (18, 31–33). An alternative view holds that the protein is confined exclusively to the organelle (9, 16). The situation has been complicated further by the recent finding of frataxin in the cytosol of a microsporidian species and in the mitosome of another (34).
We anticipated that analyzing human frataxin in a heterologous system, such as T. brucei, amenable to reverse genetics approaches, may settle this controversy. Indeed, the capacity of the 3 tested frataxin expression constructs to rescue cells with RNAi-ablated Tb-frataxin is strikingly different. A full rescue is accomplished only through efficient import of the protein into the T. brucei mitochondrion. The mitochondrial localization of frataxin is required for reversion of the low activities of the mitochondrial and cytosolic Fe-S cluster-containing proteins in the Tb-frataxin RNAi knockdowns to the near wild-type levels. Similar results have been obtained recently with frataxin of the microsporidian Encephalitozoon cuniculi that was able to restore Fe-S cluster synthesis in yeast only when endowed with a mitochondrial presequence (34).
In yeast and mammalian cells, it is well established that frataxin deficiency affects the activity of aconitase in the cytosol and the mitochondrion (3, 35, 36). The same effect was exerted on aconitase and fumarase in both compartments but only in the T. brucei cells with mitochondrially localized frataxin. Neither the cytosolic nor the mitochondrial activity of both enzymes was restored when frataxin was confined to the cytosol.
Reflecting their bacterial ancestry, it is likely that mitochondrial processes can be highly conserved. Nevertheless, it is surprising that the complex processing pattern of frataxin by MPP, well documented in human and yeast cells (10, 18, 33), was retained in trypanosomes, despite the large evolutionary distance of more than 1 billion years (37). Indeed, single genes encoding MPP subunits α and β have been found in the T. brucei genome (38), although the amino acid sequence similarity with their human homologs is rather low (25% and 33% for the α and β subunits, respectively).
The notion that the specificity of MPP is conserved to such an extent is supported further by mutagenesis of 1 or both cleavage sites of frataxin that have fully abolished the processing, as in human cells. However, in humans individual ablation of either of the processing sites still allows functional rescue in Friedreich's ataxia cells, but parallel mutations of both sites cause the deficiency. This proves that the presence of at least 1 mature form, but not of the accumulated preprocessed and intermediate forms, is sufficient for the rescue (18). Surprisingly, in trypanosomes, the absence of both mature forms was able to rescue the lethal phenotype of Tb-frataxin RNAi deficiency, in virtually the same way as the properly processed mature forms. This observation deserves further investigation, but one can preliminarily conclude that, as long as frataxin enters the T. brucei mitochondrion, even its preprocessed precursor is able to acquire a functional conformation.
Our data further indicate that, contrary to in silico-based predictions, trypanosomes are endowed with mitochondrial import machinery capable of discerning complex targeting signals. Although the specificity of MPPs has remained highly conserved throughout evolution, at least in the case of frataxin in procyclic T. brucei, a specific processing does not seem to be necessary, given that the accumulated preprocessed form is able to perform frataxin functions. All in all, our data provide evidence for a remarkable conservation of the mechanisms of mitochondrial protein import and processing.
Materials and Methods
Plasmid Constructs, Transfection, RNAi Induction, Western Blot Analysis, and Digitonin Fractionation.
Frataxin amplified from the pcDNA3.1 construct was incorporated intact or mutated into various constructs based on the pABPURO vector (39) schematized in Fig. 1A. All constructs, prepared as described in SI Text, were linearized with BstXI and stably transfected into the Tb-frataxin RNAi knockdowns. The polyclonal rabbit antibodies against Tb-frataxin MRP2 and enolase were used as described previously (14, 19). The mouse monoclonal antibody against frataxin was obtained from Mitosciences (Clone ID: 18A5DB1) and was used at the suggested titer. DAPI staining, growth curves, and digitonin fractionation were performed following protocols described elsewhere (14, 19).
Measurement of Membrane Potential, ROS, and Enzymatic Activities.
Staining with TMRE (Molecular Probes) and dihydroethidium (Sigma Aldrich) and the measurement of the activities of succinate dehydrogenase, aconitase, fumarase, and threonine dehydrogenase were performed as described elsewhere (14, 19).
Immunocytochemistry.
Cells on polylysine-coated slides were fixed with 4% paraformaldehyde for 5 min, then permeabilized by −20°C methanol for 1 h, followed by incubation with the α-frataxin and MRP2 antibodies, with dilution ratio of 1:500 in PBS + 0.05% Tween-20 + 1% goat serum. Next, the slides were washed 3 times for 10 min each in PBS + 0.05% Tween-20 and then incubated with Alexa Fluor 488 donkey α-mouse IgG and Alexa Fluor 594 donkey α-rabbit IgG (Invitrogen) with a dilution ratio of 1:250. After 30 min incubation, the slides were washed as described previously, and the cells were stained with 1 μg/ml DAPI in PBS for 30 s, followed by a wash in PBS and a mount in Vectashield.
Supplementary Material
Acknowledgments.
We thank Hélene Puccio (University of Strasbourg) for a construct containing the human frataxin cDNA and Michael L. Ginger (Lancaster University), Dmitri A. Maslov (University of California), and 2 referees for comments on the manuscript. This work was supported by Awards 204/06/1558 from the Grant Agency of the Czech Republic, A500960705 from the Grant Agency of the Czech Academy of Sciences, and LC07032, 2B06129, and 600766580 from the Czech Ministry of Education.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0806762105/DCSupplemental.
References
- 1.Lukeš J, Hashimi H, Zíková A. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet. 2005;48:277–299. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- 2.Keeling PJ, et al. The tree of eukaryotes. Trends Ecol Evol. 2005;20:670–676. doi: 10.1016/j.tree.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Bulteau A-L, et al. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 4.González-Cabo P, Vazquez-Manrique RP, García-Gimeno MA, Sanz P, Palau F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum Mol Genet. 2005;14:2091–2098. doi: 10.1093/hmg/ddi214. [DOI] [PubMed] [Google Scholar]
- 5.Bencze KZ, et al. The structure and function of frataxin. Crit Rev Biochem Mol Biol. 2006;41:269–291. doi: 10.1080/10409230600846058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campuzano V, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 7.Gakh O, et al. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum Mol Genet. 2006;15:467–479. doi: 10.1093/hmg/ddi461. [DOI] [PubMed] [Google Scholar]
- 8.Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 9.Lill R, Muhlenhoff U. Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- 10.Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G. Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J Biol Chem. 2000;275:41469–41475. doi: 10.1074/jbc.M006539200. [DOI] [PubMed] [Google Scholar]
- 11.Lesuisse E, et al. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1) Hum Mol Genet. 2003;12:879–889. doi: 10.1093/hmg/ddg096. [DOI] [PubMed] [Google Scholar]
- 12.Cook JD, et al. Monomeric yeast frataxin is an iron binding protein. Biochemistry. 2006;45:7767–7777. doi: 10.1021/bi060424r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busi MV, et al. Deficiency of Arabidopsis thaliana frataxin alters activity of mitochondrial Fe-S proteins and induces oxidative stress. Plant J. 2006;48:873–882. doi: 10.1111/j.1365-313X.2006.02923.x. [DOI] [PubMed] [Google Scholar]
- 14.Long S, et al. Ancestral roles of eukaryotic frataxin: Mitochondrial frataxin function and heterologous expression of hydrogenosomal Trichomonas homologs in trypanosomes. Mol Microbiol. 2008;69:94–109. doi: 10.1111/j.1365-2958.2008.06260.x. [DOI] [PubMed] [Google Scholar]
- 15.Dolezal P, et al. Frataxin, a conserved mitochondrial protein, in the hydrogenosome of Trichomonas vaginalis. Eukaryot Cell. 2007;6:1431–1438. doi: 10.1128/EC.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martelli A, et al. Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum Mol Genet. 2007;16:2651–2658. doi: 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- 17.Rouault TA, Tong WH. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol. 2005;6:345–351. doi: 10.1038/nrm1620. [DOI] [PubMed] [Google Scholar]
- 18.Condo I, et al. In vivo maturation of human frataxin. Hum Mol Genet. 2007;16:1534–1540. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- 19.Smíd O, et al. Knockdowns of mitochondrial iron-sulfur cluster assembly proteins IscS and IscU down-regulate the active mitochondrion of procyclic Trypanosoma brucei. J Biol Chem. 2006;281:28679–28686. doi: 10.1074/jbc.M513781200. [DOI] [PubMed] [Google Scholar]
- 20.Priest JW, Hajduk SL. In vitro import of the Rieske iron-sulfur protein by trypanosome mitochondria. J Biol Chem. 1996;271:20060–20069. doi: 10.1074/jbc.271.33.20060. [DOI] [PubMed] [Google Scholar]
- 21.Hausler T, Stierhof Y-D, Blattner J, Clayton C. Conservation of mitochondrial targeting sequence function in mitochondrial and hydrogenosomal proteins from the early-branching eukaryotes Crithidia, Trypanosoma and Trichomonas. Eur J Cell Biol. 1997;73:240–251. [PubMed] [Google Scholar]
- 22.Uboldi AD, et al. A mitochondrial protein affects cell morphology, mitochondrial segregation and virulence in Leishmania. Int J Parasitol. 2006;36:1499–1514. doi: 10.1016/j.ijpara.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Dolezal P, et al. Giardia mitosomes and trichomonad hydrogenosomes share a common mode of protein targeting. Proc Natl Acad Sci USA. 2005;102:10924–10929. doi: 10.1073/pnas.0500349102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tovar J, Fischer A, Clark CG. The mitosome, a novel organelle related to mitochondria in the amitochondrial parasite Entamoeba histolytica. Mol Microbiol. 1999;31:1013–1021. doi: 10.1046/j.1365-2958.1999.01414.x. [DOI] [PubMed] [Google Scholar]
- 25.Dolezal P, Likic V, Tachezy J, Lithgow T. Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313:314–318. doi: 10.1126/science.1127895. [DOI] [PubMed] [Google Scholar]
- 26.Burri L, Keeling PJ. Protein targeting in parasites with cryptic mitochondria. Int J Parasitol. 2007;37:265–272. doi: 10.1016/j.ijpara.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Schneider A, Bursac D, Lithgow T. The direct route: A simplified pathway for protein import into the mitochondrion of trypanosomes. Trends Cell Biol. 2008;18:12–18. doi: 10.1016/j.tcb.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Singha UK, et al. Characterization of the mitochondrial inner membrane protein translocator Tim17 from Trypanosoma brucei. Mol Biochem Parasitol. 2008;159:30–43. doi: 10.1016/j.molbiopara.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gentle IE, et al. Conserved motifs reveal details of ancestry and structure in the small TIM chaperones of the mitochondrial intermembrane space. Mol Biol Evol. 2007;24:1149–1160. doi: 10.1093/molbev/msm031. [DOI] [PubMed] [Google Scholar]
- 30.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 31.Acquaviva F, et al. Extramitochondrial localization of frataxin and its association with IscU1 during enterocyte-like differentiation of the human colon adenocarcinoma cell line Caco-2. J Cell Sci. 2005;118:3917–3924. doi: 10.1242/jcs.02516. [DOI] [PubMed] [Google Scholar]
- 32.Lu C, Cortopassi G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch Biochem Biophys. 2006;457:111–122. doi: 10.1016/j.abb.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Condo I, Ventura N, Malisan F, Tomassini B, Testi R. A pool of extramitochondrial frataxin that promotes cell survival. J Biol Chem. 2006;281:16750–16756. doi: 10.1074/jbc.M511960200. [DOI] [PubMed] [Google Scholar]
- 34.Goldberg AV, et al. Localization and functionality of microsporidian iron- sulfur cluster assembly. Nature. 2008;452:624–628. doi: 10.1038/nature06606. [DOI] [PubMed] [Google Scholar]
- 35.Stehling O, Elsasser HP, Bruckel B, Muhlenhoff U, Lill R. Iron-sulfur protein maturation in human cells: Evidence for a function of frataxin. Hum Mol Genet. 2004;13:3007–3015. doi: 10.1093/hmg/ddh324. [DOI] [PubMed] [Google Scholar]
- 36.Seznec H, et al. Friedreich ataxia: The oxidative stress paradox. Hum Mol Genet. 2005;14:463–474. doi: 10.1093/hmg/ddi042. [DOI] [PubMed] [Google Scholar]
- 37.Brinkmann H, Philippe H. The diversity of eukaryotes and the root of the eukaryotic tree. Adv Exp Med Biol. 2007;607:20–37. doi: 10.1007/978-0-387-74021-8_2. [DOI] [PubMed] [Google Scholar]
- 38.Berriman M, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 39.Rusconi F, Durand-Dubief M, Bastin P. Functional complementation of RNA interference mutants in trypanosomes. BMC Biotechnol. 2005;5 doi: 10.1186/1472-6750-5-6. Art No 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.