

Abstract
It is well known that ethanol damages the developing nervous system by augmenting apoptosis. Previously, this laboratory reported that ethanol augments apoptosis in fetal rhombencephalic neurons, and that the increased apoptosis is associated with reduced activity of the phosphatidylinositol 3’kinase pathway and downstream expression of pro-survival genes. Other laboratories have shown that another mechanism by which ethanol induces apoptosis in developing neurons is through the generation of reactive oxygen species (ROS) and the associated oxidative stress.
The present study used an in vitro model to investigate the potential neuroprotective effects of several antioxidants against ethanol-associated apoptosis in fetal rhombencephalic neurons. The investigated antioxidants included three phenolics: (-)-epigallocatechin-3-gallate (EGCG), a flavanoid polyphenol found in green tea; curcumin, found in tumeric; and resveratrol (3,5,4’-trihydroxystilbene), a component of red wine. Additional antioxidants, including melatonin, a naturally occurring indole, and α-lipoic acid, a naturally occurring dithiol, were also investigated.
These studies demonstrated that a 24-hour treatment of fetal rhombencephalic neurons with 75 mM ethanol caused a 3-fold increase in the percentage of apoptotic neurons. However, co-treatment of these cultures with any of the five different antioxidants prevented ethanol-associated apoptosis. Antioxidant treatment did not alter the extent of apoptosis in control neurons, i.e., those cultured in the absence of ethanol. These studies showed that several classes of antioxidants can exert neuroprotection against ethanol-associated apoptosis in fetal rhombencephalic neurons.
Keywords: ethanol, antioxidant, apoptosis, neuroprotection
Introduction
Fetal Alcohol Syndrome (FAS) is considered the most common preventable cause of mental retardation in the United States and affects approximately1-2 per 1,000 live births (Sampson et al., 1997). Prenatal exposure to alcohol can cause a range of disorders known as Fetal Alcohol Spectrum Disorders (FASD) of which FAS is the most severe. Children with FAS exhibit growth deficiencies, mental retardation, and specific craniofacial abnormalities (Jones and Smith, 1975; Mattson et al., 1996; Wattendorf, and Muenke, 2005). In utero ethanol exposure is also associated with impairments in learning, verbal skills, attention, visual-spatial skills, executive function, and memory (Mattson et al., 1996; Mattson et al., 1999). Human studies show that fetal exposure to alcohol can damage several developing CNS regions, including the cerebellum, basal ganglia, and corpus callosum (Roebuck et al., 1998; Riley et al., 2004); it also affects cortical thickness (Sowell et al., 2007).
In vitro and in vivo studies of animal models show that ethanol damages the developing CNS by reducing neurons in the cortex, cerebellum, hippocampus, and dorsal and median raphe (Marcussen, et al., 1994; Goodlett and Eilers, 1997; Tajuddin and Druse, 1999; 2001; Chen et al., 2001; Jacobs and Miller, 2001; Moulder, et al., 2002). Reportedly, a single high dose of ethanol, administered during a vulnerable developmental period, causes neurodegeneration in the rodent forebrain, caudate nucleus, nucleus accumbens, hippocampus, amygdala, thalamus, and cerebellum (Ikonomidou, et al., 2000; Dikranian et al., 2005. The CNS damage associated with early ethanol exposure adversely impacts several developing CNS neurotransmitter systems, e.g., those containing GABA, serotonin dopamine, noradrenaline, acetylcholine, and glutamate (reviewed by Druse, 1996; Goodlett and Horn, 2001).
Although the mechanism(s) by which ethanol damages the developing CNS are not fully elucidated, it is well established that apoptosis is involved. In fact, this laboratory finds that the loss of the developing serotonin (5-HT) neurons of the dorsal and median raphe region (Tajuddin and Druse, 1999; 2001) is caused by ethanol-associated apoptosis (Druse et al., 2004; 2005; 2007). Moreover, these effects are accompanied by a decrease in the activity of the phosphatidylinositol 3’kinase (PI-3K) → pAkt pro-survival pathway and the downstream reduction in the expression of NF-kB dependent genes that encode pro-survival/anti-apoptotic proteins (Druse et al., 2005- 2007), e.g. Bcl-2, Bcl-XL, X-inhibitor of apoptosis protein (XIAP), cIAP-1, and cIAP-2.
Another mechanism by which ethanol damages the developing CNS is through the generation of oxidative stress (Heaton et al., 2002; Ramachandran et al., 2003; Watts et al., 2005; Lee et al., 2007). In vitro ethanol exposure causes a brisk increase in reactive oxygen species (ROS) in cortical and fetal rhombencephalic neurons (Ramachandran et al., 2003; Lee et al., 2007); the ROS increase in cortical neurons is followed by mitochondrial-mediated apoptosis (Ramachandran et al., 2003). It appears that the extent of the ROS response to ethanol depends on the brain region examined and on the relative developmental vulnerability of that brain region (Heaton et al., 2003). Fetal cells are particularly vulnerable to oxidative stress because they have low levels of endogenous antioxidants (Ramachandran et al., 2003) and because ethanol alters levels of enzymatic antioxidants (Heaton et al., 2003; Watts et al., 2005).
The present study investigated the potential neuroprotective effects of several antioxidants, including three phenolics: (-)-epigallocatechin-3-gallate (EGCG), a flavanoid polyphenol found in green tea; curcumin, found in tumeric; and resveratrol (3,5,4’-trihydroxystilbene), a component of red wine (Zhuang et al., 2003). This study also evaluated potential neuroprotective effects of two other antioxidants: melatonin, a naturally occurring indole; and α-lipoic acid, a naturally occurring dithiol. Although these compounds all exert antioxidant/free radical chelating effects, they also mediate non-antioxidant functions that might contribute to their neuroprotective effects. Interestingly, it appears that the non-antioxidant functions of antioxidants involve several diverse mechanisms. Some of the non-antioxidant effects, i.e., those of α-lipoic acid (LA), dihydrolipoic acid (DHLA), melatonin, curcumin, and resveratrol, involve the maintenance of cellular levels of endogenous antioxidants and/or antioxidant enzymes (Suh et al., 2004; Shila et al., 2005; Barlow-Walden et al., 1995; Kotler et al., 1998; Zhuang et al., 2003; Juknat et al., 2005; Lin, 2007). In addition, some antioxidants, i.e., LA, curcumin, and EGCG can rapidly activate the PI-3K pro-survival pathway in certain cell types (Zhang et al., 2001; Muller et al., 2003; Koh et al., 2004; Antonio and Druse, 2006; Kang et al., 2007), suggesting that this effect may be directly linked with the action of these antioxidants. Additional pro-survival effects of antioxidants are also likely to be involved; EGCG can down-regulate the expression of several pro-apoptotic genes (Levites et al., 2002; Mandel et al., 2004).
Results
The present study investigated the potential of several antioxidants to attenuate ethanol-associated apoptosis. Cultures of fetal rhombencephalic neurons were maintained for 24 hours [day in vitro 5 (DIV5) to DIV6] in the presence of 0 mM (control) or 75 mM ethanol (ethanol) and in the presence or absence of either a single antioxidant, i.e., 1 μM EGCG, 1 μM melatonin, 1 μM curcumin, 10 μM α-lipoic acid, or 10 μM resveratrol. At the end of the treatment period, neurons were stained with Hoechst 33342 to identify and quantify neurons with fragmented/apoptotic or nonapoptotic nuclei.
Figure 1 includes pictures of neurons that were stained with Hoechst 33342, which identifies fragmented/apoptotic nuclei. Pictures of neurons treated with 0 or 75 mM ethanol are shown in Figures 1A and 1B and those of neurons co-treated with the five antioxidants are shown in Figures 1C-1L. The diverse sizes of nuclei reflect the different sizes and morphology of the mixed population of neuronal cells in these cultures. Each arrow identifies a single apoptotic nucleus or a cluster of fragments of an apoptotic nucleus. The results of analyses of Hoechst stained neurons are graphically presented in Figure 2 for clarity. Data from the antioxidant treatment groups and the associated figures follow: EGCG - Figures 1C & D, Figure 2A; curcumin - Figures 1E & 1F, Figure 2B); resveratrol - Figures 1G & 1H, Figure 2C; α-lipoic acid - Figures 1I & 1J, Figure 2D; and melatonin - Figures 1K & 1L, Figure 2E. For each “n”, all treatments with ethanol and each of the five antioxidants were conducted simultaneously.
Figure 1. Ethanol augments apoptosis in fetal rhombencephalic neurons in vitro, and antioxidants prevent the pro-apoptotic effects of 75 mM ethanol.
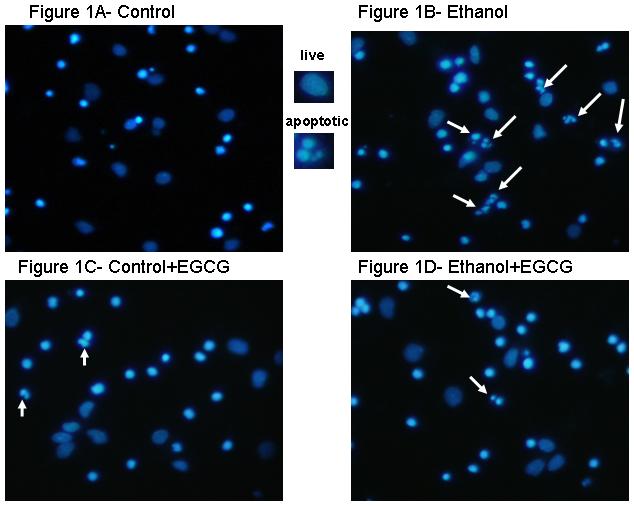
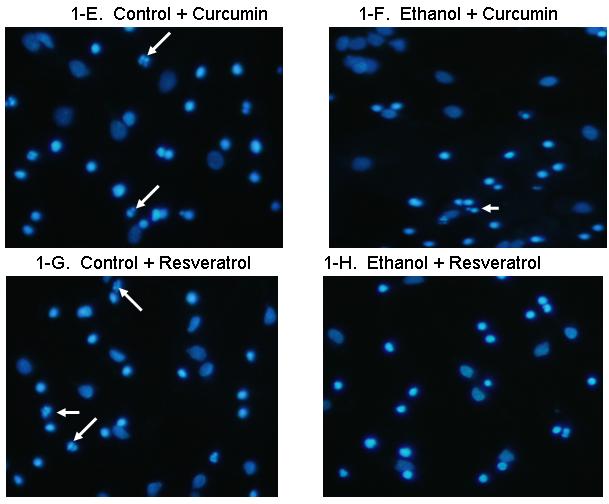
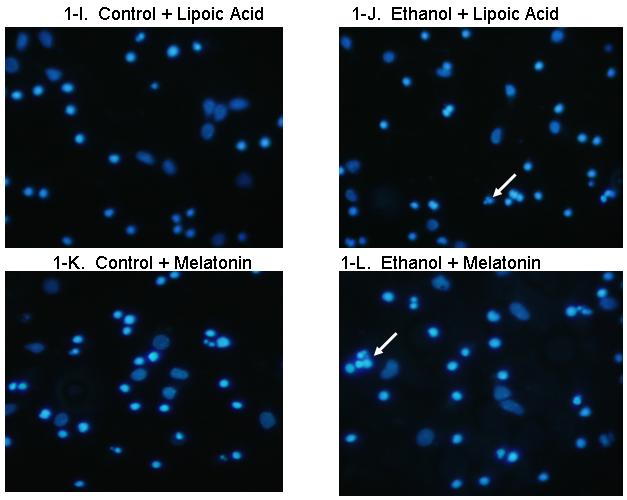
Figures 1A and 1B show fetal rhombencephalic neurons stained with Hoechst 33342 from cultures that were maintained under control conditions (no ethanol) or that were treated for the last 24 hours (DIV5 to DIV6) with 75 mM ethanol (ethanol). Control and ethanol-treated neurons treated with EGCG (1C, 1D), curcumin (1E, 1F), resveratrol (1G, 1H), LA (1I, 1J), and melatonin (1K, 1L) are also depicted. Hoechst-stained living and apoptotic neurons were identified as described. A higher magnification (see inset) was used to identify neurons that exhibit the characteristics of apoptotic cells. Arrows point to one or a cluster of fragmented/apoptotic nuclei.
Figure 2. A graphic depiction of the percentage of apoptotic neurons in cultures of fetal rhombencephalic neurons that were maintained under control conditions (no ethanol) or in the presence of 75 mM ethanol and treated with EGCG (2A), curcumin (2B), resveratrol (2C), LA (2D) or melatonin (2E).
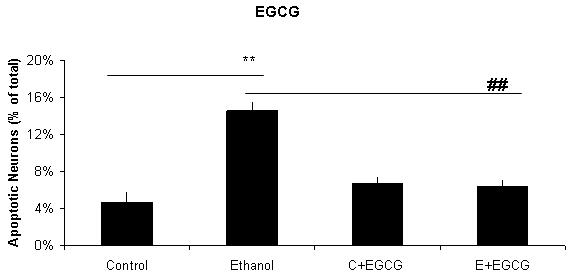
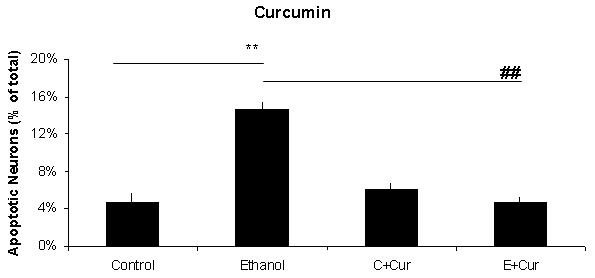
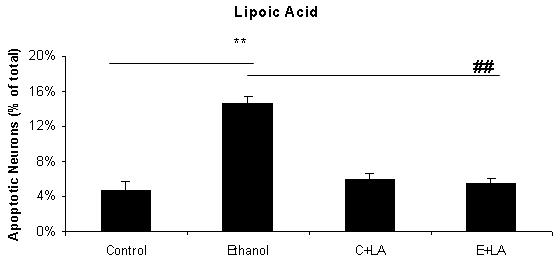
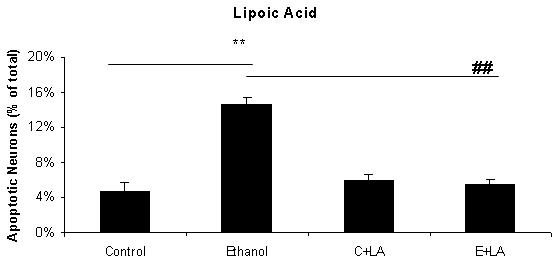
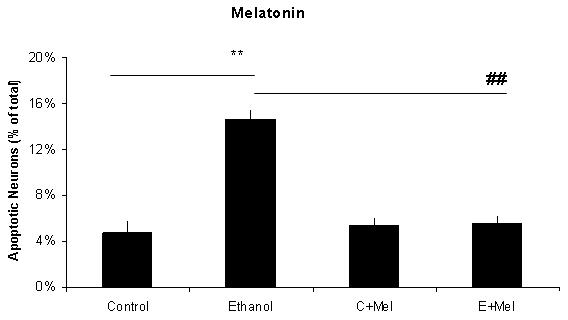
The ** indicates that ethanol treatment significantly augmented apoptosis (p < .01); this treatment tripled the number of apoptotic neurons. Each of these antioxidants exerted a significant main effect when cultures of fetal rhombencephalic neurons were co-treated with the antioxidant and ethanol [(1 μM EGCG (F(1,23) = 13.5, p < .01), 1 μM curcumin (F(1,23) = 20.2, p < .01), 10 μM resveratrol (F(1,23) = 22.3, p < .01), 10 μM α-lipoic acid (F(1,22) = 24.9, p < .01) , and 1 μM melatonin (F(1,23) = 19.4, p < .01)]. The ## indicates that the percentage of apoptotic neurons in ethanol-treated cultures was significantly decreased by co-treatment with antioxidant (p < .01). In fact, the percentage of apoptotic neurons in cultures co-treated with ethanol and each of the antioxidants was comparable to that in control cultures (p> .05). In addition, antioxidant treatment did not significantly alter the percentage of apoptotic neurons (p > .05) in control (no ethanol) cultures. In each of the 5-7 separate experiments, treatments with ethanol and each of five antioxidants were conducted simultaneously. Results are graphically depicted for each antioxidant for clarity of presentation.
The results depicted in Figures1 and 2 demonstrate that a 24-hour treatment of fetal rhombencephalic neurons with ethanol markedly augments apoptosis. In cultures maintained with no ethanol or antioxidants (control cultures), only 4.7% of fetal rhombencephalic neurons maintained under control conditions had apoptotic nuclei. However, the concentration of neurons with apoptotic nuclei tripled (p < .01) when fetal rhombencephalic neurons were maintained in the presence of 75 mM ethanol.
Co-treatment with each antioxidant provides significant neuroprotection to the ethanol-treated neurons {EGCG [F(1,23) = 13.5, p < .01], curcumin [F(1,23) = 20.2, p < .01], resveratrol [F(1,23) = 22.3, p < .01], α-lipoic acid [F(1,22) = 24.9, p < .01], and melatonin [F(1,23) = 19.4, p < .01]}. In fact, the percentage of apoptotic neurons in cultures co-treated with ethanol and antioxidant is comparable to those of control neurons (p> 0.05) as well as to control neurons treated with antioxidant (p> 0.05).
Discussion
The present study clearly demonstrates that ethanol augments apoptosis in fetal rhombencephalic neurons. This finding confirms earlier studies from this laboratory (Druse et al., 2004; 2005; 2007) and reports that detected ethanol-associated apoptosis in neurons from other brain regions. For example, previous studies found that ethanol increases apoptosis in the developing neural crest (Dunty et al., 2001) and cerebellar granule cells (Ramachandran et al., 2001) as well as in cells from cortex (Cheema et al., 2000) and forebrain (Ikonomidou et al., 2000). Very likely, the CNS damage found in children exposed to ethanol in utero is due in part to the ethanol-associated apoptotic reduction in CNS neurons.
Previously, this laboratory reported that one of the mechanisms by which ethanol caused cell death in fetal rhombencephalic neurons was likely to involve the decreased activity of the PI-3K prosurvival pathway and the reduced expression of downstream NF- κB dependent pro-survival genes, i.e., Bcl-2, Bcl-XL, XIAP, cIAP1, and cIAP2. Importantly, the neuroprotective effects of the 5-HT1A agonist ipsapirone (Druse et al., 2004-2006) and of the astroglial protein S100B (Druse et al., 2007) were accompanied by maintenance of normal to elevated activity of PI-3K-> pAkt (Druse et al., 2005; 2007). The latter effects were associated with increased expression of the genes encoding the anti-apoptotic proteins XIAP (ipsapirone or S100B treatments), Bcl-XL (ipsapirone) and/or Bcl-2 (S100B) (Druse et al., 2006; 2007) in ethanol-treated neurons.
It is significant that several laboratories (Heaton et al., 2002; Ramachandran et al., 2003; Watts et al., 2005), including this one (Lee et al., 2007), report that ethanol treatment is associated with increased production of ROS, because increased levels of ROS cause apoptotic cell death (Koh et al., 2003). The involvement of ROS in ethanol-associated apoptosis of fetal rhombencephalic neurons is emphasized by in vivo studies which show that exogenous antioxidants exert both neuroprotective (Heaton et al., 2004) and anti-teratogenic effects (Chen et al., 2004) against the damaging effects of ethanol. Moreover, the present investigation shows that all antioxidants investigated in this study, i.e. the phenols EGCG, curcumin and resveratrol; the indole melatonin; and the dithiol α-lipoic acid, prevent and/or attenuate ethanol-associated apoptosis.
The ability of the investigated antioxidants to prevent and/or attenuate ethanol associated apoptosis extends our understanding of their neuroprotective effects. Previous studies showed that LA treatment counteracts an arsenic-induced decrease in GSH levels in several brain regions (Shila et al., 2005), and that EGCG protects PC12 cells against the effects of serum withdrawal (Reznichenko et al., 2005) and oxidative stress (Koh etal., 2003). In addition, resveratrol prevents NO-related toxicity (Bastianetto et al., 2000) in hippocampal neurons and curcumin blocks toxicity from the Alzheimer’s disease related Aβ42 oligomer in neuroblastoma cells (Yang et al., 2005). Moreover, melatonin prevents H2O2 activation of the pro-apoptotic enzyme, caspase-3 (Juknat et al., 2005). Nonetheless, while low doses of these antioxidants are neuroprotective, high doses of many antioxidants exert pro-oxidant effects and augment apoptosis (Watjen et al., 2005; Williams et al., 2004).
It is likely that antioxidant/free radical scavenging properties of antioxidants make a significant contribution to their anti-apoptotic effects. However, each of these antioxidants exerts additional effects that can potentially contribute to their neuroprotective actions. Some of their non-antioxidant effects contribute to the maintenance of a healthy ROS level within the cell by modulating levels of endogenous antioxidants and the antioxidant enzymes, which facilitate the detoxification of ROS. For example, α-lipoic acid is converted to dihydrolipoic acid within the cell, and both of these compounds increase cellular levels of the endogenous antioxidant GSH (Suh et al., 2004). GSH levels can also be influenced by melatonin, which increases the activity of the GSH-associated antioxidant enzymes GSH peroxidase and GSH reductase (Barlow-Walden et al., 1995; Juknat et al., 2005). Reportedly, both resveratrol and curcumin induce the gene encoding heme oxygenase 1 (HOX1) (Zhuang et al., 2003; Lin, 2007); HOX 1 is an antioxidant enzyme that degrades the pro-oxidant heme. Antioxidants can also exert neuroprotective effects by increasing the activity of pro-survival proteins or by downregulating the expression of pro-apoptotic genes. For example, LA, curcumin, and EGCG can activate the PI-3K pro-survival pathway (Zhang et al., 2001; Muller et al., 2003; Koh et al., 2004; Antonio and Druse, 2006; Kang et al., 2007), and EGCG mediates downregulation of the expression of Bax and Bad (Levites et al., 2002; Mandel et al., 2004), which encode pro-apoptotic proteins. Moreover, the protein kinase C (PKC) pathway has been implicated with the protective effects of EGCG (Reznichenko et al.., 2005). Additional studies are needed to establish whether any or all of these alternative non-antioxidant mechanisms contributed to the neuroprotective effects of the five investigated antioxidants against ethanol-associated apoptosis in fetal rhombencephalic neurons.
Experimental Procedure
Neuronal Cultures
Dissection and culturing of fetal rhombencephalic neurons have been described by this laboratory (Druse et al., 2004-2007). Rhombencephalic tissue was dissected from gestation day 14 (G14) embryos taken from timed-pregnancy Sprague-Dawley rats, where G=0 corresponds to the day of insemination (Honegger and Monnet-Tschudi, 1997; Eriksen and Druse, 2001). Tissue from the G14 rhombencephalon was used because this brain region contains the cell bodies of developing 5-HT neurons and other CNS neurons (Shahar et al., 1989). All animal care and use procedures were reviewed and approved by the Institutional Animal Care and Use Committee at Loyola University Chicago, Stritch School of Medicine.
Using sterile techniques, rhombencephalic tissue was mechanically disaggregated (Eriksen and Druse, 2001) and the suspension was collected. Dispersed cells were seeded on the first day in vitro, i.e., DIV1, onto four-chambered slides (Nalge and Nunc, Naperville, IL) that were previously coated with poly-D-lysine at a density of ∼350,000 cells/chamber. Cells were maintained in Dulbecco’s Minimal Essential Media/F12 (DMEM/F12) media, supplemented with hydrocortisone-21 sulfate, Basal Medium Eagles Vitamin Solution, B27 serum-free medium supplement, and 0.25% fetal bovine serum. After 24 hrs (DIV2), gliogenesis was arrested using 0.4 μM cytosine arabinoside. Media was changed on DIV2 and DIV5. Neurons were maintained in culture for a total of 5 days (DIV1 to DIV6). Previous studies in this laboratory showed that by DIV6, 5-HT neurons comprised ∼20% to 30% of fetal rhombencephalic neurons and glial fibrillary acidic protein (GFAP) positive astrocytes accounted for < 5% of the cells (Druse et al., 2004).
Ethanol and Antioxidant Treatments
On DIV5, cultures were divided into groups that would be treated with 0 mM ethanol (control) or 75 mM ethanol and/or with specific antioxidants (1 μM EGCG, 1 μM melatonin, 1 μM curcumin, 10 μM α-lipoic acid, or 10 μM resveratrol. At this time the media was changed from that containing standard B27 supplement to one in which the B27 supplement lacked antioxidants. Antioxidant doses were based on the lowest dose that appeared to demonstrate ethanol-associated neuroprotection in preliminary studies which screened these antioxidants at concentrations of 1, 10, and 50 μM. This laboratory reported that use of an ethanol chamber system, used in this and prior studies, maintains the ethanol concentration in cultures at ≥85% of the initial concentration. (Eriksen and Druse, 2001).
Detection and Quantification of Apoptotic Nuclei
Neurons with unfragmented and fragmented/apoptotic nuclei were identified using Hoechst 33342, a nucleic acid stain, as described previously by this laboratory (Druse et al., 2004). Previously, we showed that Hoechst 33342, which is used to visualize fragmented nuclei, and TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling), which labels fragmented DNA in apoptotic cells, identify the identical population of apoptotic fetal rhombencephalic neurons in control and ethanol-treated cultures (Druse et al., 2004). For each “n”, ∼600 control and ethanol neurons were analyzed methodically without consideration of treatment; these neurons were counted on ∼15 fields from two chambers (7-8 fields/chamber). Five to seven separate replicate experiments were performed for each treatment group. As described previously (Druse et al., 2004), intact cells characterized as apoptotic cells contained fragmented apoptotic nuclei, while non-apoptotic cells lacked fragmented nuclei. We did not observe necrotic cells, e.g., those exhibiting signs of swelling and the presence of diffuse or finely clumped chromatin. Apparent extracellular debris, fragmented cells, and ‘dots’, were not counted.
Statistical Analyses
Data were analyzed using a 2-way ANOVA [ethanol × antioxidant] and a Tukey’s post-hoc analysis. A p < 0.05 was considered to be significant.
Acknowledgements
This research was supported by a grant from the USPHS - AA03490 and T32 AA13527.
Abbreviatons
- DHLA
dihydrolipoic acid
- DIV
day in vitro
- EGCG
(-)-epigallocatechin-3-gallate
- LA
α-lipoic acid
- ROS
reactive oxygen species
Footnotes
Please address all communications to Mary Druse Manteuffel, Ph.D., Professor of Cell Biology, Neurobiology, and Anatomy, LUSSOM, Maywood, IL 60153. Phone: 708-216-3370, Fax: 708-216-8523, email: mmanteu@lumc.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section: 2
Nervous System Development, Regeneration and Aging
Literature references
- Antonio AM, Druse MJ. Protective effects of antioxidants on ethanol-treated rhombencephalic neurons. Alcohol. Clin. Exp. Res. 2006;30:116A. [Google Scholar]
- Barlow-Walden L, Reiter RJ, Abe M, Pablos M, Menendez-Pelaez A, Chen LD, Poeggeler B. Melatonin stimulates brain glutathione peroxidase activity. Neurochem. Int. 1995;26:497–502. doi: 10.1016/0197-0186(94)00154-m. [DOI] [PubMed] [Google Scholar]
- Bastianetto S, Zheng W-H, Quirion R. Neuroprotective abilities of resveratrol and other red wine constituents against nitric oxide-related toxicity in cultured hippocampal neurons. Br. J. Pharmacol. 2000;131:711–720. doi: 10.1038/sj.bjp.0703626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema ZF, West JR, Miranda RC. Ethanol induces Fas/Apo [apoptosis]-1 mRNA and cell suicide in the developing cerebral cortex. Alcohol. Clin. Exp. Res. 2000;24:535–543. [PubMed] [Google Scholar]
- Chen WJ, Berryhill EC, West JR. Zinc supplementation does not attenuate alcohol-induced cerebellar Purkinje cell loss during the brain growth spurt period. Alcohol. Clin. Exp. Res. 2001;25:600–605. [PubMed] [Google Scholar]
- Chen SY, Dehart DB, Sulik KK. Protection from ethanol-induced limb malformations by the superoxide dismutatse/catalse mimetic EUK-134. FASEB J. 2004;18:1234–1236. doi: 10.1096/fj.03-0850fje. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol- induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Res. Dev. Brain Res. 2005;155:1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Druse MJ. Neurotransmitter function: changes associated with in utero alcohol exposure. In: Abel EL, editor. Fetal Alcohol Syndrome. From Mechanisms to Prevention. CRC Press; 1996. pp. 171–189. [Google Scholar]
- Druse MJ, Tajuddin NF, Gillespie RA, Dickson E, Atieh M, Pietrzak CA, Le PT. The serotonin-1A agonist ipsapirone prevents ethanol-associated death of total rhombencephalic neurons and prevents the reduction of fetal serotonin neurons. Brain Res. Dev. Brain Res. 2004;150:79–88. doi: 10.1016/j.devbrainres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Druse M, Tajuddin NF, Gillespie RA, Le P. Signaling pathways involved with serotonin1A agonist-mediated neuroprotection against ethanol-induced apoptosis of fetal rhombencephalic neurons. Brain Res. Dev. Brain Res. 2005;159:18–28. doi: 10.1016/j.devbrainres.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Druse MJ, Tajuddin NF, Gillespie RA, Le P. The effects of ethanol and the serotonin(1A) agonist ipsapirone on the expression of the serotonin(1A) receptor and several antiapoptotic proteins in fetal rhombencephalic neurons. Brain Res. 2006;1092:79–86. doi: 10.1016/j.brainres.2006.02.065. [DOI] [PubMed] [Google Scholar]
- Druse M, Gillespie RA, Tajuddin NF, Rich M. S100B-mediated protection against the pro-apoptotic effects of ethanol on fetal rhombencephalic neurons. Brain Res. 2007;1150:46–54. doi: 10.1016/j.brainres.2007.02.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunty WC, Chen SY, Zucker RM, Dehart DB, Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorders. Alcohol. Clin. Exp. Res. 2001;25:1523–1535. [PubMed] [Google Scholar]
- Eriksen JL, Druse MJ. Astrocyte-mediated trophic support of developing serotonin neurons: effects of ethanol, buspirone, and S100B. Brain Res. Dev. Brain Res. 2001;131:9–15. doi: 10.1016/s0165-3806(01)00240-1. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Eilers AT. Alcohol-induced Purkinje cell loss with a single binge exposure in neonatal rats: a stereological study of temporal windows of vulnerability. Alcohol. Clin. Exp. Res. 1997;21:738–744. [PubMed] [Google Scholar]
- Goodlett CR, Horn KH. Mechanisms of alcohol-induced damage to the developing nervous system. Alcohol Res. Health. 2001;25:175–184. [PMC free article] [PubMed] [Google Scholar]
- Heaton MD, Paiva M, Mayer J, Miller R. Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci. Lett. 2002;334:83–86. doi: 10.1016/s0304-3940(02)01123-0. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Madorsky I, Mayer J, Moore DB. Effects of ethanol on neurotrophic factors, apoptosis-related proteins, endogenous antioxidants, and reactive oxygen species in neonatal striatum: relationship to periods of vulnerability. Dev. Brain Res. 2003;140:237–252. doi: 10.1016/s0165-3806(02)00610-7. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Madorsky I, Paiva M, Siler-Marsiglio I. Vitamin E amelioration of ethanol neurotoxicity involves modulation of apoptosis-related protein levels in neonatal rat cerebellar granule cells. Dev. Brain Res. 2004;150:117–124. doi: 10.1016/j.devbrainres.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Honegger P, Monnet-Tschudi F. In Protocols for Neural Cell Culture. Human Press; Totowa, NJ: 1997. Aggregating Neural Cell Cultures; pp. 25–49. [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW. Proliferation and death of cultured fetal neocortical neurons: effects of ethanol on the dynamics of cell growth. J. Neurocytol. 2001;30:391–401. doi: 10.1023/a:1015013609424. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. The fetal alcohol syndrome. Teratology. 1975;12:1–10. doi: 10.1002/tera.1420120102. [DOI] [PubMed] [Google Scholar]
- Juknat AA, del Valle Armanino Méndez M, Quaglino A, Fameli CI, Meno M, Kotler ML. Melatonin prevents hydrogen peroxide-induced bax expression in cultured rat astrocytes. J. Pineal Res. 2005;38:84–92. doi: 10.1111/j.1600-079X.2004.00166.x. [DOI] [PubMed] [Google Scholar]
- Kang ES, Woo IS, Kim HJ, Eun SY, P KS, Kim HJ, Chang KC, Lee JH, Lee HT, Kim J-H, Nishinaka T, Yabe-Nishimura C, Seo HG. Up- regulation of aldose reductase expression mediated by phsophatidylinositol 3-kinase/Akt and Nrf2 is involved in the protective effect of curcumin against oxidative damage. Free Radic. Biol. Med. 2007;43:535–545. doi: 10.1016/j.freeradbiomed.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Koh S-H, Kim SH, Kwon H, Park Y, Kim KS, Song CW, Kim J, Kim M-H, Yu H-J, Henkel JS, Jung HK. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/Akt and glycogen synthase kinase-3. Mol. Brain Res. 2003;118:72–81. doi: 10.1016/j.molbrainres.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Koh S-H, Kim SH, Kwon H, Kim JG, Kim JH, Yang K-H, Kim J, Kim SU, Yu H-H, Do BR, Kim KS, Jung HK. Phosphatidylinositol-3 kinase/Akt and GSK-3 mediated cytoprotective effects of epigallocatechin gallate on oxidative stress-injured neuronal-differentiated N18D3 cells. Neurotoxicology. 2004;25:793–802. doi: 10.1016/j.neuro.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Kotler ML, Rodriguez C, Sainz RM, Antolin I, Menendez-Pelaez A. Melatonin increases gene expression for antioxidant enzymes in rat brain cortex. J. Pineal Res. 1998;24:83–89. doi: 10.1111/j.1600-079x.1998.tb00371.x. [DOI] [PubMed] [Google Scholar]
- Lee JH, Tajuddin NF, Le PT, Druse MJ. A serotonin-1A agonist provides protection against the ethanol-induced decrease of endogenous antioxidant enzymes in fetal rhombencephalic neurons. Alcohol. Clin. Exp. Res. 2007;31:37A. [Google Scholar]
- Levites Y, Amit T, Youdim MBH, Mandel S. Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (-) epigallocatechin-3-gallate neuroprotective action. J. Biol. Chem. 2002;277:30574–30580. doi: 10.1074/jbc.M202832200. [DOI] [PubMed] [Google Scholar]
- Lin JK. Molecular targets of curcumin. Adv. Exp. Med. Biol. 2007;595:227–243. doi: 10.1007/978-0-387-46401-5_10. [DOI] [PubMed] [Google Scholar]
- Mandel S, Weinreb TA, Youdim MBH. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J. Neurochem. 2004;88:1555–1569. doi: 10.1046/j.1471-4159.2003.02291.x. [DOI] [PubMed] [Google Scholar]
- Marcussen BL, Goodlett CR, Mahoney JC, West JR. Developing rat Purkinje cells are more vulnerable to alcohol-induced depletion during differentiation than during neurogenesis. Alcohol. 1994;11:147–156. doi: 10.1016/0741-8329(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Riley EP, Delis DC, Stern C, Jones KL. Verbal learning and memory in children with fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1996;20:810–816. doi: 10.1111/j.1530-0277.1996.tb05256.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Goodman AM, Caine C, Delis DC, Riley EP. Executive functioning in children with heavy prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 1999;23:1808–1815. [PubMed] [Google Scholar]
- Moulder KL, Fu T, Melbostad H, Cormier RJ, Isenberg KE, Zorumbski CF, Mennerick S. Ethanol-induced death of postnatal hippocampal neurons. Neurobiol. Dis. 2002;10:396–409. doi: 10.1006/nbdi.2002.0523. [DOI] [PubMed] [Google Scholar]
- Muller C, Dunschede F, Koch E, Vollmar AM, Kiemer AK. Alpha-lipoic acid preconditioning reduces ischemia-reperfusion injury of the rat liver via the PI3- kinase/Akt pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;285:G769–778. doi: 10.1152/ajpgi.00009.2003. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003;74:577–588. doi: 10.1002/jnr.10767. [DOI] [PubMed] [Google Scholar]
- Reznichenko L, Amit T, Youdim MBH, Mandel S. Green tea polyphenol (-)-epigallocatechin-3-gallate induces neurorescue of long-term serum-deprived PC12 cells and promotes neurite outgrowth. J. Neurochem. 2005;93:1157–1167. doi: 10.1111/j.1471-4159.2005.03085.x. [DOI] [PubMed] [Google Scholar]
- Riley EP, McGee CL, Sowell ER. Teratogenic effects of alcohol: a decade of brain imaging. Am. J. Med. Genet. C. Semin. Med. Genet. 2004;127:35–41. doi: 10.1002/ajmg.c.30014. [DOI] [PubMed] [Google Scholar]
- Roebuck TM, Mattson SM, Riley EP. A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 1998;22:339–344. doi: 10.1111/j.1530-0277.1998.tb03658.x. [DOI] [PubMed] [Google Scholar]
- Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JM., Jr. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56:317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Shahar A, Vellis JD, Vernadakis A, Haber B. A Dissection and Tissue Culture Manual of the Nervous System. Alan R. Liss, Inc.; New York: 1989. [Google Scholar]
- Shila S, Subathra M, Devi MA, Panneerselvam C. Arsenic intoxication- induced reduction of glutathione level and of the activity of related enzymes in rat brain regions: reversal by DL-alpha-lipoic acid. Arch. Toxicol. 2005;79:140–146. doi: 10.1007/s00204-004-0614-8. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Mattson SN, Kan E, Thompson PM, Riley EP, Toga AW. Abnormal cortical thickness and brain-behavior correlation patterns in individuals with heavy prenatal alcohol exposure. Cereb. Cortex. 2007 doi: 10.1093/cercor/bhm039. April 18, Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Wang H, Liu RM, Liu J, Hagen TM. (R)-alpha-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: evidence for increased cysteine requirement for GSH synthesis. Arch. Biochem. Biophys. 2004;423:126–135. doi: 10.1016/j.abb.2003.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajuddin NF, Druse MJ. In utero ethanol exposure decreased the density of serotonin neurons. Maternal ipsapirone treatment exerted a protective effect. Brain Res. Dev. Brain Res. 1999;117:91–97. doi: 10.1016/s0165-3806(99)00102-9. [DOI] [PubMed] [Google Scholar]
- Tajuddin NF, Druse MJ. A persistent deficit of serotonin neurons in the offspring of ethanol-fed dams: protective effects of maternal ipsapirone treatment. Brain Res. Dev. Brain Res. 2001;129:181–188. doi: 10.1016/s0165-3806(01)00199-7. [DOI] [PubMed] [Google Scholar]
- Watjen W, Michels G, Steffan B, Niering P, Chovolou Y, Kampkotter A, Tran-Thi QH, Proksch P, Kahl R. Low concentrations of flavonoids are protective in rat H4IIE cells whereas high concentrations cause DNA damage and apoptosis. J. Nutr. 2005;135:525–531. doi: 10.1093/jn/135.3.525. [DOI] [PubMed] [Google Scholar]
- Wattendorf DJ, Muenke M. Fetal alcohol spectrum disorders. Am. Fam. Physician. 2005;72:279–282. [PubMed] [Google Scholar]
- Watts LT, Rathinam ML, Schenker S, Henderson GI. Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J. Neurosci. Res. 2005;80:655–666. doi: 10.1002/jnr.20502. [DOI] [PubMed] [Google Scholar]
- Williams RJ, Spencer JP, Rice-Evans C. Flavonoids: antioxidants or signalling molecules? Free Radic. Biol. Med. 2004;36:838–849. doi: 10.1016/j.freeradbiomed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen P, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005;18:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xing GQ, Barker JL, Chang Y, Maric D, Ma W, Li BS, Rubinow DR. Alpha-lipoic acid protects rat cortical neurons against cell death induced by amyloid and hydrogen peroxide through the Akt signaling pathway. Neurosci. Lett. 2001;312:125–128. doi: 10.1016/s0304-3940(01)02205-4. [DOI] [PubMed] [Google Scholar]
- Zhuang H, Kim Y-S, Koehler RC, Doré S. Potential mechanism by which resveratrol, a red wine constituent, protects neurons. Ann. N.Y. Acad. Sci. 2003;993:276–286. doi: 10.1111/j.1749-6632.2003.tb07534.x. [DOI] [PubMed] [Google Scholar]