

Abstract
The synthesis of olefin metathesis catalysts containing chiral, monodentate N-heterocyclic carbenes and their application to asymmetric ring-closing metathesis (ARCM) is reported. These catalysts retain the high levels of reactivity found in the related achiral variants (1a and 1b). Using the parent chiral catalysts 2a and 2b and derivatives that contain steric bulk in the meta positions of the N-bound aryl rings (catalysts 3-5), five- through seven-membered rings were formed in up to 92% ee. The addition of sodium iodide to catalysts 2a-4a (to form 2b-4bin situ) caused a dramatic increase in enantioselectivity for many substrates. Catalyst 5a, which gave high enantiomeric excesses for certain substrates without the addition of NaI, could be used in loadings of ≤1 mol %. Mechanistic explanations for the large sodium iodide effect as well as possible mechanistic pathways leading to the observed products are discussed.
Introduction
The development of well-defined catalysts has made olefin metathesis an important, reliable, and widespread method for constructing carbon-carbon double bonds.1 Since the initial report of asymmetric olefin metathesis for small molecule synthesis,2 a variety of chiral, ruthenium- and molybdenum-based alkylidene catalysts have been developed.3-4 The molybdenum catalysts have been shown to give excellent enantioselectivities in asymmetric ring-closing metathesis (ARCM), asymmetric ring-opening/ring-closing metathesis (ARORCM), and asymmetric ring-opening/cross metathesis (AROCM).5-6 Our interest in ruthenium-based olefin metathesis catalysts stems from their increased functional group tolerance compared to the molybdenum systems, as well as their stability to air and moisture.6b Two classes of chiral ruthenium catalysts have been explored (Chart 1): those containing monodentate N-heterocyclic carbenes (NHCs) with chirality in the backbone of the carbene (2a and 2b) developed in our laboratory,3c and those containing chiral, bidentate NHC/binaphthyl ligands (6a and 6b) developed by Hoveyda et al.3a-b While catalysts of the latter type gave good enantioselectivities and yields for AROCM reactions,7 they exhibited reduced reactivity and selectivity toward ARCM relative to those of the former class.
Chart 1.

Ruthenium Olefin Metathesis Catalysts.
The ruthenium catalysts containing monodentate, chiral NHC's (2a and 2b) had the same air and moisture stability of the parent catalyst 1a, as well as a similar level of reactivity.8 Additionally, we achieved a single example of 90% ee using 2b.3c Based on this initial discovery, and because ARCM remains challenging for 6a and 6b, we decided to modify 2a and 2b to enhance enantioselectivity and expanded the substrate scope of ARCM. The synthesis of new chiral ruthenium catalysts for asymmetric olefin metathesis, as well as their reactivity and selectivity in expanding the scope of ARCM, is reported herein. Catalysts containing substitution on the aryl ring para to the ortho-isopropyl group (3a, 3b, 4a, and 4b) showed very similar enantioselectivities to that of the parent chiral catalysts 2a and 2b. However, substitution on the same side of the ring as the ortho-isopropyl group (5a and 5b) caused an increase in enantioselectivity to the extent that in a number of substrates, the dichloride catalyst 5a could be used at very low catalyst loadings to obtain high enantiomeric excesses and conversions.
Results and Discussion
Design and Synthesis of Chiral Ruthenium Catalysts
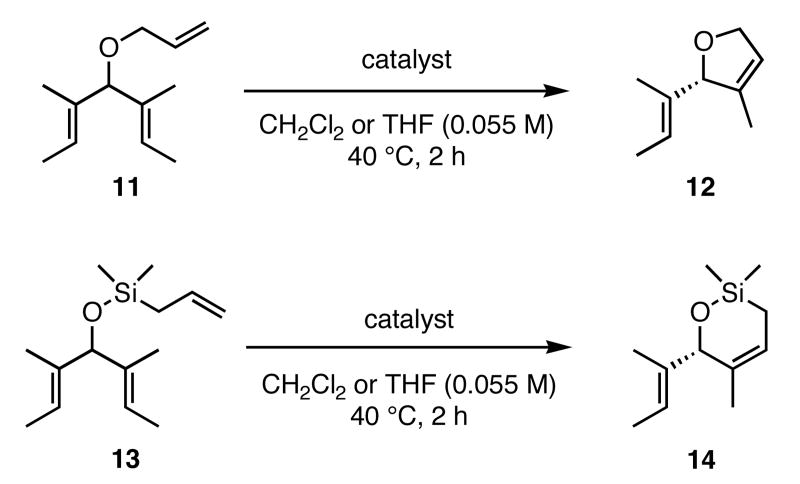
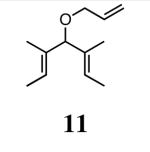
In our initial study we discovered that NHC's derived from enantioenriched 1,2-diamines could transfer the chirality of the backbone to the ortho-substituted N-aryl rings.3c This “gearing” effect, which results in the ortho-subtituent of the unsymmetrically-substituted arene rings to reside on the face opposite to the phenyl groups on the backbone, has been to shown to induce asymmetry in the ring-closing metathesis of prochiral trienes. Using the ring-closing metathesis of 11 to form 12 as a test reaction, we varied the backbone of the NHC as well as the substituent in the ortho-position of the N-aryl rings and discovered that 2b was the most successful catalyst, forming dihydrofuran 12 in 90% ee. Based on the success of varying the ortho-substitution, we explored catalysts containing substitution in the meta-position of the N-bound aryl rings.
The synthesis of the chiral dihydroimidazolium salts (9a-9d) was straight-forward and modular. Pd2(dba)3/BINAP was used to couple the substituted aryl bromides (7a-d) to commercially available (1R,2R)-diphenylethylenediamine.9-10 Compound 7b was used as a mixture of aryl bromides; thus, the diamine coupling reaction was followed by a Pd-catalyzed reduction to yield 8b. The para-methoxy group in aryl bromide 7c was present as a synthetic handle and was not expected to affect enantioselectivities due to its remote location relative to the Ru center. The chiral diamines were reacted with triethylorthoformate to form dihydroimidazolium BF4− salts 9a-d in good yields over two or three steps. Ligand substitution with bis-phosphine compound 10 gave the desired chiral ruthenium benzylidenes as brown solids.11 All of these ruthenium compounds were purified using silica gel chromatography with non-degassed solvents, and they were stored in air without significant decomposition over a period of at least three months. Conversions for the ligand substitution step were generally 90%, but challenging chromatographic separations gave varied isolated yields. The analogous catalysts with two iodides bound to the ruthenium (2b-5b) were generated in situ by dissolving the dichloride catalyst in THF in the presence of 25 equivalents of NaI.
Enantioselectivities and Reactivities of Chiral Ru Catalysts for ARCM
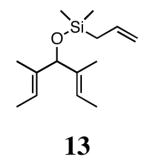
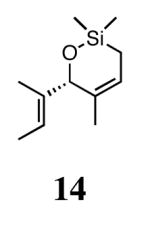
In our previous study we discovered that substrate 11, derived from (2E,5E)-3,5-dimethylhepta-2,5-dien-4-ol, gave the highest enantioselectivities and good conversions, so this substrate and a silyl ether analogue (13) were used to examine the reactivities and enantioselectivities of the new catalysts. The presence of trisubstituted alkenes was critical to obtaining high enantiomeric excess using catalyst 2b with triene 11. As can be seen in Table 1, catalysts 2a-4a gave relatively similar enantioselectivities and conversions for substrate 11. In all of these cases, the diiodide catalysts (2b-4b) caused dramatic increases in the enantioselectivity of the reaction (i.e. 35% ee with 2a, 90% ee with 2b). There is no simple pattern for selectivity based on the steric bulk in the meta-position for catalysts 2-4: increasing the size of the meta group from H to t-butyl caused a sequential decrease in the enantiomeric excess (2a-4a); but when NaI was added (2b-4b), it decreased for meta-isopropyl but increased again when the larger t-butyl group was in the meta-position. The largest difference in selectivity was seen when catalyst 5a was used. An increase of 11% ee was observed relative to 2a, and 5b gave 90% ee just as 2b. High conversions were obtained with all of the catalysts in 2 h at 40 °C.
Table 1.
Relative Reactivity and Selectivity of Asymmetric Ruthenium Catalysts.
 | ||||
|---|---|---|---|---|
| 11 to 12a | 13 to 14a | |||
| catalyst | eeb(%) | convc (%) |
eeb (%) | convc (%) |
| 2a | 35 | >98 | 83 | >98 |
| 3a | 31 | >98 | 81 | >98 |
| 4a | 30 | >98 | 75 | >98 |
| 5a | 46 | >98 | 92 | >98 |
| 2b | 90 | >98 | 86 | 68 |
| 3b | 84 | >98 | 90 | >98 |
| 4b | 87 | >98 | 85 | >98 |
| 5b | 90 | >98 | 92 | 58 |
Reactions with 2a-5a (2 mol %) run in CH2Cl2; catalysts 2b-5b generated by stirring 2a-5a (4 mol %) in THF with 25 equiv NaI.
Enantiomeric excesses determined by chiral GC; see supporting information for chromatograms and proof of absolute stereochemistry.
Determined by 1H NMR spectrum of crude reaction mixture.
When 13 was reacted with these catalysts, higher enantioselectivities were obtained without the need for NaI. As with substrate 11, selectivity decreased as steric bulk in the meta-position increased (catalysts 2a-4a). When the diiodide catalysts 2b-4b were used, enantioselectivities increased for all of the reactions relative to those obtained using 2a-4a. Introducing meta-substitution adjacent to the ortho-isopropyl group (catalyst 5a) resulted in a 92% ee for the ring closing of 13 without the need for NaI. This result was the highest enantiomeric excess for ring-closing metathesis we had observed. Moreover, since the dichloride catalysts are generally more reactive than the corresponding diiodide catalysts, complete conversion could be achieved with lower catalyst loadings of 5a! Conversions with catalysts 2b and 5b decreased relative to the reactions with the dichloride catalysts when substitution was on just one side of the N-bound aryl rings. On the other hand, conversions remained high for catalysts 3b and 4b, which possess substitution on both sides of the aryl rings. This increase in conversion may be due to the substituent in the meta-position increasing the steric bulk of the catalyst, and therefore protecting it from various bimolecular decomposition pathways.12
Expanded Substrate Scope for ARCM
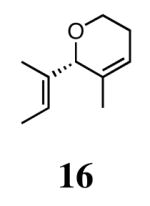
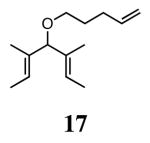
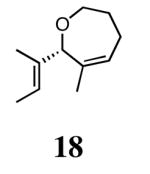
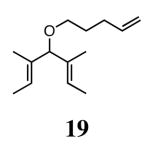
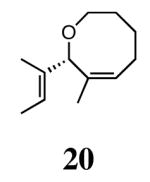
The ability of the chiral ruthenium catalysts 2-5 to give high conversions and enantioselectivities for the ARCM reactions of 11 and 13 encouraged us to examine other substrates which afford 5–8 membered rings upon ARCM. Based on the results from our initial screen, catalysts 2b and 5a were used. Two types of prochiral trienes were explored: alkenyl ethers and alkenyl silyl ethers (Table 2). Conversions >90% and 90% ees were obtained when generating 5- and 6-membered rings 12 and 16. The isolated yields were moderately reduced due to the volatility of the products during the purification procedure. The synthesis of a 7-membered ring from 17 occurred in 85% ee when 2b was used. Unfortunately the formation of the desired product was only ∼5%, with the major species being unreacted starting material and homocoupled product. The yield for the same reaction could be drastically increased when catalyst 5a was used,13 and because 5a is more selective than the other catalysts in the absence of NaI (65% ee for 18 using 2a), the desired product could be obtained in 92% yield and 76% ee. Synthesis of an 8-membered ring was more challenging, and 2b catalyzed the ARCM reaction of 19 to 20 in 85% ee, but with only a ∼2% yield. Attempts to increase the yield using 5a resulted in a similar yield and a reduced enantiomeric excess. This result was not surprising, as generating 8-membered rings via ring-closing olefin metathesis is a challenging problem.
Table 2.
ARCM Reactions with Chiral Ruthenium Catalysts.a
| triene | product | catalyst (mol %) | ee (%)b | conversion (%)c | yield (%) |
|---|---|---|---|---|---|
|
|
2b (4) | 90 | >98 | 64 |
|
|
2b (4) | 90 | >98 | 77 |
|
|
2b (4) | 85 | 5 | nd |
| 5a (2)d | 76 | 93 | 92 | ||
|
|
2b (4) | 85 | ∼2e | nd |
|
|
5a (0.8) | 92 | >98 | 77f |
|
|
5a (1) | 92 | 65 | 64 |
|
|
2b (4) | 78 | >98 | 98 |
Conditions for reactions with 2b: NaI (25 equiv relative to catalyst) and 2a in THF (0.055 M in triene) for 1h at rt, then add triene and stir for 2h at 40 °C; conditions for reactions with 5a: triene, CH2Cl2 (0.055 M in triene), and 5a for 2h at 40 °C.
Enantiomeric excesses determined by chiral GC; see supporting information for chromatograms and proof of absolute stereochemistry.
Determined by 1H NMR spectrum of crude reaction mixture.
See footnote 13.
Never isolated as a pure compound.
Reaction done on a 4 mmol (0.95 g) of 13 scale. nd = not determined
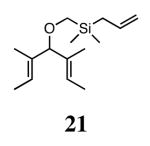
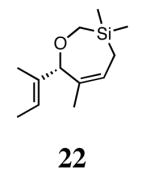
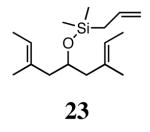
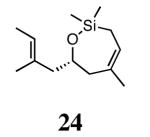
Silyl ethers proved to be excellent substrates, as both 6- and 7-membered rings (14 and 22) formed in good conversions and 92% ee's with 5a. Catalyst loadings of 1 mol % or less could be used in these reactions. The ring-closing of 13 illustrated that the high yield and enantiomeric excess were maintained when the reaction was scaled up to almost 1 g of substrate. Using the analogous diiodide catalyst 5b for ARCM of 13 and 21 resulted in 6- and 7-membered rings with the same enantiomeric excesses but lower conversions than with the dichloride catalyst 5a. Substrate 23, which placed the prochiral center further from the trisubstituted olefins, also underwent ARCM to form a 7-membered ring in good enantiomeric excess and excellent yield with a substitution pattern different than 22.
Table 2 provides the best results between catalysts 2b and 5a. In most reactions (ARCM of 11, 13, 15, 21, and 23), the yields and enantioselectivities are very similar whether catalyst 2b, 5a, or 5b is used. Because both 2b and 5a promote the ARCM of 5–7 membered rings, there is no need for specific catalyst/substrate matching.
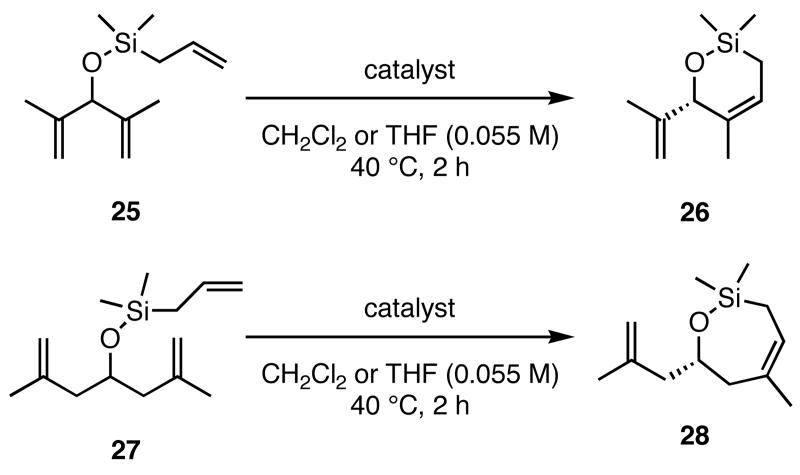
Trienes other than those discussed above were also examined in the ARCM reactions (Table 3). Substrates analogous to 13 and 23, but lacking a terminal methyl group on the olefin (25 and 27), formed cyclic products 26 and 28 in reduced enantioselectivities and conversions. When the diiodide catalysts were used, conversions were <20%. The lower conversions using 25 and 27 relative to 11 and 23 may be due to the formation of a ruthenium methylidene versus a ruthenium ethylidene, respectively. Ruthenium methylidenes are known to decompose more rapidly than other ruthenium alkylidenes, and in these cases decomposition may have occurred more quickly than the cross-metathesis that introduces the ring-closing substrate to the catalyst.14
Table 3.
Challenging ARCM Substrates.
 | ||||
|---|---|---|---|---|
| 25 to 26a | 27 to 28a | |||
| catalyst | ee b (%) | conv c (%) |
ee b (%) | conv c (%) |
| 2a | 16 | 72 | −5 | >98 |
| 3a | 45 | 92 | −5 | >98 |
| 5a | 30 | 93 | −8 | >98 |
| 2b | nd | <2 | 3 | 15 |
| 3b | nd | 6 | 15 | 18 |
Reactions with 2a-5a (2 mol %) run in CH2Cl2; catalysts 2b and 3b generated by stirring 2a and 3a (4 mol %) in THF with 25 equiv NaI.
Enantiomeric excesses determined by chiral GC; see supporting information for chromatograms and proof of absolute stereochemistry.
Determined by 1H NMR spectrum of crude reaction mixture. nd = not determined
A number of variables other than catalyst and substrate in the ARCM reactions were examined. Using a variety of solvents typical for olefin metathesis, such as CH2Cl2, THF, and benzene, had very little affect on the conversions and enantiomeric excesses (Figure 1). The slight reduction in selectivity for ARCM reactions in benzene suggests that solvent coordination to the catalyst is not involved as in the molybdenum systems.15 Lowering the temperature of the reactions in CH2Cl2 to 0 °C led to a small increase (4-5%) in enantiomeric excesses when 2a and 2b were used, but catalysts 3b and 5a formed the product with slightly reduced (2-3%) enantioselectivities (Figure 2). In all cases the conversions were reduced to no greater than 40% even when 5 mol % of the catalyst was used and the reaction was allowed to proceed for 24h. The reactions which used I− to form the diiodide catalysts had the best enantioselectivities when NaI in THF was used; NaI in CH2Cl2 and [Bu4N]I in either THF or CH2Cl2 resulted in reduced ees and reduced yields. No difference in enantioselectivity or conversion was observed when dry NaI was used in place of ACS-grade NaI stored on the bench top. This fact is a testament to the moisture stability of these ruthenium-based catalysts.
Figure 1.

Solvent Effect on Enantioselectivity of ARCM.
Figure 2.

Temperature Effect on Enantioselectivity of ARCM.
Mechanistic Considerations in Model Determination
In order to rationally design more reactive and selective chiral ruthenium catalysts for olefin metathesis, we must first develop a model which explains the enantioselectivity in these reactions. If the ARCM reaction has a degree of reversibility, a decrease in ee over time would be expected. Although this was not observed, further experiments were performed to support the irreversibility of this reaction. When enantioenriched 14 was exposed to achiral catalyst 1a, no erosion of the enantiomeric excess was observed, which suggests that once the ring is formed, it does not undergo a secondary ring-opening/ring-closing process. The same conclusion was drawn when enantioenriched 12 was heated under 60 psi of ethylene in the presence of 1a (Figure 3). Under these forcing conditions, the enantiomeric excess of unreacted 12 did not erode, and the ethylenolysis product 28 also retained the stereochemistry from the ARCM reaction.16
Figure 3.

Ethylenolysis of 12.
In the ARCM reactions of the trienes described above, most likely a ruthenium alkylidene derived from the least-substituted olefin initially forms. This species binds one of the diastereotopic olefins, and through a metallacyclobutane intermediate/transition state, forms the ring-closed product. Our initial model3c assumed olefin coordination occurred cis to the NHC (Scheme 2, lower pathway), on the face opposite the isopropyl group (structure 30), and that interaction determined the absolute stereochemistry of the product. The interaction of a halide ligand with a substituent on the ring of the cyclic intermediate was also proposed to be an important, stereo-defining interaction.17 The actual position of the coordinating olefin relative to the NHC is unclear; experimental evidence exists to support olefin binding both cis and trans to the NHC (Scheme 2).18 In the most recent report on this issue,18c Piers and coworkers provide compelling evidence for the observation of a 14-electron ruthenacyclobutane trans to the NHC. Computational studies support olefin binding trans to the NHC,19 and a recent computational study of 2b reacting with 11 calculated that the diastereotopic olefin coordinated trans to the NHC (Scheme 2, upper pathway).19a Due to the tilt of the ortho-substituted N-bound aryl ring,20 the alkylidene is positioned underneath the isopropyl group (structure 32), which was found to be the “smaller” side of the aryl ring (Figure 4), instead of underneath the ortho C—H bond (structure 31). In addition to the position of the alkylidene determining the absolute stereochemistry of the product, the cyclic intermediate formed upon olefin binding is also important. The pendent olefin not involved in the ring-closing reaction prefers to be in the pseudo-equatorial position of the forming ring. Although the discussion presented here focuses on a substrate that forms a 5-membered ring, substrates that form other ring sizes presumably have an energetically favored ring conformation once olefin binding occurs. Therefore the topics mentioned above can be extended to other ring-closing substrates. The importance of the position of the substituents on the ring in the cyclic transition state could explain the higher enantioselectivities observed for the substrates containing dimethylsilyl groups: there are more non-hydrogen substituents on the ring, and therefore the energy differences for various ring conformations are greater than those for substrates with only methylenes in the ring.
Scheme 2.

Proposed Pathways Leading to the Desired Product.
Figure 4.

Suggested Alkylidene Position in trans-Binding Transition State.
Our initial cis-binding hypothesis was supported by the fact that when a larger halide ligand was present, the enantiomeric excess of the product increased. With recent experimental and computational studies providing evidence for olefin binding trans to the NHC, we sought to relate our experimental data to the suggested trans-binding mechanism. If olefin binding occurs trans to the NHC, exchanging the chloride ligands for iodides should not have a major steric impact on the transition state.21 Instead, the iodide ligands may have an electronic effect. When the chlorides in 1a were exchanged for iodides (1b), phosphine dissociation occurred more rapidly, but the reactivity of the active species did not increase.14b In the case of the chiral catalysts 2b-4b, a lower reactivity could increase the enantioselectivity by causing the reaction to proceed through a late, more product-like transition state.22 The added steric bulk in 5a may hamper its reactivity enough to mimic the electronic deactivation present in the diiodide catalysts 2b-4b, resulting in a late transition state and higher degree of stereochemical communication between the catalyst and the substrate. Substrates 25 and 27, which have alkenes with less steric hindrance compared to substrates 13 and 23, may form products in lower enantiomeric excess due to a higher reactivity with the catalyst. Although recent studies suggest olefin binding occurs trans to the NHC, and the observed absolute stereochemistry can be justified using a proposed model based on trans binding, the experimental data does not provide enough support to rule out a cis-binding mechanism.
Conclusions
We have synthesized novel asymmetric olefin metathesis catalysts, which contain chiral, monodentate NHC ligands, and have shown their use in asymmetric ring-closing metathesis of prochiral trienes containing trisubstituted alkenes. Adding substitution to the position para to the ortho-isopropyl group of the N-bound aryl rings (3a and 4a) of the parent compound 2a had a relatively small effect on the enantioselectivities of the ARCM reactions. By exchanging the chloride ligands for iodides, an increase in enantioselectivity was observed for catalysts 2b-4b relative to 2a-4a. In the case of catalyst 5a, substitution on the same side of the aryl ring results in enantiomeric excesses in up to 92% and very high conversions with low catalyst loadings (less than 1 mol %). Two proposed models for the formation of the observed products have been discussed: if the incoming olefin binds cis to the NHC, the stereo-defining interaction is the face of the ruthenium to which the olefin binds; if the incoming olefin binds trans to the NHC, the stereo-defining interaction is the position of the alkylidene under the N-bound aryl ring. In both cases, the position of the pendent olefin in the forming ring also plays an important role in the transition state.
The general interest in developing ruthenium olefin metathesis catalysts stems from their ease of handling and their different functional group tolerance relative to that of the molybdenum catalysts. Catalysts 2a-5b, much like the achiral analogues, are also stable to air and moisture, making them simple to use. The synthesis of enantioenriched 5–7 membered rings has been illustrated, and although further development is needed to expand the substrate scope of ruthenium-catalyzed ARCM, catalysts containing chiral, monodentate NHC ligands are promising due to their high activity, low catalyst loadings, and ease of handling.
Supplementary Material
Experimental procedures and spectral data for catalysts, substrates, and products. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 1.

Synthesis of Chiral Ruthenium Olefin Metathesis Catalysts.
Acknowledgments
We thank the NIH for financial support, Dr. Steven D. Goldberg and Angela Blum for helpful discussions, and Donde Anderson, Dr. Michael Day, and Larry Henling for the crystal structure of the Rh compound.
References
- 1.For recent reviews on olefin metathesis, see: Grubbs RH, editor. Handbook of Metathesis. Wiley-VCH; Weinheim: 2003. Trnka T, Grubbs RH. Acc Chem Res. 2001;34:18–29. doi: 10.1021/ar000114f.Fürstner A. Angew Chem Int Ed. 2000;39:3012–3043.Grubbs RH, Chang S. Tetrahedron. 1998;54:4413–4450.Ivin KJ. J Mol Cat A Chem. 1998;133:1–16.
- 2.a Fujimura O, Grubbs RH. J Am Chem Soc. 1996;118:2499–2500. [Google Scholar]; b Fujimura O, de la Mata FJ, Grubbs RH. Organometallics. 1996;15:1865–1871. [Google Scholar]
- 3.Ruthenium catalysts: Van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH. J Am Chem Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228.Van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH. J Am Chem Soc. 2002;124:4954–4955. doi: 10.1021/ja020259c.Seiders TJ, Ward DW, Grubbs RH. Org Lett. 2001;3:3225–3228. doi: 10.1021/ol0165692.
- 4.Molybdenum catalysts: Totland KM, Boyd TJ, Lavoie GG, Davis WM, Schrock RR. Macromolecules. 1996;29:6114–6125.Alexander JB, La DS, Cefalo DR, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:4041–4042.Zhu SS, Cefalo DR, La DS, Jamieson JY, Davis WM, Hoveyda AH, Schrock RR. J Am Chem Soc. 1999;121:8251–8259.Alexander JB, Schrock RR, Davis WM, Hultzsch KC, Hoveyda AH, Houser JH. Organometallics. 2000;19:3700–3715.Aeilts SL, Cefalo DR, Bonitatebus PJ, Jr, Houser JH, Hoveyda AH, Schrock RR. Angew Chem Int Ed. 2001;40:1452–1456.Tsang WCP, Schrock RR, Hoveyda AH. Organometallics. 2001;20:5658–5669.Hultzsch KC, Bonitatebus PJ, Jernelius J, Schrock RR, Hoveyda AH. Organometallics. 2001;20:4705–4712.Schrock RR, Jamieson JY, Dolman SJ, Miller SA, Bonitatebus PJ, Hoveyda AH. Organometallics. 2002;21:409–417.Tsang WCP, Jernelius JA, Cortez GA, Weatherhead GS, Schrock RR, Hoveyda AH. J Am Chem Soc. 2003;125:2591–2596. doi: 10.1021/ja021204d.
- 5.a Sattely ES, Cortez GA, Moebius DC, Schrock RR, Hoveyda AH. J Am Chem Soc. 2005;127:8526–8533. doi: 10.1021/ja051330s. [DOI] [PubMed] [Google Scholar]; b Jernelius JA, Schrock RR, Hoveyda AH. Tetrahedron. 2004;60:7345–7351. [Google Scholar]
- 6.For reviews of asymmetric olefin metathesis reactions, see: Hoveyda AH. In: Handbook of Metathesis. Chapter 23 Grubbs RH, editor. Vol. 2. Wiley-VCH; Weinheim: 2003. Schrock RR, Hoveyda AH. Angew Chem Int Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576.Hoveyda AH, Schrock RR. Chem Eur J. 2001;7:945–950. doi: 10.1002/1521-3765(20010302)7:5<945::aid-chem945>3.0.co;2-3.
- 7.Gillingham DG, Kataoka O, Garber SB, Hoveyda AH. J Am Chem Soc. 2004;126:12288–12290. doi: 10.1021/ja0458672. [DOI] [PubMed] [Google Scholar]; Also see ref 7.
- 8.Scholl M, Ding S, Less CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe JP, Buchwald SL. J Org Chem. 2000;65:1144–1157. doi: 10.1021/jo9916986. [DOI] [PubMed] [Google Scholar]
- 10.Both enantiomers of 1,2-diphenylethylenediamine are commercially available. See Supporting Information for the synthesis of the aryl bromides 7b-7d and other experimental details.
- 11.Although we have been unable to obtain x-ray quality crystals of 2a-5b, the carbene derived from chiral salt 9d has been bound to [Rh(COD)Cl]2, and an x-ray crystal structure of this complex shows that the isopropyl groups are oriented anti to the phenyl rings on the NHC backbone. See Supporting Information.
- 12.Hong SH, Day MW, Grubbs RH. J Am Chem Soc. 2004;126:7414–7415. doi: 10.1021/ja0488380. [DOI] [PubMed] [Google Scholar]
- 13.1 Mol % of 5a is added at the beginning of the reaction, and then another 1 mol % is added after 2 h, and the 7-membered ring 18 is isolated in 92% yield with a 76%ee. When 2 mol % catalyst 5a is used at the beginning of the reaction, a 74% yield is obtained. Presumably all of the starting material is homocoupled first, and then a second metathesis reaction forms the ruthenium alkylidene that leads to product. The homocoupling reaction forms a ruthenium methylidene, which may decompose at roughly the same rate as it performs the desired olefin metathesis reaction. Adding fresh catalyst allows most of the remaining unreacted starting material and homocoupled product to proceed to the desired 7-membered ring.
- 14.a Ulman M, Grubbs RH. J Org Chem. 1999;64:7202–7207. [Google Scholar]; b Sanford MS, Love JA, Grubbs RH. J Am Chem Soc. 2001;123:6543–6554. doi: 10.1021/ja010624k. [DOI] [PubMed] [Google Scholar]; (c) reference 12.
- 15.Teng X, Cefalo DR, Schrock RR, Hoveyda AH. J Am Chem Soc. 2002;124:10779–10784. doi: 10.1021/ja020671s. [DOI] [PubMed] [Google Scholar]
- 16.See Supporting Information for more details.
- 17.Allowing the hydrogen instead of the non-metal bound vinyl group to interact with the iodide also puts the vinyl group in the pseudo-equatorial position of the ring (see 30).
- 18.Evidence for cis-coordination: Trnka TM, Day MW, Grubbs RH. Organometallics. 2001;20:3845–3847. Evidence for trans-coordination: Tallarico JA, Bonitatebus PJ, Jr, Snapper ML. J Am Chem Soc. 1997;119:7157–7158.Romero PE, Piers WE. J Am Chem Soc. 2005;127:5032–5033. doi: 10.1021/ja042259d.
- 19.a Costabile C, Cavallo L. J Am Chem Soc. 2004;126:9592–9600. doi: 10.1021/ja0484303. [DOI] [PubMed] [Google Scholar]; b Adlhart C, Chen P. J Am Chem Soc. 2004;126:3496–3510. doi: 10.1021/ja0305757. [DOI] [PubMed] [Google Scholar]; c Fomine S, Vargas SM, Tlenkopatchev MA. Organometallics. 2003;22:93–99. [Google Scholar]; d Cavallo L. J Am Chem Soc. 2002;124:8965–8973. doi: 10.1021/ja016772s. [DOI] [PubMed] [Google Scholar]; e Vyboishchikov SF, Bühl M, Thiel W. Chem Eur J. 2002;8:3962–3975. doi: 10.1002/1521-3765(20020902)8:17<3962::AID-CHEM3962>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 20.The x-ray crystal structure of an analogue of 2a in reference 3c shows that the plane of the N-bound aryl ring is not orthogonal to the plane of the NHC. QM/MM calculations in reference 19a concluded that the N-bound aryl rings of a substrate-bound ruthenium alkylidene also are not orthogonal to the plane of the NHC.
- 21.Computations suggest that as the halide ligand increases in size (van der Waals radii) from Cl− to I−, the X—Ru—X bond angle also increases. A larger bond angle positions the halides closer to the Ru=CHR moiety (a smaller angle means the halides are pulled away from the alkylidene), which puts the halides in closer proximity to the reacting olefin, creating a smaller pocket, and therefore a more selective reaction. See reference 19a.
- 22.Palucki M, Finney NS, Pospisil PJ, Güler ML, Ishida T, Jacobsen EN. J Am Chem Soc. 1998;120:948–954. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for catalysts, substrates, and products. This material is available free of charge via the Internet at http://pubs.acs.org.