

Abstract
The budding yeast Saccharomyces cerevisiae, a powerful model system for the study of basic eukaryotic cell biology, has been used increasingly as a screening tool for the identification of bioactive small molecules. We have developed a novel yeast toxicity screen that is easily automated and compatible with high-throughput screening robotics. The new screen is quantitative and allows inhibitory potencies to be determined, since the diffusion of the sample provides a concentration gradient and a corresponding toxicity halo. The efficacy of this new screen was illustrated by testing materials including 3,104 compounds from the NCI libraries, 167 marine sponge crude extracts, and 149 crude marine-derived fungal extracts. There were 46 active compounds among the NCI set. One very active extract was selected for bioactivity-guided fractionation resulting in the identification of crambescidin 800 as a potent antifungal agent.
There has been sustained interest in using the budding yeast Saccharomyces cerevisiae as both a model organism to study basic eukaryotic cell biology and as a system to engage in therapeutic lead discovery. Its genome has been sequenced and much is known about its fundamental biology.1,2 Many cellular processes are highly conserved between yeast and mammalian cells, and observations in yeast have often translated into the discovery of similar processes in humans.3 The use of specific yeast deletion mutants in chemical screening has been ongoing for years,4–8 but the recent availability of arrayed, genome-wide deletion libraries9 and the development of high-throughput, chip-based sensitivity screens has sparked a renaissance in yeast chemical biology.10–14 Compound sensitivity profiles across the entire set of haploid deletion strains have allowed strains to be clustered according to their compound sensitivity profiles.7,8,13 Alternatively, the method of “haploinsufficiency profiling”, by which agents are profiled against the genome-wide set of heterozygous deletion mutants, has been used to identify putative targets directly.12–14 Therefore, yeast provides a broadly applicable tool for the discovery of novel bioactive compounds and their targeted pathways.
In the current resurgence of the use of yeast as a tool in biomedical research, almost forgotten is the utility of S. cerevisiae to guide the discovery of new chemical entities with broad-spectrum antifungal activity.15 Prior to the 1970’s there was little or no demand for new-generation antifungal agents and the known drugs consisted of the polyene natural products (amphotercin B and nystatin), the synthetic azoles (fluconazole, itraconazole, or voriconazole), or 5-fluorocytosine. During the period 1981–2002, there were 23 new small-molecule antifungal agents approved for clinical use, but aside from two novel natural products of the echinocandin class as glucan synthesis inhibitors,16 the rest were azoles or squalene epoxidase inhibitors.17 There is now widespread recognition that both immune-compromised and healthy individuals are susceptible to potentially fatal fungal diseases, and that new agents are needed.18 We have begun a program of small-molecule library screening to address this problem.
The screen described in this report is a high-throughput version of the well-established halo assay for antimicrobial compounds. In part, this work extends the practices of the late Prof. Rinehart who continually maximized the use of biological analysis to further exploit the significance of natural products.19 We call this approach the high-throughput (HT) yeast halo assay, as it is fast, robust, and provides a quantitative output that correlates well with inhibitory potency data determined in liquid culture. The power of this strategy is illustrated through the evaluation of compound diversity libraries. A further proof-of-concept result includes the assay-guided isolation, from a crude sponge extract, of crambescidin 800 – a compound first described by Prof. Rinehart in 1991.20
Results and Discussion
We set out to develop a yeast toxicity screen that would be quantitative, easily automated, and compatible with high-throughput screening robotics. For typical small molecule screens performed in 384-well format, compounds are generally diluted 100- to 500-fold from DMSO stocks into the assay plate and this concentration persists for the duration of the assay. In the high-throughput (HT) yeast halo assay outlined in Figure 1, compounds are not confined to discrete wells, but are pin-transferred directly into agar plates and allowed to diffuse freely from the site of addition.
Figure 1.
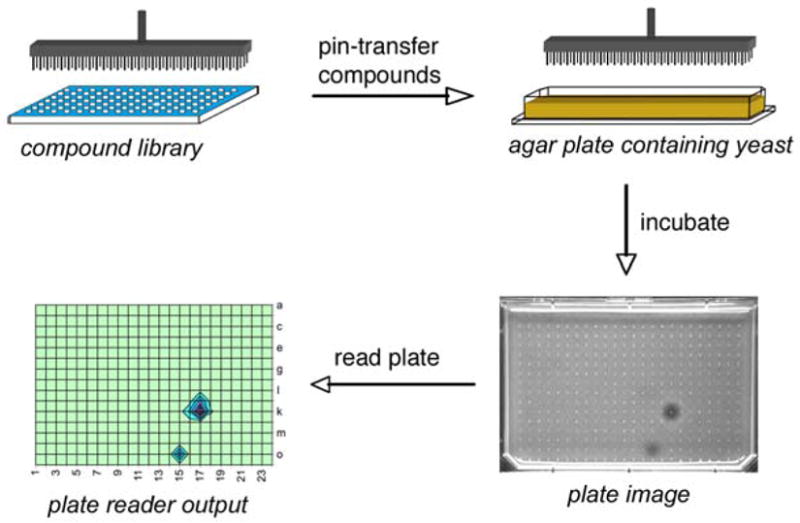
HT yeast halo assay strategy.
As a potential difficulty, some compounds of interest may require multiple cell cycles (> 90 min) to achieve lethality. In such cases fast diffusion from the site of compound addition may limit the ability to detect slower-acting, though potentially interesting, bioactive compounds. The diffusion behavior of a set of fluorescent compounds was therefore investigated, as shown in Figure 2. Three fluorophores of differing molecular weight and hydrophobicity, 3-amino-7-methyl-coumarin (AMC, 175 daltons), tetramethylrhodamine (TMR, 386 daltons), and rhodamine B-conjugated dextran (TMR-DX, average MW ~3000 daltons), were transferred into single-well agar plates using notched pins that deliver 0.2 μL (±8%) each, and the fluorescence signal was measured as a function of time using a plate reader. Since the pins were arrayed in standard 384-well format, a regular plate reader could be used to quantify fluorescence at the center of compound delivery in an area of ~1 mm2. The fluorescence intensity in agar was compared with the intensity measured in solution at the same concentration and path length, enabling the calculation of a dilution factor as a function of time for each compound. As shown in Figure 2, the compound diffusion rates correlated roughly with molecular weight, with TMR-DX diffusing the slowest, TMR diffusing at an intermediate rate, and AMC diffusing approximately three times faster than TMR-DX. Nevertheless, despite the fast diffusion, AMC maintained an effective dilution of 1:100 within the 1-mm2-detection zone for approximately 6 h, which corresponds to four cell cycles.
Figure 2.

Fluorescence diffusion experiment in agar. Compounds were pin-transferred into agar and the fluorescence was measured as a function of time.
Neither of the rhodamine derivatives diffused completely over the course of the experiment. Both TMR and its dextran conjugate (TMR-DX) remained detectable even after 10 h, and at this time point their initial diffusion appeared complete with a much more slowly diffusing species remaining within the detection zone, as seen in Figure 2 at 10 h. Despite the observed differences among the test fluorophores, the results with AMC suggest that small, drug-like molecules will persist within the zone of detection for at least 3–4 yeast cell cycles in a concentration range of 50–100 μM (given 0.2 μL of a 10 mM stock concentration).
An initial test of the yeast halo HT assay system was carried out using 3,104 compounds from the National Cancer Institute’s Diversity, Mechanistic, and Natural Products libraries. 21 The compounds were formatted into 384-well plates as 1 mM or 10 mM DMSO stock solutions. The Diversity set includes 1990 compounds selected from a larger library based on structural diversity using a calculated 3-point pharmacophore model. The Mechanistic set consists of 879 compounds selected from ~40,000 compounds that have been tested in the NCI human tumor 60 cell line screen22 and represents a diverse range of growth inhibition patterns. The Natural Products set contains 235 structurally diverse natural products. The sets include many known bioactive compounds as well as diverse synthetic and natural products with unknown biochemical mechanisms. All three compound libraries were pin-transferred from the plated DMSO stocks directly onto OmniTrays containing agar seeded with yeast, and the plates were allowed to grow overnight. Optical density readings were obtained using a plate reader, providing a quantitative measure of potency for a given toxin. Out of 3,104 compounds, 46 distinct halos were detected and were verified by visible inspection. This hit rate of 1.5% compares with a hit rate of 0.07% when we screened a library of 15,000 diverse synthetic compounds (ChemDiv) under the same conditions.
There are some interesting activity trends and structural patterns that merit discussion for the 46 active compounds. Their structures are shown in Figure 3 and Table 1 lists their known activities and/or biological targets, including their activities against NCI tumor and leukemia cell lines. Some of these compounds are fungal toxins, such as malachite green oxalate (NSC-5550), NSC-5992, fentichlor (NSC-55636) and dehydroabietylamine (NSC-65238), in which the modes of actions and targets are unknown. Other compounds are associated with biological activities in other organisms, such as NSC-12155 (trypanosomes), tricycline B (NSC-47932; bacteria), and NSC-305787 (malaria). The bioactive compounds with established biological targets or mechanisms of action fell into several general categories: (a) genotoxic agents: ellipticine (NSC-71795), bleomycin (NSC-125066), a nitrobenzofuroxan (NSC-207895), and N,N-dimethyldaunomycin (NSC-258812); (b) eukaryotic translation inhibitors: trichodermin (NSC-73846), anisomycin (NSC-76712), and verrucarin A (NSC-126728); (c) antimetabolite: 3-deazaneplanocin (NSC-617989); and (d) toxic organometallics: NSC-168597, NSC-268879, and NSC-321237. Rapamycin (NSC-226080) was identified as a potent hit and is known to inhibit the Tor pathway in yeast via its complex with the Fpr1 protein.15 One aminoalkyl phenothiazine derivative (NSC-24113) also scored as a relatively potent hit in the HT yeast halo assay. This compounds is similar to the antipsychotic drug chlorpromazine and is known to inhibit calmodulin, an essential protein in yeast.23 In addition, two polycyclic aromatic compounds (NSC-13480 and NSC-638432) were identified as possible intercalators and/or topoisomerase II inhibitors based on their chemical structures.
Figure 3.

Structures of NCI hits discovered from the HT yeast halo assay screen.
Table 1.
NCI Diversity Set: Comparison of HT Yeast Halo Assay IC20’s with Other Biological Activities
| yeast assay
|
NCI cell line activity
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| NSC #a | common nameb | originc | IC20d (μM)e | IC20 (μM)f | other activity/ target or action | leukemiag | solid tumorh | comments | ref. |
| 4623 | none | syn. | 96.9 | 53.0 | unknown | NAi | NA | n/aj | |
| 5550 | malachite green oxalate | syn. | 1.9 | antifungal /unknown | NA | NA | treatment for fish diseases | n/a | |
| 5992 | none | syn. | 32.0 | antifungal/ unknown | NA | NA | 39 | ||
| 10777 | none | syn. | 34.9 | 113.0 | unknown | NA | NA | n/a | |
| 12155 | none | syn. | 65.0 | antitrypanosomal/ unknown | NA | NA | 40 | ||
| 13239 | none | syn. | 92.0 | unknown | NA | NA | n/a | ||
| 13480 | none | syn. | 49.0 | unknown | NA | NA | n/a | ||
| 17383 | none | syn. | 10.3 | 12.1 | unknown | NA | NA | n/a | |
| 17600 | none | syn. | 39.0 | unknown | NA | NA | n/a | ||
| 22907 | none | syn. | 27.0 | unknown | NA | NA | n/a | ||
| 24113 | none | syn. | 1.4 | unknown | NA | NA | n/a | ||
| 35446 | none | syn. | 94.7 | 115.0 | unknown | NA | NA | n/a | |
| 47932 | tricycline B | syn. | 18.0 | antimicrobial/ FtsZ in E. coli | NA | NA | 41,42 | ||
| 48160 | none | syn. | 70.0 | 100.0 | unknown | NA | NA | n/a | |
| 55636 | fentichlor | syn. | 8.6 | antifungal/ unknown | NA | NA | agrochemical | 39,43 | |
| 64875 | azaspirane derivative | syn. | 6.9 | 15.4 | unknown | NA | μM | 44,45 | |
| 65238 | dehydroabietylamine | plant NP | 7.9 | 0.93 | antifungal/ unknown | NA | μM | antifouling agent | 46 |
| 71795 | ellipticine | plant NP | 16.0 | anticancer/ topoisomerase II | NA | NA | 47 | ||
| 73846 | trichodermin | bact. NP | 4.0 | eukaryotic translation inhibitor/ ribosome | NA | NA | 48–50 | ||
| 76712 | anisomycin | bact. NP | 13.0 | eukaryotic translation inhibitor/ ribosome | NA | μM | potential psychiatric drug | 51 | |
| 109509 | none | semisyn. | 15.4 | 6.9 | unknown | NA | NA | n/a | |
| 122657 | none | syn. | 4.1 | 0.9 | unknown | NA | NA | n/a | |
| 125066 | bleomycin | bact. NP | 6.4 | genotoxic/ DNA-damaging | NA | NA | 52–54 | ||
| 126728 | verrucarin A | fungal NP | 1.1 | eukaryotic translation inhibitor/ ribosome | μM | μM | 55,56 | ||
| 150117 | none | syn. | 21.2 | 12.1 | unknown | μM | NA | n/a | |
| 168597 | tributyllead chloride | syn. | 1.0 | unknown | μM | μM | toxic organometallic | n/a | |
| 168656 | none | syn. | 69.4 | 33.7 | unknown | NA | NA | n/a | |
| 180973 | tamoxifen | syn. | 25.0 | anticancer/ antiestrogen | NA | NA | breast cancer; perturbs Ca2+ metabolism in yeast | 11,57 | |
| 186063 | phenothiazine derivative | syn. | 80.0 | anti-inflammatory and others/ calmodulin | NA | NA | 58 | ||
| 207895 | nitrobenzofuroxan derivative | syn. | 3.8 | 2.3 | antitumor/ DNA-damaging | μM | NA | 59–61 | |
| 226080 | rapamycin | bact. NP | 0.01 | immunosuppressive and antiproliferative/ TOR protein | NA | NA | antifungal | 62 | |
| 258812 | N,N-dimethyldaunomycin | semisyn. | <0.9 | antitumor/ DNA intercalator | NA | μM | 63 | ||
| 268879 | none | syn. | <0.9 | unknown | NA | μM | toxic organometallic | n/a | |
| 301460 | trichopolyn I | fungal NP | 1.9 | 0.6 | immunosuppressive/ biological membranes | NA | antibiotic and antifungal | 64–67 | |
| NA | NA | ||||||||
| 305787 | none | syn. | 4.1 | 12.1 | antimalarial | NA | NA | 68 | |
| 306864 | diosgenin | plant NP | 0.9 | 2.1 | antiproliferative/ suppresses | NA | NA | cortisone, progesterone | 69 |
| Akt activation | precursor | ||||||||
| NA | NA | ||||||||
| 310342 | none | syn. | 47.3 | 57.2 | unknown | NA | NA | n/a | |
| 312033 | striatin E | fungal NP | 62.9 | 52.1 | unknown | NA | μM | parent compound, calmodulin- binding | 70 |
| NA | NA | ||||||||
| 321237 | none | syn. | 2.3 | unknown | NA | NA | toxic organometallic | n/a | |
| 322661 | none | syn. | 65.8 | 99.2 | unknown | NA | NA | n/a | |
| 371777 | none | syn. | 8.5 | 11.0 | unknown | NA | NA | n/a | |
| 617989 | 3-deazaneplanocin | syn. | 116 | antiviral/(S)-adenosyl-L- homocysteine hydrolase | NA | NA | potential Ebola virus treatment | 71,72 | |
| 620358 | 15-oxosteviol methyl ester | plant NP | 42.7 | 33.2 | anti-inflammatory/ unknown | μM | NA | natural sweetener; derivatives mutagenic | 73–75 |
| 638432 | none | syn. | 25 | 23.6 | unknown | NA | NA | 76 | |
| 641691 | vicenistatin | fungal NP | 44.0 | antitumor/ unknown | NA | μM | 27 | ||
| 670224 | none | syn. | 2.6 | unknown | NA | NA | n/a | ||
For structures of NCI compounds see Figure 3.
Syn = synthetic, semisyn .= semisynthetic, NP = natural product.
The IC20 value was defined as the concentration when (doubling time of the DMSO control)/ (doubling time with compound) was equal to 0.8.
Determined in liquid media.
Calculated based on halo density.
Leukemia LC50 activity evaluated online using NCI 60 cell line cancer screen data from September 2005. See http://dtp.nci.nih.gov/dtpstandard/cancerscreeningdata/index.jsp.
Solid tumor LC50 activity evaluated online using NCI 60 cell line cancer screen data from September 2005. See http://dtp.nci.nih.gov/dtpstandard/cancerscreeningdata/index.jsp.
No activity, “NA”, was defined as an LC50 value greater than 10.0 μM.
No references available, “n/a”.
While many of the NCI hits have known biological targets and/or mechanisms of action, the majority of the hits (33 out of 46) have unknown targets in yeast and represent potentially interesting biological probes and/or antifungal therapeutic lead structures. For example, tamoxifen (NSC-180973), a commonly used anticancer drug that inhibits estradiol binding to the estrogen receptor, is reported to have antifungal properties.24 Although its target in yeast is unknown, a recent study has shown that tamoxifen perturbs calcium homeostasis in yeast by increasing cytosolic Ca2+ to toxic levels.11 Other potentially interesting hits include structurally complex natural products such as trichopolyn (NSC-301460), diosgenin (NSC-306864), striatin E (NSC-312033), vicenistatin (NSC-641691), a steviol derivative (NSC-620358), and a terpene alkaloid (NSC-65238). Some of these compounds are likely to target biologically interesting, medicinally relevant proteins in mammalian cells whose homologs are cause for their toxicity in yeast. For example, steviol and its derivatives, natural sweeteners from Stevia rebaudiana, have been shown to possess anti-inflammatory activity25 and tumor inhibitory effects,26 although their target(s) have not been identified. Vicenistatin, an interesting macrolactam, exhibits antitumor activity via an unknown mechanism.27 The identification of candidate targets for these compounds in yeast by screening for resistant mutants or by haploinsufficiency profiling14 will likely shed light on their relevant mechanisms in higher eukaryotes.
A comparison of the HT yeast halo assay hit rates among the natural products versus the synthetics provided some noteworthy patterns. The 46 active compounds included 34 synthetics out of 2,869 (1.2%) and 12 natural products out of 235 (5.1%). The overall hit rate for the natural products was actually the greatest since they accounted for 26% of the total antifungal hits but comprised only 8.2% of the total library content.
We randomly selected 22 compounds, seven natural products and 15 synthetics, of the 46 active NCI diversity set compounds and tested their inhibitory potencies in liquid culture. The HT yeast halo assay was repeated on this subset in triplicate, and the halos were compared with the IC20 values obtained in liquid culture. A correlation exists between halo size (544 nm absorbance using a plate reader) and liquid culture IC20s (r2=0.80), as seen in Figure 4. This correlation was used to predict the IC20s for the remaining compounds (see Table 1 for experimentally determined and predicted IC20s for NCI compounds).
Figure 4.
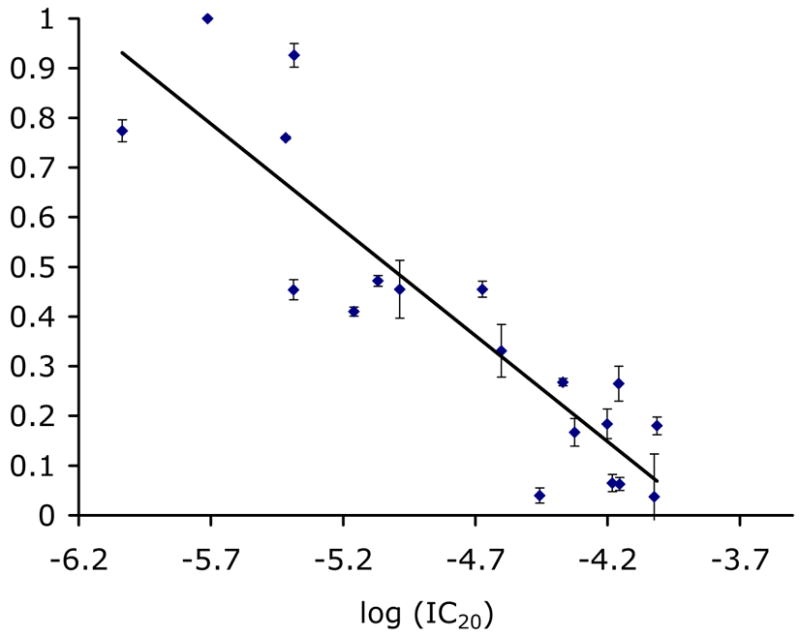
Correlation between the fraction of inhibition of yeast growth in agar and IC20 values determined in liquid culture.
The good correlation seen between the HT yeast halo assay and the IC20 values determined in liquid culture suggests that IC20 values can be estimated with high confidence from the readings in agar within a range of 2 log units, from 100 μM to 1 μM. Compounds with IC20 values higher than 100 μM were not detectable by the HT halo assay, while highly potent compounds (IC20 < 0.8 μM) yielded halos that were larger than the light beam of the plate reader. In the case of rapamycin (NSC-226080, IC20 = 10 nM), for example, the halo extended beyond the site of compound addition into surrounding “wells”, and these zones were detected as false hits in the plate reader output. Such halos are readily identified by visual inspection of the plate. Potencies can be estimated by measuring the halo diameter directly and, in principle, an algorithm could be written to detect these highly potent hits in the plate reader output. In these cases, an upper limit of 0.8 μM can be assigned to the IC20 value and the exact potency can be determined in liquid culture by standard methods.
Encouraged by the relatively higher hit rate noted above for the natural products among the NCI hits, it seemed worthwhile to next evaluate selected extracts from the (University of California, Santa Cruz) repository in the HT yeast halo assay. Each of the operations shown in Figure 5 was employed to create a screening/fractionation strategy that rapidly interrogates both crude natural product extracts and semi-pure fractions for yeast toxicity. The general scheme involves the following steps: (a) extraction of marine sponges and sponge-derived fungi cultures with organic solvents; (b) screening of the extracts in 384-well format using the HT yeast halo assay; (c) fractionation of hit extracts by reversed phase HPLC and collection of short fractions in deep-well plates; (d) reformatting of the resulting fractions as DMSO stocks in 384-well format; (e) assaying of the fractions in the HT yeast halo assay; and (f) LC-MS analysis of active wells and correlation of biological activity with mass abundance. Overall, this scheme proved to be workable and easy to implement.
Figure 5.
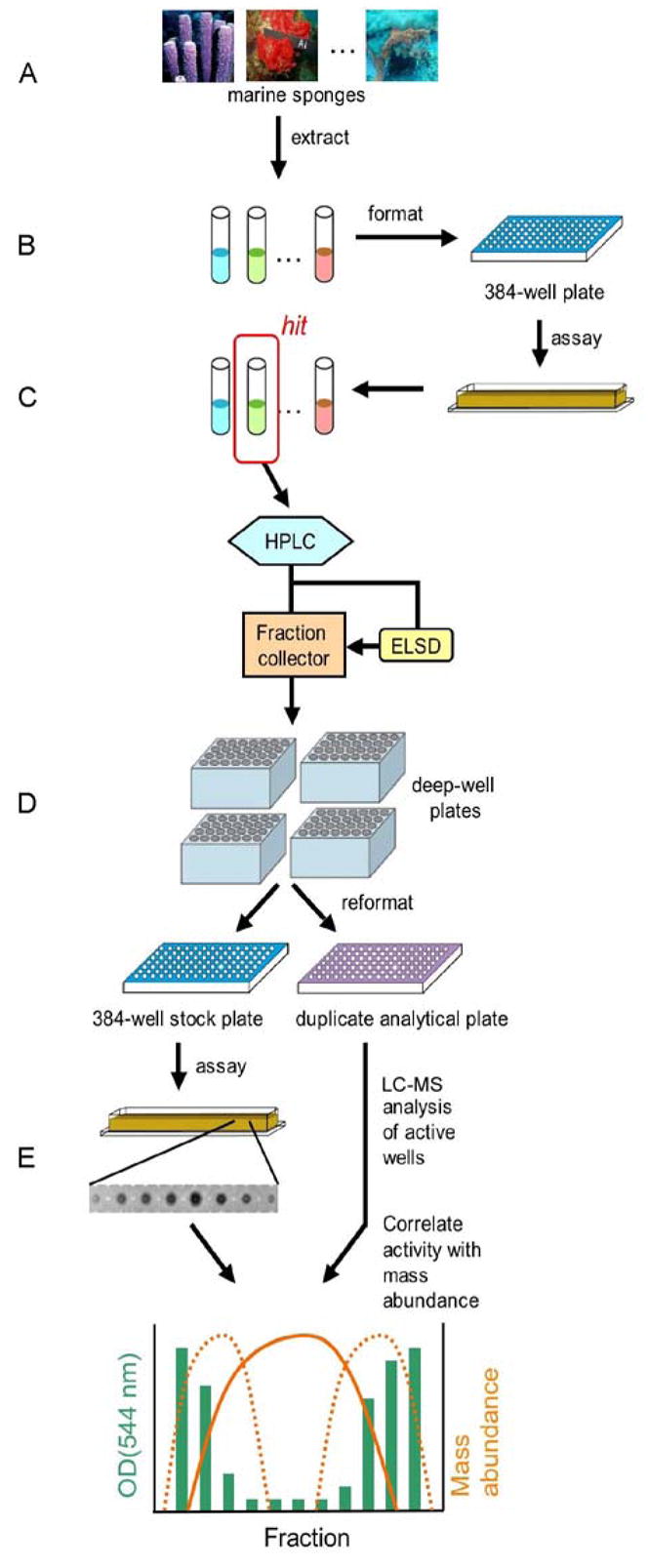
Schematic of the HT yeast halo assay-guided purification of natural products.
A single 384-well plate arrayed with 167 sponge extracts and 149 fungal extracts was screened in the HT yeast halo assay. One of these, the dichloromethane extract from the marine sponge Monanchora unguifera, showed potent activity and was selected for fractionation by reversed-phase HPLC. The resulting fractions were formatted into a 384-well plate and re-screened. A major peak of bioactivity was identified as illustrated in Figure 6. The parallel LC-MS analysis of the active wells showed a compound with a molecular weight of 800 daltons for which the abundance corresponded to the level of yeast toxicity across the peak. Further purification by HPLC afforded a pure compound with an IC20 in liquid culture of 0.83 μM, which was on a par with some of the most active compounds from the NCI Diversity set (Table 1).
Figure 6.
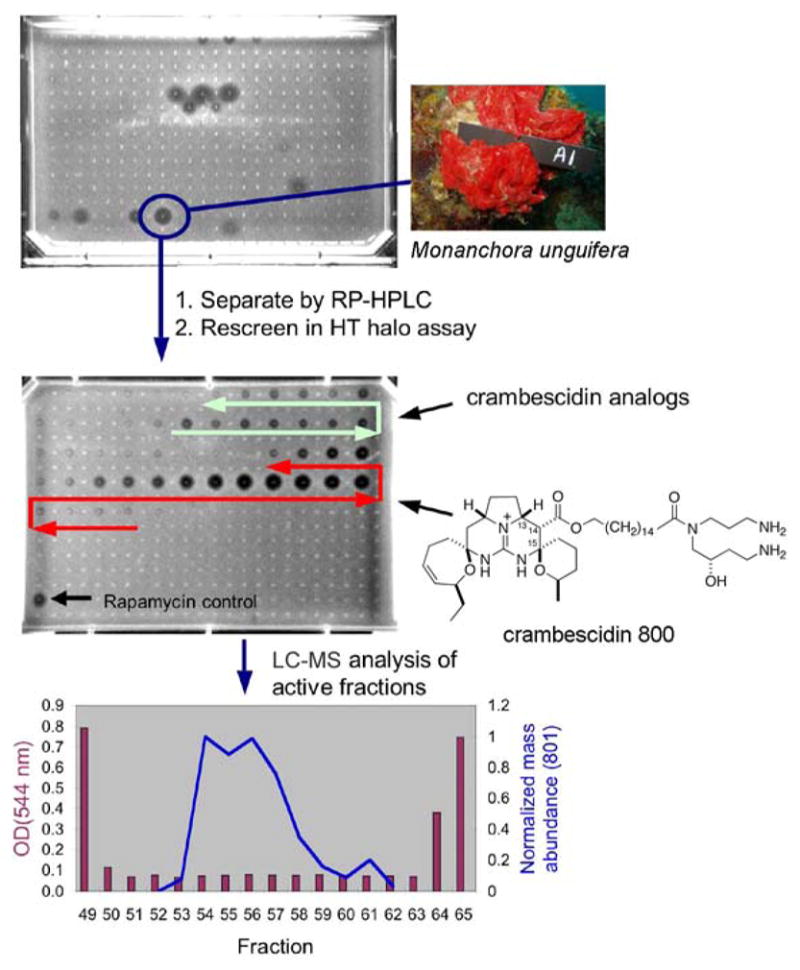
Schematic representation of HT yeast halo assay-guided purification of crambescidin 800 from the sponge Monanchora unguifera.
Dereplication using the above taxonomic information alongside the MS and NMR data showed this potent compound had the planar structure of crambescidin 800. The task of distinguishing among the 64 diastereomeric structures, based on the possible stereochemical variations at the chiral sites of the pentacyclic guanidinium core of the crambescidin ring system, was completed using input from two sources. As an important biosynthetic insight, only two series of pentacyclic guanidinium systems, and similarly, just two series of tricyclic guanidinium frameworks, have been observed. There are more than twenty such natural products reported from organisms, including sponges and sea stars (as summarized in Table S2). For both ring types the single change in stereochemistry occurs as follows: (a) the pentacyclic-containing natural products all have conserved configurations of 3S, 8S, 10S, 19R, with the compounds headed by crambescidin 800 and ptilomycalin A possessing the 13R, 14S, 15R stereochemical motif, whereas the other diastereomer series, consisting of just two compounds (one being 13,14,15-isocrambescidin 800), have the opposite stereochemistry at carbons 13, 14, and 15, and (b) the tricyclic containing natural products also have the conserved 10S configuration (ring system renumbered as shown in Table S2) but with opposite configurations at 13R as found in batzelladine B and 13S as observed in batzelladine A. We assigned the crambescidin 800 stereochemistry based on the NMR data set discussed further in the Experimental Section.
The polycyclic guanidinium core of crambescidin 800 and its congeners (Table S2) are associated with a variety of biological activities. Crambescidin 800 inhibits HIV-1 envelope-mediated fusion,28 exhibits cytotoxicity toward various cancer cell lines,20,29 demonstrates antimalarial activity,30 is active against Herpes simplex type 1 (HSV-1) virus,20 induces the differentiation of K562 cells into erythroblasts, alters the morphology of neuronal cell lines,31 and exhibits general cytotoxicity toward mammalian cells.32 In addition, crambescidin analogs have demonstrated potent and reversible blockage of Ca2+ channels,33 and synthetic analogs of crambescidin 800 have shown selective toxicity toward solid tumors vs. normal cells.34 Crambescidin analogs, including crambescidin 800, were also shown to specifically inhibit the interaction between the HIV protein Nef and its host ligands.35 The target(s) of crambescidin 800 in yeast may represent homologs of its target(s) in mammalian cells, or its antifungal activity may represent a new and distinct biological activity. The identification of its cellular target(s) in yeast may shed light on this interesting class of compounds and provide a basis for understanding its potent cytotoxicity in higher eukaryotes.
The HT yeast halo assay has several advantages over the traditional multi-well screens. First, in liquid culture, drug-resistant mutations may develop in a population, obscuring the effects of a toxic compound and leading to a false negative result. In agar the yeast are immobilized, such that sporadic mutants would remain localized in a single colony and not affect halo formation. Second, a wider range of compound potencies can be observed in the HT yeast halo assay compared with liquid assays. Discrimination between compounds of varying potencies is difficult when they are added at concentrations that far exceed their minimum inhibitory concentrations (MICs). Third, in the HT yeast halo assay, differences in halo diameter report directly on relative potencies over a wide range of concentrations, even at doses well above the MIC. This is useful especially for HT screening applications, since initial screens are usually performed at a single concentration. The wide dynamic range of the yeast HT halo assay can also be particularly useful when screening for selective toxicity between two or more mutant yeast strains. For example, a compound may exhibit selective toxicity toward a mutant strain vs. wild type, but if its lethality toward wild type is above a certain threshold level then the compound would appear nonselective in liquid culture. Finally, in the HT yeast halo assay, differential toxicity would be observed as a difference in halo diameter over a wide range of potencies with no limiting threshold.
The scheme for identifying antifungal compounds described in this paper represents a slight modification of the classic bioassay-guided purification strategy commonly employed in natural products discovery. With recent advances in laboratory automation, high-throughput screening, and highly sensitive microscale probes for NMR spectroscopy, the use of natural products as input into phenotypic assays has undergone a renaissance.36 Convergence of modern natural product isolation methods with yeast chemical genomics techniques thus promises to further advance the rapid identification of potent natural products with novel mechanisms of action. Formatting extracts into 384-well plates allows for rapid screening of thousands of extracts, in principle, against multiple yeast mutants or against wild type yeast under different growth conditions. The single fractionation step and the formatting of narrowly-cut fractions into 384-well plates has the advantage of efficiency and may lead to the identification of active compounds of low abundance that would be overlooked in a traditional multi-step assay-guided purification approach, since these compounds are significantly enriched upon fractionation.
The yeast HT halo screen is quantitative and allows IC20 values to be determined since the diffusion of the sample provides a concentration gradient and a corresponding toxicity halo. The new screen successfully identified NCI compounds and the marine natural product crambescidin, with potent and previously unrecognized yeast toxicity, as promising antifungal leads. We have also shown that this simple screen can be adapted to monitoring the bioactivity of fractionated marine natural product extracts, which allowed for the rapid identification of crambescidin 800 as being responsible for the major antifungal activity in the Monanchora unguifera dichloromethane extract. Finally, it is apparent that the majority of antifungal hits found in the NCI library are derived from natural products, further emphasizing the rich biological activity of this class of compounds. We are currently applying genetic and genomic techniques, including haploinsufficiency profiling, in order to identify their molecular targets in yeast.
Experimental Section
General Experimental Procedures
Yeast strain BY4741 was obtained from Open Biosystems. Growth media reagents were purchased from Sigma. OmniTrays were purchased from VWR (46600-638). Compounds were obtained from the National Cancer Institute’s Developmental Therapeutics Program. Crude marine sponge extracts were collected over three expeditions from 2002–2005 in Papua New Guinea and Vanuatu.
Measuring Fluorophore Diffusion in Agar
10 mM DMSO stocks of AMC, TMR, and TMR-DX, were pin-transferred into an OmniTray containing solidified agar (prepared in the absence of media). The agar was prepared such that the height of the agar in the plate was 0.5 cm. The rate of diffusion of the dye at the point of transfer was quantified using a plate reader in the fluorescence mode. Fluorescence measurements were obtained in liquid buffer (PBS) using a 96-well plate with the same volume (176 μL) that corresponded to the 0.5 cm path length used in the agar plate. Flourescence was measured every 15 min after pin transfer for 24 hours.
High-Throughput Yeast Halo Assay
A solution of 2x YPD-H (YPD media buffered with HEPES) was prepared by dissolving 10 g yeast extract, 20 g peptone, and 20 g dextrose in 500 mL 25 mM HEPES at pH 7.5. After autoclaving, the pH was readjusted to pH 7 with NaOH, and the media was filter-sterilized. A solution of 2x agar was prepared by suspending 1.5 g granulated agar (Fisher, BP1423-500) in 100 mL H2O, and the suspension was autoclaved. For each plate screened, 20 mL YPD-H-agar was prepared by mixing 10 mL 2x YPD-H at 50–55 °C with 10 mL 2x agar at 50–55 °C. The warm medium was inoculated with 400 μL of an overnight yeast culture (BY4743 diploid wt strain), poured into an OmniTray, and the tray was set on a flat surface to cool for 30 min.
Compounds were pin-transferred from DMSO stocks plated in 384-well polypropylene trays (Nunc) into the cooled agar with a pin-tool robot (VP903B, V&P Scientific) using notched pins that deliver 0.2 μL (±8%) each. Before and between applications pins were cleaned by submersion in methanol (3x), DMSO (3x), and finally methanol (3x). After each wash step the pins were applied to blotting paper (V&P Scientific) to absorb excess solvent. Immediately after compound addition, an initial absorbance reading (Absblank) was taken of the plate using a 384-well compatible plate reader at 544 nm. The plate was incubated at 30 °C for 24 h and then read again. Halos were verified by visual inspection, and the active compounds were obtained as individual samples from the NCI Developmental Therapeutics Program. Their purities and identities were verified by LC-MS, and only those compounds whose purities exceeded 95% were carried forward. DMSO stocks (10 mM) of these compounds were arrayed in a new 384-well plate with several DMSO control wells separating each compound, and the compounds were re-screened in the HT halo assay. The plate was screened in triplicate, and for each compound, inhibition was quantified using the following equation: Fi = [(Absdiagonal − Absdiagonal-blank) − (Absi − Absi-blank)]/(Absdiagonal − Absdiagonal-blank); where Fi is the fraction growth inhibition for compound i such that complete inhibition within the zone of addition equals 1 and no detectable halo equals 0. Absdiagonal is the average absorbance (Abs) of the DMSO-containing wells diagonal to each compound well (i.e., the absorbance corresponding to maximum yeast growth); Absdiagonal-blank is the agar blank for the diagonals (i.e., the average Abs of the diagonal wells taken just after compound addition and prior to yeast growth); Absi is the Abs for the well containing compound i after yeast growth; Absi-blank is the agar blank for the well containing compound y (i.e., the Abs corresponding to the well containing compound i taken just after compound addition and prior to yeast growth).
Growth Inhibition Measurement in Liquid Culture
Yeast (wild type diploid strain BY4743, 0.015 OD) were incubated with six two-fold dilutions of each compound in 100 μl cultures in 96 well plates, in addition to DMSO controls. ODs were read every 15 min using a TECAN plate reader, and the plate was agitated just prior to reading to suspend the yeast. Yeast doubling times at each concentration were calculated and compared to the doubling time in DMSO. The IC20 was defined as the concentration in which the doubling time of the DMSO control divided by the doubling time with compound was equal to 0.8.
Natural Product Purification and Structure Elucidation
Marine sponges (167) were extracted sequentially with hexanes, methanol and dichloromethane using an accelerated solvent extractor (ASE). In addition, sponge-derived fungi were cultured using malt and Czapek-Dox media and the liquid cultures were extracted with ethyl acetate. Sponge extracts and sponge-derived fungi culture extracts were dried, taken up into DMSO at 5 mg/mL, and formatted into a 384-well polypropylene plate and screened in the HT yeast halo assay described above.
Since the dichloromethane-derived crude extract from Monanchora unguifera produced a strong halo, it was selected for further fractionation and compound identification. Thus, 40 mg of crude extract was taken up into methanol and purified on a C18 column (2 × 20 cm) with a Gilson 215 robotic fraction collector triggered by a light-scattering detector (ELSD) to allow collection of minor (30 μg) components. Fractions were collected in deep-well 96-well plates (1.7 mL/well) and completely evaporated using a centrifugal evaporator (Savant). Fractions were taken up into 40 uL DMSO, reformatted into a single 384-well plate using a Hydra liquid dispenser, and screened in the HT yeast halo assay. LC-MS analysis of active wells showed an ion peak at m/z 401.3 [M+2H]++ and a parent ion peak at m/z 801.8 [M+H]+ and further investigation indicated the presence of a compound with the exact mass of m/z 801.623 and the molecular formula C45H81N6O6. Large-scale purification of crambescidin 800 was performed by reversed-phase HPLC and its growth inhibitory potency was determined in liquid culture (IC20 = 0.83 μM). NMR analysis identified crambescidin 800 as the major active constituent. The presence of 13,14,15-isocrambescidin 800 was ruled out based on 1H and 13C NMR data for position 14 (1H-14: crambescidin 800, 3.20 ppm, J = 5.6 Hz; 13,14,15-isocrambescidin 800, 3.80 ppm, J = 3.4 Hz; experimental, 3.10 ppm, J = 4.8 Hz; 13C: crambescidin 800, 49.6 ppm; 13,14,15-isocrambescidin 800, 42.5 ppm; experimental, 49.3 ppm).37 The experimental NMR data for position 14 were also consistent with data from acetylcrambescidin 80038 (1H-14: acetylcrambescidin 800, 2.94 ppm, J = 5.0 Hz; experimental, 3.10 ppm, J = 4.8 Hz; 13C: acetylcrambescidin 800, 48.9 ppm; experimental, 49.6 ppm), which bears the identical stereochemical assignment to crambescidin 800 at positions 13,14, and 15.
Supplementary Material
13C and 1H NMR spectra of crambescidin 800, isolation scheme, underwater and above water photographs of Monanchora unguifera, comparison of 13C NMR signals for several known polycyclic guanidinium alkaloids, and a table of stereochemical diversity found in naturally-derived polycyclic guanidinium alkaloids. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by grants from the National Institutes of Health CA104569-01 (R.S.L.), GM56062-6 (T.R.H.) CA47135 (P.C.), and NMR equipment grants from NSF CHE-0342912 and NIH S10-RR19918. Special thanks to the Roche Palo Alto campus for the generous donation of an LC-MS system.
Footnotes
Dedicated to the late Dr. Kenneth L. Rinehart of the University of Illinois at Urbana-Champaign for his pioneering work on bioactive natural products.
References and Notes
- 1.Kumar A, Snyder M. Nat Rev Genet. 2001;2:302–312. doi: 10.1038/35066084. [DOI] [PubMed] [Google Scholar]
- 2.See Table 1 of: Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. Science. 1996;274:546–549. doi: 10.1126/science.274.5287.546.
- 3.Hartwell LH. Biosci Rep. 2002;22:373–394. doi: 10.1023/a:1020918107706. [DOI] [PubMed] [Google Scholar]
- 4.Eng WK, Faucette L, Johnson RK, Sternglanz R. Mol Pharmacol. 1988;34:755–760. [PubMed] [Google Scholar]
- 5.Simon JA, Szankasi P, Nguyen DK, Ludlow C, Dunstan HM, Roberts CJ, Jensen EL, Hartwell LH, Friend SH. Cancer Res. 2000;60:328–333. [PubMed] [Google Scholar]
- 6.Dunstan HM, Ludlow C, Goehle S, Cronk M, Szankasi P, Evans DR, Simon JA, Lamb JR. J Natl Cancer Inst. 2002;94:88–94. doi: 10.1093/jnci/94.2.88. [DOI] [PubMed] [Google Scholar]
- 7.Tung RD, Rich DH. Tetrahedron Lett. 1987;28:1139–1142. [Google Scholar]
- 8.Simon JA, Yen TJ. Methods Mol Biol. 2003;223:555–576. doi: 10.1385/1-59259-329-1:555. [DOI] [PubMed] [Google Scholar]
- 9.Mager WH, Winderickx J. Trends Pharm Sci. 2005;26:265–273. doi: 10.1016/j.tips.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- 11.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ, Boone C. Cell. 2006;126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 12.Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, Hinshaw JC, Garnier P, Prestwich GD, Leonardson A, Garrett-Engele P, Rush CM, Bard M, Schimmack G, Phillips JW, Roberts CJ, Shoemaker DD. Cell. 2004;116:121–137. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- 13.Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Proc Natl Acad Sci U S A. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Nat Genet. 1999;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 15.(a) Sehgal SN, Baker H, Vézina CJ. Antibiot. 1975;28:727–732. doi: 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]; (b) Jacob MR, Walker LA. In: Antifungal Agents: Methods and Protocols. Ernst EJ, Rogers PD, editors. Vol. 118. Humana Press; Totowa: 2005. pp. 83–110. [Google Scholar]
- 16.Vicente MF, Basilio A, Cabello A, Pelaez F. Clin Microbiol Infect. 2003;9:15–32. doi: 10.1046/j.1469-0691.2003.00489.x. [DOI] [PubMed] [Google Scholar]
- 17.Newman DJ, Cragg GM, Snader KM. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 18.Barrett D. Biochim Biophys Acta. 2002;1587:224–233. doi: 10.1016/s0925-4439(02)00085-6. [DOI] [PubMed] [Google Scholar]
- 19.Jefford CW, Rinehart KL. In: Pharmaceuticals and the Sea. Shield LS, editor. Technomic Publishing Co; Lancaster, PA: 1988. [Google Scholar]
- 20.Jares-Erijman EA, Sakai R, Rinehart KL. J Org Chem. 1991;56:5712–5715. [Google Scholar]
- 21.http://dtp.nci.nih.gov/branches/dscb/repo_open.html.
- 22.Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 23.Davis TN, Urdea MS, Masiarz FR, Thorner J. Cell. 1986;47:423–431. doi: 10.1016/0092-8674(86)90599-4. [DOI] [PubMed] [Google Scholar]
- 24.Beggs WH. Res Commun Chem Pathol Pharmacol. 1993;80:125–128. [PubMed] [Google Scholar]
- 25.Boonkaewwan C, Toskulkao C, Vongsakul M. J Agric Food Chem. 2006;54:785–789. doi: 10.1021/jf0523465. [DOI] [PubMed] [Google Scholar]
- 26.Yasukawa K, Kitanaka S, Seo S. Biol Pharm Bull. 2002;25:1488–1490. doi: 10.1248/bpb.25.1488. [DOI] [PubMed] [Google Scholar]
- 27.Shindo K, Kamishohara M, Odagawa A, Matsuoka M, Kawai H. J Antibiot. 1993;46:1076–1081. doi: 10.7164/antibiotics.46.1076. [DOI] [PubMed] [Google Scholar]
- 28.Chang LC, Whittaker NF, Bewley CA. J Nat Prod. 2003;66:1490–1494. doi: 10.1021/np030256t. [DOI] [PubMed] [Google Scholar]
- 29.Crambescidin 800 trihydrochloride (NSC 713155) and 13,14,15-isocrambescidin 800 trihydrochloride (NSC 713157) have been screened in the NCI 60 cell lines (October 2006 Release) See: http://dtp.nci.nih.gov/dtpstandard/cancerscreeningdata/index.jsp.
- 30.Lazaro JEH, Nitcheu J, Mahmoudi N, Ibana JA, Mangalindan GC, Black GP, Howard-Jones AG, Moore CG, Thomas DA, Mazier D, Ireland CM, Concepcion GP, Murphy PJ, Diquet B. J Antibiot. 2006;59:583–590. doi: 10.1038/ja.2006.78. [DOI] [PubMed] [Google Scholar]
- 31.Aoki S, Kong DX, Matsui K, Kobayashi M. Anticancer Res. 2004;24:2325–2330. [PubMed] [Google Scholar]
- 32.Palagiano E, Demarino S, Minale L, Riccio R, Zollo F, Iorizzi M, Carre JB, Debitus C, Lucarain L, Provost J. Tetrahedron. 1995;51:3675–3682. [Google Scholar]
- 33.Berlinck RGS, Braekman JC, Daloze D, Bruno I, Riccio R, Ferri S, Spampinato S, Speroni E. J Nat Prod. 1993;56:1007–1015. doi: 10.1021/np50097a004. [DOI] [PubMed] [Google Scholar]
- 34.Aron ZD, Pietraszkiewicz H, Overman LE, Valeriote F, Cuevas C. Bioorg Med Chem Lett. 2004;14:3445–3449. doi: 10.1016/j.bmcl.2004.04.071. [DOI] [PubMed] [Google Scholar]
- 35.Olszewski A, Sato K, Aron ZD, Cohen F, Harris A, McDougall BR, Robinson WE, Overman LE, Weiss GA. Proc Natl Acad Sci U S A. 2004;101:14079–14084. doi: 10.1073/pnas.0406040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 37.Jares-Erijman EA, Ingrum AL, Carney JR, Rinehart KL, Sakai R. J Org Chem. 1993;58:4805–4808. [Google Scholar]
- 38.Braekman JC, Daloze D, Tavares R, Hajdu E, Van Soest RWM. J Nat Prod. 2000;63:193–196. doi: 10.1021/np990403g. [DOI] [PubMed] [Google Scholar]
- 39.Marsh PB, Butler ML, Clark BS. J Ind Eng Chem. 1949;41:2176–2184. [Google Scholar]
- 40.Goble FC, Hoppe JO. Antibiot Chemother. 1952;2:581–589. [PubMed] [Google Scholar]
- 41.Rajaian H, Nazer AHK. J Appl Anim Res. 1999;15:117–120. [Google Scholar]
- 42.Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchison TJ, Kirschner MW, RayChaudhuri D. Proc Natl Acad Sci U S A. 2004;101:11821–11826. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tashika Y, Ohya H, Tsubokawa H, Iwata K. Chemotherapy. 1959;7:67–70. [Google Scholar]
- 44.Mirabelli CK, Badger AM, Sung CP, Hillegass L, Sung CM, Johnson RK, Picker D, Schwartz D, Dorman J, Martellucci S. Anti-Cancer Drug Des. 1989;3:231–242. [PubMed] [Google Scholar]
- 45.Badger AM, Schwartz DA, Picker DH, Dorman JW, Bradley FC, Cheeseman EN, DiMartino MJ, Hanna N, Mirabelli CK. J Med Chem. 1990;33:2963–2970. doi: 10.1021/jm00173a010. [DOI] [PubMed] [Google Scholar]
- 46.Makarenko NG, Shchmidt EN, Raldugin VO, Dubovenko ZV. Mikrobiol Z. 1980;42:98–102. [PubMed] [Google Scholar]
- 47.Auclair C. Arch Biochem Biophys. 1987;259:1–14. doi: 10.1016/0003-9861(87)90463-2. [DOI] [PubMed] [Google Scholar]
- 48.Godtfredsen WO, Vangedal S. Acta Chem Scand. 1965;19:1088–1102. doi: 10.3891/acta.chem.scand.19-1088. [DOI] [PubMed] [Google Scholar]
- 49.Tate WP, Caskey CT. J Biol Chem. 1973;248:7970–7972. [PubMed] [Google Scholar]
- 50.Wei CM, Hansen BS, Vaughan MH, Jr, McLaughlin CS. Proc Natl Acad Sci U S A. 1974;71:713–717. doi: 10.1073/pnas.71.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schindler D, Davies J. Meth Cell Biol. 1975;12:17–38. doi: 10.1016/s0091-679x(08)60949-8. [DOI] [PubMed] [Google Scholar]
- 52.Chen J, Stubbe J. Nat Rev Cancer. 2005;5:102–112. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]
- 53.Earhart CF., Jr Antibiotics. 1979;5:298–312. [Google Scholar]
- 54.Maeda K, Kosaka H, Yagishita K, Umezawa H. J Antibiot. 1956;9:82–85. [PubMed] [Google Scholar]
- 55.Hernandez F, Cannon M. J Antibiot. 1982;35:875–881. doi: 10.7164/antibiotics.35.875. [DOI] [PubMed] [Google Scholar]
- 56.McPhail AT, Sim GA. J Chem Soc Perkin Trans 1. 1966;16:1394–1406. doi: 10.1039/j39660001394. [DOI] [PubMed] [Google Scholar]
- 57.MacGregor JI, Jordan VC. Pharmacol Rev. 1998;50:151–196. [PubMed] [Google Scholar]
- 58.Prozialeck WC, Weiss B. J Pharmacol Exper Ther. 1982;222:509–516. [PubMed] [Google Scholar]
- 59.Kessel D, Belton JG. Cancer Res. 1975;35:3735–3740. [PubMed] [Google Scholar]
- 60.Belton JG, Conalty ML, O’Sullivan JF. Proc Roy Irish Acad B. 1976;76:133–149. [PubMed] [Google Scholar]
- 61.Thompson S, Kellicutt L. Mutat Res. 1977;48:145–153. doi: 10.1016/0027-5107(77)90154-3. [DOI] [PubMed] [Google Scholar]
- 62.Raught B, Gingras AC, Sonenberg N. Proc Natl Acad Sci U S A. 2001;98:7037–7044. doi: 10.1073/pnas.121145898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tong GL, Wu HY, Smith TH, Henry DW. J Med Chem. 1979;22:912–918. doi: 10.1021/jm00194a005. [DOI] [PubMed] [Google Scholar]
- 64.Fuji K, Fujita E, Takaishi Y, Fujita T, Arita I, Komatsu M, Hiratsuka N. Experientia. 1978;34:237–239. doi: 10.1007/BF01944702. [DOI] [PubMed] [Google Scholar]
- 65.Fujita T, Takaishi Y, Okamura A, Fujita E, Fuji K, Hiratsuka N, Komatsu M, Arita I. J Chem Soc Chem Commun. 1981:585–587. [Google Scholar]
- 66.Matsuzaki K, Shioyama T, Okamura E, Umemura J, Takenaka T, Takaishi Y, Fujita T, Miyajima K. Biochim Biophys Acta Biomem. 1991;1070:419–428. doi: 10.1016/0005-2736(91)90082-j. [DOI] [PubMed] [Google Scholar]
- 67.Iida A, Mihara T, Fujita T, Takaishi Y. Bioorg Med Chem Lett. 1999;9:3393–3396. doi: 10.1016/s0960-894x(99)00621-6. [DOI] [PubMed] [Google Scholar]
- 68.Novotny J, Collins CH, Starks FW. J Pharm Sci. 1974;63:1264–1267. doi: 10.1002/jps.2600630821. [DOI] [PubMed] [Google Scholar]
- 69.Shishodia S, Aggarwal BB. Oncogene. 2006;25:1463–1473. doi: 10.1038/sj.onc.1209194. [DOI] [PubMed] [Google Scholar]
- 70.Hecht HJ, Hoefle G, Steglich W, Anke T, Oberwinkler F. J Chem Soc Chem Commun. 1978:665–666. [Google Scholar]
- 71.Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, Marquez VE. Biochem Biophys Res Commun. 1986;135:688–694. doi: 10.1016/0006-291x(86)90048-3. [DOI] [PubMed] [Google Scholar]
- 72.Hosoya M, Shigeta S, Nakamura K, De Clercq E. Antiviral Res. 1989;12:87–98. doi: 10.1016/0166-3542(89)90072-7. [DOI] [PubMed] [Google Scholar]
- 73.Pezzuto JM, Compadre CM, Swanson SM, Nanayakkara D, Kinghorn AD. Proc Natl Acad Sci U S A. 1985;82:2478–2482. doi: 10.1073/pnas.82.8.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shigematsu Y, Murofushi N, Takahashi N. Agric Biol Chem. 1982;46:2313–2318. [Google Scholar]
- 75.Compadre CM, Hussain RA, Nanayakkara NPD, Pezzuto JM, Kinghorn AD. Biomed Environ Mass Spectr. 1988;15:211–222. doi: 10.1002/bms.1200150405. [DOI] [PubMed] [Google Scholar]
- 76.DeVito SC, Stephani RA. Med Chem Res. 1991;1:47–51. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
13C and 1H NMR spectra of crambescidin 800, isolation scheme, underwater and above water photographs of Monanchora unguifera, comparison of 13C NMR signals for several known polycyclic guanidinium alkaloids, and a table of stereochemical diversity found in naturally-derived polycyclic guanidinium alkaloids. This material is available free of charge via the Internet at http://pubs.acs.org.