

Abstract
Therapies that target the synthesis of estrogen or the function of estrogen receptor(s) have been developed to treat breast cancer. While these approaches have proven to be beneficial to a large number of patients, both de novo and acquired resistance to these drugs is a significant problem. Recent advances in our understanding of the molecular mechanisms that contribute to resistance have provided a means to begin to predict patient responses to these drugs and develop rational approaches for combining therapeutic agents to circumvent or desensitize the resistant phenotype. Here, we review common mechanisms of antiestrogen resistance and discuss the implications for prediction of response and design of effective combinatorial treatments.
Keywords: Tamoxifen, antiestrogen, aromatase inhibitor, fulvestrant, estrogen receptor
1. Endocrine therapies for breast cancer
Estrogen and the steroid estrogen receptors (ERs) are critical regulators of breast epithelial cell proliferation, differentiation, and apoptosis. Mammals express two ERs, ERα and ERβ, which show distinct tissue distributions and functions (for review, see [1]). Mice with targeted deletions of one or both ER genes have established that ERα is the key regulator of mammary gland development. ERα is expressed in 15-30% of the luminal epithelial cells present in normal breast tissue; estrogen stimulation of these cell results in transcription of various genes, including many that are involved in cell cycle regulation (for review, see [2]). However, estrogen-dependent proliferation of breast epithelial cells is thought to occur in a paracrine fashion, such that ERα-containing cells produce growth factors that induce proliferation in adjacent ER-negative cells. In malignant breast tissue, the action of estrogen is deregulated, resulting in a shift to proliferation without differentiation or apoptosis. The percentage of epithelial cells expressing ERα is significantly increased under these conditions. Moreover, these cells now proliferate, marking a shift from paracrine to autocrine growth.
ERβ is more ubiquitously expressed throughout mammary tissue than is ERα [3]. ERβ is often found co-expressed with ERα in the luminal epithelial cells but has also been detected in myoepithelial cell and surrounding stromal cell nuclei. While its specific function(s) in normal and neoplastic tissues remain unknown, ERβ is essential for the fully differentiated phenotype of the normal mammary gland in mice [4]. In ERβ -/- mice, the mammary gland develops normally until puberty. However, several abnormalities are apparent during pregnancy and lactation, including incomplete penetration of the fat pad by glandular tissue, an abnormally large increase in the size of the lumen of ducts and alveoli, and a decrease in the total number of alveoli. Also, in luminal epithelial cells of these mice, the loss of ERβ disrupts the formation of tight junctions and alters the expression of β-catenin, features often associated with a malignant phenotype. It has been suggested that the function of ERβ in these processes may contribute to the protection conferred by pregnancy and lactation against breast cancer.
Several reports describe a loss of ERβ during carcinogenesis (for review see [5,6]), providing support for its possible role as a tumor suppressor. Indeed, ERβ has been reported to suppress the function of ERα as a transcriptional activator when both receptors are co-expressed [5,7] and to promote anti-proliferative and pro-apoptotic effects [8]. Several genes have been identified that are regulated by ERβ but not ERα; studies are underway to determine whether these genes may have anti-proliferative or pro-apoptotic properties that could account for the putative tumor suppressor function of ERβ [5,9,10].
A majority of invasive breast tumors express ERα, and as discussed above, ERβ is often down-regulated. These findings provide a strong rationale for therapies that disrupt the actions of estrogen and ERα. Current approaches include inhibiting the function of ERs with the use of antiestrogens such as tamoxifen, raloxifene, and ICI 182,780 (faslodex, fulvestrant), or blocking the conversion of androgens into biologically active estrogens through the action of aromatase inhibitors such as letrozole, anastrozole, and exemestane (see Section 1.1). Recent studies on ERβ have also highlighted its growing value as a diagnostic and prognostic marker in breast cancer and its potential for use as a new strategy for delaying and retarding breast tumorigenesis (see Section 1.1.1).
1.1. Classes of therapeutic agents
1.1.1. Selective Estrogen Receptor Modulators (SERMs)
SERMs have been developed as agents that function as ER antagonists in some tissues, including breast, and agonists in other tissues such as the heart and bone (for review, see [11]). The antagonist effects of tamoxifen in breast tissue are thought to result from its ability to bind to the ligand-binding domain (LBD) of the ER, effectively blocking the potential for estrogen stimulation. Tamoxifen binding further prevents critical ER conformational changes that are required for the association of coactivators. However, tamoxifen treatment is associated with an increased risk of endometrial cancer [12]. Other SERMs, such as raloxifene, have been developed with the goal of reducing some of the deleterious effects of tamoxifen.
New classes of SERMs are being developed that exhibit mixed agonist/antagonist activities that are subtype specific (“dual SERMs”) or preferentially affect one ER over the other (“subtype-selective SERMs”) (reviewed in [13]). However, it is important to note that selectivity is often assessed in the context of binding affinity relative to 17β-estradiol. Therefore, a “selective” compound may actually interact with both receptors but have a higher affinity for one versus the other. The relative binding affinities of several agents have been determined in HeLa cells engineered to express ERα or ERβ [14]. Propyl pyrazole triol (PPT) was shown to be a selective agonist for ERα in this system, while the synthetic ligands diarylpropionitrile (DPN) and 8β-VE2, and the natural products genistein and biochanin A, were preferential agonists of ERβ. In contrast, the antiprogestin RU486 was shown to be a strong ERβ antagonist with only modest agonist activity toward ERα. DPN has been shown to inhibit proliferation and induce apoptosis in the HC11 mouse mammary cell line but this compound has not been widely studied in human breast cancer cell lines [8]. It has also been reported that a modified androstenediol is a potent activator of ERβ by ligand binding assay, but its biological activity in breast cancer cells requires further investigation [15].
Dual SERM compounds that inhibit ERα while activating ERβ are particularly attractive, as ERβ often performs growth-inhibitory functions in breast tissues and cell lines (see above, Section 1). An example of this type of SERM is TAS-108, a novel, steroidal antiestrogen that inhibits ERα while acting as a partial agonist of ERβ ([16] and references therein). In vitro, TAS-108 effectively inhibits the growth of tamoxifen-resistant breast cancer cells, and Phase I studies have established that this agent is not only well-tolerated but appears to induce stable disease in women with advanced breast cancer. Where and how this particular SERM will be used in the clinic, particularly in sequence with the other classes of therapeutic agents described below, has yet to be determined. However, this and future compounds that have similar profiles will be important additions to the current repertoire of endocrine therapies.
1.1.2. Selective estrogen receptor downregulators (SERDs)
The pure ER antagonist ICI 182,780 (hereafter called fulvestrant) binds to ERα with 100-fold greater affinity than does tamoxifen and in so doing, inhibits receptor dimerization and abrogates estrogen signaling [17]. Both laboratory and clinical studies have shown a decrease in overall ER protein levels in response to fulvestrant treatment (discussed in [18]). Importantly, a majority of ERα-expressing tamoxifen-resistant breast cancers remain sensitive to fulvestrant treatment. This has led to its approval in the United States for use in treating ER-positive patients following failure to respond to first-line antiestrogen therapy.
1.1.3. Aromatase Inhibitors (AIs)
Aromatase inhibitors (AIs) represent a new class of endocrine therapy drugs that inhibit the action of aromatase, an enzyme necessary for the conversion of androgens to estrogens (for review, see [19]). Clinically, AIs have had success as a second line of therapy for post-menopausal patients who have progressed after tamoxifen treatment. They also show promise as first-line adjuvant treatment for ERα-positive breast cancers.
1.2 Resistance to endocrine therapies
While considerable progress has been made in developing strategies to target estrogen and ERs in breast cancer, approximately 30% of ERα-positive breast cancers do not respond to tamoxifen treatment (de novo resistance). In addition, the majority of tumors that initially respond to treatment develop resistance over time (acquired resistance), despite continued expression of ERα (reviewed in [11,20]). Interestingly, many of these resistant tumors still respond to fulvestrant and AIs, indicating that estrogen remains an important regulator of tumor growth under these circumstances [11]. These data provide support for the idea that endocrine-targeted therapies can lead to the activation of novel signaling pathways that circumvent the effects of antiestrogens [21]. De novo and acquired resistance to AIs have also been reported. The appearance and development of resistant tumors are therefore major obstacles in the struggle to control the growth and metastasis of breast carcinomas.
1.3. Biological effects of endocrine therapies
Homeostasis of non-cancerous cells requires a balance between proliferation, cell cycle arrest, and apoptosis; this equilibrium is disrupted during cancer progression. Endocrine therapies induce cell cycle arrest and apoptosis through a reduction in expression of cell cycle regulators such as c-Myc and cyclin D1, accumulation of hypophosphorylated Rb, induction of the cell cycle inhibitors p21WAF1/CIP1 and p27Kip1, induction of the putative tumor suppressor interferon regulatory factor-1 (IRF-1), and inhibition of pro-survival pathways through decreased Bcl-2 expression and increased Bax expression ([22-25]; for review, see [20,26]). Breast tumor cells that are resistant to the growth-inhibitory and apoptotic effects of antiestrogens such as tamoxifen overcome these influences through a variety of mechanisms that are the focus of this review.
2. Molecular mechanisms of antiestrogen resistance
Emerging data from breast tumor biopsies indicate that altered expression and/or modification of several growth factor receptors and downstream signaling molecules correlate with tamoxifen resistance (Figure 1). Epidermal growth factor receptor (EGFR), human epidermal growth factor receptor type 2 (HER2), and insulin-like growth factor-1 receptor (IGF-1R) signaling pathways are often elevated in non-responsive tumors that exhibit either de novo or acquired resistance [27-30], as is the activity of kinases that function downstream of these receptors such as extracellular-regulated kinase (ERK) 1/2, p38, AKT, and p21-activated kinase-1 (Pak1) [30-32]. Expression and/or phosphorylation of substrates of these receptor and non-receptor kinases are also elevated in tumors resistant to tamoxifen; such molecules include the cytoplasmic adapter molecule “breast cancer antiestrogen resistance 1” (BCAR1; also known as p130Cas; hereafter called Cas), the coactivator “amplified in breast cancer 1” (AIB1), and ERα itself [28,33-35]. Finally, dysregulated expression of molecules involved in cell survival, such as c-Myc, Bad, and Bcl-2, can also be predictive of a poor response to endocrine therapies such as tamoxifen [36-39]. Clinical data addressing the correlation between some of these markers and antiestrogen resistance are presented in greater detain in Section 4.1. While these studies are limited in both scope and number, however, the association between expression/activation of these molecules and resistance to endocrine therapies has been corroborated (and in fact extended) in tissue culture and animal models. These data are reviewed below.
Figure 1.

Molecular mechanisms of tamoxifen resistance. Model depicting molecules implicated in antiestrogen resistance and discussed in this review.
2.1. ERα, ERβ, and receptor co-regulatory proteins
Expression of ERα has long been considered the primary determinant of a clinical response to endocrine or antiestrogen therapy. However in 1996, the concept of estrogen signaling and ER function in breast cancer was significantly altered by the discovery of ERβ [40]. The DNA binding domain of full-length ERβ1 shares 96% identity with that of ERα, and although the affinity of this receptor for estrogen is similar to that of ERα, the response to antagonists like tamoxifen and fulvestrant is often different. Both receptors can modulate transcription, either directly from estrogen response elements (EREs), or indirectly through tethering to AP-1 and Sp1 binding sites (reviewed in [41]). However, although ERα perceives tamoxifen and fulvestrant as antagonists, it has been shown that ERβ can utilize these compounds as agonists when associated with AP-1 and Sp1 sites [42,43]. Additionally, two naturally occurring variants, ERβ2/ERβcx and ERβ5, contain carboxy-terminal deletions that prevent their binding to ligand, and it has been suggested that these receptors may function as dominant-inhibitory mutants of full-length ERβ1. Murphy et al. have recently reviewed the evidence for a role for ERβ1 and its variants in breast cancer diagnosis and prediction of endocrine therapy response [6].
The transcriptional activities of ERα, ERβ, and other steroid hormone receptors are modulated by coregulatory proteins in either a positive or negative fashion. The association of coactivators and corepressors with estrogen receptors is regulated by conformational changes induced by ligand binding, with estrogen inducing coactivator recruitment and antiestrogens leading to association with corepressors. Several excellent reviews have recently detailed the expression, function, and clinical relevance of these coregulators in breast cancer and endocrine resistance [44-47].
2.1.1. ERα expression and downregulation
In human breast tumors, the vast majority of ERα-negative tumors exhibit de novo or intrinsic resistance to tamoxifen and other antiestrogens; however, small numbers of ERα-negative but progesterone receptor-positive (PR-positive) tumors respond to antiestrogen treatment (reviewed in [48]). One possible explanation for this finding is that PR-positive tumors retain a functional ERα signaling pathway, particularly since expression of PR is an estrogen-regulated event, and that ERα is present in these tumors at levels that are below the limit of detection for ligand-binding or immunohistochemical assays. The importance of PR as a marker of antiestrogen response is substantiated by studies showing that 70% of ERα-positive/PR-positive tumors effectively respond to tamoxifen, while only 34% of ERα-positive/PR-negative patients respond to tamoxifen therapy [49].
In the context of de novo antiestrogen resistance displayed by ERα-negative breast cancer cells, several studies suggest that it may be possible to restore tamoxifen sensitivity through re-expression of ERα. Epigenetic silencing of the ER due to hypermethylation of the promoter region has been reported both in vitro and in vivo [50,51]. For example, ERα expression can be restored in the ERα-negative MDA-MB-231 breast cancer cell line by either treatment with the demethylating agent 5-aza-2′-deoxycytidine or the histone deacetylase inhibitor trichostatin A [52]. Importantly, when endogenous ERα expression was restored in this way, MDA-MB-231 cells became sensitive to growth inhibition by tamoxifen. Based on data such as these, it has been suggested that restoration of ERα expression may be a viable option for the treatment of some ER-negative breast cancers (discussed in [53]). However, a separate study showed that ectopic expression of ERα in MDA-MB-231 cells was not sufficient to render the cells sensitive to tamoxifen [54]. This suggests that the level and cellular context of ERα expression will be important factors to consider when attempting to modulate ERα levels in “ER-negative” tumors.
Although downregulation of ERα expression can contribute to de novo antiestrogen resistance, loss of the receptor during the acquisition of tamoxifen resistance is not commonly observed in vivo ([55,56]; reviewed in [57]). However, loss of ERα has been reported in a few models of acquired antiestrogen resistance. For example, the ZR-75-derived 9a1 cell line becomes ERα-negative in the presence of tamoxifen but reverts to being ERα-positive when tamoxifen is removed and estrogen is reintroduced [58].
Like tamoxifen, fulvestrant inhibits ERα transcriptional activity. In addition, fulvestrant has also been shown to accelerate ERα protein degradation via the ubiquitin-proteasome pathway, leading to its reclassification as a SERD (see above). Loss or downregulation of ERα has the potential be a viable mechanism for antiestrogen resistance in the context of acquired resistance to fulvestrant or other SERDs, but current evidence to this effect is contradictory. ERα protein is completely lacking and only minimal mRNA is detectable in several fulvestrant-resistant cell lines, including MCF-7-r, T47D-r, and ZR-75-r cells [59]. MCF-7-F resistant cells show a 90% decrease in both ERα mRNA and protein, accompanied by significant changes in estrogen-induced expression of ER target genes [60]. In contrast, the MCF-7/182R-1, -6, and -7 models of fulvestrant resistance maintain ERα expression and function, although its expression at the protein level is reduced by one-third as compared to the parental cell line [61,62]. MCF-7/LCC9 breast cancer cells, which display acquired resistance to fulvestrant and cross-resistance to tamoxifen, show no downregulation of ERα [63]. This cell line was originally derived from the estrogen-independent MCF-7/LCC1 cell line [64]; interestingly both MCF-7/LCC1 and MCF-7/LCC9 cells express higher levels of ERα mRNA and protein than the parental MCF-7 cell line [65]. MCF-7/LCC9 cells have also been shown to contain increased basal ERα transcriptional activity that is not inhibited by fulvestrant [66]. Factors that drive the differential expression and activity of ERα among these cell culture models of fulvestrant resistance are not known, but one likely explanation for these disparities may be the distinct schemes of drug selection that have been used to generate resistant cells in vitro. Clinical studies confirm that fulvestrant can reduce ERα levels following either short- (21 days) or long- (6 months) term treatments, but that expression is not completely eliminated (reviewed in [67]). Interestingly, patients who do not show a clinical response to fulvestrant treatment also do not show a decrease in ERα expression.
2.1.2. ERα mutation
Mutation of the ER may also impact endocrine therapy responsiveness. For example, one tamoxifen-resistant variant of the ERα-positive T47D cell line, T47DCO, has acquired three ERα deletion mutations that affect the hormone- and DNA-binding domains of the receptor [68]. This cell line also expresses different levels of PR [69], suggesting that these variants are functionally distinct from the wild type receptor. These and many other deletions, exon duplications or insertions, point mutations, and alternative splicing events have the potential to regulate ERα function in breast and other tissues. Herynk and Fuqua have recently published a review of known ER mutations and their connection to human disease, several of which are linked to tamoxifen response and resistance [70]. For example, duplication of exons 6 and 7 in an estrogen-independent variant of the MCF-7 cell line leads to the production of an ER that can no longer bind to estrogen or tamoxifen [71]. Short insertions in exon 6 have also been identified in tamoxifen-resistant tumors [72]. A tyrosine-to-asparagine substitution at residue 537 (Y537N), originally identified in metastatic breast cancer, confers constitutive transcriptional activity in vitro and renders the ER insensitive to both estrogen and tamoxifen, while mutation of this tyrosine to either serine or alanine results in a constitutively active receptor whose activity is still inhibited by tamoxifen and fulvestrant [73]. In contrast, a lysine-to-arginine substitution at residue 303 (K303R) that was first identified in breast hyperplasia and a number of breast tumors results in a receptor that is hypersensitive to substantially lower concentrations of estrogen than normal (10−12 vs. 10−9M) [74]. The prevalence of this mutation in patient populations is unclear, as more recent studies have not found ERα K303R in other patient groups [75]. The difficulty in identifying functional ERα mutations in vivo that affect tamoxifen therapy response may stem from the fact that such mutations are relatively rare, estimated to be present in only 1% of breast tumors [51].
2.2. Growth factor receptors
As discussed above, tumors resistant to tamoxifen often show high expression and/or activation of EGFR, its close relative HER2, IGF-R1, and proteins that function downstream of these receptors such as ERK1/2, and AKT (for review, see [28,76-78]). In vitro data corroborate a role for these growth factor receptors in mediating antiestrogen resistance, both through crosstalk with the ER and through independent pathways.
2.2.1. EGFR and HER2
Several studies have established that overexpression of EGFR or HER2 in ER-positive, antiestrogen-sensitive, breast cancer cells can confer resistance to these drugs [79-81]. Both ERK1/2 and AKT activity appear to contribute to the resistant phenotype under these conditions. Other groups have isolated antiestrogen-resistant cell lines following long-term treatment of ER-positive breast cancer cells with either tamoxifen or fulvestrant. In these studies, acquisition of resistance often coincides with overexpression of the EGFR and activation of downstream molecules such as ERK1/2, PI3K, and AKT [59,63,82,83]. Expression of one of the ligands for EGFR, transforming growth factor α (TGFα), is elevated in one of these cell models, resulting in an autocrine loop that is required for maintenance of the resistant phenotype [84]. Unlike the parental cells, which are relatively impervious to EGFR signaling pathways, the resistant clones are highly dependent on these pathways for growth and survival.
While the mechanism(s) by which these receptors promote antiestrogen resistance are not clear, several lines of evidence suggest that they may function to provide survival signals that override apoptotic programs in the cell. First, when EGFR functions are blocked by either the tyrosine kinase inhibitor Iressa or blocking antibodies, apoptosis is induced [85-88]. Second, the expression and/or activity of several anti-apoptotic molecules such as AKT, Bcl-2 and Bcl-xL are upregulated under conditions of HER2 overexpression in MCF-7 cells [89,90]. However, events considered in other contexts to be generally pro-apoptotic are also associated with overexpression of EGFR or HER2 in human tumors. For example, elevated levels of phospho-p38 have been reported in HER2-expressing tumors as well as in tamoxifen-resistant xenografts [29,30]. While its role in regulating apoptosis under these conditions is not clear, activated p38 may promote resistance to tamoxifen through its ability to enhance the nuclear functions of ER [91]. Finally, recent data from our group indicate that the EGFR may work together with the non-receptor protein tyrosine kinase c-Src and the adapter molecule Cas to promote survival [92].
In addition to the direct effect of activated growth factor receptors on cell proliferation and survival, crosstalk between these receptors and ER plays a significant role in regulating the cellular response to antiestrogens. Signaling through EGFR and HER2 can result in downregulation of ERα expression in cultured cells [93]. This inverse relationship has been corroborated in a recent clinical study by the Southwest Oncology Group [27]. Not only does growth factor signaling affect ER levels, but it can also strongly impact ER nuclear functions, at least in part through promoting phosphorylation of the ER, its coactivators, and/or its corepressors (for review, see [94]).
Conversely, ERα appears to regulate growth factor receptor activities through rapid, non-genomic functions. This may be augmented by an accumulation of cytoplasmic ERα at the expense of nuclear ERα under conditions of high HER2 expression [95]. Interestingly, it appears that tamoxifen may be able to function as an agonist for these non-genomic ER activities under some circumstances, perhaps explaining how cells expressing high levels of HER2 and the coactivator AIB1 show increased proliferation in the presence of both estrogen and tamoxifen [90]. Additional support for this crosstalk comes from preclinical studies showing enhanced efficacy and/or delays in the appearance of resistance in the presence of combined therapies that target both tyrosine kinase receptors and estrogen signaling ([87,96]; and see below, Section 4.3).
2.2.2. IGF-1R signaling
The involvement of IGF signaling in antiestrogen resistance is less clear than is the role for EGFR and HER2. While data are mixed about whether IGF-1R levels are elevated or decreased in resistant tumors and cell lines [28,97,98], evidence for a role of IGF-1R signaling in acquired resistance is emerging. This may be mediated at least in part through crosstalk with EGFR, HER2, and/or ERα and likely involves the scaffolding molecule insulin receptor substrate 1 (IRS-1) [77,99,100].
2.3. Cas/c-Src/BCAR3
Numerous cytoplasmic proteins whose functions are coupled to growth factor receptors have been linked to antiestrogen resistance, further supporting a role for these pathways. Two of these molecules, Cas and “Breast Cancer Anti-Estrogen Resistance 3” (BCAR3, also known as AND-34 and Nsp2), were identified in a screen designed to isolate proteins capable of inducing antiestrogen resistance in sensitive MCF-7 and ZR-75-1 cells [101,102]. Interestingly, both of these molecules are expressed and interact with one another in breast cancer cells (Schrecengost and Bouton, unpublished data), supporting a role for this molecular pathway in the promotion of resistance.
2.3.1. Cas and c-Src
Cas functions in a wide variety of cellular processes, including proliferation, survival, cell adhesion, and migration [103,104]. Cas overexpression promotes increased proliferation and inhibition of apoptosis in MCF-7 breast cancer cells treated with tamoxifen [92,105]. This requires the association of Cas with c-Src, as documented by the finding that, unlike wildtype Cas, overexpression of a Cas mutant that does not interact with c-Src does not promote growth in the presence of tamoxifen [92]. Interactions between c-Src and Cas result in activation of c-Src catalytic activity, promotion of serum- and anchorage-independent growth, and enhancement of cellular migration [106,107]. Interestingly, two substrates of c-Src, Tyrosine 845 (Tyr-845) on the EGFR and signal transducer and activator of transcription (STAT) 5b, have been implicated in Cas-dependent tamoxifen resistance [92]. These data suggest a mechanism whereby c-Src activation by Cas results in the phosphorylation of a number of c-Src substrates and subsequent activation of various pro-proliferative and survival pathways that allow the cells to grow in the presence of tamoxifen. Interestingly, ER nuclear functions remain unaffected by this pathway [92], suggesting that therapies that target the Src-Cas-EGFR-STAT5b signaling axis may be able to synergize with tamoxifen for the treatment of “resistant” tumors.
Several other studies provide additional insight into how c-Src and Cas may contribute to tamoxifen resistance. First, ERα and phosphatidylinositol 3-kinase (PI3K) have been found in the c-Src/Cas complex in T47D breast cancer cells following rapid estrogen stimulation [108]. While it is clear that these complexes help regulate estrogen-dependent cell proliferation and survival, however, it is not yet known whether they are important for promoting resistance to antiestrogens. Second, pharmacological inhibition of c-Src in MCF-7 cells enhances the growth-inhibitory and apoptotic-inducing effects of tamoxifen, further strengthening a role for c-Src in this process [109]. Finally, cells treated over a period of two weeks with tamoxifen or fulvestrant show a c-Src-dependent increase in Cas phosphorylation that correlates with activation of a survival pathway involving AKT [110].
Together, these studies point to a key role for Cas and c-Src in tamoxifen resistance in cell culture. Whether these pathways also play a role in clinical resistance remains to be determined. c-Src/Cas interactions are favored under conditions in which both proteins are expressed at high levels [106]. Thus it is intriguing that a large percentage of human breast tumors contain high levels of both c-Src and Cas, and elevated c-Src kinase activity [33,111,112]. Moreover, c-Src/Cas complexes can be isolated from a majority of breast cancer cell lines (Schrecengost and Bouton, personal communication). This suggests that one of the mechanisms by which c-Src activity may be elevated in breast cancer cells is through its association with Cas. Cas overexpression in human tumors correlates with poor overall and relapse-free survival and a poor intrinsic response to tamoxifen treatment [33]. The majority of the Cas-overexpressing tumors were shown to be ER-positive, suggesting that Cas promotes de novo resistance to tamoxifen through mechanisms that do not involve downregulation of the ER. In contrast, Cas expression levels do not appear to correlate with acquired tamoxifen resistance [113]. c-Src and the EGFR are also frequently (∼30%) co-overexpressed in breast tumors, while 20-30% co-overexpress HER2 and c-Src [114,115]. Based on these data, it is reasonable to postulate that, in at least a subset of tumors, tamoxifen resistance may be mediated through a coordinated mechanism involving these receptor tyrosine kinases, c-Src, and Cas.
2.3.2. BCAR3/AND-34
BCAR3/AND-34 (hereafter called BCAR3) is a Cas binding partner that was identified in a genetic screen along with Cas as a molecule whose overexpression conferred antiestrogen resistance [102]. Since its discovery, BCAR3 has been implicated in altering the activity, expression, and intracellular location of many proteins, including Cas, Ras and Rho family GTPases, and Pak1 ([107,116-118]; for review, see [119]). BCAR3 overexpression induces antiestrogen resistance through a process that involves PI3K and Rac1 [118]. Estrogen-independent growth can also be induced by BCAR3 expression, a process that requires R-Ras activation of AKT [120]. While these data suggest that BCAR3 overexpression may promote antiestrogen resistance in cultured cells, however, it is yet to be determined whether this molecule contributes to resistance in the clinical setting.
2.4. PI3K/AKT
Downstream of growth factor receptor and adapter protein activation, signaling intermediates such as AKT play an important role in cell proliferation, survival, and endocrine resistance. AKT, also known as protein kinase B, is activated by PI3K, leading to substrate phosphorylation and cell survival. AKT activity is subsequently counter-balanced by the tumor suppressor PTEN. Historically, all three isoforms of AKT have been considered oncogenic, although recent studies have reported that different isoforms may play distinct roles in invasion and cell migration (discussed in [121]). AKT is most often considered to be a survival factor, preventing apoptosis under conditions of cell stress. However, it has become increasingly clear that AKT also participates in cell cycle regulation (reviewed in [122]). Cell cycle proteins with established connections to antiestrogen resistance, including those that are AKT targets, will be detailed in Section 2.6.
In ER-positive breast cancer cell lines, PI3K-mediated AKT activation is linked to rapid phosphorylation of ERα and ligand-independent receptor activation [123]. AKT can also stimulate ERβ transcriptional activity and enhance coactivator recruitment to this receptor [124]; whether this has an impact on antiestrogen resistance has yet to be determined.
Overexpression of constitutively active AKT in breast cancer cell lines can induce estrogen independence and resistance to tamoxifen and fulvestrant [125], while inhibition of PI3K or AKT restores tamoxifen sensitivity [126]. In addition to endocrine agents, activated AKT can also confer resistance to conventional chemotherapeutics such as doxorubicin [127]. The link between AKT and tamoxifen resistance is not solely observed in cell lines engineered to overexpress this kinase; MCF-7 cells selected for resistance to tamoxifen exhibit upregulated endogenous AKT activity that is required for antiestrogen resistance [128,129]. However, a separate study showed that an aromatase-expressing MCF-7 cell line (MCF-7/Ca), when grown as xenografts in nude mice, did not upregulate the PI3K/AKT pathway in the process of acquiring resistance to the aromatase inhibitor letrozole [130].
The mechanisms by which AKT may confer antiestrogen resistance are varied. Phosphorylation of the ER on serine 167 is discussed below (Section 2.5). AKT can also activate the mammalian target of rapamycin (mTOR), which controls cap-dependent translation, cell growth, and survival (recently reviewed in [131]). This can occur in response to activation of AKT by growth factor receptors such as EGFR and IGF-1R; AKT then phosphorylates and inactivates the tuberous sclerosis complex, which normally keeps mTOR kinase activity in check. Rapamycin and its analog CCI-779 are able to restore tamoxifen sensitivity in MCF-7 cells overexpressing constitutively active AKT [132]. Another mTOR inhibitor (RAD-001) has also been shown to inhibit the growth of wild type MCF-7 cells, as well as MCF-7/Aro cells that stably over-express aromatase [133]. Moreover, a combination of the aromatase inhibitor letrozole with RAD-001 was shown to more effectively inhibit MCF-7/Aro proliferation than either drug alone. However, given that MCF-7/Ca cells with acquired resistance to letrozole do not contain elevated levels of activated AKT [130], it would seem that a definitive role for the PI3K/AKT/mTOR pathway in aromatase inhibitor resistance remains to be determined. Nonetheless, mTOR and rapamycin-like drugs that target mTOR are considered to be attractive molecular targets and therapeutic agents, respectively, for treating breast cancer (reviewed by [134,135]; see Section 4.3).
2.5. ER phosphorylation
The estrogen receptor is a target of serine/threonine as well as tyrosine phosphorylation (reviewed in [136]). While ER phosphorylation is a key element of non-genomic or rapid estrogen actions, several studies have shown that it can also affect conventional, transcription-mediated events. Reported ER phosphorylation sites include tyrosine 537, serines 104, 106, 118, 167, 236, and 305, and threonine 311 (Figure 2, and discussed in [137]).
Figure 2.
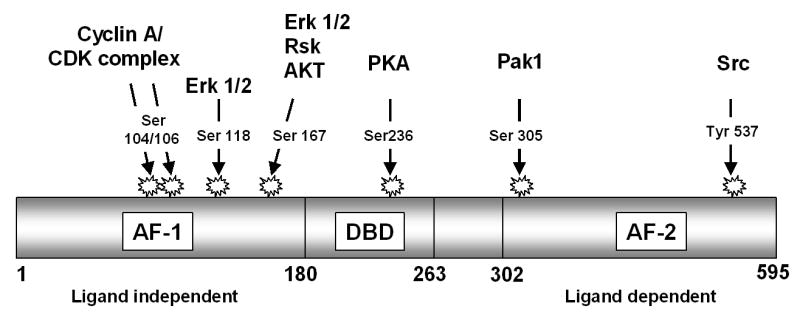
ERα phosphorylation. Domain structure of ERα showing phosphorylation sites and the kinases that mediate these phosphorylations.
Tyr-537, which is located in the activation function-2 (AF-2) domain, is phosphorylated by Src family kinases [138]. Mutation of Tyr-537 to phenylalanine increases basal ER transcriptional activity without changing the DNA binding capacity of the receptor [139]. Mutation of Tyr-537 to either serine or alanine also results in estrogen-independent transcription, but both 4-hydroxytamoxifen and fulvestrant inhibit this activity [140,141]. Although the full function(s) of Tyr-537 phosphorylation are still under investigation, the evidence that “constitutively active” Tyr-537 mutants are still inhibited by antiestrogens argues against a critical role for this residue in endocrine therapy resistance. It has also yet to be determined whether Tyr-537 phosphorylation is observed in breast tumors.
The effect of serine phosphorylation on ER functions is better established than is tyrosine phosphorylation and it is more likely to contribute to the resistant phenotype. Ser-104 and Ser-106 are located in the AF-1 domain and are targets of the cyclin A/CDK2 complex; mutation of these sites to alanine prevents cyclin A-mediated enhancement of ER transcriptional activity [142]. Conversely, cyclin A enhances ER transcriptional activity in the presence of tamoxifen. Together, these data suggest that inappropriate cyclin A/CDK2-mediated phosphorylation of Ser-104 and Ser-106 may play a role in endocrine resistance in breast tumors. Cyclin A has been implicated as an independent prognostic factor for poor outcome in tamoxifen-treated breast cancer patients [143].
Ser-167, also located within the AF-1 domain, was initially proposed to be phosphorylated in response to estrogen binding [144]. However, subsequent studies have revealed that its phosphorylation is ligand-independent, and that this site is a major target of ERK, RSK, and AKT kinases (reviewed in [136]). Overexpression of either EGFR or AKT increases Ser-167 phosphorylation and reduces tamoxifen sensitivity, whereas RNAi-mediated inhibition of AKT abrogates Ser-167 phosphorylation and restores tamoxifen sensitivity [145]. In vitro, AKT-mediated phosphorylation of Ser-167 can increase ER binding to DNA and enhances the interaction of ER with the coactivator SRC3 in the presence of estrogen [146]. Despite the evidence that Ser-167 phosphorylation appears to play an important role in promoting antiestrogen resistance in cell culture, however, clinical evidence of a definitive role for phosphorylated Ser-167 in antiestrogen resistance is less convincing (see Section 4.1).
Ser-236, located within the DNA binding domain of the receptor, has been shown to be phosphorylated by protein kinase A (PKA) [147]. While phosphorylation at other sites seems to positively regulate ER transcriptional activity, PKA phosphorylation at Ser-236 appears to inhibit receptor dimerization in the absence of ligand. This suggests that Ser-236 represents a negative regulatory site that has yet to be implicated in the process of tamoxifen resistance.
Ser-305 is located in the hinge region of the receptor, and its phosphorylation by Pak1 has been shown to increase ER transcriptional activity [148]. Substitution of glutamic acid for serine at this site recapitulates increased receptor activation and induces expression of the ER target gene cyclin D1 [149]. Pak1 is a well-known mediator of cell survival and migration, and has itself recently been shown to contribute to tamoxifen resistance. Pak1 overexpression in MCF-7 cells enhances cyclin D1 expression and prevents tamoxifen-mediated inhibition of ER activity [150,151]. Conversely, lower expression of Pak1 in primary breast tumors is associated with improved disease-free survival in patients treated with tamoxifen [32]. Pak1 co-immunoprecipitation with ER is enhanced by tamoxifen treatment of resistant cell lines [150,151]. Pak1-mediated stimulation of ER transcriptional activity through Ser-305 is reported to require processive phosphorylation of Ser-118, suggesting that these two sites work together to regulate ER activity. Interestingly, Ser-305 can also be phosphorylated by PKA. While PKA-mediated phosphorylation of Ser-236 appears to negatively regulate receptor activities (see above), phosphorylation of Ser-305 by PKA is thought to prevent tamoxifen from inducing the inactive conformation of the ER [152].
Ser-118 is perhaps the best studied site of ER phosphorylation and is widely considered to be a target of ERK1/2, although other kinases such as the TFIIH cyclin-dependent kinase, cyclin-dependent kinase 7 (CDK7), and glycogen synthase kinse-3 (GSK-3) may also phosphorylate this site [153-156]. Interestingly, estrogen-induced phosphorylation of Ser-118 occurs within 10 minutes of stimulation, and this can be inhibited by 4-hydroxytamoxifen but not fulvestrant [155]. Recent studies by Likhite et al. have helped to clarify the function of Ser-118 phosphorylation in tamoxifen-sensitive and resistant cells with respect to hormone, DNA, and coregulator binding [146]. Stable overexpression of HER2 in MCF-7 cells, which induces tamoxifen resistance, also results in increased basal phosphorylation of Ser-118 that is no longer enhanced by estrogen stimulation. Using purified wild type and serine-mutated ER in in vitro kinase assays, this group subsequently demonstrated that phosphorylation of Ser-118 by ERK1/2 specifically enhances recruitment of the coactivator SRC3 to estrogen-bound receptor, but has no effect on the affinity of ER for estrogen or DNA. In contrast, phosphorylation at Ser-118 reduced the association of ER with trans-hydroxytamoxifen, which has the potential to attenuate the growth inhibitory effects of this antiestrogen.
The role of Ser-118 phosphorylation in antiestrogen resistant cell culture models is not fully understood. Kuske et al. have recently reported that estrogen-induced Ser-118 phosphorylation levels are comparable in parental MCF-7 cells and the estrogen-independent MCF-7/LCC1 cell line, but that basal and estrogen-induced levels of phospho-Ser-118 are significantly reduced in the antiestrogen-resistant MCF-7/LCC9 cell line [65]. In studies by other investigators, baseline phosphorylation of Ser-118 is increased in tamoxifen-resistant MCF-7 cells as compared to the parental cell line and is reduced by MEK and EGFR tyrosine kinase inhibitors, which also inhibit cell growth [157]. Finally, Weitsman et al. suggest that, although both HER2 overexpression and activation of the MAPK pathway can induce antiestrogen resistance, neither has any effect on Ser-118 phosphorylation of ER in MCF-7 cells [158]. Instead, they suggest that the activity of IkB kinase-α (IKKα) is involved. A potential role for IKKα in ER phosphorylation has only recently been suggested [159], and further studies are needed to clarify its role in antiestrogen resistance.
2.6. Cell cycle regulators
Cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors are the major regulators of cell cycle progression. Antiestrogens like tamoxifen induce cell cycle arrest during the G1 phase; therefore, genes that control cell cycle progression have the potential to significantly impact drug sensitivity and resistance. In breast cancer, normal expression and function of cyclins D1 and E, and the CDK inhibitors p21 and p27, are often specifically disrupted. Caldon et al. have recently published a comprehensive review of cell cycle control in breast cancer that summarizes how cyclins and CDK inhibitors influence cancer progression and may serve as markers of treatment outcome [160]. Butt et al. have also published a summary of the literature concerning the role of cyclins D1 and E in endocrine resistance [161]. Given the timeliness of these reviews, we will only summarize key information about these molecules and focus on the most recent literature in this area.
2.6.1. Cyclin D1
Cyclin D1 is a well-studied contributor to antiestrogen resistance in breast cancer. Essential for early progression through the G1 phase, cyclin D1 both modulates CDK4/6-dependent phosphorylation of the tumor suppressor Rb and sequesters the CDK inhibitors p21 and p27 [160,162]. Cyclin D1 is a direct transcriptional target of estrogen signaling, where its induction is mediated by ER/AP-1 and ER/Sp1 transcription complexes; conversely, antiestrogens like tamoxifen inhibit cyclin D1 expression (discussed in [161]). AKT can indirectly affect cyclin D1 expression by phosphorylating and inhibiting the activity of GSK3β, which normally induces cyclin D1 degradation [163]. In cell culture, stable overexpression of cyclin D1 can confer resistance to tamoxifen as well as fulvestrant [164]. Moreover, some cells that have acquired resistance to tamoxifen show increased expression of cyclin D1 [165]. However, these cells are still growth-inhibited by fulvestrant.
Recent studies have shed some light on potential mechanisms of cyclin D1 upregulation in the context of antiestrogen resistance, some of which involve crosstalk between growth factor receptors and other signaling molecules. Tamoxifen-resistant MCF-7 cells overexpressing HER2 upregulate cyclin D1 [90]. Similarly, the tamoxifen-resistant phenotype of MCF-7 cells overexpressing Pak1 is partly due to the increased expression of cyclin D1 [32]. BCAR3 overexpression in MCF-7 also induces cyclin D1 promoter activity, coincident with the adoption of antiestrogen resistance [117]. In some instances, tamoxifen has been reported to stimulate breast cancer, although this has been observed more widely in cell culture and xenograft studies than in the clinic. Under conditions in which tamoxifen or raloxifene functions as an ER agonist, cyclin D1 regulates cell cycle progression much as it does in parental MCF-7 cells stimulated by estrogen. For example, cyclin D1 expression was found to be significantly elevated in raloxifene-stimulated MCF-7 xenografts [166]. Treatment with the HER2 inhibitor trastuzumab decreased cyclin D1 expression in these xenografts and concomitantly inhibited tumor growth. Similarly, cyclin D1 was shown to be essential for cell cycle progression induced by tamoxifen in resistant MCF-7 cells grown in tissue culture [167].
Evidence for a role of cyclin D1 in clinical endocrine resistance is still emerging (reviewed in [160]). Cyclin D1 is overexpressed in ∼50% of breast cancers. Some studies have reported that cyclin D1 overexpression is more often observed in highly differentiated, slower growing tumors than in highly proliferative tumors. Other studies report that cyclin D1 amplification is more frequent in ER-positive tumors, whereas overexpression in the absence of amplification is more common in ER-negative tumors [168]. Not surprisingly, it is in the ER-positive cases where cyclin D1 amplification is associated with a worse outcome. How these findings will affect the clinical management of ER-positive breast tumors and future use of tamoxifen therapy remains to be determined.
2.6.2. Cyclin E
In response to inhibition of Rb by cyclin D1 early in G1 phase, the transcription factor E2F strongly induces the expression of cyclin E, which associates with CDK2 to form an active complex that promotes entry into S phase [160]. Cyclin E/CDK2 activity is subsequently counterbalanced by the CDK inhibitors p21 and p27. While not a direct target of the ER, estrogen-dependent induction of c-Myc can activate cyclin E complexes by stimulating release of the inhibitor p21. CDC25A can also participate in estrogen-mediated cyclin E activation [169].
The contribution of cyclin E to tamoxifen resistance is less clear than for cyclin D1. This may be due to the presence of multiple low molecular weight forms of cyclin E that are generated by post-translational proteolytic cleavage events (reviewed in [170]). Low molecular weight cyclin E variants, which exhibit greater activity than full-length cyclin E, can induce antiestrogen resistance in MCF-7 cells [171]. Further studies are required to clarify the mechanism(s) by which low molecular weight cyclin E is induced in breast cancer, and its function in antiestrogen resistance. Other groups have reported that full-length cyclin E can also confer resistance to tamoxifen and fulvestrant [164,172].
2.6.3. p21 and p27
The CDK inhibitors p21 and p27 are important negative regulators of cell cycle progression that counteract the activities of cyclin D1 and cyclin E. p27 is largely regulated in a post-transcriptional manner, while p21 is primarily controlled at the level of transcription (reviewed in [160]). In MCF-7 cells, both tamoxifen and fulvestrant increase the expression of p21 and p27 during cell cycle arrest [173]. p27 induction by tamoxifen induces quiescence in breast cancer cells and prevents a positive proliferative response following re-stimulation by growth factors such as IGF-I and EGF [174]. Combined treatment with tamoxifen and the farnesyl transferase inhibitor FTI-277, which causes a potent G1 arrest, maintains the expression and association of p21 and p27 with cyclin E/CDK2 complexes [23]. These two studies suggest that preservation of p27 and p21 is critical for the inhibitory effects of antiestrogens like tamoxifen in controlling breast cancer cell growth. In contrast, downregulation of these CDK inhibitors prevents the growth inhibitory effects of these drugs [173].
p21 and p27 functions are regulated in part by AKT through phosphorylation of residues within their nuclear localization sequences, leading to sequestration in the cytoplasm where they are unable to interact with cyclin E and CDK2 (discussed in [175]). c-Myc also plays a role in regulating p21, although the mechanism is less clear. Most significantly, c-Myc appears to block tamoxifen- and fulvestrant-induced expression of p21 [176]. In MCF-7/LCC9 breast cancer cells that have acquired resistance to fulvestrant and cross-resistance to tamoxifen, p21 expression is no longer regulated by estrogens and antiestrogens [176]. Studies of other models of antiestrogen resistance also support the idea that p21 and/or p27 play important roles in antiestrogen sensitivity and resistance. Unlike parental MCF-7 cells in which tamoxifen or fulvestrant treatment increases p27 binding to cyclin E, antiestrogens do not alter p27 binding in the tamoxifen-resistant LY-2 variant [177]. These LY-2 cells display constitutive activation of MEK and ERK1/2, and inhibition of these kinases partially restores antiestrogen sensitivity and p27 function. Other cell culture models of resistance, such as MCF-7/LCC2 and MCF-7/HER2-18 cells, exhibit increased MEK/ERK activity, although whether this affects antiestrogen resistance through modulating p27 remains to be determined [177]. These data supporting a role for p21 and p27 in antiestrogen responses and resistance are corroborated by clinical evidence (see Section 4.1).
3. Endocrine therapies; effect on motility/invasion/metastasis
Metastatic breast cancer is ultimately the greatest cause of disease mortality. Tamoxifen is beneficial for decreasing secondary disease occurrence, both at local and distant sites [178]. However, the major phenotype of endocrine resistance is cancer recurrence. While clinical data have not definitively linked antiestrogen resistance with metastasis, many of the proteins/pathways discussed above in Section 2 that are involved in circumventing antiestrogen-induced blocks in cell cycle progression and survival are also implicated in increased migration and metastasis of breast cancer cells.
Hiscox et al. have begun to tackle the molecular relationship between antiestrogen resistance and invasion/metastasis [179]. They found that MCF-7 cells selected for tamoxifen resistance showed enhanced basal motility and an invasive phenotype. Treatment of these cells with the pharmacological inhibitors gefitinib and AZD0530 inhibited migration and invasion, indicating that the invasive phenotype of these resistant cells was dependent on EGFR and c-Src, respectively. The ability of tumor cells to metastasize requires multiple genotypic and phenotypic changes. Signaling perturbations associated with the epithelial to mesenchymal transition (EMT) contribute significantly to invasive and metastatic disease [180]. These include increased expression of extracellular matrix proteins, downregulation of E-cadherin, disruption of adherens junctions complexes, accumulation of β-catenin and NF-kB in the nucleus, and transcription of target genes such as c-Myc and cyclin D1 [181,182]. Interestingly, the tamoxifen-resistant MCF-7 cells investigated by Hiscox et al. displayed an altered morphology reminiscent of an EMT, marked by an elongated, fibroblast-like phenotype that lacked characteristic cell-cell contacts [179]. Tyrosine phosphorylation of β-catenin was elevated in the tamoxifen-resistant cells, coincident with accumulation of the protein, loss of its association with E-cadherin, increased interactions with EGFR, and increased transcription of c-Myc and cyclin D1. These effects were reversed in the presence of gefitinib, supporting a mechanism whereby the EGFR drives phosphorylation of β-catenin and activation of c-Src to initiate EMT-mediated metastatic progression.
Many of the other molecules that have been mentioned above in the context of promoting antiestrogen resistance also play a role in promoting invasiveness. For example, Pak1 is functionally linked to regulation of the actin cytoskeleton through Rho family GTPases [150]. The Pak1 gene is closely linked to CCND1 (cyclin D1) at 11q13, a region that is often amplified in breast cancers [183]. Similarly, amplification of the gene encoding c-Myc in breast tumors appears to correlate with the transition from in situ to invasive carcinoma [38,184]. Finally, in a study of matched primary and metastatic breast tumors, Bcl-2 expression was found to be elevated in metastatic tumors coincident with a retention of ERα expression [38]. These results conflicted with those from a similar study performed on paired primary and recurrent tamoxifen-resistant tumors, which found that the recurrent tumors did not show a correlation between ER status and Bcl-2 expression [30]. However, there was a correlation between ERα and phosphorylated p38 and ERK in these relapsed specimens. These discrepancies may be due to differences in inclusion criteria and the source of recurrent tissue between studies. The former study evaluated mostly tumors at distant sites after discontinuation of hormonal therapy, while the latter study evaluated locoregional recurrences while the patients continued therapy.
4. Antiestrogen resistance: implications for treatment
4.1. Prediction of response
It is clear from the discussion above that resistance to endocrine therapy likely occurs through diverse mechanisms that vary from patient to patient. Consequently, it is imperative to develop molecular signatures that can predict the likelihood of response and potential for relapse of individual tumors. Initially, predictive markers for tamoxifen resistance included expression of ER and receptor tyrosine kinases such as EGFR and HER2. As our understanding of antiestrogen resistance has evolved, these markers have broadened. Currently, the overall expression of proteins, protein modifications, and intracellular localization are all being analyzed for predictive value of response to endocrine therapy.
As mentioned above, survival pathways regulated by PI3K, AKT, PTEN, and Bcl-2 family members play an important role in antiestrogen resistance. Studies are currently underway to determine whether increased activation and/or expression of AKT, decreased activity of PTEN, or altered expression of Bad could be predictive markers for response to anti-hormonal treatments. For example, a retrospective analysis of 402 ERα-positive breast carcinoma biopsies treated with tamoxifen for a median of 5 years revealed that high expression of AKT2, as measured by immunohistochemistry, was predictive of improved overall survival [31]. Patients who relapsed following tamoxifen treatment were more likely to have low AKT2 expression and high cytoplasmic phospho-AKT (Ser-473). Interestingly, expression of AKT1 and AKT3 did not have a significant predictive correlation. Bad expression could also predict tamoxifen-treatment outcome. Patients in this same clinical study whose tumors had high levels of Bad expression had improved disease-free survival rates compared to patients whose tumors had low levels of Bad [36]. There was no association between phospho-Bad (Ser-112), Bax, Bcl-2 or Bcl-xL expression and response to tamoxifen treatment in this group of patients. A related study examined 252 primary ERα human breast carcinomas to determine whether activated AKT or loss of heterozygosity (LOH) of PTEN was predictive of response to tamoxifen [185]. Phospho-AKT correlated with HER2 status in this study, as did PTEN LOH. From this patient cohort, 36 patient samples were used to examine the relationship between activated AKT and the efficacy of endocrine therapy for metastatic breast cancer. Patients with phospho-AKT-positive tumors received less benefit from endocrine therapy than did patients with phospho-AKT-negative tumors. A third study evaluated PTEN expression in 100 patients with ERα breast carcinomas who had been treated with tamoxifen [186]. High levels of PTEN were recorded for 81% of patients who had non-recurrent disease and only 41% of patients with recurrent disease. Conversely, low PTEN expression was associated with a shorter relapse free survival and reduced disease-specific survival. PTEN expression was an even stronger predictor of relapse and survival when the analysis was restrained to only stage I breast cancer patients.
Like the PI3K/AKT survival pathway, cyclin D1 expression is associated with multiple mechanisms of antiestrogen resistance. Patients with ERα-positive tumors and high levels of cyclin D1 mRNA had a worse prognosis for overall survival [187]. In a small subset of these patients who were treated with endocrine therapy, an apparent association between high cyclin D1 mRNA and decreased duration of response was made. Similar results were obtained by evaluating protein levels of cyclin D1 in 248 post-menopausal patients who had been randomized into 2-year tamoxifen treatment or no-treatment groups [188]. Interestingly, the 55 strongly ERα-positive patients with tumors showing moderate or low cyclin D1 levels responded to tamoxifen treatment, whereas the 46 patients with both high ERα and cyclin D1 expression in their tumors showed no difference in survival between tamoxifen and no treatment.
The association between cyclin E levels and antiestrogen response is even less clear. Some of the clinical studies focused on cyclin E have shown a correlation between the presence of low molecular weight and/or full-length cyclin E and poor survival, while others have not found such an association ([189] and discussed in [161]). A recent study examining the expression of cyclin E and two proteases implicated in the generation of low molecular weight cyclin E (proteinase 3 and neutrophil elastase) in 205 breast cancer patients revealed that cyclin E was significantly associated with poor disease-free survival in the entire cohort, and with tamoxifen resistance in the 110 treated patients [190]. However, expression of neutrophil elastase was not associated with poor outcome or treatment resistance. Further studies are required to clarify the role of these variants in clinical endocrine resistance.
Coincident with cyclin D1 and cyclin E expression, p21 and p27 levels may also prove to be predictive for clinical response to antiestrogen resistance. High levels of p27 expression in a group of 512 patients with premenopausal breast cancer patients treated with tamoxifen and the gonadotropin-releasing hormone agonist goserelin were strongly associated with improved overall and disease-free survival [191]. Data for p21 are less consistent, perhaps because they may be confounded by the complex regulation of p21 expression and contributions from other proteins such as p53 and IRF-1 that may independently affect breast cancer outcome (discussed in [20,161]). It may also be that subcellular localization of p21, rather than overall expression levels, will prove to be a better marker of therapeutic response and treatment outcome. In line with this suggestion, Perez-Tenorio et al. have recently reported that cytoplasmic p21 is associated with increased AKT activity and poor tamoxifen response in a cohort of 280 women [192].
As mentioned above, serine phosphorylation of ERα may also prove to be useful for predicting therapeutic response (see Section 2.5). Recent studies have centered on the role of Ser-118 phosphorylation in predicting clinical outcome in breast cancer patients. In a series of 117 lymph node-negative primary breast tumors from patients treated with tamoxifen following surgery with or without radiation, Murphy et al. observed Ser-118 phosphorylation only in tumors with positive estrogen binding activity [193]. Phosphorylated Ser-118 was also observed more frequently in PR-positive tumors than PR-negative tumors, and patients with tumors positive for Ser-118 phosphorylation showed increased disease-free survival. Bergqvist et al. reported that increased MAPK activation and expression of phosphorylated Ser-118 correlated with one another and also predicted better survival [194]. However, overexpression of HER2 was inversely correlated with activated ERK1/2 and phosphorylated Ser-118, suggesting that at least in this cohort, HER2 activation does not drive phosphorylation and hyperactivation of ER. Moreover, this apparent improved outcome observed in patients with Ser-118 phosphorylation appears to contradict some of the cell culture experiments that showed a link between Ser-118 phosphorylation and antiestrogen resistance (see Section 2.5). This discrepancy may be explained by the possibility that Ser-118 phosphorylation of the ER may have distinct functions in different phases of breast cancer; there is some evidence to suggest that it may be associated with a better outcome prior to endocrine therapy but a worse outcome following acquisition of tamoxifen resistance [35].
The correlation between Ser-167 phosphorylation and clinical response to antiestrogens is equally complex. Kirkegaard et al. found that high levels of active AKT correlated with reduced survival and increased Ser-167 phosphorylation in tamoxifen-treated patients [31]. However, Yamashita et al. reported that phosphorylated Ser-167, along with expression of ER and PR, were associated with an improved response to endocrine therapy and survival [195]. The fact that Ser-167 can be phosphorylated by kinases other than AKT may suggest that it is AKT rather than phosphorylated Ser-167 that is responsible for the poor outcome in the Kirkegaard study [31].
In addition to the ERα modulation, PR status has also been suggested to be predictive of response to antiestrogens. As discussed above (Section 2.1), the presence of PR is reflective of a functional ER pathway because PR synthesis is initiated by ER-induced transcription. Bardou et al. analyzed two databases containing information on ER and PR status of a large number of breast cancer patients [196]. They found that PR expression was associated with disease-free survival independently of ERα status. However, ERα-positive/PR-positive tumors received the greatest benefit from adjuvant hormonal therapy when both ERα and PR were taken into consideration, while loss of expression of either receptor reduced responsiveness by nearly half. Additional studies have also found that ER/PR-positive breast tumors have significant improvement in prognosis after either adjuvant tamoxifen or letrozole treatments [197,198]. This supports the hypothesis that ERα positive tumors that lack PR expression are less likely to respond to anti-hormonal therapy because they are no longer dependent on estrogen for survival. Conversely, ERβ expression appears to be predictive of a positive response to endocrine therapy and better overall survival [6]. This is potentially due to the putative tumor suppressor functions of ERβ discussed above (see Section 1), and provides the rationale for developing SERMs that exhibit selective activities to ER subtypes (see Section 1.1.1).
4.2. Molecular profiling studies of antiestrogen resistance
Gene expression analysis and molecular profiling have added to our understanding of the mechanisms of antiestrogen response and resistance. Several groups have used these approaches on tumor samples to develop gene signatures that can predict clinical responses to antiestrogen treatment [199,200]. However, in some instances, these profiles have not been validated by independent studies [201]. Expression profiling of cell culture and xenograft models has also been extensively utilized to help clarify the molecular actions of antiestrogens and aromatase inhibitors. Several of these studies have confirmed biological evidence indicating that SERMs like tamoxifen and raloxifene can behave as both antagonists and agonists of the ER, whereas fulvestrant is a pure antagonist. For example, expression of cell cycle-associated genes such as cyclin A1 and CDC25B was unaffected by estrogen, tamoxifen, or raloxifene but strongly regulated by fulvestrant [202]. The effects of tamoxifen and raloxifene were also similar to estrogen with respect to expression of Bcl-2-interacting killer (BIK), an estrogen-inhibited, pro-apoptotic gene that has been demonstrated to play a critical role in fulvestrant-induced breast cancer cell death [203].
Other studies have examined regulation of gene expression by letrozole and anastrazole as compared to tamoxifen [204]. As might be expected, the effects of these aromatase inhibitors were more similar to each other than they were to tamoxifen. However, the major functional classes of genes regulated by all three drugs were comparable, and included apoptosis, Wnt, and MAPK signaling pathways. This suggests that, rather than looking at individual genes, these approaches may be most useful for identifying functional groupings of genes that work together in specific signaling pathways to regulate responses to antiestrogens. A recent study in MCF-7 cells comparing acquired resistance to tamoxifen and fulvestrant found that tamoxifen resistance was strongly associated with changes in the PKA pathway, MAPK phosphatases, and calcium binding proteins. In contrast, downregulation of ER and major alterations in growth factor receptor and interferon signaling were observed in cells resistant to fulvestrant [60].
Interferon-inducible genes have also been found to be differentially regulated in the MaCa 3366/TAM xenograft model of acquired tamoxifen resistance when compared to the tamoxifen-sensitive MaCa 3366 counterpart [205]. Interestingly, loss of the putative tumor suppressor interferon regulatory factor 1 (IRF-1) has been specifically implicated in fulvestrant resistance [25] and is required for tamoxifen-induced cell death of normal mammary epithelial cells [206]. Another interferon-induced gene, calgranulin A (S100A8) [207], is upregulated in MCF-7, T47D, and ZR-75-1-derived models of fulvestrant resistance [59], providing additional support for a role of interferon-inducible genes in antiestrogen resistance.
While expression profiling has provided insight into molecular pathways that may contribute to antiestrogen resistance, there are some challenges associated with these approaches that must be carefully considered. Multiple expression array platforms and technologies can make comparing the findings of different investigators difficult, especially at the level of individual genes. Statistical analysis of these data is also very complex, and the risk of excluding biologically relevant genes while including others that don't contribute to the phenotype is a significant one. Unfortunately, these challenges are greatly magnified in the context of analysis of tumor tissues [208-210]. Nonetheless, these approaches can be quite powerful, particularly when one considers functional grouping of genes rather than individual molecules.
4.3. Combinatorial therapies
Strategies for treating breast tumors have significantly broadened in recent years because of a growing appreciation for the fact that resistance to endocrine therapies can and will invariably arise through a variety of distinct mechanisms. Our increasing understanding of the molecular basis for these failures has brought to the forefront the possibility of using combination therapies for treating breast cancer. There are two separate rationales for using combination therapies. First, targeting multiple pathways at once can potentially delay or halt the acquisition of a resistant phenotype. Alternatively, treatments can be administered to effectively resensitize tumors to endocrine therapies for which they are resistant. In many cases, the practical strategies for addressing these distinct phenomena are the same. For example, as discussed above, growth factor receptor signaling is often increased in antiestrogen-resistant breast tumors, leading to upregulation of proliferation and survival pathways that may be distinct from estrogen signaling per se. Thus, treatment with gefitinib or trastuzumab to inhibit the EGFR or HER2, respectively, along with hormonal therapies would potentially block both the pro-proliferative activity of the ER and the proliferation/survival pathways activated by the growth factor receptors. Some of the current trials evaluating the efficacy of these combinations include a Phase II randomized trial (Translational Oncology Research International trial) that is investigating fulvestrant and trastuzumab in combination as a first-line treatment for patients with ERα /HER2-positive breast tumors, a comparative study evaluating anastrozole versus fulvestrant with the addition of gefitinib in women with ERα-positive tumors who have metastatic disease and have failed chemotherapy and/or endocrine therapy, and a combination of fulvestrant with the dual inhibitor lapatinib in postmenopausal women with ERα-positive and EGFR-and/or HER2-postive tumors who have had previous exposure to an aromatase inhibitor (reviewed in [211,212]).
In addition to targeting growth factor receptors, several clinical trials have been initiated to investigate the effect of farnesyl transferase inhibitors such as tipifarnib in combination with endocrine therapies. However, these trials have been somewhat disappointing in that they have not thus far corroborated pre-clinical models showing that tipifarnib in combination with tamoxifen resulted in greater tumor regression than either treatment alone [213]. Additional studies are underway that examine the efficacy of combining tipifarnib with AIs (reviewed in [214]).
Clinical studies with mTOR antagonists in combination with hormonal therapies are also ongoing. As a single agent, CCI-779 has been somewhat effective in Phase II studies of metastatic breast cancer [215]. Currently, Phase II and Phase III trials are testing either CCI-779 or RAD-001 in combination with letrozole, as compared to letrozole alone. Interim results show that the combination of CCI-779 and letrozole translates into a clinical benefit rate of 77% versus 66% for letrozole alone, although these values have not yet reached statistical significance [135].
Finally, extensive efforts are being made to determine whether pre-clinical data showing that tumors resistant to one class of hormonal therapy may still retain sensitivity to alternate endocrine therapies hold true in the clinic. Results of the Phase II Swiss Group for Clinical Research Trial (SAKK 21/00), which compared the efficacy of fulvestrant treatment in AI-sensitive versus AI-resistant patients, were recently published (reviewed in [216]). Overall fulvestrant yielded a clinical benefit (measured as an objective response or stable disease >24 weeks) in 30% of patients, irrespective of whether they had previously been treated with AIs. There are currently multiple planned or ongoing clinical trials combining AIs with antiestrogens. These include Phase II and Phase III trials evaluating use as first line and neoadjuvant treatment (reviewed in [214]).
Acknowledgments
The authors would like to acknowledge support of our research from the Susan G. Komen Breast Cancer Foundation (PDF0503551) and Department of Defense Breast Cancer Research Program (BC051851) to RBR, the Department of Defense Breast Cancer Research Program (BC050339) to RSS, the National Cancer Institute institutional training grant (T32 CA009109) for RSS and MSG; and the National Institutes of Health (R01 CA 096846) to AHB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol Aspects Med. 2006;27:299–402. doi: 10.1016/j.mam.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Anderson E, Clarke RB. Steroid receptors and cell cycle in normal mammary epithelium. J Mammary Gland Biol Neoplasia. 2004;9:3–13. doi: 10.1023/B:JOMG.0000023584.01750.16. [DOI] [PubMed] [Google Scholar]
- 3.Speirs V, Skliris GP, Burdall SE, Carder PJ. Distinct expression patterns of ER alpha and ER beta in normal human mammary gland. J Clin Pathol. 2002;55:371–374. doi: 10.1136/jcp.55.5.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forster C, Makela S, Warri A, Kietz S, Becker D, Hultenby K, et al. Involvement of estrogen receptor beta in terminal differentiation of mammary gland epithelium. Proc Natl Acad Sci USA. 2002;99:15578–15583. doi: 10.1073/pnas.192561299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saji S, Hirose M, Toi M. Clinical significance of estrogen receptor beta in breast cancer. Cancer Chemother Pharmacol. 2005;56 1:21–26. doi: 10.1007/s00280-005-0107-3. [DOI] [PubMed] [Google Scholar]
- 6.Murphy LC, Watson PH. Is oestrogen receptor-beta a predictor of endocrine therapy responsiveness in human breast cancer? Endocr Relat Cancer. 2006;13:327–334. doi: 10.1677/erc.1.01141. [DOI] [PubMed] [Google Scholar]
- 7.Matthews J, Wihlen B, Tujague M, Wan J, Strom A, Gustafsson JA. Estrogen receptor (ER) beta modulates ER alpha-mediated transcriptional activation by altering the recruitment of c-fos and c-jun to estrogen-responsive promoters. Mol Endocrinol. 2006;20:534–543. doi: 10.1210/me.2005-0140. [DOI] [PubMed] [Google Scholar]
- 8.Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA. Estrogen receptors alpha (ER alpha) and beta (ER beta) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24:6605–6616. doi: 10.1038/sj.onc.1208807. [DOI] [PubMed] [Google Scholar]
- 9.Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) alpha or ER beta in human osteosarcoma cells: Distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- 10.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 11.Lewis JS, Jordan VC. Selective estrogen receptor modulators (SERMs): Mechanisms of anticarcinogenesis and drug resistance. Mutat Res. 2005;591:247–263. doi: 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 12.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 13.Veeneman GH. Non-steroidal subtype selective estrogens. Curr Med Chem. 2005;12:1077–1136. doi: 10.2174/0929867053764662. [DOI] [PubMed] [Google Scholar]
- 14.Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, et al. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem Pharmacol. 2006;71:1459–1469. doi: 10.1016/j.bcp.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Blizzard TA, Gude C, Morgan JD, 2nd, Chan W, Birzin ET, Mojena M, et al. Androstenediol analogs as ER-beta-selective SERMs. Bioorg Med Chem Lett. 2006;16:834–838. doi: 10.1016/j.bmcl.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Buzdar AU. TAS-108: A novel steroidal antiestrogen. Clin Cancer Res. 2005;11:906S–908S. [PubMed] [Google Scholar]
- 17.Howell A. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr Relat Cancer. 2006;13:689–706. doi: 10.1677/erc.1.00846. [DOI] [PubMed] [Google Scholar]
- 18.Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90 1:S2–S6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brueggemeier RW. Update on the use of aromatase inhibitors in breast cancer. Expert Opin Pharmacother. 2006;7:1919–1930. doi: 10.1517/14656566.7.14.1919. [DOI] [PubMed] [Google Scholar]
- 20.Riggins RB, Bouton AH, Liu MC, Clarke R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitam Horm. 2005;71:201–237. doi: 10.1016/S0083-6729(05)71007-4. [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Masri S, Wang X, Phung S, Yuan YC, Wu X. What do we know about the mechanisms of aromatase inhibitor resistance? J Steroid Biochem Mol Biol. 2006;102:232–240. doi: 10.1016/j.jsbmb.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carroll JS, Prall OW, Musgrove EA, Sutherland RL. A pure estrogen antagonist inhibits cyclin E-CDK2 activity in MCF-7 breast cancer cells and induces accumulation of p130-E2F4 complexes characteristic of quiescence. J Biol Chem. 2000;275:38221–38229. doi: 10.1074/jbc.M004424200. [DOI] [PubMed] [Google Scholar]
- 23.Doisneau-Sixou SF, Cestac P, Faye JC, Favre G, Sutherland RL. Additive effects of tamoxifen and the farnesyl transferase inhibitor FTI-277 on inhibition of MCF-7 breast cancer cell-cycle progression. Int J Cancer. 2003;106:789–798. doi: 10.1002/ijc.11263. [DOI] [PubMed] [Google Scholar]
- 24.Thiantanawat A, Long BJ, Brodie AM. Signaling pathways of apoptosis activated by aromatase inhibitors and antiestrogens. Cancer Res. 2003;63:8037–8050. [PubMed] [Google Scholar]
- 25.Bouker KB, Skaar TC, Fernandez DR, O'Brien KA, Riggins RB, Cao D, et al. Interferon regulatory factor-1 mediates the proapoptotic but not cell cycle arrest effects of the steroidal antiestrogen ICI 182,780 (faslodex, fulvestrant) Cancer Res. 2004;64:4030–4039. doi: 10.1158/0008-5472.CAN-03-3602. [DOI] [PubMed] [Google Scholar]
- 26.Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer. 2003;10:179–186. doi: 10.1677/erc.0.0100179. [DOI] [PubMed] [Google Scholar]
- 27.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, et al. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: A Southwest Oncology Group Study. Clin Cancer Res. 2004;10:5670–5676. doi: 10.1158/1078-0432.CCR-04-0110. [DOI] [PubMed] [Google Scholar]
- 28.Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, et al. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer. 2005;12 1:S99–S111. doi: 10.1677/erc.1.01005. [DOI] [PubMed] [Google Scholar]
- 29.Dowsett M, Johnston S, Martin LA, Salter J, Hills M, Detre S, et al. Growth factor signalling and response to endocrine therapy: The Royal Marsden Experience. Endocr Relat Cancer. 2005;12 1:S113–117. doi: 10.1677/erc.1.01044. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: Relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23:2469–2476. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 31.Kirkegaard T, Witton CJ, McGlynn LM, Tovey SM, Dunne B, Lyon A, et al. AKT activation predicts outcome in breast cancer patients treated with tamoxifen. J Pathol. 2005;207:139–146. doi: 10.1002/path.1829. [DOI] [PubMed] [Google Scholar]
- 32.Holm C, Rayala S, Jirstrom K, Stal O, Kumar R, Landberg G. Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst. 2006;98:671–680. doi: 10.1093/jnci/djj185. [DOI] [PubMed] [Google Scholar]
- 33.van der Flier S, Brinkman A, Look MP, Kok EM, Meijer-van Gelder ME, Klijn JG, et al. BCAR1/p130Cas protein and primary breast cancer: Prognosis and response to tamoxifen treatment. J Natl Cancer Inst. 2000;92:120–127. doi: 10.1093/jnci/92.2.120. [DOI] [PubMed] [Google Scholar]
- 34.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 35.Sarwar N, Kim JS, Jiang J, Peston D, Sinnett HD, Madden P, et al. Phosphorylation of ER alpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ER alpha phosphorylation in breast cancer progression. Endocr Relat Cancer. 2006;13:851–861. doi: 10.1677/erc.1.01123. [DOI] [PubMed] [Google Scholar]
- 36.Cannings E, Kirkegaard T, Tovey SM, Dunne B, Cooke TG, Bartlett JM. Bad expression predicts outcome in patients treated with tamoxifen. Breast Cancer Res Treat. 2006 doi: 10.1007/s10549-006-9323-8. in press. [DOI] [PubMed] [Google Scholar]
- 37.Linke SP, Bremer TM, Herold CD, Sauter G, Diamond C. A multimarker model to predict outcome in tamoxifen-treated breast cancer patients. Clin Cancer Res. 2006;12:1175–1183. doi: 10.1158/1078-0432.CCR-05-1562. [DOI] [PubMed] [Google Scholar]
- 38.Planas-Silva MD, Bruggeman RD, Grenko RT, Smith JS. Overexpression of c-Myc and Bcl-2 during progression and distant metastasis of hormone-treated breast cancer. Exp Mol Pathol. 2007;82:85–90. doi: 10.1016/j.yexmp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Rahko E, Blanco G, Bloigu R, Soini Y, Talvensaari-Mattila A, Jukkola A. Adverse outcome and resistance to adjuvant antiestrogen therapy in node-positive postmenopausal breast cancer patients-the role of p53. Breast. 2006;15:69–75. doi: 10.1016/j.breast.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonnell DP. The molecular determinants of estrogen receptor pharmacology. Maturitas. 2004;48 1:S7–S12. doi: 10.1016/j.maturitas.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ER alpha and ER beta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 43.Zou A, Marschke KB, Arnold KE, Berger EM, Fitzgerald P, Mais DE, et al. Estrogen receptor beta activates the human retinoic acid receptor alpha-1 promoter in response to tamoxifen and other estrogen receptor antagonists, but not in response to estrogen. Mol Endocrinol. 1999;13:418–430. doi: 10.1210/mend.13.3.0253. [DOI] [PubMed] [Google Scholar]
- 44.Smith CL, O'Malley BW. Coregulator function: A key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 45.Hall JM, McDonnell DP. Coregulators in nuclear estrogen receptor action: From concept to therapeutic targeting. Mol Interv. 2005;5:343–357. doi: 10.1124/mi.5.6.7. [DOI] [PubMed] [Google Scholar]
- 46.Kumar R, Gururaj AE, Vadlamudi RK, Rayala SK. The clinical relevance of steroid hormone receptor corepressors. Clin Cancer Res. 2005;11:2822–2831. doi: 10.1158/1078-0432.CCR-04-1276. [DOI] [PubMed] [Google Scholar]
- 47.Girault I, Bieche I, Lidereau R. Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas. 2006;54:342–351. doi: 10.1016/j.maturitas.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53:25–71. [PubMed] [Google Scholar]
- 49.Honig SF. Treatment of metastatic disease: Hormonal therapy and chemotherapy. In: Harris JR, Lippman ME, Morrow M, Helman S, editors. Diseases of the Breast. Lippincott-Raven; Philadelphia: 1996. pp. 669–734. [Google Scholar]
- 50.Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–2555. [PubMed] [Google Scholar]
- 51.Roodi N, Bailey LR, Kao WY, Verrier CS, Yee CJ, Dupont WD, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst. 1995;87:446–451. doi: 10.1093/jnci/87.6.446. [DOI] [PubMed] [Google Scholar]
- 52.Sharma D, Saxena NK, Davidson NE, Vertino PM. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: Tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006;66:6370–6378. doi: 10.1158/0008-5472.CAN-06-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giacinti L, Claudio PP, Lopez M, Giordano A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist. 2006;11:1–8. doi: 10.1634/theoncologist.11-1-1. [DOI] [PubMed] [Google Scholar]
- 54.Garcia M, Derocq D, Freiss G, Rochefort H. Activation of estrogen receptor transfected into a receptor-negative breast cancer cell line decreases the metastatic and invasive potential of the cells. Proc Natl Acad Sci USA. 1992;89:11538–11542. doi: 10.1073/pnas.89.23.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, et al. Changes in estrogen receptor, progesterone receptor, and PS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995;55:3331–3338. [PubMed] [Google Scholar]
- 56.Encarnacion CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SA, Osborne CK. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat. 1993;26:237–246. doi: 10.1007/BF00665801. [DOI] [PubMed] [Google Scholar]
- 57.Clarke R, Skaar TC, Bouker KB, Davis N, Lee YR, Welch JN, et al. Molecular and pharmacological aspects of antiestrogen resistance. J Steroid Biochem Mol Biol. 2001;76:71–84. doi: 10.1016/s0960-0760(00)00193-x. [DOI] [PubMed] [Google Scholar]
- 58.van den Berg HW, Lynch M, Martin J, Nelson J, Dickson GR, Crockard AD. Characterisation of a tamoxifen-resistant variant of the ZR-75-1 human breast cancer cell line (ZR-75-9a1) and ability of the resistant phenotype. Br J Cancer. 1989;59:522–526. doi: 10.1038/bjc.1989.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sommer A, Hoffmann J, Lichtner RB, Schneider MR, Parczyk K. Studies on the development of resistance to the pure antiestrogen faslodex in three human breast cancer cell lines. J Steroid Biochem Mol Biol. 2003;85:33–47. doi: 10.1016/s0960-0760(03)00139-0. [DOI] [PubMed] [Google Scholar]
- 60.Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006;66:11954–11966. doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 61.Larsen SS, Madsen MW, Jensen BL, Lykkesfeldt AE. Resistance of human breast-cancer cells to the pure steroidal anti-estrogen ICI 182,780 is not associated with a general loss of estrogen-receptor expression or lack of estrogen responsiveness. Int J Cancer. 1997;72:1129–1136. doi: 10.1002/(sici)1097-0215(19970917)72:6<1129::aid-ijc31>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 62.Jensen BL, Skouv J, Lundholt BK, Lykkesfeldt AE. Differential regulation of specific genes in MCF-7 and the ICI 182780-resistant cell line MCF-7/182R-6. Br J Cancer. 1999;79:386–392. doi: 10.1038/sj.bjc.6690061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brunner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, et al. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997;57:3486–3493. [PubMed] [Google Scholar]
- 64.Brunner N, Boulay V, Fojo A, Freter CE, Lippman ME, Clarke R. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer Res. 1993;53:283–290. [PubMed] [Google Scholar]
- 65.Kuske B, Naughton C, Moore K, Macleod KG, Miller WR, Clarke R, et al. Endocrine therapy resistance can be associated with high estrogen receptor α (ERα) expression and reduced ERα phosphorylation in breast cancer models. Endocr Relat Cancer. 2006;13:1121–1133. doi: 10.1677/erc.1.01257. [DOI] [PubMed] [Google Scholar]
- 66.Riggins RB, Zwart A, Nehra R, Clarke R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther. 2005;4:33–41. [PubMed] [Google Scholar]
- 67.Howell A. Fulvestrant (‘faslodex’): Current and future role in breast cancer management. Crit Rev Oncol Hematol. 2006;57:265–273. doi: 10.1016/j.critrevonc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 68.Graham ML, 2nd, Krett NL, Miller LA, Leslie KK, Gordon DF, Wood WM, et al. T47DCO cells, genetically unstable and containing estrogen receptor mutations, are a model for the progression of breast cancers to hormone resistance. Cancer Res. 1990;50:6208–6217. [PubMed] [Google Scholar]
- 69.Graham ML, 2nd, Smith JA, Jewett PB, Horwitz KB. Heterogeneity of progesterone receptor content and remodeling by tamoxifen characterize subpopulations of cultured human breast cancer cells: Analysis by quantitative dual parameter flow cytometry. Cancer Res. 1992;52:593–602. [PubMed] [Google Scholar]
- 70.Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25:869–898. doi: 10.1210/er.2003-0010. [DOI] [PubMed] [Google Scholar]
- 71.Pink JJ, Wu SQ, Wolf DM, Bilimoria MM, Jordan VC. A novel 80 kDa human estrogen receptor containing a duplication of exons 6 and 7. Nucleic Acids Res. 1996;24:962–969. doi: 10.1093/nar/24.5.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karnik PS, Kulkarni S, Liu XP, Budd GT, Bukowski RM. Estrogen receptor mutations in tamoxifen-resistant breast cancer. Cancer Res. 1994;54:349–353. [PubMed] [Google Scholar]
- 73.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997;57:1244–1249. [PubMed] [Google Scholar]
- 74.Fuqua SA, Wiltschke C, Zhang QX, Borg A, Castles CG, Friedrichs WE, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. 2000;60:4026–4029. [PubMed] [Google Scholar]
- 75.Zhang Z, Yamashita H, Toyama T, Omoto Y, Sugiura H, Hara Y, et al. Estrogen receptor alpha mutation (A-to-G transition at nucleotide 908) is not found in different types of breast lesions from Japanese women. Breast Cancer. 2003;10:70–73. doi: 10.1007/BF02967628. [DOI] [PubMed] [Google Scholar]
- 76.Navolanic PM, Steelman LS, McCubrey JA. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy. Int J Oncol. 2003;22:237–252. [PubMed] [Google Scholar]
- 77.Nicholson RI, Hutcheson IR, Knowlden JM, Jones HE, Harper ME, Jordan N, et al. Nonendocrine pathways and endocrine resistance: Observations with antiestrogens and signal transduction inhibitors in combination. Clin Cancer Res. 2004;10:346S–354S. doi: 10.1158/1078-0432.ccr-031206. [DOI] [PubMed] [Google Scholar]
- 78.Nicholson RI, Hutcheson IR, Britton D, Knowlden JM, Jones HE, Harper ME, et al. Growth factor signalling networks in breast cancer and resistance to endocrine agents: New therapeutic strategies. J Steroid Biochem Mol Biol. 2005;93:257–262. doi: 10.1016/j.jsbmb.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 79.Kurokawa H, Lenferink AE, Simpson JF, Pisacane PI, Sliwkowski MX, Forbes JT, et al. Inhibition of HER2/neu (ErbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000;60:5887–5894. [PubMed] [Google Scholar]
- 80.Kurokawa H, Arteaga CL. Inhibition of ErbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer. Clin Cancer Res. 2001;7:4436S–4442S. discussion 4411S-4412S. [PubMed] [Google Scholar]
- 81.Kurokawa H, Arteaga CL. ErbB (HER) receptors can abrogate antiestrogen action in human breast cancer by multiple signaling mechanisms. Clin Cancer Res. 2003;9:511S–515S. [PubMed] [Google Scholar]
- 82.McClelland RA, Barrow D, Madden TA, Dutkowski CM, Pamment J, Knowlden JM, et al. Enhanced epidermal growth factor receptor signaling in MCF7 breast cancer cells after long-term culture in the presence of the pure antiestrogen ICI 182,780 (faslodex) Endocrinology. 2001;142:2776–2788. doi: 10.1210/endo.142.7.8259. [DOI] [PubMed] [Google Scholar]
- 83.Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, et al. Elevated levels of epidermal growth factor receptor/c-ErbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003;144:1032–1044. doi: 10.1210/en.2002-220620. [DOI] [PubMed] [Google Scholar]
- 84.Hutcheson IR, Knowlden JM, Madden TA, Barrow D, Gee JM, Wakeling AE, et al. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat. 2003;81:81–93. doi: 10.1023/A:1025484908380. [DOI] [PubMed] [Google Scholar]
- 85.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 86.Yarden RI, Wilson MA, Chrysogelos SA. Estrogen suppression of EGFR expression in breast cancer cells: A possible mechanism to modulate growth. J Cell Biochem. 2001;81:232–246. doi: 10.1002/jcb.1142. [DOI] [PubMed] [Google Scholar]
- 87.Gee JM, Harper ME, Hutcheson IR, Madden TA, Barrow D, Knowlden JM, et al. The antiepidermal growth factor receptor agent gefitinib (ZD1839/Iressa) improves antihormone response and prevents development of resistance in breast cancer in vitro. Endocrinology. 2003;144:5105–5117. doi: 10.1210/en.2003-0705. [DOI] [PubMed] [Google Scholar]
- 88.Okubo S, Kurebayashi J, Otsuki T, Yamamoto Y, Tanaka K, Sonoo H. Additive antitumour effect of the epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (Iressa ZD1839) and the antioestrogen fulvestrant (faslodex, ICI 182,780) in breast cancer cells. Br J Cancer. 2004;90:236–244. doi: 10.1038/sj.bjc.6601504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB. Overexpression of HER2 modulates Bcl-2 Bcl-xL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res. 1996;2:1215–1219. [PubMed] [Google Scholar]
- 90.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 91.Lee H, Bai W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002;22:5835–5845. doi: 10.1128/MCB.22.16.5835-5845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Riggins RB, Thomas KS, Ta HQ, Wen J, Davis RJ, Schuh NR, et al. Physical and functional interactions between Cas and c-Src induce tamoxifen resistance of breast cancer cells through pathways involving epidermal growth factor receptor and signal transducer and activator of transcription 5b. Cancer Res. 2006;66:7007–7015. doi: 10.1158/0008-5472.CAN-05-3952. [DOI] [PubMed] [Google Scholar]
- 93.Holloway JN, Murthy S, El-Ashry D. A cytoplasmic substrate of mitogen-activated protein kinase is responsible for estrogen receptor-alpha down-regulation in breast cancer cells: The role of nuclear factor-kappa B. Mol Endocrinol. 2004;18:1396–1410. doi: 10.1210/me.2004-0048. [DOI] [PubMed] [Google Scholar]
- 94.Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, et al. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: Implicated role of growth factor signaling and estrogen receptor coregulators. Cancer Chemother Pharmacol. 2005;56 1:10–20. doi: 10.1007/s00280-005-0108-2. [DOI] [PubMed] [Google Scholar]
- 95.Yang Z, Barnes CJ, Kumar R. Human epidermal growth factor receptor 2 status modulates subcellular localization of and interaction with estrogen receptor alpha in breast cancer cells. Clin Cancer Res. 2004;10:3621–3628. doi: 10.1158/1078-0432.CCR-0740-3. [DOI] [PubMed] [Google Scholar]
- 96.Massarweh S, Osborne CK, Jiang S, Wakeling AE, Rimawi M, Mohsin SK, et al. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006;66:8266–8273. doi: 10.1158/0008-5472.CAN-05-4045. [DOI] [PubMed] [Google Scholar]
- 97.Brockdorff BL, Heiberg I, Lykkesfeldt AE. Resistance to different antiestrogens is caused by different multi-factorial changes and is associated with reduced expression of IGF receptor 1 alpha. Endocr Relat Cancer. 2003;10:579–590. doi: 10.1677/erc.0.0100579. [DOI] [PubMed] [Google Scholar]
- 98.Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-1 receptor signaling in tamoxifen-resistant breast cancer: A supporting role to the epidermal growth factor receptor. Endocrinology. 2005;146:4609–4618. doi: 10.1210/en.2005-0247. [DOI] [PubMed] [Google Scholar]
- 99.Santen RJ, Song RX, Zhang Z, Kumar R, Jeng MH, Masamura A, et al. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer. 2005;12 1:S61–S73. doi: 10.1677/erc.1.01018. [DOI] [PubMed] [Google Scholar]
- 100.Cesarone G, Garofalo C, Abrams MT, Igoucheva O, Alexeev V, Yoon K, et al. RNAi-mediated silencing of insulin receptor substrate 1 (IRS-1) enhances tamoxifen-induced cell death in MCF-7 breast cancer cells. J Cell Biochem. 2006;98:440–450. doi: 10.1002/jcb.20817. [DOI] [PubMed] [Google Scholar]
- 101.Dorssers LC, van Agthoven T, Dekker A, van Agthoven TL, Kok EM. Induction of antiestrogen resistance in human breast cancer cells by random insertional mutagenesis using defective retroviruses: Identification of BCAR-1, a common integration site. Mol Endocrinol. 1993;7:870–878. doi: 10.1210/mend.7.7.8413311. [DOI] [PubMed] [Google Scholar]
- 102.van Agthoven T, van Agthoven TL, Dekker A, van der Spek PJ, Vreede L, Dorssers LC. Identification of BCAR3 by a random search for genes involved in antiestrogen resistance of human breast cancer cells. EMBO J. 1998;17:2799–2808. doi: 10.1093/emboj/17.10.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bouton AH, Riggins RB, Bruce-Staskal PJ. Functions of the adapter protein Cas: Signal convergence and the determination of cellular responses. Oncogene. 2001;20:6448–6458. doi: 10.1038/sj.onc.1204785. [DOI] [PubMed] [Google Scholar]
- 104.Defilippi P, Di Stefano P, Cabodi S. p130cas: A versatile scaffold in signaling networks. Trends Cell Biol. 2006;16:257–263. doi: 10.1016/j.tcb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 105.Brinkman A, van der Flier S, Kok EM, Dorssers LC. BCAR1, a human homologue of the adapter protein p130Cas, and antiestrogen resistance in breast cancer cells. J Natl Cancer Inst. 2000;92:112–120. doi: 10.1093/jnci/92.2.112. [DOI] [PubMed] [Google Scholar]
- 106.Burnham MR, Bruce-Staskal PJ, Harte MT, Weidow CL, Ma A, Weed SA, et al. Regulation of c-Src activity and function by the adapter protein Cas. Mol Cell Biol. 2000;20:5865–5878. doi: 10.1128/mcb.20.16.5865-5878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Riggins RB, Quilliam LA, Bouton AH. Synergistic promotion of c-Src activation and cell migration by Cas and AND-34/BCAR3. J Biol Chem. 2003;278:28264–28273. doi: 10.1074/jbc.M303535200. [DOI] [PubMed] [Google Scholar]
- 108.Cabodi S, Moro L, Baj G, Smeriglio M, Di Stefano P, Gippone S, et al. p130cas interacts with estrogen receptor alpha and modulates non-genomic estrogen signaling in breast cancer cells. J Cell Sci. 2004;117:1603–1611. doi: 10.1242/jcs.01025. [DOI] [PubMed] [Google Scholar]
- 109.Planas-Silva MD, Hamilton KN. Targeting c-Ssrc kinase enhances tamoxifen's inhibitory effect on cell growth by modulating expression of cell cycle and survival proteins. Cancer Chemother Pharmacol. 2007 doi: 10.1007/s00280-006-0398-z. in press. [DOI] [PubMed] [Google Scholar]
- 110.Cowell LN, Graham JD, Bouton AH, Clarke CL, O'Neill GM. Tamoxifen treatment promotes phosphorylation of the adhesion molecules, p130Cas/BCAR1, Fak and Src, via an adhesion-dependent pathway. Oncogene. 2006;25:7597–7607. doi: 10.1038/sj.onc.1209747. [DOI] [PubMed] [Google Scholar]
- 111.Ottenhoff-Kalff AE, Rijksen G, van Beurden EA, Hennipman A, Michels AA, Staal GE. Characterization of protein tyrosine kinases from human breast cancer: Involvement of the c-Src oncogene product. Cancer Res. 1992;52:4773–4778. [PubMed] [Google Scholar]
- 112.Verbeek BS, Vroom TM, Adriaansen-Slot SS, Ottenhoff-Kalff AE, Geertzema JG, Hennipman A, et al. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol. 1996;180:383–388. doi: 10.1002/(SICI)1096-9896(199612)180:4<383::AID-PATH686>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 113.van der Flier S, Chan CM, Brinkman A, Smid M, Johnston SR, Dorssers LC, et al. BCAR1/p130Cas expression in untreated and acquired tamoxifen-resistant human breast carcinomas. Int J Cancer. 2000;89:465–468. doi: 10.1002/1097-0215(20000920)89:5<465::aid-ijc11>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 114.Biscardi JS, Belsches AP, Parsons SJ. Characterization of human epidermal growth factor receptor and c-Src interactions in human breast tumor cells. Mol Carcinog. 1998;21:261–272. doi: 10.1002/(sici)1098-2744(199804)21:4<261::aid-mc5>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 115.Belsches-Jablonski AP, Biscardi JS, Peavy DR, Tice DA, Romney DA, Parsons SJ. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene. 2001;20:1465–1475. doi: 10.1038/sj.onc.1204205. [DOI] [PubMed] [Google Scholar]
- 116.Gotoh T, Cai D, Tian X, Feig LA, Lerner A. p130cas regulates the activity of AND-34, a novel Ral, Rap1, and R-Ras guanine nucleotide exchange factor. J Biol Chem. 2000;275:30118–30123. doi: 10.1074/jbc.M003074200. [DOI] [PubMed] [Google Scholar]
- 117.Cai D, Felekkis KN, Near RI, O'Neill GM, van Seventer JM, Golemis EA, et al. The GDP exchange factor AND-34 is expressed in B cells, associates with HEF1, and activates Cdc42. J Immunol. 2003;170:969–978. doi: 10.4049/jimmunol.170.2.969. [DOI] [PubMed] [Google Scholar]
- 118.Felekkis KN, Narsimhan RP, Near R, Castro AF, Zheng Y, Quilliam LA, et al. AND-34 activates phosphatidylinositol 3-kinase and induces anti-estrogen resistance in a SH2 and GDP exchange factor-like domain-dependent manner. Mol Cancer Res. 2005;3:32–41. [PubMed] [Google Scholar]
- 119.Felekkis K, Quilliam LA, Lerner A. Characterization of AND-34 function and signaling. Methods Enzymol. 2005;407:55–63. doi: 10.1016/S0076-6879(05)07006-0. [DOI] [PubMed] [Google Scholar]
- 120.Yu Y, Feig LA. Involvement of R-Ras and Ral GTPases in estrogen-independent proliferation of breast cancer cells. Oncogene. 2002;21:7557–7568. doi: 10.1038/sj.onc.1205961. [DOI] [PubMed] [Google Scholar]
- 121.Toker A, Yoeli-Lerner M. AKT signaling and cancer: Surviving but not moving on. Cancer Res. 2006;66:3963–3966. doi: 10.1158/0008-5472.CAN-06-0743. [DOI] [PubMed] [Google Scholar]
- 122.Kim D, Chung J. AKT: Versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–115. doi: 10.5483/bmbrep.2002.35.1.106. [DOI] [PubMed] [Google Scholar]
- 123.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 124.Duong BN, Elliott S, Frigo DE, Melnik LI, Vanhoy L, Tomchuck S, et al. AKT regulation of estrogen receptor β transcriptional activity in breast cancer. Cancer Res. 2006;66:8373–8381. doi: 10.1158/0008-5472.CAN-05-3845. [DOI] [PubMed] [Google Scholar]
- 125.Faridi J, Wang L, Endemann G, Roth RA. Expression of constitutively active AKT-3 in MCF-7 breast cancer cells reverses the estrogen and tamoxifen responsivity of these cells in vivo. Clin Cancer Res. 2003;9:2933–2939. [PubMed] [Google Scholar]
- 126.DeGraffenried LA, Friedrichs WE, Fulcher L, Fernandes G, Silva JM, Peralba JM, et al. Eicosapentaenoic acid restores tamoxifen sensitivity in breast cancer cells with high AKT activity. Ann Oncol. 2003;14:1051–1056. doi: 10.1093/annonc/mdg291. [DOI] [PubMed] [Google Scholar]
- 127.Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible AKT activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–717. [PubMed] [Google Scholar]
- 128.Jordan NJ, Gee JM, Barrow D, Wakeling AE, Nicholson RI. Increased constitutive activity of PKB/AKT in tamoxifen resistant breast cancer MCF-7 cells. Breast Cancer Res Treat. 2004;87:167–180. doi: 10.1023/B:BREA.0000041623.21338.47. [DOI] [PubMed] [Google Scholar]
- 129.Frogne T, Jepsen JS, Larsen SS, Fog CK, Brockdorff BL, Lykkesfeldt AE. Antiestrogen-resistant human breast cancer cells require activated protein kinase B/AKT for growth. Endocr Relat Cancer. 2005;12:599–614. doi: 10.1677/erc.1.00946. [DOI] [PubMed] [Google Scholar]
- 130.Brodie A, Sabnis G, Jelovac D. Aromatase and breast cancer. J Steroid Biochem Mol Biol. 2006;102:97–102. doi: 10.1016/j.jsbmb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 132.DeGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant AKT activity. Clin Cancer Res. 2004;10:8059–8067. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 133.Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O'Reilly T, Evans DB, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11:5319–5328. doi: 10.1158/1078-0432.CCR-04-2402. [DOI] [PubMed] [Google Scholar]
- 134.Carraway H, Hidalgo M. New targets for therapy in breast cancer: Mammalian target of rapamycin (mTOR) antagonists. Breast Cancer Res. 2004;6:219–224. doi: 10.1186/bcr927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Johnston SR. Targeting downstream effectors of epidermal growth factor receptor/HER2 in breast cancer with either farnesyltransferase inhibitors or mTOR antagonists. Int J Gynecol Cancer. 2006;16 2:543–548. doi: 10.1111/j.1525-1438.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- 136.Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- 137.Murphy LC, Weitsman GE, Skliris GP, Teh EM, Li L, Peng B, et al. Potential role of estrogen receptor alpha (ER alpha) phosphorylated at serine(118) in human breast cancer in vivo. J Steroid Biochem Mol Biol. 2006;102:139–146. doi: 10.1016/j.jsbmb.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 138.Arnold SF, Obourn JD, Jaffe H, Notides AC. Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by Src family tyrosine kinases in vitro. Mol Endocrinol. 1995;9:24–33. doi: 10.1210/mend.9.1.7539106. [DOI] [PubMed] [Google Scholar]
- 139.Yudt MR, Vorojeikina D, Zhong L, Skafar DF, Sasson S, Gasiewicz TA, et al. Function of estrogen receptor tyrosine 537 in hormone binding, DNA binding, and transactivation. Biochemistry. 1999;38:14146–14156. doi: 10.1021/bi9911132. [DOI] [PubMed] [Google Scholar]
- 140.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–1398. doi: 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 141.Tremblay GB, Tremblay A, Labrie F, Giguere V. Ligand-independent activation of the estrogen receptors alpha and beta by mutations of a conserved tyrosine can be abolished by antiestrogens. Cancer Res. 1998;58:877–881. [PubMed] [Google Scholar]
- 142.Rogatsky I, Trowbridge JM, Garabedian MJ. Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J Biol Chem. 1999;274:22296–22302. doi: 10.1074/jbc.274.32.22296. [DOI] [PubMed] [Google Scholar]
- 143.Michalides R, van Tinteren H, Balkenende A, Vermorken JB, Benraadt J, Huldij J, et al. Cyclin A is a prognostic indicator in early stage breast cancer with and without tamoxifen treatment. Br J Cancer. 2002;86:402–408. doi: 10.1038/sj.bjc.6600072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Arnold SF, Obourn JD, Jaffe H, Notides AC. Serine 167 is the major estradiol-induced phosphorylation site on the human estrogen receptor. Mol Endocrinol. 1994;8:1208–1214. doi: 10.1210/mend.8.9.7838153. [DOI] [PubMed] [Google Scholar]
- 145.Glaros S, Atanaskova N, Zhao C, Skafar DF, Reddy KB. Activation function-1 domain of estrogen receptor regulates the agonistic and antagonistic actions of tamoxifen. Mol Endocrinol. 2006;20:996–1008. doi: 10.1210/me.2005-0285. [DOI] [PubMed] [Google Scholar]
- 146.Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA. Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol Endocrinol. 2006;20:3120–3132. doi: 10.1210/me.2006-0068. [DOI] [PubMed] [Google Scholar]
- 147.Chen D, Pace PE, Coombes RC, Ali S. Phosphorylation of human estrogen receptor alpha by protein kinase A regulates dimerization. Mol Cell Biol. 1999;19:1002–1015. doi: 10.1128/mcb.19.2.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang RA, Mazumdar A, Vadlamudi RK, Kumar R. p21-activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and promotes hyperplasia in mammary epithelium. EMBO J. 2002;21:5437–5447. doi: 10.1093/emboj/cdf543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Balasenthil S, Barnes CJ, Rayala SK, Kumar R. Estrogen receptor activation at serine 305 is sufficient to upregulate cyclin D1 in breast cancer cells. FEBS Lett. 2004;567:243–247. doi: 10.1016/j.febslet.2004.04.071. [DOI] [PubMed] [Google Scholar]
- 150.Rayala SK, Molli PR, Kumar R. Nuclear p21-activated kinase 1 in breast cancer packs off tamoxifen sensitivity. Cancer Res. 2006;66:5985–5988. doi: 10.1158/0008-5472.CAN-06-0978. [DOI] [PubMed] [Google Scholar]
- 151.Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, et al. p21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006;66:1694–1701. doi: 10.1158/0008-5472.CAN-05-2922. [DOI] [PubMed] [Google Scholar]
- 152.Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 153.Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273:13317–13323. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- 154.Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, et al. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell. 2000;6:127–137. [PubMed] [Google Scholar]
- 155.Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. 2002;21:4921–4931. doi: 10.1038/sj.onc.1205420. [DOI] [PubMed] [Google Scholar]
- 156.Medunjanin S, Hermani A, De Servi B, Grisouard J, Rincke G, Mayer D. Glycogen synthase kinase-3 interacts with and phosphorylates estrogen receptor alpha and is involved in the regulation of receptor activity. J Biol Chem. 2005;280:33006–33014. doi: 10.1074/jbc.M506758200. [DOI] [PubMed] [Google Scholar]
- 157.Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, et al. Bidirectional cross talk between ER alpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. 2006;96:131–146. doi: 10.1007/s10549-005-9070-2. [DOI] [PubMed] [Google Scholar]
- 158.Weitsman GE, Li L, Skliris GP, Davie JR, Ung K, Niu Y, et al. Estrogen receptor-alpha phosphorylated at ser118 is present at the promoters of estrogen-regulated genes and is not altered due to HER-2 overexpression. Cancer Res. 2006;66:10162–10170. doi: 10.1158/0008-5472.CAN-05-4111. [DOI] [PubMed] [Google Scholar]
- 159.Park KJ, Krishnan V, O'Malley BW, Yamamoto Y, Gaynor RB. Formation of an IKK alpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 160.Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem. 2006;97:261–274. doi: 10.1002/jcb.20690. [DOI] [PubMed] [Google Scholar]
- 161.Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer. 2005;12 1:S47–S59. doi: 10.1677/erc.1.00993. [DOI] [PubMed] [Google Scholar]
- 162.Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 163.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 164.Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002;62:6916–6923. [PubMed] [Google Scholar]
- 165.Kilker RL, Hartl MW, Rutherford TM, Planas-Silva MD. Cyclin D1 expression is dependent on estrogen receptor function in tamoxifen-resistant breast cancer cells. J Steroid Biochem Mol Biol. 2004;92:63–71. doi: 10.1016/j.jsbmb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 166.O'Regan RM, Osipo C, Ariazi E, Lee ES, Meeke K, Morris C, et al. Development and therapeutic options for the treatment of raloxifene-stimulated breast cancer in athymic mice. Clin Cancer Res. 2006;12:2255–2263. doi: 10.1158/1078-0432.CCR-05-2584. [DOI] [PubMed] [Google Scholar]
- 167.Kilker RL, Planas-Silva MD. Cyclin D1 is necessary for tamoxifen-induced cell cycle progression in human breast cancer cells. Cancer Res. 2006;66:11478–11484. doi: 10.1158/0008-5472.CAN-06-1755. [DOI] [PubMed] [Google Scholar]
- 168.Sutherland RL, Musgrove EA. Cyclins and breast cancer. J Mammary Gland Biol Neoplasia. 2004;9:95–104. doi: 10.1023/B:JOMG.0000023591.45568.77. [DOI] [PubMed] [Google Scholar]
- 169.Foster JS, Henley DC, Ahamed S, Wimalasena J. Estrogens and cell-cycle regulation in breast cancer. Trends Endocrinol Metab. 2001;12:320–327. doi: 10.1016/s1043-2760(01)00436-2. [DOI] [PubMed] [Google Scholar]
- 170.Akli S, Keyomarsi K. Low-molecular-weight cyclin E: The missing link between biology and clinical outcome. Breast Cancer Res. 2004;6:188–191. doi: 10.1186/bcr905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Akli S, Zheng PJ, Multani AS, Wingate HF, Pathak S, Zhang N, et al. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res. 2004;64:3198–3208. doi: 10.1158/0008-5472.can-03-3672. [DOI] [PubMed] [Google Scholar]
- 172.Dhillon NK, Mudryj M. Ectopic expression of cyclin E in estrogen responsive cells abrogates antiestrogen mediated growth arrest. Oncogene. 2002;21:4626–4634. doi: 10.1038/sj.onc.1205576. [DOI] [PubMed] [Google Scholar]
- 173.Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci USA. 2000;97:9042–9046. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Carroll JS, Lynch DK, Swarbrick A, Renoir JM, Sarcevic B, Daly RJ, et al. p27(Kip1) induces quiescence and growth factor insensitivity in tamoxifen-treated breast cancer cells. Cancer Res. 2003;63:4322–4326. [PubMed] [Google Scholar]
- 175.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (AKT) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 176.Mukherjee S, Conrad SE. c-Myc suppresses p21WAF1/CIP1 expression during estrogen signaling and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2005;280:17617–17625. doi: 10.1074/jbc.M502278200. [DOI] [PubMed] [Google Scholar]
- 177.Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276:40888–40895. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- 178.Dowsett M, Houghton J, Iden C, Salter J, Farndon J, A'Hern R, et al. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according oestrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann Oncol. 2006;17:818–826. doi: 10.1093/annonc/mdl016. [DOI] [PubMed] [Google Scholar]
- 179.Hiscox S, Morgan L, Barrow D, Dutkowskil C, Wakeling A, Nicholson RI. Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: Inhibition by gefitinib (‘Iressa’, ZD1839) Clin Exp Metastasis. 2004;21:201–212. doi: 10.1023/b:clin.0000037697.76011.1d. [DOI] [PubMed] [Google Scholar]
- 180.Kang Y, Massague J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 181.Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat. 2006;97:263–274. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- 182.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schraml P, Schwerdtfeger G, Burkhalter F, Raggi A, Schmidt D, Ruffalo T, et al. Combined array comparative genomic hybridization and tissue microarray analysis suggest Pak1 at 11q13.5-q14 as a critical oncogene target in ovarian carcinoma. Am J Pathol. 2003;163:985–992. doi: 10.1016/S0002-9440(10)63458-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Robanus-Maandag EC, Bosch CA, Kristel PM, Hart AA, Faneyte IF, Nederlof PM, et al. Association of c-Myc amplification with progression from the in situ to the invasive stage in c-Myc-amplified breast carcinomas. J Pathol. 2003;201:75–82. doi: 10.1002/path.1385. [DOI] [PubMed] [Google Scholar]
- 185.Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, et al. Activation of PI3K/AKT signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13:137–144. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- 186.Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. 2005;18:250–259. doi: 10.1038/modpathol.3800296. [DOI] [PubMed] [Google Scholar]
- 187.Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, Nicholson RI, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. 1999;5:2069–2076. [PubMed] [Google Scholar]
- 188.Stendahl M, Kronblad A, Ryden L, Emdin S, Bengtsson NO, Landberg G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br J Cancer. 2004;90:1942–1948. doi: 10.1038/sj.bjc.6601831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Span PN, Tjan-Heijnen VC, Manders P, Beex LV, Sweep CG. Cyclin-E is a strong predictor of endocrine therapy failure in human breast cancer. Oncogene. 2003;22:4898–4904. doi: 10.1038/sj.onc.1206818. [DOI] [PubMed] [Google Scholar]
- 190.Desmedt C, Ouriaghli FE, Durbecq V, Soree A, Colozza MA, Azambuja E, et al. Impact of cyclins E, neutrophil elastase and proteinase 3 expression levels on clinical outcome in primary breast cancer patients. Int J Cancer. 2006;119:2539–2545. doi: 10.1002/ijc.22149. [DOI] [PubMed] [Google Scholar]
- 191.Pohl G, Rudas M, Dietze O, Lax S, Markis E, Pirker R, et al. High p27Kip1 expression predicts superior relapse-free and overall survival for premenopausal women with early-stage breast cancer receiving adjuvant treatment with tamoxifen plus goserelin. J Clin Oncol. 2003;21:3594–3600. doi: 10.1200/JCO.2003.02.021. [DOI] [PubMed] [Google Scholar]
- 192.Perez-Tenorio G, Berglund F, Esguerra Merca A, Nordenskjold B, Rutqvist LE, Skoog L, et al. Cytoplasmic p21WAF1/CIP1 correlates with AKT activation and poor response to tamoxifen in breast cancer. Int J Oncol. 2006;28:1031–1042. [PubMed] [Google Scholar]
- 193.Murphy LC, Niu Y, Snell L, Watson P. Phospho-serine-118 estrogen receptor-alpha expression is associated with better disease outcome in women treated with tamoxifen. Clin Cancer Res. 2004;10:5902–5906. doi: 10.1158/1078-0432.CCR-04-0191. [DOI] [PubMed] [Google Scholar]
- 194.Bergqvist J, Elmberger G, Ohd J, Linderholm B, Bjohle J, Hellborg H, et al. Activated ERK1/2 and phosphorylated oestrogen receptor alpha are associated with improved breast cancer survival in women treated with tamoxifen. Eur J Cancer. 2006;42:1104–1112. doi: 10.1016/j.ejca.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 195.Yamashita H, Nishio M, Kobayashi S, Ando Y, Sugiura H, Zhang Z, et al. Phosphorylation of estrogen receptor alpha serine 167 is predictive of response to endocrine therapy and increases postrelapse survival in metastatic breast cancer. Breast Cancer Res. 2005;7:R753–764. doi: 10.1186/bcr1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bardou VJ, Arpino G, Elledge RM, Osborne CK, Clark GM. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol. 2003;21:1973–1979. doi: 10.1200/JCO.2003.09.099. [DOI] [PubMed] [Google Scholar]
- 197.Ferno M, Stal O, Baldetorp B, Hatschek T, Kallstrom AC, Malmstrom P, et al. Results of two or five years of adjuvant tamoxifen correlated to steroid receptor and S-phase levels. South Sweden Breast Cancer Group, and South-East Sweden Breast Cancer Group. Breast Cancer Res Treat. 2000;59:69–76. doi: 10.1023/a:1006332423620. [DOI] [PubMed] [Google Scholar]
- 198.Ellis MJ, Coop A, Singh B, Tao Y, Llombart-Cussac A, Janicke F, et al. Letrozole inhibits tumor proliferation more effectively than tamoxifen independent of HER1/2 expression status. Cancer Res. 2003;63:6523–6531. [PubMed] [Google Scholar]
- 199.Jansen MP, Foekens JA, van Staveren IL, Dirkzwager-Kiel MM, Ritstier K, Look MP, et al. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J Clin Oncol. 2005;23:732–740. doi: 10.1200/JCO.2005.05.145. [DOI] [PubMed] [Google Scholar]
- 200.Goetz MP, Suman VJ, Ingle JN, Nibbe AM, Visscher DW, Reynolds CA, et al. A two-gene expression ratio of homeobox 13 and interleukin-17B receptor for prediction of recurrence and survival in women receiving adjuvant tamoxifen. Clin Cancer Res. 2006;12:2080–2087. doi: 10.1158/1078-0432.CCR-05-1263. [DOI] [PubMed] [Google Scholar]
- 201.Reid JF, Lusa L, De Cecco L, Coradini D, Veneroni S, Daidone MG, et al. Limits of predictive models using microarray data for breast cancer clinical treatment outcome. J Natl Cancer Inst. 2005;97:927–930. doi: 10.1093/jnci/dji153. [DOI] [PubMed] [Google Scholar]
- 202.Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004;64:1522–1533. doi: 10.1158/0008-5472.can-03-3326. [DOI] [PubMed] [Google Scholar]
- 203.Hur J, Chesnes J, Coser KR, Lee RS, Geck P, Isselbacher KJ, et al. The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc Natl Acad Sci USA. 2004;101:2351–2356. doi: 10.1073/pnas.0307337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Itoh T, Karlsberg K, Kijima I, Yuan YC, Smith D, Ye J, et al. Letrozole-, anastrozole- and tamoxifen-responsive genes in MCF-7aro cells: A microarray approach. Mol Cancer Res. 2005;3:203–218. doi: 10.1158/1541-7786.MCR-04-0122. [DOI] [PubMed] [Google Scholar]
- 205.Becker M, Sommer A, Kratzschmar JR, Seidel H, Pohlenz HD, Fichtner I. Distinct gene expression patterns in a tamoxifen-sensitive human mammary carcinoma xenograft and its tamoxifen-resistant subline MaCa 3366/TAM. Mol Cancer Ther. 2005;4:151–168. [PubMed] [Google Scholar]
- 206.Bowie ML, Dietze EC, Delrow J, Bean GR, Troch MM, Marjoram RJ, et al. Interferon-regulatory factor-1 is critical for tamoxifen-mediated apoptosis in human mammary epithelial cells. Oncogene. 2004;23:8743–8755. doi: 10.1038/sj.onc.1208120. [DOI] [PubMed] [Google Scholar]
- 207.Xu K, Geczy CL. IFN-gamma and TNF regulate macrophage expression of the chemotactic S100 protein S100A8. J Immunol. 2000;164:4916–4923. doi: 10.4049/jimmunol.164.9.4916. [DOI] [PubMed] [Google Scholar]
- 208.Jeffrey SS, Lonning PE, Hillner BE. Genomics-based prognosis and therapeutic prediction in breast cancer. J Natl Compr Canc Netw. 2005;3:291–300. doi: 10.6004/jnccn.2005.0016. [DOI] [PubMed] [Google Scholar]
- 209.Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, et al. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 210.Paik S. Methods for gene expression profiling in clinical trials of adjuvant breast cancer therapy. Clin Cancer Res. 2006;12:1019S–1023S. doi: 10.1158/1078-0432.CCR-05-2296. [DOI] [PubMed] [Google Scholar]
- 211.Dowsett M, Nicholson RI, Pietras RJ. Biological characteristics of the pure antiestrogen fulvestrant: Overcoming endocrine resistance. Breast Cancer Res Treat. 2005;93 1:S11–S18. doi: 10.1007/s10549-005-9037-3. [DOI] [PubMed] [Google Scholar]
- 212.Johnston SR. Clinical efforts to combine endocrine agents with targeted therapies against epidermal growth factor receptor/human epidermal growth factor receptor 2 and mammalian target of rapamycin in breast cancer. Clin Cancer Res. 2006;12:1061S–1068S. doi: 10.1158/1078-0432.CCR-05-2125. [DOI] [PubMed] [Google Scholar]
- 213.Head J, Johnston SR. New targets for therapy in breast cancer: Farnesyltransferase inhibitors. Breast Cancer Res. 2004;6:262–268. doi: 10.1186/bcr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Leary A, Dowsett M. Combination therapy with aromatase inhibitors: The next era of breast cancer treatment? Br J Cancer. 2006;95:661–666. doi: 10.1038/sj.bjc.6603316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–5322. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 216.Perey L, Paridaens R, Hawle H, Zaman K, Nole F, Wildiers H, et al. Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: Final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00) Ann Oncol. 2007;18:64–69. doi: 10.1093/annonc/mdl341. [DOI] [PubMed] [Google Scholar]