

Abstract
Major obstacles in the development of new therapeutic anticancer drugs are the low bioavailability of hydrophilic substances as well as the nonspecific toxicity towards healthy tissues. As such cell-targeting oligopeptides have emerged as attractive drug delivery vehicles for a variety of different types of cargo. The recently identified peptide Pep42 binds to the glucose-regulated protein 78 (GRP78), which is overexpressed on the cell surface of human cancer cells and internalizes into these cells. Herein, we demonstrate how Pep42 can be utilized as a carrier for different types of cytotoxic drugs to specifically target human cancer cell lines in vitro in a strictly GRP78-dependent manner. Furthermore, the mechanism of internalization of Pep42 was elucidated and found to involve clathrin-mediated endocytosis. Pep42 subsequently co-localizes within the lysosomal compartment. Importantly, we also provide evidence that Pep42 conjugated quantum dots have the ability to selectively enrich in tumor tissue in a xenograft mouse model. Our results suggest that the highly specific GRP78-Pep42 interaction can be utilized for the generation of Pep42-drug conjugates as a powerful anticancer drug delivery system that could substantially enhance the efficacy of chemotherapy by increasing the drug-tumor specificity; thus, minimizing the adverse side effects associated with conventional cancer therapeutics.
Keywords: Drug delivery, tumor targeting, GRP78, heat shock protein, cyclic peptide
INTRODUCTION
The lack of specificity seen with anticancer drugs in chemotherapy towards tumor tissues can result in adverse side effects, thereby limiting their clinical applications.1 To overcome this obstacle, methods of delivering cytotoxic drugs specifically into the tumor tissue have to be investigated.2 A promising therapeutic approach is the conjugation of a cytotoxic drug to a ligand that recognizes a well characterized cell surface marker, which is exclusively or at least preferentially expressed on tumor cells. Oligopeptides are candidates for such drug delivery purposes and have been employed successfully as targeting ligands.3–6 They can be chemically synthesized in large quantities, have low innate toxicity, are readily amenable to derivatization and are able to penetrate solid tumors.7 A number of studies have shown that peptides are able to deliver a wide range of bioactive molecules such as proteins, oligonucleotides, siRNAs, nanoparticles and other large particles such as liposomes to a variety of cell lines both in vitro and in vivo.3–5, 8, 9
The panning of phage display peptide libraries against cells, often referred to as biopanning, has proven successful for the isolation of peptidic ligands.10–12 However, these identified peptides typically did not possess any intrinsic cell type specificity and only recently has the identification of cell type-specific peptides been reported.13, 14 These peptides display certain characteristics of cell-penetrating peptides (CPPs) but, in contrast to all other previously described CPPs, demonstrate a restricted cell recognition pattern. Our laboratory has developed phage display biopanning protocols to identify peptides of constrained origin that internalize into tumor cells.15 Recently, we reported the selection and characterization of Pep42, a cyclic 13-mer oligopeptide,
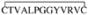 , that specifically binds to glucose-regulated protein 78 (GRP78), internalizes into highly metastatic melanoma cells, and eventually remains inside the cell.16 In cancer cells, the overexpression of GRP78, an intracellular chaperone and member of the heat shock protein 70 (HSP70) family, provides a protective cellular response against stress conditions. However, this GRP78 overexpression represents a potentially attractive target as it results in the presence of GRP78 molecules on the cell surface of these cancer cells.17–19 Indeed, the presence of GRP78 on the cell surface has been demonstrated for a number of tumors, e.g. malignant human breast and lung cancers, thus making GRP78 a pan-neoplastic antigen.20–23
, that specifically binds to glucose-regulated protein 78 (GRP78), internalizes into highly metastatic melanoma cells, and eventually remains inside the cell.16 In cancer cells, the overexpression of GRP78, an intracellular chaperone and member of the heat shock protein 70 (HSP70) family, provides a protective cellular response against stress conditions. However, this GRP78 overexpression represents a potentially attractive target as it results in the presence of GRP78 molecules on the cell surface of these cancer cells.17–19 Indeed, the presence of GRP78 on the cell surface has been demonstrated for a number of tumors, e.g. malignant human breast and lung cancers, thus making GRP78 a pan-neoplastic antigen.20–23
Herein, we provide mechanistic investigations into a cancer cell-specific drug delivery approach exploiting tumor cell-specific expression of GRP78 using our recently identified GRP78 ligand, Pep42. In total, 5 cancer cell lines were evaluated for their surface expression level of GRP78. The specificity of the Pep42-GRP78 binding interactions were also examined using GRP78 siRNA experiments, which demonstrated that GRP78 is the singular cellular receptor for Pep42. In order to increase the efficacy of the Pep42 drug conjugates, the mechanism of internalization as well as the intracellular fate of Pep42 was investigated using live cell confocal fluorescence microscopy. These experiments established that Pep42 after binding to GRP78 is taken up via receptor-mediated endocytosis and ultimately locates into the lysosomal compartment of the cell. To evaluate the potential of Pep42 for the delivery of different cargo types into cancer cells, two cytotoxic Pep42-drug conjugates were generated, one based on the chemical fusion of Pep42 to the apoptosis-inducing oligopeptide D-(KLAKLAK)2, while the other consisted of a photosensitizer, hematoporphyrin, chemically conjugated to Pep42. Excitingly, both drug conjugates were able to exert their cytotoxic effects on a number of cancer cells in a GRP78-dependent manner, thus again underscoring the specificity of the Pep42-GRP78 interaction. Importantly, the ability of Pep42-nanoparticle conjugates to home into tumor tissue was demonstrated in an in vivo xenograft mouse model, which further confirmed the potential of GRP78 ligands as new cancer drug delivery vehicles.
Experimental Methods
Cell Lines and Culture Conditions
Human melanoma cells Me6652/4 and SJSA-1 osteosarcoma cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Invitrogen). HepG2 hepatoma cells and a normal human lung fibroblast cell line, CCD-11Lu (both from ATCC, Manassas, VA) were grown in Eagle’s Minimum Essential Medium (EMEM, Invitrogen) supplemented with 10% FBS, 1.0 mM sodium pyruvate, 2 mM L-glutamine, 1.5g/L sodium bicarbonate and 0.1 mM non-essential amino acids. A549 lung adenocarcinoma cells (ATCC) were maintained in Ham’s F12K medium (ATCC) supplemented with 10% FBS, 2 mM L-glutamine and 1.5 g/L sodium bicarbonate. Normal human dermal fibroblasts (HDFa) from adult skin (Cascade Biologics, Portland, OR) were cultured in Medium 106 supplemented with low serum growth supplement (LSGS). All cell cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 in air.
Synthesis of Peptides and Peptide Conjugates
All peptides were prepared by stepwise solid phase protocols for Fmoc chemistry using DIC/HOBt as previously described.16, 24 Pep42-(Ac-HN-CTVALPGGYVRKC-CONH2)-fluorescein isothiocyanate (Pep42-FITC) and Pep42-Bpa-Biotin syntheses have been described; while Pep42-D-(KLAKLAK)2 and D-(KLAKLAK)2 were synthesized in a similar manner.16 A scrambled peptide with the sequence Ac-HN-CGVTPVGARYLKC-CONH2 and a linear Pep42 with the sequence Ac-HN-MTVALPGGYVRKM-CONH2were also synthesized. Unrelated sequences (H2N-ACDEYIRVWYARHC AhxK (FITC)-CONH2 or H2N-ACGWDGVDYMSDWVC AhxK (FITC) -CONH2) from the same pIX cyclic peptide library were used as negative controls. Pep42-dPEG6- hematoporphyrin were prepared by conjugating hematoporphyrin (Sigma, St. Louis, MO) with PEG linkers (Fmoc-Amido-dPEG6-Acid from Quanta BioDesign, Ltd., Powell, OH) on the free Lysine side chain of Pep42 using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) and DMSO for 24 h at RT. RP-HPLC was performed employing binary gradients of solvents A and B, where A is 0.1% TFA in water and B is 0.09% TFA in acetonitrile. Analytical RP-HPLC was performed using a Vydac 214TP5415 column at a flow rate of 1 mL/min, with detection at 214 nm during a linear gradient of 20–80 %B over 30 min. Preparative RP-HPLC was performed using a Vydac 214TP101522 column at a flow rate of 10 mL/min, with detection at 220 nm during a linear gradient of 25–55%B over 30 min. In all cases, fractions were analyzed off-line using an ABI/Sciex 150EX single quadruple mass spectrometer and judged for purity after a consistent summing of 20 scans in multichannel analysis (MCA) mode, using the [M + 2]2+ charged species. For preparative purification purposes, fractions that contained no single [M + 2]2+ charged species which accounted for more than 10% of the total ion intensity were designated “pure” and pooled; the homogeneity of this pool was verified by analytical RP-HPLC and was >96%. Concentrations of peptides or peptide conjugates were determined using a UV-Vis absorption spectrometer. Pep42-FITC: 490 nm (ε = 68,000); Pep42 or Pep42-Bpa-Biotin: 280 nm (ε = 1280); Pep42-D-(KLAKLAK)2: 280 nm (ε = 1400); Pep42-dPEG6- hematoporphyrin or hematoporphyrin: 390 nm (ε = 79,400).
Cell-Free GRP78 Binding Assay
GRP78 (Stressgen, San Diego, CA), HSP70 (Stressgen) and BSA (Bio-Rad, Richmond, CA) (10 μg/mL, 25 μL/well) were immobilized on 96-well microtiter wells overnight at 4 °C. After blocking with 1% skim milk, 109 colony forming unit (cfu) of peptide-phage (Pep42-phage or an unrelated peptide-phage as negative control) in 25 μL of PBS/ 1% skim milk was added into each well for 1 h at room temperature (RT) with slow agitation. For detection of peptide-phage, wells were washed with PBS and bound phage clones were recovered by infection with host bacterial (log phase ER2537, New England Biolabs, Ipswich, MA). Aliquots of the bacterial culture were seeded on a SOBAG plate and incubated overnight at 37 °C.16 The Phage cfu was determined in triplicates. For Pep42 binding assays, increasing concentrations of Pep42 or a scrambled Pep42 were added to wells coated with GRP78 and incubated for 30 min at 37 °C, followed by an incubation step with 109 cfu of Pep42-phage for 1h at RT.
Identification of GRP78 on the Cell Surface of Human Cancer Cells
Expression of surface GRP78 on cells was assessed by flow cytometry. Cells were dissociated with Versene 1:5,000 (Invitrogen) and resuspended in Opti-MEM I Reduced-Serum Medium (Invitrogen). Approximately 1x106 cells were harvested, washed and incubated with a saturating amount (10 μg/mL) of mouse anti-GRP78 or a monoclonal anti-β-actin antibody (Sigma) as a negative control in staining buffer (PBS pH 7.4), 4% FBS and 0.1% NaN3) for 1 h at 4 °C. Cells were washed and stained with FITC conjugated anti-mouse IgG secondary antibody for 45 min at 4° C. Additional washing (2×), with cold FCM washing buffer (Santa Cruz Biotechnology, Santa Cruz, CA) were followed by propidium iodide (Invitrogen) as a counterstain. Fluorescence intensity of the samples was measured on a FACSort (BD Biosciences, San Diego, CA) using Cellquest and FlowJo software and at the least, 5x103 cells were analyzed for each sample.
Cell Surface Binding Analysis using Flow Cytometry
Cultured cells were dissociated and washed in culture medium and aliquoted (~1x105–106 cells/tube) into culture tubes. Opti-MEM I reduced Serum Medium was renewed 1 h at 4 °C prior to the addition of the peptide. To the cells was added either 50 nM FITC-conjugated Pep42 or a FITC-conjugated unrelated peptide as negative control and incubated for 25 min at 4 °C. The cells were washed (2x) with a cold FCM washing buffer; this was followed by a propidium iodide counterstain and FACS analysis. A goat polyclonal anti-GRP78 antibody (Santa Cruz Biotechnology) was used to evaluate the competitive inhibition of Pep42-FITC or control peptide-FITC binding.
Western Blot Analysis
Cells were lysed in RIPA buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 5 mM EDTA, 1% Triton X-100), supplemented with 1:100 protease inhibitor cocktail (Sigma) and 1:100 phosphatase inhibitor cocktail set II (Calbiochem, San Diego, CA). Cell lysates were centrifuged at 10,000g for 20 min. The protein concentration in the supernatant was determined by Bradford assay (Bio-Rad). Aliquots containing 20–50 μg of protein were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by electrotransfer to a nitrocellulose membrane. Immunodetection was carried out using Streptavidin-horseradish peroxidase (HRP) (Amersham Biosciences, Piscataway, NJ), Hsp70/Hsc70 antibody (Stressgen) or Bip/GRP78 antibody (BD Biosciences).
Transfection of Cells with Small Interfering RNA (siRNA) Oligomers
The siRNA used against human GRP78 (Santa Cruz Biotechnology) was a pool of 3 target-specific 20–25 nt siRNAs that were designed to knock down gene expression. Cells were thus transfected with GRP78 siRNA or control siRNA (Santa Cruz) using Lipofectamine 2000 reagent (Invitrogen), and the transfection protocol was provided with the kit. After 48 hours, Western blot analysis and cell viability assays were carried out.
Fluorescence Microscopy
Quantum Dots (QDots 525, Quantum Dot Corp., Hayward, CA) coupled to Pep 42 were accomplished as previously detailed.16 For treatment with fluorescent proteins, A549 cells were grown on a 35-mm glass bottom dish to approximately 70% confluence. After a 30 min incubation period at 37 °C in serum-free Ham’s F12K medium, 6 μM of Pep42-Quantum Dots (QDots) were added 1 h before the addition of the fluorescent proteins. Final concentrations of fluorescent materials were 10 μg/mL Alexa Fluor (AF)-594-conjugated transferrin, 10 μg/mL AF-594 labeled cholera toxin subunit B (recombinant), 25 nM Mitotracker Red, 20 nM ERtracker Red and 12 nM Lysotracker Red. All reagents except the QDots were obtained from Invitrogen. At different time points as indicated in each figure, media were changed with PBS and co-localization experiments in live cells were imaged using laser scanning confocal microscope (MRC1024, Bio-Rad, Hercules, CA). Weighted Co-localized coefficients were estimated using Zeiss LSM Image examiner software. For inhibitor-treated experiments, Chlorpromazine, Nystatin and Methyl-β-cyclodextrin (MCD) were purchased from Sigma-Aldrich. A549 cells were pretreated with 25 μM Nystatin, 30 μM Chlorpromazine, or 5 mM Methyl-β-cyclodextrin (MCD) for 10 min, and then Pep42-QDots, AF-594-transferrin or AF-594- cholera toxin subunit B were added, respectively.
Cell Viability Assay
Cell suspensions of 1x104 cells per well were seeded in 96-well plates for 18 h in medium containing 10% FBS. Before carrying out the assay culture conditions were changed to low-serum medium (1% FBS) after which the cells were incubated with increasing concentrations of the selected peptides including Pep42-D-(KLAKLAK)2 or (Pep42 and free D-(KLAKLAK)2) in 100 μL for the indicated time at 37 °C. Cell viability was measured with a cell proliferation reagent (Promega, St. Luis Obispo, CA). For the evaluation of the porphyrin phototoxicity, cells were prepared as described (vide supra) and treated with varying concentrations of Pep42- hematoporphyrin or free hematoporphyrins at 37 °C, respectively. After the indicated time as designated in each figure, the medium was replaced with 50 mM HEPES (pH 7.4) and the cells were exposed to visible light for 1 h at RT 24. Upon completing of photodynamic treatment the cells were incubated for 24 h at 37 °C, and the phototoxicity was detected as described (vide supra).
Apoptosis Assay using Flow Cytometry
Apoptotic cells were stained with Annexin V-FITC and propidium iodide (Invitrogen) according to the manufacturer’s instructions and analyzed by flow cytometry. Cell debris was excluded from analysis by appropriate forward light scatter threshold settings. Cells that stained with Annexin V-FITC, but not propidium iodide, were regarded as early apoptotic cells.25
Establishment of Tumor-Bearing Mice
The ability of peptide-coated quantum dots to home to their targets in vivo was tested in xenograft mice. To establish tumor xenografts, 106 exponentially growing Melanoma Me6652/4 tumor cells were injected intradermally in the flank of CB17Scid male mice (Taconic Farms, Germantown, NY). The mice were used for in vivo targeting experiments 4 weeks after the tumor cell inoculation. Peptide-coated quantum dots preparations (100–200 μg in 0.1–0.2 mL PBS) were injected into the lateral tail vein of a mouse and allowed to circulate for 120 min. While still under anesthesia, the mouse was perfused with 4% paraformaldehyde through the heart. The organs were harvested and frozen in Tissue Tek OCT embedding medium (Sakura Finetek, Torrance, CA) before sectioning. The sections were mounted with Vectashield mounting medium with or without DAPI (Vector Laboratories, Burlingame, CA) to visualize cell nuclei before examination under an inverted fluorescent or confocal microscope. The anti-CD31 antibody (10 μg/ml; BD Biosciences) applied on the tissue sections was detected with Alexa-594–conjugated secondary antibodies.
Results
GRP78 is the exclusive cell surface receptor for Pep42
Applying phage display technology our laboratory has identified the cyclic peptide Pep42,
that specifically recognizes GRP78 present on the cell surface of melanoma Me6652/4 cells, and is subsequently internalized.16 For tumor site-specific therapy it is of paramount importance that the targeted molecular feature is indeed unique, or primarily restricted to cancer cells and absent on normal cells. It had been previously shown that a number of human primary and cancer cell lines overexpress GRP78, resulting in the cell membrane localization of this protein; thus, we reasoned that GRP78 might represent an attractive target for tumor cell-specific cytotoxic therapy.17, 26
In order to study the mechanism of internalization and the in vivo localization of Pep42, we have synthesized the peptide by replacing the valine at position 12 with a lysine residue to facilitate the coupling of conjugates 16. We note that since no functional differences between this lysine-bearing derivative and the original Pep42 sequence were observed, we have termed this sequence “Pep42” for the sake of simplicity (Supporting Information, Figure S1).
To unequivocally probe the exclusivity of the binding interactions between Pep42 and GRP78, we evaluated its ability to discriminate between recombinant GRP78 protein and HSP70, another member of the HSP family that shares a high degree of protein sequence homology with GRP78, as well as to bovine serum albumin (BSA) as a surrogate for serum proteins in a cell-free system. The Pep42-phage indeed bound specifically only to immobilized GRP78 while no binding was observed to either control protein (Fig. 1A). Furthermore, the binding of Pep42 to GRP78 was inhibited in a dose-dependent manner by the addition of free Pep42, while the addition of scrambled control peptide did not affect the binding of phage to GRP78 (Fig. 1B). These results demonstrated that Pep42 specifically recognizes and binds to GRP78.
Figure 1.
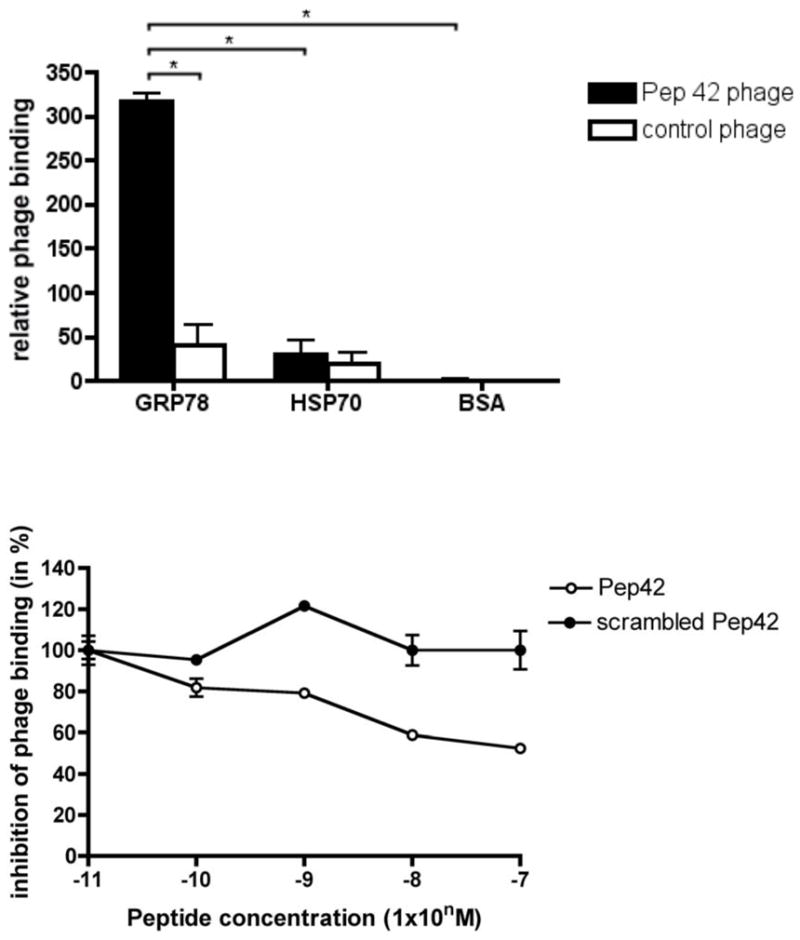
Pep42-displaying phage particles bind specifically to GRP78. A, GRP78, HSP70 and BSA were coated on microtiter wells at 10 μg/mL and incubated with either Pep42-phage or control phage. The control phage displays an unrelated sequence. Phage input was 109 cfu per well. Results are expressed as mean ± standard error of the mean (SEM) of triplicate wells. B, The inhibition of binding of Pep42-phage to immobilized GRP78 was obtained by the addition of increasing concentrations of Pep42 or a scrambled Pep42. *Student’s t test, p<0.05.
The binding of Pep42 to GRP78 overexpressed on the surface of cancer cells was also analyzed. The presence of GRP78 on the cancer cell surface has only recently been appreciated, while GRP 78 is typically absent on the cell membrane of normal cells.20 Indeed, a global profiling approach of the cell surface proteome of cancer cells uncovered that GRP78 is a cell membrane protein on a number of cancer cells, thus making GRP78 a pan-neoplasmic antigen.26 To evaluate Pep42’s ability to bind to human cancer cell lines other than Me6652/4, we analyzed the expression of surface GRP78 in a range of cancer cell lines including lung adenocarcinoma cells (A549), osteosarcoma cells (SJSA-1), hepatoma cells (HepG2) and two normal control fibroblast cell lines via flow cytometry using a monoclonal antibody (mAb) directed against human GRP78. A shift of fluorescence was observed for HepG2, SJSA-1 and A549 cells, while no shift could be seen using the normal skin fibroblast cell line, HDFa and a normal lung fibroblast cell line, CCD-11Lu (Supporting Information, Figure S2), indicating the low expression of GRP78 on the surface of these two control cell lines.
The ability of Pep42 to selectively bind to the positively identified GRP78-expressing cell lines was investigated with Me6652/4, HepG2, A549 and SJSA-1 cells as well as control HDFa cells. The cells were incubated with 50 nM FITC-labeled Pep42 or a FITC-labeled control peptide for 25 min at 4 °C, and cell fluorescence was measured by flow cytometry. As shown in Fig. 2A, Pep42 bound to GRP78 on the surface of Mel6652/4, HepG2, A549 and SJSA-1 cells, while only minimal recognition was observed for HDFa cells. Based on these GRP78 expression data and Pep42 binding results, the cancer cell lines were used interchangeably.
Figure 2.

Pep42 binding to tumor cells is strictly dependent on GRP78 cell surface expression. A, Cells were incubated with either 50 nM FITC-conjugated Pep42 (red curve) or FITC-conjugated control peptide (green curve) for 25 min. at 4 °C with slow rotation. The black curves correspond to cells incubated in the absence of any peptide. B, Pep42 binding to tumor cells was estimated using photoaffinity-labeling. Western blot of Pep42 with its interacting proteins is shown. Here, cells were incubated with 50 μM Pep42-Bpa-Biotin for 1 h at 4 °C in the dark. The Pep42 protein interactions found on the cell surface were purified by avidin-agarose affinity chromatography, separated by SDS-PAGE, and transferred onto a nitrocellulose membrane. Upon incubation with streptavidin-HRP the membrane was developed with luminal and analyzed by chemoluminescence. C, Inhibition of binding of Pep42 to HepG2 cells was evaluated by the addition of a monoclonal anti-GRP78 antibody and the subsequent incubation of cells with Pep42 or a control peptide. **Difference of cells treated with Pep42 only, Student’s t-test, p<0.01. D, Effect of silencing the GRP78 gene expression on binding of Pep42 to Me6652/4 cells. After 48 h of transfection with GRP 78 siRNA or control siRNA, cells were incubated with Pep42-Bpa-Biotin. The detection was carried out as described for Fig. 2B.
A photoaffinity labeling experiment generally used to analyze ligand-receptor interactions was also conducted in analogy to our previous report that lead to the identification of the Pep42 receptor on Me6652/4 cells but now using HepG2 cells and HDFa cells.16 The data clearly demonstrated that Pep42 interacts with the same protein on HepG2 cells as it does on Me6652/4, but does not bind to any other surface protein on the control HDFa cells (Fig. 2B).
An anti-GRP78 monoclonal antibody was utilized to block Pep42 on the cell surface of HepG2 cells, which were chosen due to their high level of GRP78 surface expression, and the binding of Pep42 was subsequently examined. The pretreatment of cells with the mAb significantly decreased the binding of Pep42 to HepG2 cells (Fig. 2C). Lastly, GRP78 expression in Me6652/4 cells was suppressed by transfection with GRP78-specific siRNA. As shown in Fig. 2D, the endogenous GRP78 protein level was significantly reduced in cells transfected with GRP78 siRNA but not in those treated with control siRNA. The GRP78-specific siRNA had no effect on the expression levels of control proteins. Importantly, Pep42 binding was notably reduced in cells treated with the GRP78-specific siRNA. In total, these results underscore that the binding of Pep42 to cells is strongly dependent on the cell surface expression level of GRP78, thus granting cancer cell-targeting specificity for Pep42.
Selective induction of apoptosis in GRP78-overexpressing tumor cells mediated by Pep42-D-(KLAKLAK)2
To evaluate the drug delivery capabilities of Pep42, in vitro cell killing assays were conducted utilizing conjugates of Pep42 chemically-fused to the apoptosis-inducing peptide D-(KLAKLAK)2. 27, 28 As previously noted the amphipathic peptide D-(KLAKLAK)2 does not interact with the zwitterionic plasma membrane of eukaryotic cells, however, it can disrupt the negatively charged mitochondrial membrane and, thus, subsequently induces apoptosis after internalization.27
Pep42-D-(KLAKLAK)2-mediated cytotoxicity was observed in the two GRP78-positive tumor cell lines analyzed, Me6652/4 and SJSA-1 (Fig. 3A).29 In order to reduce non-specific background and increase sensitivity the assays were carried out in low serum containing media. Importantly, no cytotoxic effects were seen when these two cell lines were incubated with an equimolar mixture of Pep42 and free D-(KLAKLAK)2 (Fig. 3B). The GRP78-negative cell line was completely resistant to the peptide conjugate-mediated cytotoxicity. Interestingly, the kinetics of the induction of apoptosis were slightly different depending on the cell line suggesting a varying degree of susceptibility to toxicity in these cells (Supporting Information, Figure S3).
Figure 3.

Selective killing and induction of apoptosis in surface GRP78 expressing cancer cells mediated by Pep42-D-(KLAKLAK)2. A, Cells were incubated with increasing concentrations of the peptides Pep42-D-(KLAKLAK)2 or B, a mixture of free Pep42 and D-(KLAKLAK)2 in 100 μL 1% FBS or supplemented medium for 24 h at 37 °C. The viability of the cells was assessed by a MTT assay. Results are expressed as percentages of cells still viable relative to non-treated cells. C, SJSA-1 cells cultured in the presence of Pep42-D-(KLAKLAK)2 or negative controls (free D-(KLAKLAK)2, free Pep42 or scrambled Pep42) for 6 h at 37 °C were washed in cold PBS and resuspended in staining buffer prior to the addition of FITC-labeled Annexin V and propidium iodide for 15 min. at room temperature. Flow cytometry was performed on SJSA-1 cells gated on the basis of their forward and side light scatter with any cell debris excluded from the analysis. Cell staining with Annexin V-FITC, but no propidium iodide were regarded as early apoptotic cells. Early apoptotic cells (1) Pep42-D-(KLAKLAK)2 35.8%; (2) Free Pep42 2.97%; (3) Free D-(KLAKLAK)2 1.82%; (4) Medium 1.35%; (5) scrambled Pep42-D-(KLAKLAK)2 2.78%. D, The role of surface GRP78 siRNA or control siRNA using Lipofectamine 2000 reagent. After 48 hours, cells were treated with 30 μM Pep42-D-(KLAKLAK)2 for 3 h at 37 °C. *Difference from Pep42-D-(KLAKLAK)2 treated cells, student’s t test, p<0.05.
The evaluation of the extent of the apoptotic cell death induced by Pep42-D-(KLAKLAK)2 in SJSA-1 cells was undertaken. Flow cytometric analysis using propidium iodide (PI) and Annexin V-FITC revealed that Pep42-D-(KLAKLAK)2 induced an increase of Annexin V-positive cells, which represent cells in the early stages of apoptosis (Fig. 3C). Moreover, we investigated the importance of surface GRP78 in the cell killing by Pep42-D-(KLAKLAK)2. Indeed, the knockdown of GRP78 protein using siRNA methodology resulted in a significant reduction in the extent of cell death induced by Pep42-D-(KLAKLAK)2, providing further evidence that surface GRP78 is required for the delivery of D-(KLAKLAK)2 by Pep42 (Fig. 3D). This siRNA experiment also indicated that the cell membrane presence of GRP78 on human cancer cells is solely dependent on the overexpression of the gene in cancer cells and, thus, unlikely to occur to a great extent on normal cells. Taken together the data strongly suggest that Pep42 could be an effective tumor cell-targeting drug delivery system.
Pep42/GRP78 complex is internalized by clathrin-mediated endocytosis
To take advantage of the full potential of Pep42 as a novel carrier for cancer cell targeting, it is important to elucidate the underlying mechanism that enables the cell-targeting peptide to internalize into cells and localize into a specific intracellular compartment. Endocytosis can occur through a number of mechanisms including clathrin-mediated, caveolae/lipid raft-mediated and clathrin- and caveolae-independent pathways.5 Transferrin is known to be internalized via clathrin-coated invaginations on the plasma membrane that detach to form clathrin-coated vesicles.30 On the other hand, bacterial toxins, such as cholera toxin, utilize caveolae-mediated endocytosis to enter cells.30
To assess the intracellular fate of Pep42 endosomes, we added either Alexa Fluor (AF) 594-labeled transferrin and Pep42-Quantum Dots (QDots) or AF594-labeled cholera toxin subunit B (CTB) and Pep42-QDots to cultured A549 cells. As seen in Fig. 4A, Pep42-QDots co-localized with transferrin and only to a lesser extent with CTB. The co-localization coeffients are also shown (Fig. 4B).
Figure 4.

Pep42 partially co-localizes with known markers of endocytosis. A, A549cells were preincubated with Pep42-QDots 525 for 1 h at 37 °C and then added 10 μg/mL AF-594 transferrin or 10 μg/mL AF-594 cholera toxin subunit B for 30 min. at 37 °C. The cells were then washed with PBS and analyzed by laser scanning confocal microscopy. B, Weighted co-localization coefficients were measured with Zeiss LSM Image examiner software. Red in the legend means the co-localized coefficient that red channel co-localizes with the green channel. Green in the legend means the co-localized coefficient that the green channel co-localizes with the red channel.
To further elucidate the uptake mechanism, we next studied the effects of several inhibitors of endocytosis on the cellular uptake of Pep42 by A549 cells. Chlorpromazine, a known inhibitor of clathrin-mediated endocytosis, had the expected inhibitory effect on the uptake of Pep42 or transferrin, but no difference was observed for CTB. In contrast, the uptake of CTB was inhibited by the pretreatment of cells with Nystatin or Methyl-β-cyclodextrin (MCD), both inhibitors of caveolae-mediated endocytosis. As anticipated, neither Nystatin nor MCD had any effect on the uptake of Pep42 or transferring. There was a minor effect on cholera toxin internalization by chlorpromazine, however, this effect was very weak (Fig. 5). The above results strongly indicate that the Pep42 uptake into A549 cells is clathrin-mediated but not caveolae-mediated endocytosis.
Figure 5.

Inhibition of Pep42 uptake using known endocytosis inhibitors. A549 cells were pretreated with 25 μM Nystatin, 30 μM Chlorpromazine or 5 mM methyl-β-cyclodextrin (MCD) for 10 min. and then Pep42-Qdots 525, AF-594-transferrin or AF-594- cholera toxin subunit B was added, respectively.
Endocytosis of clathrin-coated vesicles is also known to involve progressive endosome maturation and eventual fusion with the cell lysosome.30 We assessed the co-localization of internalized Pep42-QDots with the organelle-specific fluorescent markers Lysotracker red (lysosomes), Mitotracker red (mitochondria) and ERtracker Red (endoplasmic reticulum) (Fig. 6A). Thus, 88% Pep42-QDots co-localized with lysosomes after 1 h incubation of Pep42-QDots. At the same time, approximately 73% lysosomes were targeted by internalized Pep42-QDots (Fig. 6B) demonstrating that Pep42 preferentially localizes into the lysosomal compartment.
Figure 6.

Pep42-QDot conjugates co-localize with the lysosomal compartment. A, Co-localization of Pep42-Qdots with Lysotracker Red in A549 cells. Cells were treated with 12 nM Lysotracker Red for 30 min, 20 nM ER-tracker Red for 20 min or 25 nM Mitotracker Red for 15 min after incubation of the peptide for 1 h at 37 °C. B, Weighted co-localization coefficients were measured with Zeiss LSM Image examiner software. Red in the legend means the co-localized coefficient that red channel co-localizes with the green channel. Green in the legend means the co-localized coefficient that the green channel co-localizes with the red channel.
Pep42-mediated subcellular transport of porphyrin conjugates
The biological efficacy of porphyrin-based sensitizers for photodynamic therapy of tumors relies mainly on their efficient translocation across cellular membranes and their delivery into specific organelles of cancer cells.31 It has been demonstrated that functionalized porphyrin conjugates bearing a fusogenic peptide localize mainly into lysosomes.31 Based on the co-localization data, it was important to prove that Pep42 can specifically target and effectively carry a drug into lysosomes of tumor cells, thus, a carboxylic acid functionalized hematoporphyrin was designed and chemically conjugated to Pep42. The phototoxicity of the Pep42-hematoporphyrin conjugate was evaluated using HepG2 and A549, cell lines that were not susceptible to Pep42-D-(KLAKLAK)2 conjugate (Supporting Information, Figure S4). Cells were exposed to increasing concentrations (0–20 μM) of Pep42-hematoporphyrin conjugate for 18 h, as shown in Fig. 7A. The conjugate showed no toxicity at concentrations up to 10 μM in the dark on A549 cells (data not shown). However, the conjugate showed phototoxicity upon exposure to visible light for 1 h on HepG2 and A549 cells, while the surface GRP78-negative cell lines, HDFa and CCD-11Lu, were not affected by the conjugate. The IC50 values for the conjugate were 9 μM on HepG2, and 8 μM on A549 cells, while HepG2 and A549 cells were unaffected by 10 μM of an equimolar mixture of Pep42 and hematoporphyrin (Fig. 7B).
Figure 7.

Phototoxicity of Pep42-hematoporphyrin conjugates toward surface expressing GRP78 cancer cells. The viability of the cells was assessed by a MTT assay. Results are expressed as percentages of cells still viable relative to non-treated control cells. A, Phototoxicity of Pep42-hematoporphyrin conjugate on A549 cells and HepG2 cells. HDFa and CCD-11Lu cells served as controls. B, An equimolar mixture of 10 μM hematoporphyrin and Pep42 had no effect on HepG2 and A549 cells.
The peptide without any cargo displayed maximum internalization levels after 0.5–1 h of incubation and the maximum co-localization with lysosomes occurred after 4 h; after 6 h the peptide was degraded by enzymes of the lysosomal compartment (Supporting Information, Figure S5). These observations clearly suggest that the peptide is stable enough to reach its target site in order to deliver a putative cargo. Furthermore, Pep42 can be cleared from lysosomes without toxic effects. At the same time, when Pep42 was conjugated to hematoporphyrin, the conjugate reached a maximum internalization 3–6 h after incubation. Similar results have been reported for other peptide-cargo conjugates.32, 33 The observations indicate that the kinetics of cellular uptake are also dependent on the physicochemical character of the cargo.
Quantum dot conjugated Pep42 site-directed transport in xenograft mice
Finally, in order to evaluate the ability of Pep42 to find its target and carry a payload into the tumor tissue in vivo, cyclic Pep42 was conjugated to QDots and injected i.v. in the tail vein of tumor bearing mice. Our results demonstrate that cyclic Pep42-QDots are able to home into the tumor tissue of living mammals (Fig. 8A) while linear Pep42-QDots did not accumulate in the tumor (Fig. 8D). The Pep42-QDots showed homing specificity for the relevant tumor tissue as accumulation of fluorescence within the targeted tissue was observed. Significantly less homing of cyclic Pep42-QDots was seen in relevant control organs, i.e. liver and spleen (Fig. 8B and 8C) and could also not be observed for linear Pep42-QDots (Fig. 8E, F). These data provide evidence that Pep42 indeed has the ability to deliver cargo, in this case of significant size, specifically into the tumor tissue in vivo without accumulation in other organs, which is one prerequisite for any successful new approach in cancer chemotherapy.
Figure 8.
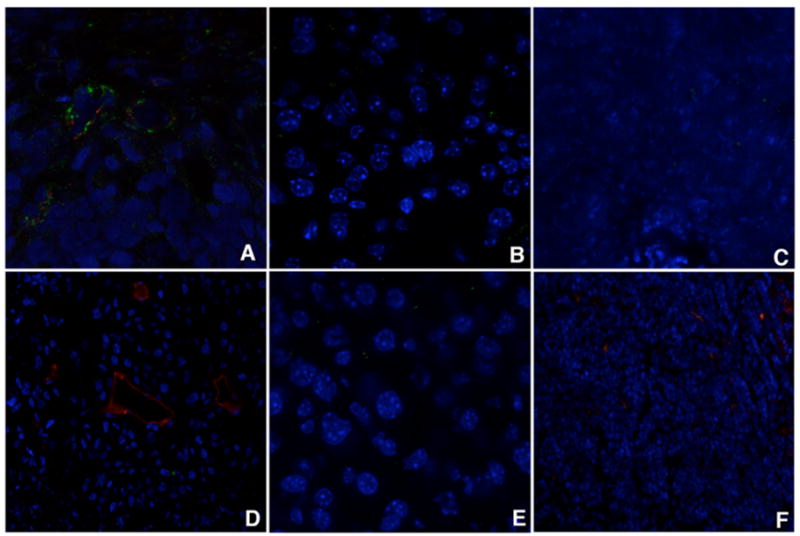
In vivo targeting of Pep42-QDot conjugates. Peptide-coated QDots were injected into the tail vein of tumor bearing CB17Scid male mice, and the presence of QDots in the tumor and control tissue was assessed by examining sections under a confocal microscope. Accumulation of conjugate was observed in the tumor tissue with cyclic peptide QDots (A), less material was found in the liver (B) and the spleen (C). Linear peptide coated QDots did not accumulate in the tumor (D), and also not in the control organs liver (E) and spleen (F). Blood vessels were stained with anti-CD31 (red).
Discussion
In this study, we report how a previously identified oligopeptide Pep42 binds specifically to overexpressed GRP78 on the surface of a number of human cancer cells. This specificity is then utilized to deliver cytotoxic drugs into malignant cells in vitro and also used as a tumor tissue homing beacon in vivo.16 We note that several linear chimeric peptides composed of GRP78 binding motifs have been identified to date, however, Pep42 is a cyclic 13-mer peptide that possesses a unique sequence, which does not share any known motifs with other GRP78-binding or otherwise tumor homing peptides.17 Moreover, our screen was conducted using a phage display biopanning protocol on intact cells, thus, ensuring that the selected ligands bound to the cellular receptor in its native context.34
Small peptidic ligands are being considered as promising new tools for the delivery of cytotoxic drugs.35 Although the mechanism of cellular uptake of canonical cell-penetrating peptides and other small peptidic ligands has not been fully understood in detail yet, it is believed that these types of peptides enter cells rapidly in a receptor-independent fashion.36 In contrast our study provides strong evidence that receptor-mediated translocation is involved in the internalization of Pep42 and that clathrin-mediated endocytosis is the main mechanism of uptake. This would be in accordance with recent data that assume more than one mechanism for peptide-mediated intracellular delivery of various types of molecules and particulate structures.3
Pep42-QDots in combination with inhibitors of endocytosis and specific organelle markers for live cells were used for the investigation of the mechanism of endocytosis and co-localization experiments. The uptake mechanism of Pep42 into cancer cells involves clathrin-coated vesicles, which then transport the internalized peptide into the lysosomal compartment of the cell. Interestingly, the co-localization data presented here amends our previous findings suggesting that Pep42 is able to co-localize with the endoplasmic reticulum. Although the exact mechanism of endocytosis and subsequent intracellular fate may in fact depend on the particular cell type, the study presented here reveals two key steps of Pep42’s cellular internalization, namely the absolutely required binding to the surface GRP78 and uptake via endocytosis.16 Importantly, artifacts of cellular peptide internalization induced by fixation, which are known in the literature 37, 38, were avoided by only using live cells for the uptake studies of the fluorescent Pep42 into cells via confocal microscopy.
To evaluate the drug delivery of Pep42, we chose drugs with vastly different biochemical properties as well as different mechanisms of action. One drug was the amphipathic peptide D-(KLAKLAK)2, an apoptosis-inducing drug, whose primary action site is the mitochondrial membrane, while the other drug consisted of hematoporphyrin, a photosensitizer that is active in the lysosomal compartment. Our data illustrate that the two drugs are not imported into any cell line tested without the appropriate drug delivery system, namely Pep42. Furthermore, conjugation of D-(KLAKLAK)2 to Pep42 generated a reagent that selectively killed Me6652/4 and SJSA-1 in a surface level GRP78-dependent manner.
Clear evidence is presented of apoptosis induction in the cells treated with the conjugate, which strongly indicates that Pep42 allowed for the intracellular delivery and subsequent mitochondrial membrane targeting of D-(KLAKLAK)2. The resistance observed of A549 and HepG2 cells to the D-(KLAKLAK)2-induced killing might be the result of a cell type-dependent inability of the internalized peptide to access the cytoplasm and/or activate cell death-triggering mechanisms. Nevertheless, these cell lines were found to be susceptible to the Pep42-hematoporphyrin conjugate. To harness its full phototoxic activity, a localization into the lysosomal compartment of cancer cells is required.31 Applying the data gathered from the co-localization experiments of Pep42, this strategy resulted in successful killing of the two tumor cell lines, while no adverse effects were observed for the control cells.
Nanoparticles are thought to have potential as novel intracellular probes for both imaging and drug delivery.39, 40 Our results suggest that Pep42-QDots possess homing specificity for the relevant tumor tissue and significantly less homing in the control organs. Although the nanoparticles used in this study are rather simple, the use of more sophisticated systems could be envisioned for the future. Such devices may, as an example, deliver a drug to the site of action, release the drug, and report back its delivery. These findings highlight that Pep42 may serve as a promising candidate for future anti-cancer drug delivery platforms.
In conclusion, Pep42 can be considered as a promising tumor-targeting drug delivery system as it possesses the ability to deliver drugs with vastly different mechanisms of action into human cancer cells. We have shown that GRP78 is the exclusive receptor for Pep42, its mechanism for internalization and site directed transport in xenograft mice. We believe the data obtained warrant future evaluation of Pep42’s drug delivery capabilities in vivo which will likely be more to be representative of clinically relevant aspects for the treatment of disease in human cancer.17 Overexpressed GRP78 on cancer cells can be selectively targeted by a cell internalizing peptide, studies demonstrating the selective delivery of payloads for the killing of tumor cells will be the subject of future investigations.
Supplementary Material
Acknowledgments
This work was supported by The Skaggs Institute for Chemical Biology and the National Institutes of Health (C A094193). We thank David Kujawa for help with the cell cultures, Dr. William B. Kiosses for help and training at the core microscopy facility at TSRI and Dr. Alan Saluk and Cheryl Kim for assistance with FACS analysis at the flow cytometry core facility at TSRI.
Supporting Information is available.
Abbreviations used
- siRNAs
small interfering ribonucleic acids
- GRP78
glucose-regulated protein 78
- HSP 70
heat shock protein 70
- QDots
quantum dot
- DMEM
Dulbecco’s modified Eagle’s medium
- Me6652/4
human melanoma cell line
- A549
human lung adenocarcinoma cell line
- SJSA-1
human osteosarcoma cell line
- HepG2
human hepatoma cell line
- CCD-11Lu
human lung fibroblast cell line
- EMEM
Eagle’s modified minimum essential medium
- HDFa
human dermal fibroblasts
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- EDCI
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- DMSO
dimethlylsulfoxide
- EDTA
ethylene diamine tetraacetate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- HRP
horseradish peroxidase
- BSA
bovine serum albumin
- PBS
phosphate-buffered saline
- MCD
methyl-β-cyclodextrin
- mAb
monoclonal antibody
- CTB
cholera toxin subunit B
- Ahx
Aminohexanoic acid
- Bpa
Benzoylphenylalanine
- SOBAG
super optimal broth ampicillin glucose
- Fmoc
9-Fluorenylmethyl carbamate
- dPEG6
discrete polyethylene glycol 6
References
- 1.Langerak AD. Chemotherapy Regimens and Cancer Care. Landes Bioscience; Georgetown, TX: 2001. p. 209. [Google Scholar]
- 2.Zhou X, Chang YC, Oyama T, McGuire MJ, Brown KC. J Am Chem Soc. 2004;126(48):15656–7. doi: 10.1021/ja0446496. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin VP, Levchenko TS. Curr Protein Pept Sci. 2003;4(2):133–40. doi: 10.2174/1389203033487298. [DOI] [PubMed] [Google Scholar]
- 4.Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Nat Biotechnol. 2000;18(4):410–4. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 5.El-Andaloussi S, Holm T, Langel U. Curr Pharm Des. 2005;11(28):3597–611. doi: 10.2174/138161205774580796. [DOI] [PubMed] [Google Scholar]
- 6.Nahrendorf M, Jaffer FA, Kelly KA, Sosnovik DE, Aikawa E, Libby P, Weissleder R. Circulation. 2006;114(14):1504–11. doi: 10.1161/CIRCULATIONAHA.106.646380. [DOI] [PubMed] [Google Scholar]
- 7.De J, Chang YC, Samli KN, Schisler JC, Newgard CB, Johnston SA, Brown KC. Molecular Biosystems. 2005;1(2):149–157. doi: 10.1039/b504572j. [DOI] [PubMed] [Google Scholar]
- 8.Jarver P, Langel U. Drug Discov Today. 2004;9(9):395–402. doi: 10.1016/S1359-6446(04)03042-9. [DOI] [PubMed] [Google Scholar]
- 9.Jarver P, Langel U. Biochim Biophys Acta. 2006;1758(3):260–3. doi: 10.1016/j.bbamem.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Moss JA, Lillo A, Kim YS, Gao C, Ditzel H, Janda KD. J Am Chem Soc. 2005;127(2):538–9. doi: 10.1021/ja0443171. [DOI] [PubMed] [Google Scholar]
- 11.Landon LA, Deutscher SL. J Cell Biochem. 2003;90(3):509–17. doi: 10.1002/jcb.10634. [DOI] [PubMed] [Google Scholar]
- 12.McGuire MJ, Samli KN, Johnston SA, Brown KC. J Mol Biol. 2004;342(1):171–82. doi: 10.1016/j.jmb.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Vives E. J Control Release. 2005;109(1–3):77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 14.Ruoslahti E, Duza T, Zhang L. Curr Pharm Des. 2005;11(28):3655–60. doi: 10.2174/138161205774580787. [DOI] [PubMed] [Google Scholar]
- 15.Gao C, Mao S, Ditzel HJ, Farnaes L, Wirsching P, Lerner RA, Janda KD. Bioorg Med Chem. 2002;10(12):4057–65. doi: 10.1016/s0968-0896(02)00340-1. [DOI] [PubMed] [Google Scholar]
- 16.Kim Y, Lillo AM, Steiniger SC, Liu Y, Ballatore C, Anichini A, Mortarini R, Kaufmann GF, Zhou B, Felding-Habermann B, Janda KD. Biochemistry. 2006;45(31):9434–9444. doi: 10.1021/bi060264j. [DOI] [PubMed] [Google Scholar]
- 17.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, Pasqualini R. Cancer Cell. 2004;6(3):275–84. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman RJ. Genes Dev. 1999;13(10):1211–33. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 19.Lee AS. Trends Biochem Sci. 2001;26(8):504–10. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 20.Misra UK, Deedwania R, Pizzo SV. J Biol Chem. 2005;280(28):26278–86. doi: 10.1074/jbc.M414467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, Kennedy KA, Patierno SR. Breast Cancer Res Treat. 2000;59(1):15–26. doi: 10.1023/a:1006332011207. [DOI] [PubMed] [Google Scholar]
- 22.Bini L, Magi B, Marzocchi B, Arcuri F, Tripodi S, Cintorino M, Sanchez JC, Frutiger S, Hughes G, Pallini V, Hochstrasser DF, Tosi P. Electrophoresis. 1997;18(15):2832–41. doi: 10.1002/elps.1150181519. [DOI] [PubMed] [Google Scholar]
- 23.Uramoto H, Sugio K, Oyama T, Nakata S, Ono K, Yoshimastu T, Morita M, Yasumoto K. Lung Cancer. 2005;49(1):55–62. doi: 10.1016/j.lungcan.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 24.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press; 2000. [Google Scholar]
- 25.Katsuta M, Takigawa Y, Kimishima M, Inaoka M, Takahashi R, Shiohara T. J Immunol. 2006;176(12):7736–44. doi: 10.4049/jimmunol.176.12.7736. [DOI] [PubMed] [Google Scholar]
- 26.Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, Zhao R, Puravs E, Tra J, Michael CW, Misek DE, Hanash SM. J Biol Chem. 2003;278(9):7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 27.Marks AJ, Cooper MS, Anderson RJ, Orchard KH, Hale G, North JM, Ganeshaguru K, Steele AJ, Mehta AB, Lowdell MW, Wickremasinghe RG. Cancer Res. 2005;65(6):2373–7. doi: 10.1158/0008-5472.CAN-04-2594. [DOI] [PubMed] [Google Scholar]
- 28.Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, Arap MA, Hong WK, Troncoso P, Logothetis CJ, Pasqualini R, Arap W. Nat Biotechnol. 2003;21(1):57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 29.Elmquist A, Langel U. Biol Chem. 2003;384(3):387–93. doi: 10.1515/BC.2003.044. [DOI] [PubMed] [Google Scholar]
- 30.Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. J Biol Chem. 2003;278(36):34141–9. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 31.Sibrian-Vazquez M, Jensen TJ, Hammer RP, Vicente MG. J Med Chem. 2006;49(4):1364–72. doi: 10.1021/jm050893b. [DOI] [PubMed] [Google Scholar]
- 32.Silhol M, Tyagi M, Giacca M, Lebleu B, Vives E. Eur J Biochem. 2002;269(2):494–501. doi: 10.1046/j.0014-2956.2001.02671.x. [DOI] [PubMed] [Google Scholar]
- 33.De Coupade C, Fittipaldi A, Chagnas V, Michel M, Carlier S, Tasciotti E, Darmon A, Ravel D, Kearsey J, Giacca M, Cailler F. Biochem J. 2005;390(Pt 2):407–18. doi: 10.1042/BJ20050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oyama T, Rombel IT, Samli KN, Zhou X, Brown KC. Biosens Bioelectron. 2006;21(10):1867–75. doi: 10.1016/j.bios.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 35.O’Connor KA, Roth BL. Nat Rev Drug Discov. 2005;4(12):1005–14. doi: 10.1038/nrd1900. [DOI] [PubMed] [Google Scholar]
- 36.Rhee M, Davis P. J Biol Chem. 2006;281(2):1233–40. doi: 10.1074/jbc.M509813200. [DOI] [PubMed] [Google Scholar]
- 37.Lundberg M, Johansson M. Biochem Biophys Res Commun. 2002;291(2):367–71. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 38.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. J Biol Chem. 2003;278(1):585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 39.Steiniger SC, Kreuter J, Khalansky AS, Skidan IN, Bobruskin AI, Smirnova ZS, Severin SE, Uhl R, Kock M, Geiger KD, Gelperina SE. Int J Cancer. 2004;109(5):759–67. doi: 10.1002/ijc.20048. [DOI] [PubMed] [Google Scholar]
- 40.Kelly KA, Nahrendorf M, Yu AM, Reynolds F, Weissleder R. Mol Imaging Biol. 2006;8(4):201–7. doi: 10.1007/s11307-006-0043-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.