

Abstract
Introducing 2′-fluoro substitution on the 2′,3′-double bond in carbocyclic nucleosides has provided biologically interesting compounds with potent anti-HIV activity. As an extension of our previous works in the discovery of anti-HIV agents, d- and l-2′,3′-unsaturated 3′-fluoro carbocyclic nucleosides were synthesized and evaluated against HIV-1 in human peripheral blood mononuclear (PBM) cells. Among the synthesized l-series nucleosides, compounds 18, 19, 26, 28 exhibited moderate antiviral activity (EC50 7.1 μM, 6.4 μM, 10.3 μM and 20.7 μM, respectively), while among the d-series, the guanosine analogue (35, d-3′-F-C-d4G) exhibited the most potent anti-HIV activity (EC50 0.4 μM, EC90 2.8 μM). However, the guanosine analogue 35 was cross-resistant to the lamivudine-resistant variants (HIV-1M184V). Molecular modeling studies suggest that hydrophobic interaction as well as hydrogen bonding stabilize the binding of compound 35 in the active site of wild type HIV reverse transcriptase (HIV-RT). In the case of l-nucleosides, these two effects are opposite which results in a loss of binding affinity. According to the molecular modeling studies, cross-resistance of d-3′-F-C-d4G (35) to M184V mutant may be caused by the realignment of the primer and template in the HIV-RTM184V interaction, which destabilizes the RT-inhibitor triphosphate complex, resulting in a significant reduction in anti-HIV activity of the d-guanine derivative 35.
Introduction
Nucleoside reverse transcriptase inhibitors (NRTI) have played important role in the treatment of HIV infections.1 However, major drawbacks of NRTI include the emergence of drug resistant variants and toxicity.2-5 Therefore, conservative efforts have been made to improve the antiviral efficacy as well as to reduce the toxicity by modifying the structure.
The structures of several potent NRTIs, such as stavudine (d4T), abacavir, reverset (d-d4FC) and elvucitabine (l-d4FC) highlight the important role of a 2′,3′-double bond to enhance the antiviral activity (Figure 1).1 Additionally, carbocyclic nucleosides such as abacavir, have attracted considerable attention due to their potent antiviral activity as well as the stability toward metabolic degradation.6 In view of these facts, it was of interest to incorporate these structural features into nucleoside analogues. Several interesting compounds with potent anti-HIV activity have been discovered as the results of this concept.7-13 In connection to these efforts, our laboratory recently reported the stereo-selective synthesis and anti-HIV activity of d- & l-2′,3′-didehyhydro-2′,3′-dideoxy-2′-fluoro-carbocyclic nucleosides.14 Among the series, the adenosine analogue with l-configuration showed the most potent anti-HIV activity (EC50 0.77 μM). Based on molecular modeling studies, it was found that both the double bond on the carbocyclic ring as well as the 2′-fluoro substitution contribute to the favorable binding affinity between the inhibitor and the HIV-RT. Hence, further exploring the antiviral activity of 3′-fluorine congeners was of interesting to expand our knowledge on the structure activity relationships of the same class of nucleosides.
Figure 1.

Several potent NRTIs with 2′,3′-double bond.
For the synthesis of 2′-fluoro carbocyclic nucleosides, the fluorine substituted unsaturated carbocyclic ring was first constructed followed by coupling with various heterocyclic bases. However, this method was found to be unsuccessful in the synthesis for the 3′-fluoro congeners due to the instability of the final products under the same conditions used. Therefore, condensation of a gem-3′,3′-difluoro sugar 8 or 29 with base moieties followed by an elimination reaction in the last step was successful to obtain the target purine nucleosides. Pyrimidine nucleosides were, however, synthesized via the linear method using the intermediate 11 or 30. Herein, synthesis, anti-HIV activity and molecular modeling studies of d- & l-2′,3′-didehyhydro-2′,3′-dideoxy-3′-fluoro-carbocyclic nucleosides are reported.
Results and Discussion
Chemistry
Both d- & l-nucleosides were synthesized, however, the following descriptions are mainly based on the l-series according to the Scheme 1, 2 and 3, unless otherwise indicated. The epoxide 1 was synthesized from d-ribose by the known method in our laboratory.14 The desired regio-isomer was obtained by reductive ring opening of the epoxide with judicious selection of reducing reagents (Scheme 1). The preliminary study suggested that compound 4 was the major product when treated the epoxide 2 with LAH. The selective opening of the epoxide by the hydride may be due to the steric hindrance of the bulky trityl group adjacent to the 3′-position. Based on this result, several other reducing reagents were investigated (Table 1). Among them, Super-Hydride® gave predominantly compound 4 (compound 4 : compound 3 = 15.3 : 1) in excellent yield (98 %). Oxidation of the alcohol 4 with PDC gave the ketone 5 as an unstable compound. It was interesting to note that introducing a di-fluorine group at the 3′-position to compound 5 was quite difficult in comparison to the 2′-fluoro isomer.14 A harsh condition of neat diethylaminosulfur trifluoride (DAST) with reaction temperature at 40 °C for 36 h was needed to convert the ketone 5 to difluoro compound 6 in 68 % yield. The following elimination reaction also gave significant problems: Treating compound 6 with potassium tert-butoxide (tBuOK) in THF at 50 °C did not produce the desired 3′-fluorovinyl moiety and only the starting material was recovered. Hence, a modified synthetic sequence was adopted as illustrated in Scheme 1 (6 to 11). Both trityl and benzyl groups were removed using iodotrimethylsilane (TMSI) in 70 % yield. The resulting diol 7 was selectively protected by tert-butyldiphenylsilyl chloride (TBDPSCl) to give the key intermediate 8 which was converted to amine 11 in three steps. For the pyrimidine nucleosides, the linear synthetic methodology reported by Shealy et al.15, 16 was used, as a direct coupling reaction using the alcohol 8 under the Mitsunobu conditions resulted in the decomposition of the starting material. The amine 11 was coupled with substituted isocynate to give the corresponding urea 12 or 16 (Scheme 2). Reaction of 12 with conc. ammonium hydroxide/ethanol/1,4-dioxane in a steel bomb gave the uridine analogue 13 in 41 % yield. Amination of 13 followed by deprotection afforded the cytidine analogue 15. For the thymidine analogue 17, ring closure and deprotection were accomplished in one step under acidic condition from compound 16. Lastly, the 2′,3′-double bond was obtained under basic elimination conditions to afford the target cytidine 18 and thymidine 19 analogues in 35 % and 46 % yield, respectively. To synthesize adenosine analogues, triphenylphosphine (TPP) and diisopropyl azodicarboxylate (DIAD) were first mixed in the THF:1,4-dioxane co-solvent at 0 °C, and then further cooled to -78 °C. The key intermediate 8 and the 6-chloropurine were added sequentially and the reaction was allowed to gradually warm up to room temperature until all the starting material was consumed (Scheme 3). The crude product 20, which was contaminated with reduced DIAD species, was directly treated with methanolic ammonia in a steel bomb at 100 °C to give the adenosine analogue 21 in 37 % yield in two steps. After removing the silyl group under acidic condition, compound 22 was treated with tBuOK in THF at 90 °C to furnish the adenine derivative 26 in 50 % yield. The compound 21 could also be converted to inosine analogue 23 by treating with formic acid followed by ammonium hydroxide in 39 % yield in two steps. After the elimination reaction similar to the method described for the adenosine analogue, the final inosine analogue 27 was obtained in 49 % yield. Condensation of alcohol 8 with 2-amino-6-chloropurine or 6-chloro-N2-isobutyrylpurine, under the Mitsunobu condition as described above, failed to give the corresponding nucleoside. However, when a mixture of TPP, 6-chloro-N2-isobutyrylpurine and alcohol 8 in dry THF was treated with DIAD at 0 °C, the desired product 24 was able to be isolated. The compound 24 was converted to the guanosine analogue 25 using formic acid followed by ammonium hydroxide in 18 % yield from 8 in two steps. The nucleoside 25 was further subjected to an elimination reaction using tBuOK in DMF at 70 °C to give the guanosine analogue 28 in 45 % yield.
Scheme 1.

Reagents and conditions: (a) i) Ref. 14, ii) α-AIBBr, CH3CN iii) K2CO3, MeOH; (b) TrCl, DMAP, Et3N, CH2Cl2; (C) Super-Hydride® (1.0 M in THF), 0 °C to room temp.; (d) PDC, AcOH, 4 Å molecular sieve, CH2Cl2; (e) Neat DAST, 40 °C; (f) TMSI, CH2Cl2; (g) TBDPSCl, imidazole, CH2Cl2; (h) MsCl, Et3N, CH2Cl2, room temp.; (i) NaN3, DMF, 130 °C; (j) H2/Pd/C, 30 psi, room temp.
Scheme 2.

Reagents and conditions: (a) (i) β-methoxyacryloyl isocyanate, THF, -30 °C to room temp. (for 12) or β-methoxy-α-methacryloyl isocyanate, THF, -30 °C to room temp.(for 16); (b) NH4OH, 1,4-dioxane/EtOH, steel bomb, 90-100 °C; (c) (i) 2,4,6-triisopropylbenzenesulfonyl chloride, DMAP, Et3N, CH3CN, room temp. (ii) NH4OH or NH3/MeOH, room temp.; (d) HCl/ MeOH, room temp.; (e) 3N HCl/1,4-dioxane, reflux 3h; (f) tBuOK, THF/1,4-dioxane, 90 °C.
Scheme 3.

Reagents and conditions: (a) DIAD, Ph3P, purines, THF or THF/1,4-dioxane; (b) NH3/MeOH, steel bomb, 110 °C; (c) 3 N HCl, MeOH; (d) (i) Formic acid, (ii) NH4OH or NH3/MeOH, room temp.; (e) tBuOK, THF/1,4-dioxane, 90 °C or tBuOK, DMF, 70 °C.
Table 1.
Ring-opening reaction of epoxide 2
 | |||
|---|---|---|---|
| Entry | Reducing agent | 4:3 | yield |
| 1 | Super-Hydride® | 15.3 : 1 | 98 % |
| 2 | LAH | 5.1:1 | 88 % |
| 3 | DIBAL-H | - | 0 % |
| 4 | Red-Al | - | 0 % |
| 5 | LiAl(t-butoxy)3 | - | 0 % |
Similar procedures were conducted to synthesize the d-series. As the difluoro-substituted nucleosides on the carbohydrate moiety exhibit interesting biological activity,17 the difluoro-nucleosides 31-33, 37 and 38 in the d-series were also evaluated against HIV-1 (Table 3). It is noteworthy that the elimination reaction in the last step proceeded with significant difficulties. First of all, the starting material and product have almost identical Rf values on a silica gel plate as well as on a column, which makes it extremely difficult to identify and separate. Secondly, the difluoro compounds are inert to the conventional methods of elimination using tBuOK in aprotic solvent, and therefore, the reaction mixture has to be heated for long periods of time to convert all the starting material to the product (Table 3, entries 1 and 3). Unfortunately, under these conditions, the newly formed target nucleosides decomposed, which resulted in low yields. Fortunately, in the search for better conditions for the elimination step during the synthesis of the d-compounds, it was found that the microwave-assisted method had several advantages over the traditional method. Upon irradiation of the difluoro-nucleosides in the microwave synthesizer with maximum output power of 300 W, the elimination reaction was completed within 5-10 min at 70 °C and gave 70-80 % yield, while a lower yield were obtained after a longer time in the traditional thermally-assisted conditions (Table 2). This methodology may provide an efficient way for preparing molecules which have a fluorovinyl moiety. Assignment of the structures of newly synthesized nucleosides was accomplished by NMR, elemental analysis, mass, UV and IR spectroscopy.
Table 3.
In vitro anti-HIV-1 activity and toxicity of d-3′,3′-difluoro-2′,3′-dideoxy-carbocyclic nucleosides and d- & l- 3′-fluoro-2′,3′-didehydro-carbocyclic nucleosides.
 | ||||||
|---|---|---|---|---|---|---|
| B | Configuration | Anti-HIV-1 activity (μM)a | Cytotoxicity (μM) | |||
| EC50 | EC90 | PBM | CEM | Vero | ||
| Adenine 31 | D | >100 | >100 | 95.1 | >100 | >100 |
| Guanosine 32 | D | >100 | >100 | >100 | >100 | >100 |
| Hypoxanthine 33 | D | >100 | >100 | >100 | >100 | >100 |
| Cytosine 37 | D | >100 | >100 | >100 | >100 | >100 |
| Thymine 38 | D | >100 | >100 | >100 | >100 | >100 |
| Cytosine 18 | L | 7.1 | 72.0 | >100 | >100 | >100 |
| Thymine 19 | L | 6.4 | >100 | >100 | >100 | >100 |
| Adenine 26 | L | 10.3 | 33.5 | >100 | >100 | >100 |
| Hypoxanthine 27 | L | >100 | >100 | >100 | >100 | >100 |
| Guanine 28 | L | 20.7 | >100 | >100 | >100 | >100 |
| Adenine 34 | D | 14.8 | 40.6 | >100 | >100 | >100 |
| Guanine 35 | D | 0.41 | 2.8 | 21.1 | >100 | >100 |
| Hypoxanthine 36 | D | >100 | >100 | >100 | >100 | >100 |
| Cytosine 39 | D | >100 | >100 | >100 | >100 | >100 |
| Thymine 40 | D | 68.8 | >100 | >100 | >100 | >100 |
| Carbovir | D | 0.087b | 0.27b | N/A | N/A | N/A |
Anti-HIV activity evaluated in PBM cells against HIV-1LAI unless otherwise indicated.
Anti-HIV activity evaluated in PBM cells against HIV-1xxBRU.
Table 2.
Elimination reactions using traditional or microwave (MW)-assisted methods
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Temperature | Reaction time | Isolated yield (%) | Methods |
| 1 | l-cytidine analogue | 90 °C | 9 h | 35 | traditional oil bath heating |
| 2 | d-cytidine analogue | 70 °C | 6 min | 84 | MW-assisted |
| 3 | l-guanosine analogue | 70 °C | 24 h | 45 | traditional oil bath heating |
| 4 | d-guanosine analogue | 70 °C | 10 min | 72 | MW-assisted |
Anti-HIV Activity
All the synthesized pyrimidine (18, 19 and 37-40) and purine (26-28, 31-36) nucleosides were evaluated against HIV-1 in human PBM cells. The EC50, EC90 and toxicity data are listed in Table 3. In the d-3′,3′-difluoro series, none of the compounds showed any antiviral activity nor cytotoxicity when tested up to 100 μM. Among the d- and l-3′-fluoro-2′,3′-unsaturated nucleosides, some of them exhibited moderate to potent anti-HIV activity. The cytidine 18, thymidine 19, adenosine 26 and guanosine 28 analogues in the l-series inhibited HIV-1 in PBM cells with EC50 ranged from 6.4 μM to 20.7 μM. The guanosine analogue 35, bearing d-configuration, is the most active compound among all the synthesized nucleosides (EC50 0.41 μM, EC90 2.8 μM), although it exhibited moderate cytotoxicity in PBM cells (IC50 21.1 μM). The antiviral activities of 3′-fluoro-2′,3′-unsaturated carbocyclic nucleosides in the current studies were generally maintained or enhanced, in comparison to that of 2′-fluoro congeners.14 Thus, the role of the fluorine substitution on the 3′-position of the double bond may have significant effects (vide infra for molecular modeling studies).
Antiviral Activity against Lamivudine-Resistant (HIV-1M184V) Mutant Strain
One of the drawbacks of the NRTI is the emergence of drug-resistant mutant strains during the extended treatment period, which may significantly compromise the clinical efficacy. Lamivudine, an important component of the highly active antiretroviral therapy (HAART), confers a single mutation at residue 184 (M184V), which caused at least a 1,000 fold decrease in its antiviral activity.18, 19 Discovery of novel NRTI agents against lamivudine-resistant mutant strain is of great interest. Unfortunately, all the potent NRTI with l-configuration against wild type HIV-1, are always cross-resistant to the lamivudine resistant mutant (M184V), which may be due to the steric hindrance between the bulky side chain of Val184 and the adjacent NRTIs' sugar ring.20 The situation is generally better in d-nucleosides, as their sugar rings project far away from the residue 184. However, the M184V mutant has also been isolated after using the abacavir, a prodrug of carbovir, which is a d-nucleoside.21-26 In view of the structural similarity between the compound 35 and carbovir, it was of interest to understand its resistance profile. Hence, we further evaluated the compound 35 against HIVM184V using carbovir (Table 4) as well as 3TC/AZT (data not shown) as control. Based on this study, the compound 35 appears to confer resistance to HIV-1M184V (Table 4). Molecular modeling was thus performed to understand the potential mechanism of the cross-resistance (vide infra).
Table 4.
Activity of d-3′-F-C-d4G against lamivudine-resistant virus (HIV-1M184V) in human PBM Cells using carbovir as control
| Compounds | xxBRU | M184V | FIa | ||
|---|---|---|---|---|---|
| EC50 (μM) | EC90 (μM) | EC50 (μM) | EC90 (μM) | ||
| d-3′-F-C-d4G 35 | 0.098 | 0.58 | 3.8 | 14.9 | 38.8 |
| Carbovirb | 0.087 | 0.27 | 0.20 | 1.1 | 2.3 |
FI is the fold increase (EC50 HIV-1M184V / EC50 HIV-1xxBRU).
We also performed the experiment using AZT and 3TC as control, in which AZT is not but 3TC is highly cross-resistant to M184V mutant, and compound 35 is cross-resistant to M184V mutant (data not shown).
Molecular Modeling Studies
Among all the synthesized nucleosides, d-3′-F-C-d4G is the most active compound, while its l-form exhibited only marginal activity against HIV-1. From the studies of drug resistant mutant, it was found that the anti-HIV activity of d-3′-F-C-d4G to HIV-RTM184V significantly decreased in comparison to the wild type virus. To understand the molecular basis of antiviral activity as well as the drug-resistance, molecular modeling studies were conducted on the interactions between the NRTI and HIV-RT. Our previous modeling studies have qualitatively demonstrated the relationship between the binding affinity and the antiviral activity.10-14 From present studies, the most active compound d-3′-F-C-d4G 35 has the most favorable relative binding energy (-24.4 kcal/mol, Table 5), which is significantly higher than that of less activity l-counterpart 28 (+18.8 kcal/mol, Table 5).
Table 5.
In vitro anti-HIV activity of selected 3′-F-C-d4Ns and carbovir against HIV wild type virus and correlation with calculated energy of complex (Inhibitor-TP) / HIV-RT
| Compound | EC50 (μM)a | Erel (Kcal/mol)c |
|---|---|---|
| d-3′-F-C-d4G 35 | 0.41 | −24.4 |
| l-3′-F-C-d4G 28 | 20.7 | +18.8 |
| Carbovir | 0.087b | -10.8 |
EC50 in PBM cells against HIV-1LAI unless otherwise indicated.
EC50 in PBM cells against HIV-1xxBRU.
Erel = (Binding energy of inhibitor-TP) – (Binding energy of natural 2′-dNTP).
The minimized structure showed that d-3′-F-C-d4G was bound tightly in the well defined binding pocket inside the wild type HIV-RT (Figure 2a). The triphosphate moiety is stabilized by the extensive hydrogen bonds with amino acids Arg65, Lys70, Lys72, Asp113 and Ala114. The carbocyclic ring stacks right over the phenyl ring of Tyr115 forming a favorable hydrophobic π-π interaction, which has been observed in our previous reports.12-14 Also, the 3′-fluorine is strongly interacting with the backbone amide of Tyr115 (Figure 2a). Stabilized by the combined effects of hydrophobic interaction and hydrogen bonding with Tyr115, d-3′-F-C-d4G is thus bound tightly with HIV-RT, reflecting a higher level of anti-HIV activity, although the initial kinase might have also played a significant role in determining the observed anti-HIV potency. In the case of l-3′-F-C-d4G, the 3′-fluorine is at a reasonable distance (2.0 Å) to interact in a hydrogen bond with backbone amide of Asp185 in lieu of Tyr115. However, we noticed that this interaction pulls the carbocyclic ring away from the Tyr115, which decreases the hydrophobic π-π interaction (Figure 2b), leads to a lower binding affinity (Table 5).
Figure 2.

(a) Binding mode of d-3′-F-C-d4G-TP/HIV-RTWT complex. The triphosphate moiety is stabilized by hydrogen bonding with residues Lys65, Arg72, Lys70, Asp113 and Ala114. The other strong hydrogen bond is detected between the 3′-fluoro and backbone amide of Tyr115. Also, the sugar ring is located right over the phenyl ring of Tyr115 forming a favorable hydrophobic interaction. (b) Comparing the binding mode of d- and l- 3′-F-C-d4G-TP, a decreased hydrophobic interaction is observed for the latter due to the hydrogen bond of its 3′-fluoro with backbone amide of Asp185 pulls the sugar ring away from the top of Tyr115 (indicated by the blue arrow).
It has been well understood that the M184V mutation causes serious problem in positioning the l-nucleoside triphosphate at the active site by interfering the sugar ring with the bulky side chain of Val184. However, reports for the d-nucleosides, which confer significant cross-resistant to M184V, are rare. The antiviral activity of d-3′-F-C-d4G in the current report showed a marked decrease in the HIV-1M184V in comparison to the HIV-1WT (Table 4, 6). To understand the underlying mechanism, we further conducted the molecular dynamics studies of d-3′-F-C-d4G-TP, carbovir-TP and dGTP binding with the wild type HIV-RT as well as M184V mutant. According to our model, it is unlikely that the resistance of M184V mutant is caused by the steric hindrance as we observed for the l-nucleosides. However, a steric clash was noticed between the Val184 side chain and the sugar ring of the final residue of the primer. In the d-3′-F-C-d4G/HIV-RTM184V complex, Val184 pushes the last residue of primer away and changes the conformation of adjacent Asp185. The conformational change of Asp185 propagates to magnesium atoms, Asp110 and triphosphate moiety of d-3′-F-C-d4G, through the strong electrostatic interactions between two magnesium atoms and nearby negative charged residues such as Arg72 (Figure 3a). Although the d-3′-F-C-d4G still maintains the hydrogen bond between the 3′-fluorine and backbone amide of Tyr115, inhibitor's sugar ring was lifted away from the surface of the Ty115 aromatic ring, resulting in the loss of hydrophobic stacking interaction which may decrease the relative binding energy (Figure 3b right, Table 6). Furthermore, the primer/template reposition results in the significant increase of the catalytic distance (3.9 Å in the wild type vs. 5.1 Å in the M184V mutant) between the 3′-OH (last residue of the primer) and α-phosphate (d-3′-F-C-d4G-TP) (Figure 3b). Consequently, the incorporation of d-3′-F-C-d4G-MP into viral DNA chain in HIV-1M184V would be expected to be more difficult than in the HIVWT, reflecting its decreased antiviral activity against the mutant. In the case of carbovir-TP, the relative binding energy to the M184V mutation also decreased, but to a less extent than the d-3′-F-C-d4G-TP, which is in accordance with the biological data (Table 6). Binding mode analysis of carbovir-TP/HIV-RTM184V complex revealed that the hydrophobic interaction was almost maintained and catalytic distance experienced only a small increase (3.2 Å in wild type vs. 3.8 Å in M184V mutant, Figure 3c). These changes may not significantly affect the incorporation of carbovir-MP into viral DNA.
Table 6.
In vitro anti-HIV-1 activity of d-3′-F-C-d4G against wild type (WT) and M184V virus in human PBM cells using carbovir as positive control and correlation with calculated energy of complex (Inhibitor-TP) / HIV-RT after molecular dynamics simulations.
| Compound | xxBRU (WT) | M184V | FIb | ΔErelc | ||
|---|---|---|---|---|---|---|
| EC50 (μM) |
Erela (Kcal/mol) |
EC50 (μM) |
Erela (Kcal/mol) |
|||
| d-3′-F-C-d4G 35 | 0.098 | -270.2d | 3.8 | 84.9d | 38.8 | -355.1d |
| Carbovir | 0.087 | -310.0d | 0.20 | -63.4d | 2.3 | -246.6d |
Erel = (Binding energy of inhibitor-TP) – (Binding energy of natural 2′-dNTP)
FI is the Fold Increase (EC50 HIV-1M184V / EC50 HIV-1xxBRU).
ΔErel = Erel (WT) – Erel (M184V).
These values were calculated based on the molecular dynamics results (refer to experimental section)
Figure 3.
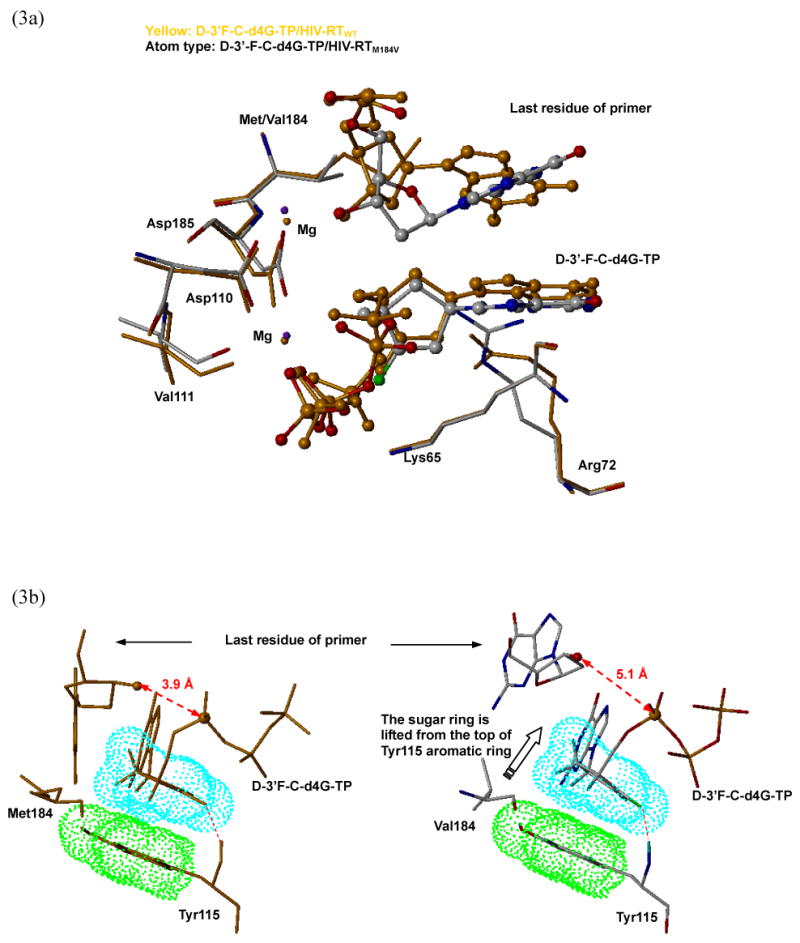

(a) Mutation of methionine to valine at the position 184 induces conformational changes of the key residues inside the active site, such as Asp185, Asp110, Val111 and Arg72. (b) The comparison of the bind modes of d-3′-F-d4G-TP with HIV-RTWT (yellow color, left) and HIV-RTM184V (atom type, right). The propagated effected from the mutation on the codon 184 lifted the d-3′-F-d4G-TP from the surface of Tyr115 causes a decrease of the hydrophobic interaction, resulting in a loss of binding affinity. Further more, the catalytic distance lengthened significantly (3.9 Å to 5.1 Å). (c) The comparison of the bind modes of carbovir-TP with HIV-RTWT (yellow color, left) and HIV-RTM184V (atom type, right). The binding mode is almost maintained in the mutant enzyme compared with the wild type enzyme.
In summary, molecular modeling studies illustrated the important roles of π-π interaction and the additional hydrogen bond in the binding affinity of d- and l-nucleosides in the HIV-RT activity site. The cross-resistance of compound 35 to HIV-RTM184V may be partially due to the primer/template repositioning and resultant increased catalytic distance and the loss of hydrophobic interaction.
Experiment Section
General Methods
Melting points were determined on a Mel-temp II apparatus and were uncorrected. Nuclear magnetic resonance spectra were recorded on a Varian Mercury 400 spectrometer at 400 MHz for 1H NMR and 100 MHz for 13C NMR or Varian Inova 500 spectrometer at 500 MHz for 1H NMR and 125 MHz for 13C NMR with tetramethylsilane as the internal standard. Chemical shifts (δ) are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or bs (broad singlet). UV spectra were recorded on a Beckman DU-650 spectrophotometer. Optical rotations were measured on a Jasco DIP-370 digital polarimeter. High resolution mass spectra were recorded on a Micromass Autospec high-resolution mass spectrometer. TLC was performed on Uniplates (silica gel) purchased from Analtech Co. Column chromatography was performed using either silica gel-60 (220-440 mesh) for flash chromatography or silica gel G (TLC grade, >440 mesh) for vacuum flash column chromatography. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA.
Microwave-Assisted Synthesis
Reactions were run in the Discover™ reactor module (CEM Corporation) of focused microwaves with a magnetron operating at a frequency at 2.45 GHz and a maximum power output of 300 W. The thick-wall tube was heated in a closed cavity located inside the instrument with continuous stirring. The temperature was measured by an IR pyrometer inside the reactor.
(+)-(1R,2S,3S,4R)-2,3-Anhydro-1-O-benzyloxy-4-(O-triphenylmethyloxymethyl)-cyclopentane (2)
To a suspension of epoxide 1 14 (16.0 g, 72.6 mmol) in anhydrous CH2Cl2, DMAP (4.4 g, 36.3 mmol), triethylamine (15.1 ml, 109 mmol) and trityl chloride (30.4 g, 109 mmol) were added at room temperature. The reaction mixture was stirred for 24 h at room temperature and concentrated in vacuo. EtOAc/H2O was added to the residue and the organic layer was collected, dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:40 to 1:20) to give 2 as a white solid (30.6 g, 91 %). mp 102-103 °C [α]24D +46.71° (c 1.47, CHCl3) 1H NMR (CDCl3, 500 MHz) δ 7.39-7.24 (m, 20H), 4.59 (d, J = 2.5 Hz, 2H), 4.17 (t, J = 7.5 Hz, 1H), 4.19-4.16 (m, 1H), 3.54 (s, 1H), 3.45 (s, 1H), 3.14-3.15 (m, 1H), 2.96-2.99 (m, 1H), 2.57 (d, J = 7.0 Hz, 1H), 1.66-1.55 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 143.9, 138.4, 128.7, 128.4, 127.9, 127.8, 127.7, 127.1, 86.8, 79.1, 71.7, 64.2, 57.9, 56.7, 39.2, 28.7. Anal. Calcd for (C32H30O3): C, H.
(+)-(1R,2S,4R)-1-O-Benzyloxy-2-hydroxyl-4-(O-triphenylmethyloxymethyl)-cyclopentane (3) and (+)-(1R,3S,4R)-1-O-Benzyloxy-3-hydroxyl-4-(O-triphenylmethyloxymethyl)-cyclopentane (4)
Epoxide 2 (26.0 g, 56.2 mmol) was dissolved in a 1.0 M THF solution of Super-Hydride® (180 mL, 180 mmol) at 0 °C. The suspension was allowed to warm up to room temperature and stirred for 30 min and EtOAc/H2O was added to quench the reaction. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:8) to give 3 (1.5 g, 6 %) as a colorless oil and 4 (24.0 g, 92 %) as a colorless oil. Compound 3: 1H NMR data is identical to the literature; 14 Compound 4: [α]25D +18.98° (c 0.93, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.43−7.22 (m, 20H), 4.48 (s, 2H), 4.01-3.96 (m, 2H), 3.25-3.22 (m, 1H), 2.96 (t, J = 8.5 Hz, 1H), 2.82 (d, J = 6.0 Hz, 1H), 2.49-2.44 (m, 1H), 2.17-2.04 (m, 2H), 1.94-1.89 (m, 1H), 1.44-1.25 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 144.1, 138.4, 128.7, 128.5, 127.9, 127.7, 127.6, 127.1, 86.8, 79.1, 76.6, 70.7, 66.2, 46.9, 40.7, 34.0. Anal. Calcd. for (C32H32O3) C, H.
(-)-(2R,4R)-4-O-Benzyloxy-2-(O-triphenylmethyloxymethyl)-cyclopentan-1-one (5)
To a solution of alcohol 4 (23.5 g, 50.6 mmol) in anhydrous CH2Cl2 (300 mL), 4 Å molecular sieve (40.5 g), pyridinium dichromate (37.6 g, 101.2 mmol) and acetic acid (4.4 mL, 76.0 mmol) were added. After stirred at room temperature for 3 h, Celite was added and stirred for another 30 min. The resulting brown slurry mixture was filtered over a Celite pad. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:50 to 1:20) to give ketone 5 (19.0 g, 81 %) as a white solid. mp: 106-108 °C; [α]25D −59.32° (c 0.62, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.38-7.20 (m, 20H), 4.52 (s, 2H), 4.35 (t, J = 5.0 Hz, 1H), 3.48-3.45 (m, 1H), 3.21-3.19 (m, 1H), 2.67-2.62 (m, 1H), 2.57-2.38 (m, 3H), 2.17-2.10 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 184.7, 143.9, 138.1, 128.7, 128.5, 127.8, 127.7, 127.6, 127.0, 86.6, 74.8, 70.6, 62.1, 46.6, 45.6, 33.0. Anal. Calcd. for (C32H30O3) C, H.
(+)-(2R,4R)-4-O-Benzyloxy-1,1-difluoro-2-(O-triphenylmethyloxymethyl)-cyclopentane (6)
Ketone 5 (19.0 g, 41.1 mmol) was dissolved in neat diethyl aminosulfur trifluoride (DAST, 86.0 mL, 656.2 mmol) at room temperature. After stirred at 40 °C for 36 h, the reaction mixture was diluted with 300 mL CH2Cl2 and then slowly added into saturated NaHCO3 (600 mL) solution. The organic layer was collected and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:100) to give 6 (13.5 g, 68 %) as a pale yellow syrup. [α]27D +3.00° (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.48-7.21 (m, 20H), 4.47 (s, 2H), 4.05 (s, 1H), 3.37-3.34 (m, 1H), 3.11-3.08 (m, 1H), 2.83-2.78 (m, 1H), 2.44-2.32 (m, 2H), 2.22-2.18 (m, 1H), 1.75-1.69 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 144.0, 138.1, 128.9, 130.7 (t, J = 252.5 Hz), 128.8, 128.5, 127.9, 127.8, 127.7, 127.6, 127.0, 86.8, 75.0, 70.8, 61.0 (d, J = 7.6 Hz), 44.7 (t, J = 21.5 Hz), 43.0 (t, J = 11.2 Hz), 34.3 (d, J = 6.1 Hz). Anal. Calcd. for (C32H30 F2O2) C, H.
(-)-(2R,4R)-1,1-Difluoro-4-hydroxy-2-hydroxymethyl-cyclopentane (7)
To a solution of 6 (12.7 g, 26.2 mmol) in anhydrous CH2Cl2 (300 mL), iodotrimethylsilane (11.2 mL, 78.6 mmol) was added at -20 °C. The reaction mixture was allowed to warm up to room temperature and stirred for 6 h. Additional portion of iodotrimethylsilane (5.6 mL, 39.3 mmol) was added and stirred for another 8 h. The reaction was quenched with MeOH at -20 °C and carefully neutralized with solid NaHCO3. The resulting brown mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:100 to 1:10) to give diol 7 (2.8 g, 70 %) as a pale brown oil. [α]25D −21.53° (c 0.90, MeOH); 1H NMR (500 MHz, CDCl3) δ 4.46-4.43 (m, 1H), 3.83-3.76 (m, 2H), 2.82-2.70 (m, 1H), 2.48-2.38 (m, 1H), 2.28-2.19 (m, 1H), 2.02-1.90 (m, 2H), 1.77 (bs, 2H); 13C NMR (125 MHz, CDCl3) δ 131.8 (dd, J = 252.4 and 248.0 Hz) 68.6 (t, J = 5.2 Hz), 60.5 (d, J = 8.6 Hz), 46.2 (t, J = 21.5 Hz), 45.7 (t, J = 23.9 Hz), 36.3 (d, J = 5.8 Hz). Anal. Calcd for (C6H10 F2O2) C, H.
(-)-(2R,4R)-2-(O-tert-Butyldiphenylsilyloxymethyl)-1,1-difluoro-4-hydroxy-cyclopentane (8)
To a solution of diol 7 (2.6 g, 17.1 mmol) and imidazole ( 1.9 g, 27.4 mmol) in anhydrous CH2Cl2 (100 mL) tert-butyldiphenylsilane chloride (4.8 mL, 18.8 mmol) was slowly added at 0 °C during 1 h. The resulting mixture was stirred at 0 °C for another 45 min and quenched with MeOH. The mixture was concentrated in vacuo and the residue was dissolved in EtOAc/H2O. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic layer was dried over MgSO4 and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:20 to 1:4) to give 8 (4.9 g, 74 %) as a colorless oil. [α]27D −14.62° (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.67-7.37 (m, 10H), 4.44 (s, 1H), 3.76 (ddd, J = 450.0, 10.5 and 5.5Hz, 2H), 2.81-2.69 (m, 1H), 2.50-2.40 (m, 1H), 2.24-2.16 (m, 1H), 2.04-1.95 (m, 2H), 1.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 135.6, 133.4, 133.4, 131.1, 129.7, 127.7, 68.7 (d, J = 6.7 Hz), 61.5 (d, J = 8.1 Hz), 46.4 (t, J = 22.4 Hz), 45.8 (t, J = 23.9 Hz), 37.1 (d, J = 5.2 Hz), 26.8, 19.2. Anal. Calcd. for (C22H28F2O2Si) C, H.
(-)-(2R,4R)-2-(O-tert-Butyldiphenylsilyloxymethyl)-1,1-difluoro-4-[(methylsulfonyl)oxy]cyclopentane (9)
Methanesulfonyl chloride (0.4 mL, 5.12 mmol) in 10 mL anhydrous CH2Cl2 (10 mL) was slowly added to a solution of alcohol 8 (1.00 g, 2.56 mmol) and triethylamine (1.4 mL, 10.2 mmol) in anhydrous CH2Cl2 (10 mL) at 0 °C. The reaction mixture was allowed to warm up to room temperature and kept for 4 h. After removing the solvent in vacuo, the residue was dissolved in EtOAc and washed with water. The organic layer was dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:20 to 1:10) to give 9 (1.17 g, 98 %) as a colorless oil. [α]25D −19.66° (c 0.32, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.67-7.38 (m, 10H), 5.26 (m, 1H), 3.76 (d, J = 5.5Hz, 2H), 3.03 (s, 3H), 2.74-2.50 (m, 3H), 2.36-2.32 (m, 1H), 2.18-2.12 (m, 1H), 1.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 135.6, 133.0 (d, J = 2.0 Hz), 129.9, 129.8 (t, J = 250.9 Hz), 127.8, 77.8 (dd, J = 3.75 and 7.6 Hz), 61.0 (dd, J = 2.9 and 4.8 Hz), 46.4 (dd, J = 21.5 and 23.9 Hz), 43.7 (t, J = 26.8 Hz), 34.8, 34.7 (t, J = 4.8 Hz), 26.8, 19.2. HR-MS Calcd. for (C23H30NF2O4SSi+H)+ 469.1680, found 469.1662.
(+)-(2R,4S)-4-Azido-2-(O-tert-butyldiphenylsilyloxymethyl)-1,1-difluoro-cyclopentane (10)
Compound 9 (1.17 g, 2.5 mmol) was dissolved in anhydrous DMF (45 mL) and heated at 130 °C for 1.5 h. After removing the solvent in vacuo, the residue was dissolved in EtOAc and washed with water. The organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:200 to 1:100) to give 10 (0.97 g, 93 %) as a colorless oil. [α]26D +4.2° (c 0.51, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.67-7.38 (m, 10H), 3.96 (m, 1H), 3.78 (ddd, J = 38.0, 11.0 and 5.5Hz, 2H), 2.56-2.34 (m, 1H), 2.21-2.10 (m, 1H), 1.82-1.75 (m, 1H), , 2.04-1.95 (m, 2H), 1.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 135.6, 133.2 (d, J = 3.4 Hz), 129.8, 128.9 (dd, J = 248.5 and 254.2 Hz), 127.8, 61.2 (dd, J = 7.1 and 2.9 Hz), 56.4 (dd, J = 6.1 and 3.8 Hz), 47.2 (dd, J = 23.4 and 21.0 Hz), 41.9 (t, J = 24.8 Hz), 32.9 (t, J = 1.5 Hz), 26.8, 19.2. HR-MS Calcd. for (C22H27F2N3OSi+H)+ 416.1970, found 416.2022.
(-)-(2R,4S)-2-(O-tert-Butyldiphenylsilyloxymethyl)-1,1-difluoro-cyclopentanamine (11)
A suspension of azido compound 10 (0.97 g, 2.33 mmol) and 10 % Pd/C (330 mg) in absolute EtOH was shaken under 30 psi of H2 at room temperature for 2.0 h. Celite was added into the solution and the slurry was filtered through a Celite pad. The volatile was removed in vacuo and the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:50 to 1:10) to give 11 (0.86 g, 95 %) as a colorless oil. [α]26D −9.98° (c 0.79, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.68-7.36 (m, 10H), 3.78 (ddd, J = 55.0, 10.0 and 5.0 Hz, 2H), 3.46-3.39 (m, 1H), 2.51-2.28 (m, 3H), 1.93-1.82 (m, 1H), 1.45-1.39 (m, 3H), 1.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 135.6 (d, , J = 2.4 Hz), 133.4 (d, , J = 1.2 Hz), 130.0 (dd, , J = 252.8 and 3.2 Hz), 129.7, 127.8, 61.6 (dd, J = 7.6 and 1.9 Hz), 48.0 (dd, J = 23.4 and 20.2 Hz), 47.9 (dd, J = 6.1 and 4.2 Hz), 45.8 (t, J = 22.4 Hz), 37.3 (d, J = 4.8 Hz) 26.8, 19.3. HR-MS Calcd. for (C22H29F2NOSi+H)+ 390.2065, found 390.2030. Anal. Calcd. for (C22H29F2NOSi) C, H, N.
(+)-(1′S,4′R)-9-[6′-(O-tert-Butyldiphenylsilyloxymethyl)-2′,3′-dideoxy-3′,3′-difluoro-cyclopentanyl]uracil (13)
To a suspension of silver cyanate (810 mg, 5.4 mmol) in anhydrous benzene (20 mL), β–methoxyacryloyl chloride (650 mg, 5.4 mmol) was added. The mixture was heated under reflux for 30 min and cooled to room temperature. The supernatant solution was added into the solution of amine 11 (700 mg, 1.8 mmol) in anhydrous THF (30 mL) at −30 °C during 15 min. The mixture was allowed to gradually warmed up to room temperature and kept overnight. After removing the solvent in vacuo, the residue was purified by column chromatography on a silica gel (EtOAc:hexanes = 1:3 to 1:1) to give crude 12 (600 mg) as a yellow syrup which was directly used for the next step. Crude compound 12 (600 mg) was dissolved in 1,4-dioxane/ethanol (20 mL/20 mL) and treated with 28 % solution of ammonium hydroxide (20 mL) in a steel bomb at 90 -100 °C for 17 h. After removing the solvent in vacuo, the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:200 to 1:100) to give 13 (360 mg, 41 %) as a pale yellow syrup. [α]26D +13.12° (c 0.31, CHCl3); UV (H2O) λmax 266.0 (MeOH); 1H NMR (500 MHz, CDCl3) δ 9.22 (s, 1H), 7.67-7.38 (m, 10H), 7.22 (d, J = 8.5 Hz, 1H), 5.68 (d, J = 8.0 Hz, 1H), 5.26-5.19 (m, 1H), 3.88 (dtd, J = 11.0, 10.0 and 5.0 Hz, 2H), 2.70-2.60 (m, 1H), 2.2.52-2.44 (m, 2H), 2.27-2.16 (m, 1H), 1.79-1.70 (m, 1H), 1.07 (s, 9H); 13C NMR (125 MHz, CD3OD) δ 162.8, 150.9, 140.0 (d, J = 2.0 Hz), 135.6, 133.1, 133.0, 129.9, 128.3 (t, J = 250.9 Hz), 127.9, 103.6, 61.0 (d, J = 5.2 Hz), 49.6, 47.0 (t, J = 21.9 Hz), 40.7 (t, J = 24.9 Hz), 32.3 (d, J = 4.2 Hz), 26.8, 19.3. HR-MS Calcd. for (C26H30F2N2O3Si+H)+ 485.2072, found 485.2169 Anal. Calcd. for (C26H30F2N2O3Si) C, H, N.
(+)-(1′S,4′R)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-(O-tert-butyldiphenylsilyloxymethyl)-cyclopentanyl]cytosine (14)
To a solution of uracil derivative 13 (360 mg, 0.74 mmol) in anhydrous acetonitrile (25 mL), 2,4,6-triisopropyl benzenesulfonyl chloride (450 mg, 1.48 mmol), 4-(dimethylamino)pyridine (90.4 mg, 0.74 mmol) and triethylamine (0.42 mL, 3.0 mmol) were added at 0 °C. After stirred at room temperature for 12 h, 28 % solution of ammonium hydroxide (15 mL) was added to the brown mixture and stirred at room temperature for another 12 h. The reaction mixture was concentrated in vacuo and the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:30) to give 14 (300 mg, 84 %) as a white solid. mp 250-252 °C [α]24D +15.86° (c 0.31, MeOH); UV (MeOH) λmax 272.0 nm; 1H NMR (500 MHz, CD3OD) δ 7.59-7.30 (m, 10H), 7.44 (d, J = 7.0 Hz, 1H), 5.75 (d, J = 7.5 Hz, 1H), 5.00-4.92 (m, 1H), 3.88 (ddd, J = 56.0, 10.5 and 6.0 Hz, 2H), 2.59-2.45 (m, 2H), 2.30-2.19 (m, 2H), 1.78-1.71 (m, 1H), 0.96 (s, 9H); 13C NMR (125 MHz, CD3OD) δ 165.8, 157.4, 142.0, 135.4, 133.1, 133.0, 129.7, 128.8 (t, J = 250.9 Hz), 127.5, 95.2, 61.1 (d, J = 8.1 Hz), 52.0 (d, J = 5.8 Hz ), 40.2 (t, J = 24.8 Hz), 32.0 (d, J = 4.2 Hz), 25.9, 18.6. HR-MS Calcd. for (C26H31F2N3O2Si+H)+ 484.2232, found 484.2212 Anal. Calcd. for (C26H31F2N3O2Si) C, H, N.
(+)-(1′S,4′R)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]cytosine (15)
To a solution of 14 (300 mg, 0.62 mmol) in MeOH (2 mL), 3 N HCl (2 mL) was added. After stirred at room temperature for 17 h, the resulting mixture was co-evaporated with EtOH and the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:15) to give 15 (110 mg, 72%) as a white solid. mp 134-136 °C; [α]23D +4.07° (c 0.22, MeOH); UV (H2O) λmax 275.0 nm (MeOH), 1H NMR (400 MHz, CD3OD) δ 7.72 (d, J = 8.0 Hz, 1H), 5.95 (d, J = 7.0 Hz, 1H), 5.14-5.07 (m, 1H), 3.88 (ddd, J = 47.0, 11.5 and 5.5 Hz, 2H), 2.70-2.35 (m, 4H), 1.97-1.90 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 165.8, 157.4, 142.0, 128.8 (t, J = 249.2 Hz), 95.2, 59.0 (d, J = 7.6 Hz), 52.0 (t, J = 7.6 Hz ), 40.1 (t, J = 25.1 Hz), 32.0 (d, J = 3.8 Hz). HR-MS Calcd. for (C10H13F2N3O2+H)+ 246.1054, found 246.0975 Anal. Calcd. for (C10H13F2N3O2·0.65H2O) C, H, N.
(+)-(1′S,4′R)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′,3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]cytosine (18)
To a suspension of 15 (110 mg, 0.45 mmol) in anhydrous THF:1,4-dioxane (10 mL:10 mL) co-solvent, potassium tert-butoxide (121 mg, 1.0 mmol) was added. The reaction mixture was stirred at 90 °C for 9 h. The yellow suspension was filtered through a short silica gel pad and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:30 to 1:15) to give 18 (35 mg, 35 %) as a white solid. mp 244-248 °C; [α]25D +127.49° (c 0.36, MeOH); UV (H2O) λmax 284.0 nm (ε 17115, pH 2), 274.0 nm (ε 11975, pH 7), 274.0 nm (ε 11680, pH 11); 1H NMR (400 MHz, CD3OD) δ 7.83 (d, J = 7.5 Hz, 1H), 5.90 (d, J = 7.5 Hz, 1H), 5.62-5.61 (m, 1H), 5.19 (s, 1H), 3.70 (ddd, J = 132.5, 11.5 and 3.5 Hz, 2H), 2.95-2.93 (m, 1H), 2.86-2.79 (m, 1H), 1.72-1.69 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 166.1, 166.0 (d, J = 282.8 Hz), 157.6, 142.5, 103.3 (d, J = 13.0 Hz), 94.3, 59.8, 55.9 (d, J = 12.2 Hz), 42.9 (d, J = 19.0 Hz), 31.7 (d, J = 6.1 Hz). Anal. Calcd. for (C10H12F1N3O2) C, H, N.
(+)-(1′S,4′R)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]thymine (17)
To a suspension of silver cyanate (400 mg, 2.7 mmol) in anhydrous benzene (8 mL), β-methoxy-α-methacryloyl chloride (360 mg, 2.7 mmol) was added. The mixture was heated under reflux for 30 min and cooled to room temperature. The supernatant solution was added into the solution of amine 11 (350 mg, 0.89 mmol) in anhydrous THF (8 mL) at −30 °C during 15 min. The mixture was allowed to gradually warmed up to room temperature and kept overnight. After removing the solvent in vacuo, the residue was purified by column chromatography on a silica gel (EtOAc:hexanes = 1:3 to 1:1) to give crude 16 (360 mg) as a yellow syrup which was directly used for the next step. Crude compound 16 (360 mg) was dissolved in 1,4-dioxane (60 mL) and treated with 3 N HCl (15 mL) at the refluxed temperature for 2.5 h. After removing the solvent in vacuo, the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:50 to 1:20) to give 17 (145 mg, 63 %) as a white solid. mp 140-142 °C; [α]23D +3.9° (c 0.15, MeOH); UV (H2O) λmax 270.0 nm; 1H NMR (400 MHz, CD3OD) δ 7.52 (s, 1H), 5.07-4.98 (m, 1H), 3.76 (ddd, J = 33.2, 10.8 and 5.2 Hz, 2H), 2.63-2.32 (m, 4H), 1.98-1.89 (m, 4H); 13C NMR (100 MHz, CD3OD) δ 164.8, 151.4, 137.6, 128.7 (dd, J = 250.8 and 248.5 Hz), 110.6, 58.9 (dd, J = 7.6 and 2.3 Hz), 50.8 (t, J = 6.8 Hz), 39.5 (t, J = 25.1 Hz), 31.3 (d, J = 3.8 Hz), 11.0. Anal. Calcd. for (C11H14F2N2O3) C, H, N.
(+)-(1′S,4′R)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]thymine (19)
Compound 17 (140 mg, 0.54 mmol) was converted to thymine derivative 19 (60 mg, 46 %) as a white solid using the same procedure as for 18. mp 182-184 °C (dec.); [α]26D +23.99° (c 0.26, MeOH); UV (H2O) λmax 272.0 nm (ε 14258, pH 2), 272.0 nm (ε 14240, pH 7), 271.0 nm (ε 11651, pH 11); 1H NMR (400 MHz, CD3OD) δ 7.69 (s, 1H), 5.58-5.53 (m, 1H), 5.15 (s, 1H), 3.69 (ddd, J = 121.6, 11.6 and 3.2 Hz, 2H), 2.91-2.90 (m, 1H), 2.78-2.70 (m, 1H), 1.76-1.70 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 165.2, 166.0 (d, J = 282.7), 151.6, 138.2, 109.7, 103.4 (d, J = 13.7 Hz), 59.6 (d, J = 1.5 Hz), 54.7 (d, J = 12.2 Hz), 42.7 (d, J = 18.3 Hz), 30.9 (d, J = 6.1 Hz), 11.0. Anal. Calcd. for (C11H13FN2O3•0.1H2O) C, H, N.
(-)-(1′S,4′R)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-(O-tert-butyldiphenylsilyloxymethyl)-cyclopentanyl]adenine (21)
Triphenylphosphine (2.12 g, 8.1 mmol) and diisopropyl azodicarboxylate (1.59 mL, 8.1 mmol)) were dissolved in anhydrous THF:1,4-dioxane (14 mL:7 mL) co-solvent and cooled to 0 °C. The resulting yellowish suspension was further cooled to −78 °C. 6-Chloropurine (1.25 g, 8.1 mmol) and a solution of alcohol 8 (630 mg, 1.61 mmol) in THF (14 mL) were added successively. The resulting mixture was kept at −78 °C for 0.5 h and then stirred at room temperature for 24 h. MeOH was added to quench the reaction and the mixture was evaporated in vacuo. The residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:10) to give 20 as a crude product, which was treated with methanolic ammonia in a steel bomb at 100 °C for 24 h. After evaporation in vacuo, the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:50 to 1:20) to give 21 as a colorless syrup (305 mg, 37 % from 8). [α]25D −10.89° (c 0.45, CHCl3); UV (MeOH) λmax 260.0; 1H NMR (500 MHz, CDCl3) δ 8.34 (s, 1H), 7.84 (s, 1H), 7.68-7.38 (m, 10H), 5.07 (m, 1H), 3.92 (ddd, J = 31.0, 10.5 and 5.0 Hz, 2H), 2.91-2.81 (m, 1H), 2.73-2.56 (m, 3H), 2.28-2.22 (m, 1H), 2.06 (bs, 1H), 1.08 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.6, 153.0, 150.2, 138.2, 135.6, 135.6, 133.2, 133.1, 129.9, 128.4 (dd, J = 252.4 and 250.0 Hz), 127.8, 120.0, 61.1 (d, J = 5.8 Hz), 49.8 (t, J = 4.8 Hz), 47.5 (t, J = 21.5 Hz), 33.7 (d, J = 17.0 Hz), 26.8, 19.3. Anal. Calcd. for (C27H31F2N5OSi) C, H, N.
(-)-(1′S,4′R)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]adenine (22)
Compound 21 (300 mg, 0.59 mmol) was dissolved in MeOH (5 mL) and treated with 3N HCl (10 mL) at room temperature for 16 h. After neutralizing with solid NaHCO3, the resulting suspension was filtered and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:50 to 1:20) to give 22 (140 mg, 88 %) as a white solid. mp 156-158 °C; [α]25D −9.86° (c 0.56, MeOH); UV (H2O) λmax 260.0 (MeOH); 1H NMR (500 MHz, CD3OD) δ 8.28 (s, 1H), 8.24 (s, 1H), 5.13 (m, 1H), 3.86 (ddd, J = 50.0, 11.5 and 6.0 Hz, 2H), 2.89-2.77 (m, 2H), 2.71-2.57 (m, 2H), 2.36-2.30 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 156.0, 152.3, 149.4, 139.5, 128.9 (dd, J = 251.8 and 248.0 Hz), 119.0, 59.1 (dd, J = 8.1 and 1.9 Hz), 50.4 (dd, J = 7.6 and 4.4 Hz), 41.1 (t, J = 25.8 Hz), 33.0 (d, J = 3.9 Hz). Anal. Calcd. for (C11H13F2N5O) C, H, N.
(-)-(1′S,4′R)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]adenine (26)
Compound 22 (60 mg, 0.22 mmol) was converted to adenine derivative 26 (27 mg, 49 %) as a white solid using the same procedure as for 18. mp 225 °C (dec.); [α]29D −43.80° (c 0.18, DMSO); UV (MeOH) λmax 261.0 nm (ε 17931, pH 2), 261.0 nm (ε 17780, pH 7.4), 261.0 nm (ε 18556, pH 11); 1H NMR (500 MHz, CD3OD) δ 8.31 (s, 1H), 8.24 (s, 1H), 5.69-5.64 (m, 1H), 5.44 (s, 1H), 3.75 (ddd, J = 126.5, 11.0 and 4.0 Hz, 2H), 3.09-3.04 (m, 1H), 3.02-3.00 (m, 1H), 2.05-2.00 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 166.3 (d, J = 283.2 Hz), 156.0, 152.2, 148.8, 139.8, 118.9, 103.2 (d, J = 13.8 Hz), 60.1, 54.0 (d, J = 12.4 Hz), 43.2 (d, J = 18.6 Hz), 32.2 (d, J = 6.2 Hz). Anal. Calcd. for (C11H12FN5O) C, H, N.
(-)-(1′S,4′R)-9-[2′,3′-Dideoxy-2′,2′-difluoro-6-hydroxymethylcyclopentanyl] hypoxanthine (23)
A crude 6-chloropurine analogue 20 (850 mg) was treated with 85 % formic acid (40 mL) at 90 °C for 3 h. After completely removing the volatile in vacuo, the residue was dissolved in concentrated ammonium hydroxide (35%) and stirred at room temperature overnight. The solution was evaporated in vacuo and the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:30 to 1:15) to give 23 (93 mg, 39 % from 8) as a white solid. mp 244-246 °C; [α]24D −10.08° (c 0.23, MeOH); UV (MeOH) λmax 248.0 nm; 1H NMR (500 MHz, CD3OD) δ 8.22 (s, 1H), 8.09 (s, 1H), 5.18-5.10 (m, 1H), 3.84 (ddd, J = 53.5, 11.5 and 6.0 Hz, 2H), 2.93-2.76 (m, 2H), 2.70-2.58 (m, 2H), 2.36-2.29 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 157.6, 148.8, 145.0, 139.0, 128.8 (dd, J = 251.9 and 248.0 Hz), 124.3, 59.1 (d, J = 6.2 Hz), 50.7 (dd, J = 7.1 and 4.2 Hz), 41.3 (t, J = 25.8 Hz), 33.1 (d, J = 4.2 Hz). Anal. Calcd. for (C11H12F2N4O2) C, H, N.
(-)-(1′S,4′R)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]hypoxanthine (27)
Compound 23 (22 mg, 0.081 mmol) was converted to inosine derivative 27 (10 mg, 49 %) as a white solid using the same procedure as for 18. mp 226-230 °C; [α]27D −33.05° (c 0.20 MeOH); UV (H2O) λmax 249.0 nm (ε 12220, pH 2), 249.0 nm (ε 12820, pH 7), 255.0 nm (ε 13552, pH 11); 1H NMR (500 MHz, CD3OD) δ 8.26 (s, 1H), 8.08 (s, 1H), 5.66-5.72 (m, 1H), 5.43 (s, 1H), 3.75 (ddd, J = 124.5, 11.5 and 4.0 Hz, 2H), 3.03-3.09 (m, 1H), 2.97 (dt, J = 14.0 and 9.5 Hz, 1H), 2.05-2.00 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 166.4 (d, J = 283.9 Hz), 157.6, 148.4, 145.1, 139.2, 123.9, 103.1 (d, J = 14.8 Hz), 60.0, 54.2 (d, J = 12.9 Hz), 43.3 (d, J = 18.6 Hz), 32.4 (d, J = 5.8 Hz). Anal. Calcd. for (C11H11FN4O2·0.1H2O) C, H, N.
(-)-(1′S,4′R)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]guanine (25)
A solution of alcohol 8 (600 mg, 1.54 mmol), tripheylphosphine (1.61 g, 6.1 mmol) and 6-chloro-N2-isobutyrylpurine (1.47 g, 6.1 mmol) in anhydrous THF (20 mL) was cooled to 0 °C and then diisopropyl azodicarboxylate (1.2 mL, 6.1 mmol) was slowly added during 2 h. The reaction mixture was allowed to warm up to room temperature and stirred for 4 h. The clear yellowish solution was concentrated in vacuo and the residue was purified by column chromatography on a silica gel (EtOAc:Hexanes = 1:10 to 1:1) to give the corresponding nucleoside 24 as a crude product, which was treated with 85% formic acid (30 mL) at 90 °C for 4 h. After completely removing the volatile, the residue was further treated with methanolic ammonia at room temperature for 24 h. After the concentration in vacuo, the residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:20 to 1:10) to give 25 (80 mg, 18 % from 8) as a white solid. mp: 253 °C (dec.); [α]27D −9.30° (c 0.25, MeOH); UV (MeOH) λmax 253.0 nm; 1H NMR (500 MHz, CD3OD) δ 7.86 (s, 1H), 3.83 (ddd, J = 50.5, 11.5 and 5.5 Hz, 2H), 2.81-2.72 (m, 2H), 2.61-2.51 (m, 2H), 2.30-2.22 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 158.0, 153.7, 151.8, 136.4, 128.9 (dd, J = 252.2 and 248.0 Hz), 116.5, 59.1 (dd, J = 8.1 and 2.4 Hz), 50.0 (dd, J = 7.1 and 3.4 Hz), 41.1 (t, J = 25.8 Hz), 32.8 (d, J = 3.4 Hz). HR-MS Calcd. for (C11H13N5F2O2+H)+ 286.1116, found 286.1137.
(+)-(1′S,4′R)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]guanine (28)
To a suspension of 25 (70 mg, 0.25 mmol) in anhydrous DMF (8 mL), potassium tert-butoxide (120 mg, 1.0 mmol) was added. The reaction mixture was stirred at 70 °C for 24 h. The yellow suspension was filtered through a short silica gel pad and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:20 to 1:10) to give 28 (30 mg, 45 %) as a white solid. mp 224-227 °C (dec.); [α]27D +40.13° (c 0.11, MeOH); UV (H2O) λmax 253.0 nm (ε 13343, pH 2), 251.0 nm (ε 13635, pH 7), 260.0 nm and 268.0 nm (ε 11470 and 11893, respectively, pH 11); 1H NMR (500 MHz, CD3OD) δ 7.92 (s, 1H), 5.52-5.46 (m, 1H), 5.37 (s, 1H), 3.74 (ddd, J = 114.0, 11.0 and 4.0 Hz, 2H), 3.05-3.00 (m, 1H), 2.90 (dt, J = 14.0 and 9.0 Hz, 1H), 2.01-1.96 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 165.9 (d, J = 282.9 Hz), 158.0, 153.8, 151.2, 136.6, 116.2, 103.1 (d, J = 13.9 Hz), 60.1, 53.2 (d, J = 12.9 Hz), 43.2 (d, J = 19.1 Hz), 32.2 (d, J = 5.6 Hz). Anal. Calcd. for (C11H12FN5O2·1.1H2O) C, H, N.
(+)-(2S,4S)-2-(O-tert-Butyldiphenylsilyloxymethyl)-1,1-difluoro-4-hydroxy-cyclopentane (29)
[α]24D +14.97° (c 0.83, CHCl3); 1H NMR (500 MHz, CD3OD) δ 7.67-7.66 (m, 4H), 74.5-7.37 (m, 11H), 4.44 (bs, 1H), 3.80 (dd, J = 5.0 and 10.5 Hz, 1H), 3.72 (dd, J = 6.0 and 10.5 Hz, 1H), 2.75 (m, 1H), 2.45 (m, 1H), 2.20 (q, J = 14.0 Hz, 1H), 2.02-1.98 (m, 2H), 1.05 (s, 9H); 13C NMR (125 MHz, CD3OD) δ 135.6, 133.3 (d, J = 3.9 Hz), 133.2, 131.2, 129.8 (d, J = 1.4 Hz), 129.1, 127.8 (d, J = 1.4 Hz), 68.8, 61.5, 46.4 (t, J = 21.4 Hz), 46.0 (t, J = 28.1 Hz), 37.1 (d, J = 5.3 Hz), 26.8, 19.3. Anal. Calcd. for (C22H28F2O2Si) C, H, N.
(+)-(2S,4R)-2-(O-tert-Butyldiphenylsilyloxymethyl)-1,1-difluoro-cyclopentanamine (30)
[α]26D +10.02° (c 0.68, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.68-7.37 (m, 10H), 3.86-3.70 (m, 2H), 3.49-3.39 (m, 1H), 2.52-2.28 (m, 3H), 1.94-1.79 (m, 1H), 1.45-1.39 (m, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.6 (d, , J = 2.4 Hz), 133.4, 130.0 (dd, , J = 253.1 and 249.2 Hz), 129.7, 127.7, 61.6 (dd, J = 7.7 and 2.3 Hz), 48.1 (m), 48.0 (dd, J = 22.9 and 21.3 Hz), 46.1 (t, J = 22.9 Hz), 37.3 (d, J = 4.6 Hz) 26.8, 19.2. Anal. Calcd. for (C22H29F2NOSi•0.2H2O) C, H, N.
(+)-(1′R,4′S)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]adenine (31)
mp 158-160 °C; [α]25D +9.50° (c 0.33, MeOH); UV (H2O) λmax 260.0 (MeOH); 1H NMR (500 MHz, CD3OD) δ 8.24 (s, 1H), 5.12 (m, 1H), 3.92 (ddd, J = 50.0, 11.5 and 5.5 Hz, 2H), 2.85 (m, 2H), 2.65 (m, 2H), 2.32 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 155.9, 152.3, 149.3, 139.5, 128.7 (dd, J = 251.6 and 248.0 Hz), 119.0, 59.2 (t, J = 5.8 Hz), 50.3, 41.1 (t, J = 25.8 Hz), 32.9. Anal. Calcd. for (C11H13F2N5O) C, H, N.
(+)-(1′R,4′S)-9-[2′,3′-Dideoxy-3′-difluoro-6′-hydroxymethylcyclopentanyl]guanine (32)
mp 258 °C (dec.); [α]28D +9.29° (c 0.15, MeOH); UV (H2O) λmax 254.0 nm (ε 10631, pH 2), 252.0 nm (ε 11008, pH 7), 256.0 nm (ε 9234, pH 11); mp >250 °C; 1H NMR (500 MHz, CD3OD) δ 7.83 (s, 1H), 3.79 (ddd, J = 51.0, 14.5 and 6.5 Hz, 2H), 2.77-2.70 (m, 2H), 2.57-2.54 (m, 2H), 2.32-2.21 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 158.0, 153.7, 151.8, 136.4, 128.9 (dd, J = 251.5 and 246.9 Hz), 116.5, 59.1 (d, J = 8.4 Hz), 49.9 (d, J = 3.8 Hz), 41.0 (t, J = 25.9 Hz), 32.8 (d, J = 3.8 Hz). Anal. Calcd. for (C11H13F2N5O2•0.3H2O) C, H, N.
(+)-(1′R,4′S)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6-hydroxymethylcyclopentanyl]hypoxanthine (33)
mp 227 °C; [α]23D +9.97° (c 0.45, MeOH); UV (MeOH) λmax 247.0 nm; 1H NMR (500 MHz, CD3OD) δ 8.25 (s, 1H), 8.09 (s, 1H), 5.14 (m, 1H), 3.84 (ddd, J = 55.0, 11.5 and 6.0 Hz, 2H), 2.82 (m, 2H), 2.65 (m, 2H), 2.32 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 157.6, 148.8, 145.0, 139.0, 128.7 (dd, J = 251.5 and 247.5 Hz), 124.3, 59.0 (dd, J = 8.0 and 2.4 Hz), 50.7, 41.3 (t, J = 25.9 Hz), 33.1 (d, J = 4.8 Hz). Anal. Calcd. for (C11H12F2N4O2) C, H, N.
(+)-(1′R,4′S)-9-[2′,3′-Dideoxy-2′3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]adenine (34)
mp 218-220 °C (dec.); [α]22D +41.00° (c 0.13, DMSO); UV (MeOH) λmax 260.0 nm (ε 10925, pH 2), 261.0 nm (ε 10807, pH 7), 261.0 nm (ε 11780, pH 11); 1H NMR (500 MHz, CD3OD) δ 8.31 (s, 1H), 8.24 (s, 1H), 5.66 (br, 1H), 5.44 (s, 1H), 3.75 (ddd, J = 130.0, 11.0 and 3.5 Hz, 2H), 3.05 (br, 1H), 2.98 (m, 1H), 2.00 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 166.2 (d, J = 287.5 Hz), 155.9, 152.1, 148.8, 139.8, 118.8, 103.2 (d, J = 14.3 Hz), 60.0, 53.9 (t J = 12.9 Hz), 43.2 (d, J = 18.6 Hz), 32.2 (d, J = 5.8 Hz); MS: m/z 250 (M+1); Anal. Calcd. for (C11H12FN5O) C, H, N.
(-)-(1′R,4′S)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]guanine (35)
To a suspension of 33 (60 mg, 0.21 mmol) in anhydrous DMF (5 mL), potassium tert-butoxide (82 mg, 0.69 mmol) was added. The reaction mixture in the thick-walled tube was placed in a microwave synthesizer and irradiated at maximum output power of 300 W with air-cooling at 70 °C for 10 min. The brown suspension was filtered through a short silica gel pad and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on a silica gel (MeOH:CH2Cl2 = 1:20 to 1:10) to give 35 (40 mg, 72 %) as a white solid. mp 220 °C (dec.); [α]25D −44.12° (c 0.11, MeOH); UV (H2O) λmax 253.0 nm (ε 13553, pH 2), 252.0 nm (ε 14393, pH 7), 256.0 nm and 268.0 (ε 11186 and 11829, respectively, pH 11); 1H NMR (500 MHz, CD3OD) δ 7.92 (s, 1H), 5.49 (m, 1H), 5.37 (s, 1H), 3.74 (ddd, J = 113.5, 11.5 and 4.0 Hz, 2H), 3.02 (m, 1H), 2.90 (dt, J = 14.0 and 9.0 Hz, 1H), 2.00-1.97 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 165.9 (d, J = 282.0 Hz), 158.0, 153.8, 151.2, 136.6, 116.2, 103.1 (d, J = 13.7 Hz), 60.0, 53.2 (d, J = 12.9 Hz), 43.2 (d, J = 18.3 Hz), 32.2 (d, J = 5.4 Hz). Anal. Calcd. for (C11H12FN5O2•0.9H2O) C, H, N.
(+)-(1′R,4′S)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]hypoxanthine (36)
mp 254-256 °C; [α]27D +34.01° (c 1.51, MeOH); UV (H2O) λmax 249.0 nm (ε 10354, pH 2), 248.5 nm (ε 13925, pH 7), 254.0 nm (ε 10142, pH 11); 1H NMR (500 MHz, CD3OD) δ 8.15 (s, 1H), 8.08 (s, 1H), 5.68 (m, 1H), 5.44 (s, 1H), 3.86 (dd, J = 10.5 and 4.5 Hz, 1H), 3.60 (m, 1H), 2.96-2.85 (m, 2H), 1.97 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 166.4 (d, J = 283.9 Hz), 157.6, 148.4, 145.1, 139.2, 123.9, 103.1 (d, J = 14.8 Hz), 60.0, 54.2 (d, J = 12.9 Hz), 43.3 (d, J = 18.6 Hz), 32.4 (d, J = 5.8 Hz). Anal. Calcd. for (C11H11FN4O2) C, H, N.
(-)-(1′R,4′S)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]cytosine (37)
mp 132-134 °C; [α]26D −4.62° (c 0.3, MeOH); UV (H2O) λmax 282.0 nm (ε 16381, pH 2), 274.0 nm (ε 10363, pH 7), 273.0 nm (ε 11099, pH 11); 1H NMR (500 MHz, CD3OD) δ 7.72 (d, J = 7.5 Hz, 1H), 5.95 (d, J = 7.0 Hz, 1H), 5.14-5.07 (m, 1H), 3.81 (ddd, J = 47.0, 11.5 and 5.5 Hz, 2H), 2.70-2.35 (m, 4H), 1.97-1.90 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 165.8, 157.5, 142.2, 128.9 (dd, J = 250.9 and 249.0 Hz), 95.2, 59.0 (t, J = 5.6 Hz), 52.0 (t, J = 6.2 Hz ), 40.1 (t, J = 25.2 Hz), 32.0 (d, J = 4.4 Hz). Anal. Calcd. for (C10H13F2N3O2•0.6H2O) C, H, N.
(-)-(1′R,4′S)-9-[2′,3′-Dideoxy-3′,3′-difluoro-6′-hydroxymethylcyclopentanyl]thymine (38)
[α]26D −3.2° (c 0.25, MeOH); mp 142-144 °C; UV (H2O) λmax 271.0 nm (ε 8478, pH 2), 271.0 nm (ε 8509, pH 7), 270.0 nm (ε 7587, pH 11); 1H NMR (400 MHz, CD3OD) δ 7.56 (s, 1H), 5.08-5.02 (m, 1H), 3.80 (ddd, J = 33.2, 9.2 and 4.8 Hz, 2H), 2.65-2.37 (m, 4H), 2.01-1.92 (m, 4H); 13C NMR (125 MHz, CD3OD) δ 164.9, 151.4, 137.7, 128.8 (dd, J = 250.9 and 248.0 Hz), 110.7, 58.9 (dd, J = 8.0 and 2.4 Hz), 50.8 (t, J = 4.2 Hz), 39.5 (t, J = 23.6 Hz), 31.4 (d, J = 3.8 Hz), 11.0. Anal. Calcd. for (C11H14F2N2O3) C, H, N.
(-)-(1′R,4′S)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]cytosine (39)
Compound 37 (60 mg, 0.24 mmol) was converted to cytosine derivative 39 (46 mg, 84 %) as a white solid using the same procedure as for 35. mp 220-230 °C; [α]25D −124.63° (c 0.33, MeOH); UV (H2O) λmax 283.0 nm (ε 15638, pH 2), 274.0 nm (ε 10805, pH 7), 274.0 nm (ε 10512, pH 11); 1H NMR (500 MHz, CD3OD) δ 7.83 (d, J = 7.5 Hz, 1H), 5.90 (d, J = 7.5 Hz, 1H), 5.63-5.59 (m, 1H), 5.19 (s, 1H), 3.70 (ddd, J = 132.5, 10.5 and 3.5 Hz, 2H), 2.95-2.93 (m, 1H), 2.82 (td, J = 14.0 and 9.0 Hz, 1H), 1.73-1.68 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 166.1, 166.0 (d, J = 283.2 Hz), 157.5, 142.6, 103.3 (d, J = 13.4 Hz), 94.3, 59.8 (d, J = 1.9 Hz), 55.9 (d, J = 8.1 Hz), 42.9 (d, J = 18.6 Hz), 31.7 (d, J = 6.1 Hz). Anal. Calcd. for (C10H12FN3O2) C, H, N.
(-)-(1′R,4′S)-9-[2′,3′-Dideoxy-2′,3′-didehydro-3′-fluoro-6′-hydroxymethylcyclopent-2-enyl]thymine (40)
mp 184-186 °C (dec.); [α]25D −24.17° (c 0.15, MeOH); UV (H2O) λmax 272.0 nm (ε 15633, pH 2), 273.0 nm (ε 15750, pH 7), 271.0 nm (ε 12548, pH 11); 1H NMR (400 MHz, CD3OD) δ 7.69 (d, J = 1.2 Hz, 1H), 5.57-5.54 (m, 1H), 5.15 (s, 1H), 3.69 (ddd, J = 121.6, 11.6 and 3.2 Hz, 2H), 2.93-2.90 (m, 1H), 2.78-2.70 (td, J = 14.4 and 9.2 Hz, 1H), 1.76-1.70 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 165.2, 166.0 (d, J = 282.7), 151.6, 138.2, 109.7, 103.4 (d, J = 13.7 Hz), 59.6 (d, J = 2.3 Hz), 54.7 (d, J = 12.2 Hz), 42.7 (d, J = 18.3 Hz), 30.9 (d, J = 6.1 Hz), 11.0. Anal. Calcd. for (C11H13FN2O3) C, H, N.
Antiviral and Cytotoxicity Assay
HIV drug susceptibility assays were performed as previously described.28 Cytotoxicity assays in PBM, CEM and Vero cells were conducted as previously described.29
Molecular Modeling Study
(a) Conformational analysis
The initial conformations of inhibitors were constructed by builder module in MACROMODEL®, version 8.5 (Schrodinger, Inc.) based on the crystal structure of carbovir. The Monte Carlo conformational search was performed in 5,000-step, in the presence of GB/SA water model using MMFFs force field in MACROMODEL.
(b) Binding affinity study to HIV-1 reverse transcriptase
All molecular modeling studies of the enzyme-substrate complexes were performed using Sybyl® 7.0 (Tripos Associates, St. Louis, MO) on a Silicon Graphics Tezro® workstation or a SGI Origin 300 workstation. The enzyme site of the enzyme-ligand complex was built based on the X-ray structure of the covalently trapped catalytic complex of HIV-1 RT with TTP and primer-template duplex (PDB entry 1rtd).30 A model of the NRTI binding site was built, which consisted of residues between Lys1 and Pro243 in the p66 subunit, and a 7:4 (template-primer) duplex. The conformationally optimized structures of carbocyclic nucleosides were used to define the initial Cartesian coordinates. The heterocyclic moiety of the n+1th nucleotide in the template overhang was modified to the base complementary to the incoming NRTIs if needed, i.e. the adenine moiety which was in the original X-ray structure (1rtd)30 was modified to guanine. The inhibitor triphosphates were manually docked to the active site of the enzyme by adjusting the torsional angles to those found in the X-ray structure.30 Gästeiger-Hückel charges were then given to the nucleoside triphosphate with formal charges (+2) to the two Mg atoms in the active site and Kollman-All-Atom charges were loaded to the enzyme site using the biopolymer module in Sybyl. Fluorine parameters were obtained from literature31, 32 and MM2 parameters were entered into the parameter files. In order to eliminate local strains resulting from merging inhibitors and/or point mutations, residues inside 6 Å from the merged inhibitors and mutated residues were annealed until energy change from one iteration to the next was less than 0.05 Kcal/mol. The annealed enzyme-inhibitor complexes were minimized by using Kollman-All-Atom force field until iteration number reached 5,000.
The structures (d-3′-F-C-d4G-TP/HIV-RTWT, d-3′-F-C-d4G-TP/HIV-RTM184V, carbovir-TP/HIV-RTWT, carbovir-TP/HIV-RTM184V, GTP/HIV-RTWT and GTP/HIV-RTM184V) were further confirmed by the molecular dynamics studies using MACROMODEL®, version 9.1 (Schrodinger, Inc.). The complex was minimized until there was no significant movement in atomic coordinates using MMFF94s force field in the presence of GB/SA continuum water model before performing molecular dynamics simulations. A conjugate gradient, Polak-Ribiere 1st derivative method was used for energy minimization.
Molecular dynamics simulations on nucleoside-TP/RT complex was performed with MMFF94s in the presence of GB/SA continuum water model on a SGI Origin 300 workstation running the IRIX 6.5 operating system by heating from 0 to 300K over 5 ps and equilibrating at 300K for an additional 10 ps. Production dynamics simulations were carried out for 500 ps with a step size of 1.5 fs at 300 K. A shake algorithm was used to constrain covalent bonds to hydrogen atoms. A distance constrain was used to constrained the two magnesium atoms with Asp110, Val111, Asp185, α- and β-phosphate of nucleotide. For simulation of the nucleoside-TP/RT complex, the residues further away than 15 Å from the active site were not considered and the residues from 6 to 15 Å were constrained by harmonic constraints. Only residues inside 6 Å sphere from the bound nucleoside-TP were allowed to move freely.
Supplementary Material
Elemental analysis data for compounds 2-40. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 4.

Reagents and conditions: (a) tBuOK, THF/1,4-dioxane, 90 °C, conventional oil bath heating for 34, 36; tBuOK, DMF, 70 °C, microwave-assisted for 35; (b) tBuOK, THF/1,4-dioxane, 60 °C, traditional oil bath heating for 40; tBuOK, DMF, 70 °C, microwave-assisted for 39.
Acknowledgments
This research was supported by the U.S. Public Health Service Grants (AI25899, AI32351 & AI41980) from the National Institute of Allergy and Infectious Diseases Emory's CFAR Grand 5-P30-AI-50409 and the Department of Veterans Affairs.
References
- 1.De Clercq E. New approaches toward anti-HIV chemotherapy. J Med Chem. 2005;48(5):1297–1313. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]
- 2.Richman DD, Fischl MA, Grieco MH, Gottlieb MS, Volherding PA, Laskin OL, Leedom JM, Groopman JE, Mildvan D, Hirsch MS, Jackson GG, Durack DT, Phil D, Lusinoff-Lehrman S. The AZT collaborative working group. The toxicity of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. N Engl J Med. 1987;317:192–197. doi: 10.1056/NEJM198707233170402. [DOI] [PubMed] [Google Scholar]
- 3.Yarchoan R, Pluda JM, Thomas RV, Mitsuya H, Brouwers P, Wyvill KM, Hartman N, Johns DG, Broder S. Long term toxicity/activity profiles of 2′,3′-dideoxyinosine in AIDS or AIDS-related complex. Lancet. 1990;336:526–529. doi: 10.1016/0140-6736(90)92085-v. [DOI] [PubMed] [Google Scholar]
- 4.Strames MC, Cheng YC. Celluar metabolism of 2′,3′-dideoxycytidine, a compound active against human immunodeficiency virus in vitro. J Biol Chem. 1987;252:988–991. [PubMed] [Google Scholar]
- 5.Peterson EA, Ramirez-Ronda CH, Hardy WD, Schwartz R, Sacks HS, Follansbee S, Peterson DM, Cross A, Anderson RE, Dunkle LM. Dose-related activity of stavudine in patients infected with human immunodeficiency virus. J Infect Dis. 1995;171(Suppl 2):S131–S139. doi: 10.1093/infdis/171.supplement_2.s131. [DOI] [PubMed] [Google Scholar]
- 6.Crimmins MT. New development in the enantioselective synthesis of cyclopentyl carbocyclic nucleosides. Tetrahedron. 1998;54:9229–9272. [Google Scholar]
- 7.Choi Y, Lee K, Hong JH, Schinazi RF, Chu CK. Synthesis and anti-HIV activity of l-2′-fluoro-2′,3′-unsaturated purine nucleosides. Tetrahedron Lett. 1998;39:4437–4440. [Google Scholar]
- 8.Lee K, Choi Y, Gullen E, Schlueter-Wirtz S, Schinazi RF, Cheng YC, Chu CK. Synthesis and anti-HIV and anti-HBV activities of 2′-fluoro-2′,3′-unsaturated L-nucleosides. J Med Chem. 1999;42:1320–1328. doi: 10.1021/jm980651u. [DOI] [PubMed] [Google Scholar]
- 9.Lee K, Choi Y, Gumina G, Zhou W, Schinazi RF, Chu CK. Structure-activity relationships of 2′-fluoro-2′,3′-unsaturated d-nucleosides as anti-HIV agents. J Med Chem. 2002;45:1313–1320. doi: 10.1021/jm010418n. [DOI] [PubMed] [Google Scholar]
- 10.Chong Y, Choo H, Choi Y, Mathew J, Schinazi RF, Chu CK. Stereoselective synthesis and antiviral activity of d-2′,3′-didehydro-2′,3′-dideoxy-2′-fluoro-4′-thionucleosides. J Med Chem. 2002;45:4888–4898. doi: 10.1021/jm020246+. [DOI] [PubMed] [Google Scholar]
- 11.Choo H, Chong Y, Choi Y, Mathew J, Schinazi RF, Chu CK. Synthesis, anti-HIV activity and molecular mechanism of drug resistance of l-2′,3′-didehydro-2′,3′-dideoxy-2′-fluoro-4′-thionucleosides. J Med Chem. 2003;46:389–398. doi: 10.1021/jm020376i. [DOI] [PubMed] [Google Scholar]
- 12.Zhu W, Chong Y, Choo H, Mathews J, Schinazi RF, Chu CK. Synthesis, structure-activity relationships, and mechanism of drug resistance of D- and L-β-3′-fluoro-2′,3′-unsaturated-4′-thionucleosides as anti-HIV agents. J Med Chem. 2004;47:1631–1640. doi: 10.1021/jm0303148. [DOI] [PubMed] [Google Scholar]
- 13.Zhou W, Gumina G, Chong Y, Wang J, Schinazi RF, Chu CK. Synthesis, structure-Activity relationships and drug resistance of β-D-3′-fluoro-2′,3′-unsaturated nucleosides as anti-HIV agents. J Med Chem. 2004;47:3399–3408. doi: 10.1021/jm040027j. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Jin YH, Rapp KL, Bennett M, Schinazi RF, Chu CK. Synthesis, antiviral activity and mechanism of drug resistance of D- and L- 2′,3′-didehydro-2′,3′-dideoxy-2′-fluoro-carbocyclic nucleosides. J Med Chem. 2005;48:3736–3748. doi: 10.1021/jm050096d. [DOI] [PubMed] [Google Scholar]
- 15.Shealy YF, O'Dell CA, Thorpe MC. carbocyclic analogues of thymidine nucleosides and related 1-substituted thymines. J Heterocycl Chem. 1981;18:383–389. [Google Scholar]
- 16.Shealy YF, O'Dell CA. Synthesis of the carbocyclic analogues of uracil nucleosides. J Heterocycl Chem. 1976;13:1015–1020. [Google Scholar]
- 17.Polpin EA, Corbett T, Flaherty L, Tarasoff P, Redman BG, Valdivieso M, Baker L. Difluorodeoxycytidine (dFdC, gemcitabine): A phase I study. Invest New Drugs. 1992;10:165–170. doi: 10.1007/BF00877241. [DOI] [PubMed] [Google Scholar]
- 18.Schinazi RF, Lloyd RM, Jr, Nguyen MH, Cannon DL, McMillan A, Ilksoy N, Chu CK, Liotta DC, Bazmi HZ, Mellors JW. Characterization human immunodeficiency viruses resistant to oxthiolane-cytosine nucleosides. Antimicrob Agents Chemother. 1993;37:875–881. doi: 10.1128/aac.37.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naeger LK, Margot NA, Miller MD. Increased drug susceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovudine-associated mutations: analysis of enzyme processivity, chain-terminator removal and viral replication. Antiviral Res. 2001;6:115–126. [PubMed] [Google Scholar]
- 20.a Gao H, Boyer PL, Sarafianos SG, Arnold E, Hughes SH. The role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J Mol Biol. 2000;300:403–418. doi: 10.1006/jmbi.2000.3823. [DOI] [PubMed] [Google Scholar]; b Schinazi RF, Schlueter-Wirtz S, Stuyver L. Early detection of mixed mutations selected by antiretroviral agents in HIV-infected primary human lymphocytes. Antimicrob Agents Chemother. 2001;12(Suppl 1):61–65. [PubMed] [Google Scholar]
- 21.Vince R, Hua M, Brownell J, Daluge S, Fangchen Lee, Shannon WM, Lavelle GC, Qualls J, Weislow OS, Kiser R, Canonico PG, Schultz RH, Narayanan VL, Mayo JG, Shoemaker RH, Boyd MR. Potent and selective activity of a new carbocyclic nucleoside analog (carbovir: NSC 614846) against human immunodeficiency virus in vitro. Biochem Biophys Res Commun. 1988;156:1046–1053. doi: 10.1016/s0006-291x(88)80950-1. [DOI] [PubMed] [Google Scholar]
- 22.Smith MS, Kessler JA, Rankin CD, Pagano JS, Kurtzberg J, Carter SG. Evaluation of synergy between carbovir and 3′-azido-2′,3′-deoxythymidine for inhibition of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 1993;37:144–147. doi: 10.1128/aac.37.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daluge SM, Good SS, Faletto MB, Miller WH, St Clair MH, Boone LR, Tisdale M, Parry NR, Reardon JE, Dornsife RE, Averett DR, Krenitsky TA. 1592U89, a novel carbocyclic nucleoside analog with potent selective anti-human immunodeficiency virus activity. Antimicrob Agents Chemother. 1997;41:1082–1093. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrigan PR, Stone C, Griffin P, Najera I, Bloor S, Kemp S, Tisdale M, Larder B. Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. J Infect Dis. 2000;181:912–920. doi: 10.1086/315317. [DOI] [PubMed] [Google Scholar]
- 25.Tisdale M, Alnadaf T, Cousens D. Combination of mutations in human immunodeficiency virus type 1 reverse transcriptase required for resistance to the carbocyclic nucleoside 1592U89. Antimicrob Agents Chemother. 1997;41:1094–1098. doi: 10.1128/aac.41.5.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ray AS, Yang Z, Shi J, Hobbs A, Schinazi RF, Chu CK, Anderson KS. Insights into the molecular mechanism of inhibition and drug resistance for HIV-1 RT with carbovir triphosphate. Biochemistry. 2002;41:5150–5162. doi: 10.1021/bi0121858. [DOI] [PubMed] [Google Scholar]
- 27.Sarafinos SG, Das K, Clark AD, Jr, Ding J, Boyer PL, Hughes SH, Arnold E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with β–branched amino acids. Proc Natl Acad Sci USA. 1999;96:10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MY, Hart GC, Hahn EF. Activity of 3′-azido-3′-deoxythymidine nucleotide dimers in primary lymphocytes infected with human immunodeficiency virus type 1. Antimicrob Agents Chemother. 1990;34:1061–1067. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuyer LJ, Lostia S, Adams M, Mathew JS, Pai BS, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antiviral activities and cellular toxicities of modified 2′,3′-dideoxy-2′,3′-didehydrocytidine analogues. Antimicrob Agents Chemother. 2002;46:3854–2860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 31.a Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]; b http://www.amber.uscf.edu/amber/Questions/fluorine.html.
- 32.Dunitz JD, Taylor R. Organic fluorine hardly ever accepts hydrogen bonds. Chem Eur J. 1997;3:89–98. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data for compounds 2-40. This material is available free of charge via the Internet at http://pubs.acs.org.