

Abstract
The cyanobacterium Synechocystis sp. PCC 6803 (S6803) expresses a two-on-two globin in which His46 (distal side) and His70 (proximal) function as heme iron axial ligands. His46 can be displaced by O2, CO, and CN−, among others, whereas His70 is not labile under native conditions. The residue preceding the proximal histidine has been implicated in controlling globin axial ligand reactivity; the details of the mechanism, however, are not well understood, and little information exists for bis-histidyl hexacoordinate proteins. In many vertebrate hemoglobins and in the Synechocystis protein, the position is occupied by an alanine whereas, in myoglobins, it is a serine involved in an intricate hydrogen bond network. We examined the role of Ala69 in S6803 hemoglobin through the effects of an Ala → Ser replacement. The substitution resulted in minor structural perturbations, but the holoprotein's response to temperature-, urea-, and acid-induced denaturation was measurably affected. Enhanced three-state behavior was manifested in the decoupling of heme binding and secondary structure formation. Urea-gradient gel experiments revealed that the stability of the apoprotein was unchanged by the replacement and that a slight alteration of the folding kinetics occurred in the holoproteins. Cyanide-binding experiments were performed to assess trans effects. The apparent rate constant for association decreased two-fold upon Ala69Ser replacement. This deceleration was attributed to a change in the lifetime of a state containing a decoordinated His46. The results demonstrated that, as in vertebrate globins and leghemoglobin, proximal influences operate to determine fundamental dynamic and thermodynamic properties of the protein.
Keywords: truncated hemoglobin, 2-on-2 globin, hexacoordinate hemoglobin, urea-gradient gel, heme binding, thermodynamic stability
Iron protoporphyrin IX (Fe-PPIX)1 serves as a cofactor in a large number of essential proteins. In the absence of covalent attachment to the protein matrix via protoporphyrin substituents, the contact between Fe(II)-PPIX (or b heme) and the protein is limited to ligation bond(s) to the iron, hydrogen bonding, van der Waals and electrostatic interactions, and hydrophobic forces. The affinity for the heme group and its chemical properties, such as redox potential, ability to bind various ligands, and propensity for generating reactive oxygen species, are strictly controlled by the protein environment.
In many b hemoproteins the iron is endogenously hexacoordinated, with the axial positions of its octahedral geometry filled by two histidine side chains (1). A well-known example is cytochrome b5, an electron-transfer protein that does not bind exogenous ligands readily. Bis-histidine coordination of the heme group is also observed in a small number of proteins belonging to the hemoglobin (Hb) superfamily. Recently discovered instances include Drosophila Hb (2), certain cyanobacterial globins (3-5), neuroglobin (6), and cytoglobin (7-9). The α subunit of adult human Hb is also capable of bis-histidine coordination of the ferric iron, particularly when in complex with a protective protein (10, 11). In these hexacoordinate globins, the axial histidines belong to the F helix (proximal side of the heme) and the E helix (distal side). It has been proposed that hexacoordination in globins facilitates reduction of the iron (12), moderates the reactivity of the heme (11), and enhances thermal stability (13).
Despite much effort applied to the study of proteins and model compounds (1), the mechanism by which iron redox potential and axial bond strength are regulated is not entirely understood. Likewise, the role of the coordination bonds in stabilizing the protein fold is not generally predictable. The globins mentioned above offer new ground for detailed analysis. In each case, coordination of the distal histidine is reversible, and an exogenous ligand can bind with measurable affinity to the heme. This property brings into focus the accessibility of the iron, the relative axial bond strength, and the respective role of proximal and distal effects in controlling heme reactivity. In this work, we used a cyanobacterial globin to explore the effects of a conservative substitution in the F helix in a bis-histidine protein.
Proximal effects have been investigated in pentacoordinate globins, and several studies provide context for the cyanobacterial system presented here. In pentacoordinate globins, substitutions at positions other than the proximal histidine (labeled F8 in the Perutz nomenclature), for example at F4 and F7, generally do not change the coordination and spin state of the iron or the fold of the protein. These residues, however, are often involved in packing interactions (14, 15) and hydrogen-bonding networks (16) that adjust essential characteristics of the protein, such as proximal Fe-N bond length and heme reactivity (17, 18). Proximal residues control solvent access (19) and contribute to the positioning of the proximal histidine and the iron relative to the heme plane, which, in turn, relate to the affinity for exogenous ligands (20) and the affinity for the heme group (21). The latter property has been shown to control the thermodynamic stability of holomyoglobin (holoMb) (22). In comparison to distal effects, proximal effects are subtle (23), but complex (21). This is well illustrated by F-helix swapping experiments in Hb and Mb and their consequences on heme spectroscopic properties (24, 25); whereas Mb can be converted into an α-type globin by insertion of α-Hb residues, such conversion does not take place in β-Hb. Thus, proximal interactions, coupled or not with subunit interactions, play a role in dictating the behavior of pentacoordinate globins.
Among the many differences in sequence observed throughout the globin family, the identity of the amino acid at the position preceding the proximal histidine (corresponding to F7 in vertebrate globins) is of particular interest. In most Mbs, the wild-type residue is a serine. The neutron diffraction structure of sperm whale Mb establishes that the NδH atoms of the proximal histidine are within H-bonding distance of the seryl oxygen at F7 and the backbone oxygen of leucine F4 (16). Furthermore, the F7 hydroxyl group forms a hydrogen bond with the 7-propionate. This region of the Mb structure is shown in Figure 1A. Replacement of the serine has diverse outcomes depending on the source organism of the protein. It is capable of altering the geometry of the proximal histidine with respect to the heme group (26-29), increasing the affinity for small ligands in a modest trans effect (26), promoting heme loss (26, 27), and facilitating solvent access in the heme pocket (26). In vertebrate Hbs, residues at F7 possess side chains that are unable to participate in H-bonding interactions. Such is also the case in leghemoglobin (Lb) A, which contains a valine at that position; introduction of a serine at F7 does not increase the resemblance to Mb and speaks to the correlation of interactions on the proximal side of the heme (21).
Figure 1.
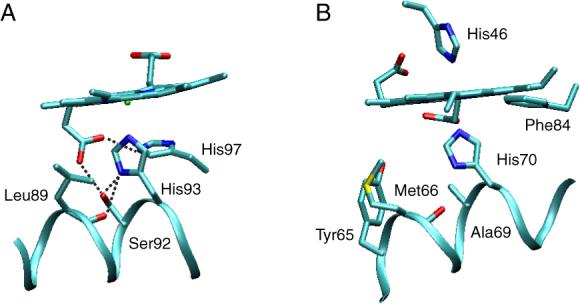
The environment of the proximal histidine in sperm whale Mb (5mbn, A) and Synechocystis rHb-R (1mwb, B). In A, the NδH group of His93 forms a hydrogen bond with Ser92 Oγ and Leu89 O. In B, the NδH group of His93 forms a hydrogen bond with Met66 O. The replacement of Ala69 with a Ser brings an O acceptor in the proximity of His70 NδH.
The hexacoordinate globin of interest to this study is encoded in the genome of Synechocystis sp. PCC 6803 (or S6803), a cyanobacterium incapable of nitrogen fixation. The protein belongs to the subfamily of “truncated globins” (trHbs). TrHbs are distantly related to the better-known vertebrate globins and can be organized into three groups according to phylogeny (30, 31). Thus far, a few structures have been solved for Group I and Group II trHbs. They present unique features: a distinctive helical topology referred to as the 2-on-2 fold (32), a heme pocket accessible via an apolar tunnel, and an elaborate network of H-bonds on the distal side of the heme (33).
Many Group I trHbs, including that from Synechocystis, contain an alanine immediately before the proximal residue (Figure 1B). Whether an exogenous ligand is bound or not, the proximal histidine of S6803 Hb assumes a staggered orientation with respect to the heme pyrrole nitrogens (Figure 2) (34-36), whereas the orientation of the analogous residue is eclipsed in Mb. S6803 rHb also has unusual ligand binding properties (37-39), in part because of endogenous hexacoordination. To examine the role of residue F7, a serine was introduced at this position. In vitro, S6803 rHb can attach the heme covalently through the formation of a bond between His117 and the heme 2-vinyl (40). In the absence of information on the physiologically relevant form(s) of the protein, we first focused on the protein in which this cross-link, extremely rare among Hbs, was not formed (rHb-R). NMR structural characterization, denaturation studies, and cyanide-binding experiments were performed to establish proximal influences in determining the globin properties. We report on the subtle changes induced by the H-OH substitution and compare these to observations made in selected pentacoordinate globins.
Figure 2.
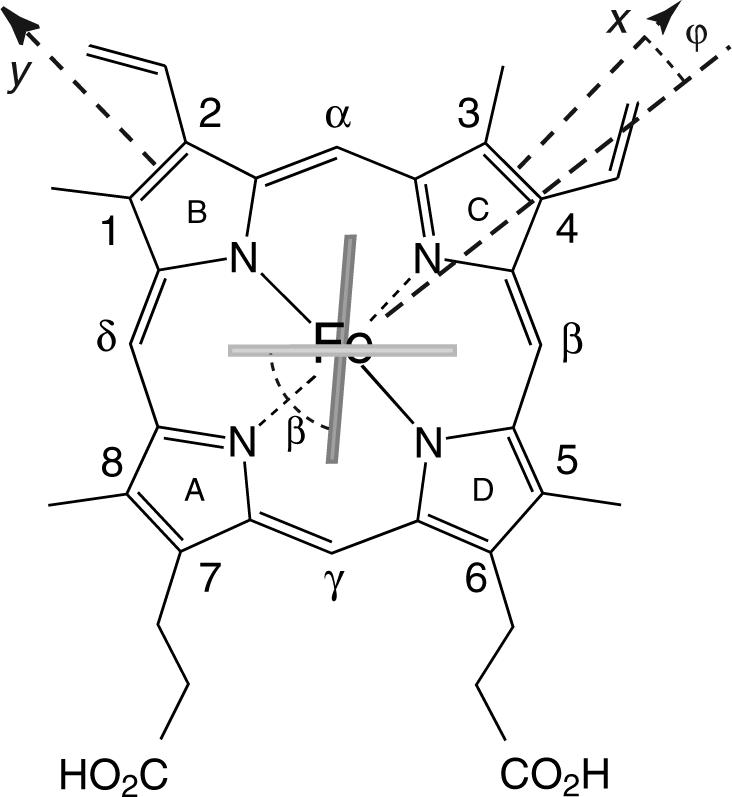
Structure of the b heme group with the nomenclature used in this work. The light and dark rectangles indicate the orientation of the proximal His70 and distal His46, respectively, in S6803 rHb-R. The ϕ and β angles characterize the geometry of the axial ligands: β is the acute angle between the two imidazole planes, and ϕ is the angle between the bisector of the two planes and the x axis. The proximal His93 in Mb is oriented along the NB–ND axis (not shown).
MATERIALS AND METHODS
Protein Preparation
The A69S S6803 Hb gene was generated from that encoding the wild-type protein with the Stratagene Site-Directed Mutagenesis protocol (La Jolla, CA). The Hbs (wild-type S6803 rHb-R and A69S S6803 rHb-R) were prepared from apoprotein inclusion bodies following the procedure established previously (41, 42). The identity of the protein was verified by sequencing of the gene (Penn State Nucleic Acid Facility) and by mass spectrometry (electrospray ionization) of the gene product. The A69S replacement had no consequence on the behavior of the protein throughout preparation and purification. The cross-linked proteins (S6803 rHb-A and A69S S6803 rHb-A) were prepared by treatment with dithionite followed by passage through a G-25 column as previously described (42). Protein concentrations were determined on a ferric heme basis with extinction coefficients of 100 mM−1 cM−1 at 410 nm (rHb-R, 41) and 75 mM−1 cM−1 at 409 nm (rHb-A) obtained with the hemochromogen assay (5).
NMR Spectroscopy
NMR samples were prepared in 20 mM phosphate buffer (95:5 1H2O:2H2O). Protein concentration was ∼1.5 mM. 1H NMR spectra were collected on a Bruker DRX-600 spectrometer operating at a proton frequency of 600.05 MHz. 1H chemical shifts were referenced to DSS through the water line with correction for pH and temperature (43). 15N NMR data were collected on an AMX2−500 spectrometer operating at a 15N frequency of 50.68 MHz. The cyanide complex of A69S S6803 rHb-R was prepared by addition of an excess of KC15N (Cambridge Isotopes), incubation, and adjustment of pH. A total of 10,000 to 100,000 transients were collected with a 100-kHz spectral width, 8 k complex data points, and 600-ms recycling time. 15N chemical shifts were verified with a solution of 4 M ammonium nitrate in 2 M nitric acid. Only chemical shift differences are reported in the text.
DQF-COSY, NOESY, TOCSY, and 1H-13C HMQC data were collected for A69S S6803 rHb-R as for the wild-type protein. The parameters for the NMR experiments (acquisition and processing) have been reported elsewhere (4). NMR data were processed with NMRpipe (44) or XWIN-NMR (Bruker BioSpin). Analysis was performed with XWIN-NMR and Sparky (45).
Optical spectroscopy
CD data were collected on either an AVIV Instruments Model 62 DS spectropolarimeter or a Jasco J-810 spectropolarimeter. Absorbance data were collected on an AVIV Instruments Model 14D spectrophotometer. All instruments were equipped with a Peltier device for temperature control and an automated titrator.
Thermal Denaturation
Thermal denaturation was monitored by CD and absorbance spectroscopy. For the absorbance experiments, ∼5−10 μM holoprotein (all holoprotein concentration measurements are reported on a per ferric heme basis) in 20 mM phosphate buffer (pH 7.2) was heated from 25 to 95 °C in 2-°C steps; spectra were collected between 260 and 700 nm after a 5-min equilibration period. Reversibility was inspected by refolding the protein from 95 to 25 °C, collecting spectra at 3-°C intervals with 7-min equilibration period. The reversibility of the holoprotein unfolding reaction was limited, likely owing to heme and protein damage at high temperature. Absorbance data at 412 nm (bound heme) and 365 nm (free heme) were used for fitting purposes. For the CD experiments, the protein concentration was 4 to 5 μM. Data were collected between 200 and 270 nm in 2-°C increments from 25 to 95 °C. Data at 209 and 222 nm were used for analysis. Thermal denaturation data were fit with the program Savuka (46) according to a two-state Gibbs-Helmholtz equation adapted for the dependence of the fully folded and fully unfolded signals on temperature (47):
| (1) |
where ΔH°Tm is the enthalpy of denaturation at the midpoint temperature (Tm). ΔG°(T) was obtained via –RT ln KU(T) from the fractional population of the native state, FN(T):
| (2) |
The observed signal, Y(T), is a function of FN(T), of the fractional population in the unfolded state (FU(T) = 1 − FN(T)), and of the spectral properties of these limiting states (YN(T) and YU(T)):
| (3) |
The ΔC°p parameter in Equation 1 was fixed to a value typical for a protein of this size (48). Adjusting the value of ΔC°p within reasonable limits did not change the results of the fit.
Chemical Denaturation
Ultra-pure urea was purified further on a mixed-bed resin column before use. For the titration experiments, the unfolded protein stock solution was 4 to 5 μM (per heme basis) in 9.5 to 10 M urea, 20 mM phosphate buffer, pH 7.2; the folded protein solution (identical protein concentration) was in 20 mM phosphate buffer (pH 7.2). The extent of secondary structure at 25 °C was monitored throughout the titration by measuring the ellipticity in the range from 200 to 270 nm. After automated combination of the folded and unfolded protein solutions, the mixture was left to equilibrate for 5 min with stirring prior to data collection. The stock and final urea concentrations were determined by measurement of the index of refraction (Leica Abbe Mark II Refractometer). Reversibility of the chemical unfolding reaction was determined by dilution into buffer of a highly concentrated solution of protein in 8 to 9 M urea. Both the A69S variant and the wild-type protein exhibited 100 % reversibility. Absorbance data (412 and 370 nm) and CD data (209 and 222 nm) were analyzed with Savuka to obtain the free energy of unfolding and the dependence of ΔG° on denaturant concentration [D]. A linear dependence was assumed:
| (4) |
.
Although evidence was found for the population of at least one equilibrium unfolding intermediate (see results), individual denaturation curves were fit satisfactorily with a two-state model.
Heme Release upon Solution Acidification
Release of the heme group from the protein matrix was monitored by absorbance spectroscopy. Hydrochloric acid was used to adjust the pH of a 10-μM holoprotein solution (5 mM phosphate buffer) from 7.4 to 2.6 in steps of 0.1 to 0.3 pH units. To minimize volume loss, the pH was determined without removing the solution from the cuvette. Spectra were collected between 260 and 700 nm at each pH. Refolding of the protein was inspected by returning the solution to neutrality with potassium hydroxide at the end of the titration. The absorbance data at 410 nm were fit with Nfit (University of Texas, Galveston) to a modified Henderson-Hasselbalch equation accounting for two distinct transitions:
| (5) |
where Y0 is the absorbance of the protein at the basic limit, Yi is the absorbance value of protein having gone through the ith protonation transition, and ni is the Hill coefficient for that transition.
Urea-gradient Gel Electrophoresis
As a second method of chemical unfolding, the proteins were subjected to urea denaturation within the confines of an acrylamide matrix (49). A mixing chamber and a peristaltic pump set at 1 mL min−1 were used to create a linear gradient ranging from 0 to 8 M urea perpendicular to the direction of electrophoresis. The gradient accounted for 6 cm of the 10-cm width of the gel, with 2-cm regions of 0 and 8 M urea at the appropriate edges. To counteract the increased viscosity of the gel matrix as a function of increasing urea concentration, a reverse gradient of acrylamide (11 to 7 %) was applied. To prevent polymerization of the acrylamide solutions before the gels were completely poured, APS and TEMED were replaced by methylene blue, sodium p-toluene sulfinate, and diphenyliodonium chloride. Gels were poured in the dark, polymerized by exposure to a 15-W light bulb for ∼30 min, and stored at 4 °C for 12 to 24 h prior to use. Gels were pre-electrophoresed at 100 V for 30 to 60 min to remove urea contaminates. After sample loading, electrophoresis was allowed to proceed for ∼4 h at 100 V. Samples contained ≥ 50 μg protein, bromophenol blue and xylene cyanol FF (used for tracking the progress of the electrophoresis), and 8 M urea (denatured samples only). Results were visualized with either Coomassie brilliant blue dye for protein or a heme-staining protocol (50, 51).
Kinetics of Cyanide Binding
Cyanide binding was monitored by visible spectroscopy. Data were collected with protein concentrations of ∼11 μM and a 200-, 500- or 1000-fold excess of cyanide. The absorbance at 423 nm (wavelength of maximum difference between free and bound states) was monitored throughout the reaction at 30-s intervals up to a total time affording an absorbance estimate for the bound state. This manual mixing method had a dead-time of 30 s at the shortest. Data were fit to a sum of exponential functions with Nfit. An approximate pseudo-first-order rate constant was obtained for the phase that accounted for the highest fraction of the population, typically > 70%. The values at the three cyanide concentrations were used to obtain an apparent bimolecular rate constant as in previous work on the wild-type and H117A rHb-Rs (37). All measurements were performed in triplicate at pH 7.2 (100 mM phosphate) and were repeated in D2O (pH 7.2, uncorrected for isotope effect). A triplicate data set was also collected on wild-type S6803 rHb-R with a 200-fold excess of cyanide at pH 8.2. The existence of multiple phases complicated data analysis and increased the uncertainty on the fitted values. This was manifested in extrapolated pseudo-first-order rate constants in the absence of cyanide that were negative in some cases. For this reason, the data were used in a semi-quantitative fashion.
RESULTS
Characterization of A69S S6803 rHb-R in the Ferric State
The hemichrome state of the protein (ferric, bis-histidine coordination) is a paramagnetic, S = ½ complex. The chemical shifts of the heme, axial histidines, and heme pocket residues were therefore expected to be sensitive reporters of structural and electronic perturbations arising from the A69S replacement. Figure 3 presents the one-dimensional spectrum of ferric A69S S6803 rHb-R along with that of the wild-type protein. The similarities are evident and support that the A69S replacement caused only minor three-dimensional and electronic changes to rHb-R. Resonance assignments for the heme group and for nearby residues were readily derived from an analysis of the homonuclear NOESY and TOCSY data and comparisons to data collected on H117A and wild-type rHb-Rs. Figure S1 in the supplementary information illustrates a section of the NOESY data that was used for axial histidine identification. The chemical shift differences between wild-type and A69S rHb-Rs were modest and localized on the proximal side of the heme, within a few residues of the replacement. On the distal side of the heme group, only His46 displayed chemical shift deviations larger than 0.1 ppm. Table 1 summarizes the chemical shift information for the axial residues and the heme group.
Figure 3.

1H spectrum of (A) ferric wild-type S6803 rHb-R, (B) ferric A69S S6803 rHb-R, and (C) cyanomet A69S S6803 rHb-R. Data were collected at 25 °C, pH 7.2. In traces A and B, the labels are as previously used (4, 41): a, heme 5-CH3; b, heme 2-α vinyl; c, His70 NδH; d, heme 1-CH3; e, His46 NδH; x, y, 2-β vinyls; z, His46 CεH. The resonance at 22.5 ppm in trace C arises from Tyr22 OηH hydrogen-bonded to the cyanide ligand.
Table 1.
Selected 1H NMR chemical shifts for S6803 rHba
| Assignmentb | WTc | A69S | Δ | |
|---|---|---|---|---|
| heme | 1-methyl | 15.03 | 15.21 | +0.18 |
| 3-methyl | 9.99 | 9.44 | −0.55 | |
| 5-methyl | 21.28 | 21.95 | +0.67 | |
| 8-methyl | 10.33 | 9.68 | −0.65 | |
| 2-α-vinyl | 15.56 | 15.86 | +0.30 | |
| trans-2-β-vinyl | −4.57 | −4.77 | −0.20 | |
| cis-2-β-vinyl | −5.22 | −5.45 | −0.23 | |
| 4-α-vinyl | 6.80 | 6.34 | −0.46 | |
| trans-4-β-vinyl | −1.53 | −1.40 | +0.13 | |
| cis-4-β-vinyl | −2.05 | −1.91 | +0.14 | |
| 6-α-propionate | 8.32 | 8.64 | +0.32 | |
| 6-α′-propionate | 9.67 | 9.97 | +0.30 | |
| 6-β-propionate | 1.41 | 1.51 | +0.10 | |
| 6-β′-propionate | 0.67 | 0.67 | 0.00 | |
| 7-α-propionate | 3.76 | 3.66 | −0.10 | |
| 7-α′-propionate | 1.74 | 1.18 | −0.51 | |
| 7-β-propionate | −0.44 | −0.64 | −0.20 | |
| 7-β′-propionate | −0.81 | −0.95 | −0.14 | |
| α-meso | 1.61 | 1.56 | −0.05 | |
| β-meso | 0.21 | 0.64 | +0.43 | |
| γ-meso | −1.1 | −1.03 | +0.07 | |
| δ-meso | 0.43 | 0.67 | +0.24 | |
| His70 | NH | 9.90 | 9.90 | 0.00 |
| CαH | 6.75 | 6.90 | +0.15 | |
| CβH | 9.62 | 10.15 | +0.53 | |
| CβH′ | 8.92 | 8.98 | +0.06 | |
| NδH | 15.0 | 14.5 | −0.5 | |
| His46 | NH | 10.71 | 10.65 | −0.06 |
| CαH | 7.70 | 7.60 | −0.10 | |
| CβH | 10.82 | 10.79 | −0.03 | |
| CβH′ | 9.20 | 9.08 | −0.12 | |
| NδH | 13.2 | 13.4 | +0.2 | |
| CεH | −11.6 | −11.3 | +0.3 | |
| axial His | m | 11.7d | 11.8 | |
| n | 5.4d | |||
| o | −1.7d | −2.8 |
The geometry of the heme cavity is defined by a set of NOEs. The proximal histidine (His70) NδH was found to be in dipolar contact with Met66 CαH, as observed for the wild-type and H117A hemichromes. Met66 assumed the same position with respect to the heme group and the flanking Tyr65, as evidenced by contacts involving Met66 CεH3, Tyr65 CεHs, and the heme 8-CH3. Compared with the wild-type protein, Met66 CεH3 was shifted upfield by ∼ 0.2 ppm, a displacement that is also observed in H117A rHb-R and, therefore, cannot be attributed to H-bonding of Ser69 to His70 or the 7-propionate with certainty. NOEs between the heme 8-CH3 and Tyr65, heme 2-vinyl and Phe84, heme 2-vinyl and Tyr53, heme 4-vinyl and Phe34, heme 3-CH3 and Leu79, and heme 3-CH3 and Val87 all confirmed similar placement of the heme. The relative proportions of native protein populating two states that differ by a 180° rotation about the heme α-γ meso axis is 95:5 in the wild-type hemichrome (4). The ratio shifted slightly in favor of the minor form in the variant. The proximal helix was traced by NHi-NHi+1 NOEs from Arg64 to Asn76, except at the level of Arg67 and Glu68 because of overlap in the 1H data. Helical conformation was also identified throughout the F helix with a number of resolved Hβi−NHi+1 NOEs. The CβH2 group of Ser69, however, was not identified. An absence of NOE is not necessarily meaningful, but since strong effects are observed between Ala69 CβH3 and His70 NδH, M66 CεH3, and Tyr65 CεHs in the wild-type and H117A proteins, this suggested that the serine side chain was not rigidly held in a single rotameric state and argued against the formation of a side chain hydrogen bond. No changes were observed in the few NOEs of the heme 7-propionate.
Characterization of A69S S6803 rHb-R in the Cyanide-bound State
Binding of cyanide to the wild-type protein results in a broadening of the 1H spectrum. The proximal-side substitution H117A has the remarkable consequence of sharpening the heme resonances (37). The A69S replacement did not have the same effect (Figure 3C); the lines for the cyanide complex remained as broad as for the wild-type, but the shifts were affected to some extent. A natural abundance 1H-13C HMQC spectrum (not shown) identified the signals from His70 CαH and CβHs. In the cyanomet form, the average axial histidine Hβ shift is indicative of imidazolate character. The average values for cyanomet H117A and A69S rHb-Rs were 6.5 ppm and 7.1 ppm, respectively. For comparison, wild-type sperm whale metMbCN and S92A metMbCN have shifts of 9.1 ppm and 8.5 ppm, respectively (29), and peroxidases display values between 14 ppm and 22 ppm (52). The moderate change in the average chemical shift observed in Mb and S6803 rHb-R is consistent with some reorganization of the proximal site geometry.
The 15N chemical shift of the bound cyanide can also be used as a probe of hydrogen bonding and imidazolate character of the axial histidine in trans. At 32 °C and neutral pH, the difference in chemical shift between free and bound cyanide was determined to be 692 ppm in the wild-type protein (37). This difference increased to 701 ppm in the A69S variant. Evidence for H-bonding of the bound cyanide is provided by the 22-ppm exchangeable signal attributed to Tyr22 (B10) Oη1H in Figure 3C (37). Thus, this aspect of the structure appeared conserved. The difference of less than 10 ppm in C15N shift between the two proteins did not indicate a significant change in imidazolate character, as the deviation was a small fraction of the total range of shifts observed for globins and peroxidases (53).
Thermal Denaturation of Ferric A69S S6803 rH-R
The effect of the replacement on the thermodynamic stability of the protein was inspected with equilibrium unfolding experiments. Thermal denaturation data were collected by measuring absorbance and ellipticity as functions of temperature. The first method yielded the apparent fraction of natively bound heme and the second the apparent fraction of folded polypeptide. Both fractions were calculated with a two-state model; they are shown in Figure 4.
Figure 4.
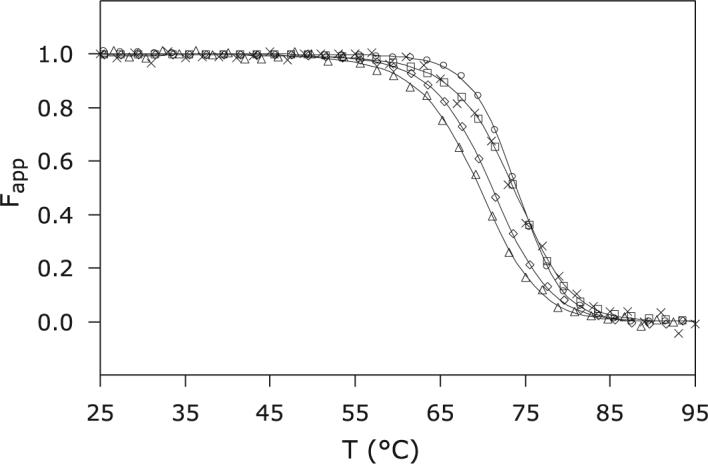
Thermal denaturation of S6803 rHb-R. CD and visible spectroscopy data were collected at pH 7.2 on the wild-type and A69S proteins in the ferric state. ×: Ferric wild-type S6803 rHb-R, CD; □: ferric wild-type S6803 rHb-R, visible; Δ ferric A69S S6803 rHb-R, CD; △: ferric A69S S6803 rHb-R, visible; ○: cyanomet A69S rHb-R, visible.
The near coincidence of the two curves for the wild-type protein supported that heme release occurred concomitantly with loss of secondary structure. In the thermal denaturation of b hemoproteins, the interpretation of denaturation curves is complicated by the solubility properties of the heme group. When the thermal energy is sufficient, the heme is released from its binding site, but remains associated, presumably non-specifically, with the protein matrix. Thus, the experiment measures the difference in free energy between specific and non-specific binding of heme to the protein. The stability of the apoprotein also factors into the response as a function of external conditions (22, 54). When the A69S rHb-R data were compared with those obtained for the wild-type protein, it appeared that the midpoint of the thermal transition occurred at a slightly lower temperature in the variant (Table 2). Assuming that the non-specific affinity and the apoprotein stability were unchanged by the replacement, the result implied a lower specific affinity for the heme group, translating into an apparent lower stability of the holoprotein. More striking than the lowering of Tm, however, was the non-coincidence of the curves obtained by monitoring the CD (backbone) and Soret (heme) signals. The discrepancy was small but reproducible, even though the thermal denaturation process was only partially reversible. The non-coincident curves were indicative of three-state behavior at a minimum.
Table 2.
Thermal denaturation of ferric wild-type and A69S rHb-R
| Protein | TM (°C)a | ΔH° (kJ mol−1)b | ΔCp (kJ mol−1 K−1) |
|---|---|---|---|
| wt (vis and CD) | 73.6 ± 0.2 | 288 ± 10 | 7.1c |
| A69S (vis) | 71.15 ± 0.04 | 284 ± 3 | 7.1c |
| A69S (CD) | 69.62 ± 0.13 | 271 ± 6 | 7.1c |
| CN-A69S (vis) | 73.99 ± 0.05 | 368 ± 4 | 5.8 ± 1.8 |
Midpoint of thermal denaturation in pH 7.2, 20 mM phosphate buffer. Errors reported are standard deviations from global fitting of multiple data sets.
Change in enthalpy evaluated at the midpoint of the thermal unfolding transition.
Value fixed during fitting.
Chemical Denaturation of A69S S6803 rHb-R
To verify that heme affinity was decreased by the substitution, urea-induced denaturation of the wild-type and variant proteins was performed. At neutral pH, the process was fully reversible and lent itself to thermodynamic analysis. Figure 5 presents the apparent fraction of folded protein as a function of urea concentration. Non-coincidence of the signals arising from heme release and secondary structure loss was observed for both wild-type and A69S rHb-Rs, with the midpoint of the transition corresponding to heme release occurring at a higher urea concentration. Thermodynamic parameters obtained from two-state analysis are listed in Table 3. The difference in the free energy of unfolding for the two proteins was small. In further support that the experimental methods were monitoring slightly different transitions, the m values obtained for the CD and absorbance data were not within error of each other. In addition, comparison of the ΔG° values obtained on the two hemichromes via the same method indicated that the substitution resulted in a change in the balance of free energy associated with heme ligation and secondary structure formation.
Figure 5.
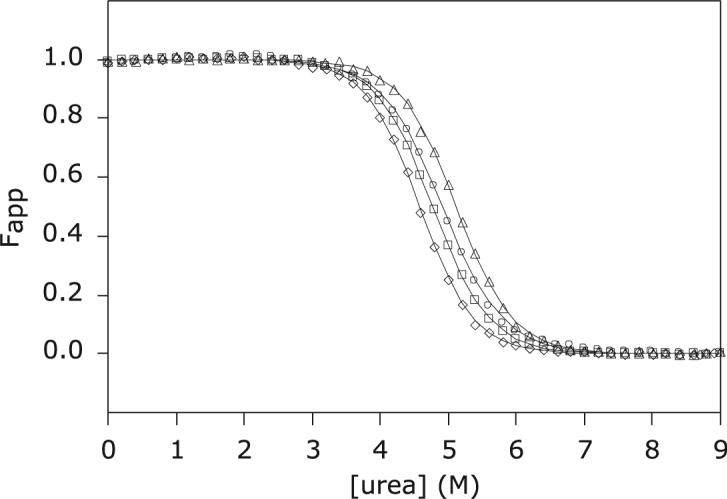
Urea-induced denaturation of S6803 rHb-R. CD and visible spectroscopy data were collected at pH 7.2 and 25 °C on the wild-type and A69S proteins in the ferric state. ○: Ferric wild-type S6803 rHb-R, CD; Δ ferric wild-type S6803 rHb-R, visible; △: ferric A69S S6803 rHb-R, CD; □: ferric A69S S6803 rHb-R, visible.
Table 3.
Urea denaturation of ferric wild-type and A69S rHb-R
| ΔGo (kJ mol−1) a | m (kJ mol−1 M−1) b | Cm (M) c | |
|---|---|---|---|
| wt (vis) | 30.6 ± 0.3 | 5.99 ± 0.05 | 5.11 |
| wt (CD) | 27.4 ± 0.3 | 5.57 ± 0.06 | 4.9 |
| A69S (vis) | 28.38 ± 0.17 | 5.94 ± 0.04 | 4.78 |
| A69S (CD) | 28.6 ± 0.4 | 6.25 ± 0.08 | 4.6 |
Free energy of unfolding at 25 °C in pH 7.2, 20 mM phosphate buffer. Errors reported are standard deviations from global fitting of multiple data sets.
Dependence of free energy of unfolding on denaturant concentration.
Midpoint of urea denaturation, calculated from ΔGo and m.
Heme Release from A69S S6803 rHb-R upon Solution Acidification
S6803 rHb-R can undergo a chemical modification by which the b heme becomes covalently attached to His117 via the 2-vinyl group, yielding “rHb-A” (40). When the cross-link is absent, lowering the pH results in the release of the heme group from its pocket in addition to the protonation of His46 and His70. These coupled processes can be detected optically as the cofactor spin state changes from low to high. pH titrations of the wild-type rHb-R display a sharp transition with a midpoint at pH 3.6 and a Hill coefficient of ∼ 3.4 (42). A second transition with a higher pKa (∼ 5) is observed. The titration curve is shown in Figure 6, which also contains A69S S6803 rHb-R data. In the variant, the midpoint for heme release occurred at pH 3.8, and the Hill coefficient was ∼ 3. The higher pKa of the main transition obscured the second transition. In both proteins, the Hill coefficient suggested that release was coupled to at least three protonation events, which would arise from His46, His70, and a third group, possibly the heme 7-propionate as a hydrogen bond was noted to Lys42 (34).
Figure 6.
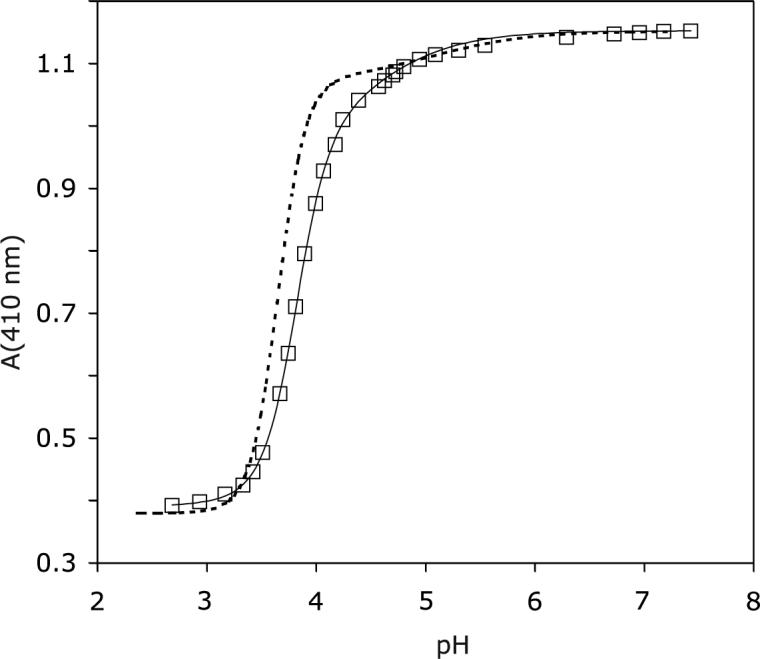
pH titration of ferric A69S rHb-R. Data were collected at 25 °C. The solid line represents the fit to a two-pKa model. The main transition has pKa = 3.8 and Hill coefficient ∼ 3. The dashed line represents the results of the titration of ferric wild-type rHb-R (42), scaled to attain identical intensity at pH 7.2.
Urea Gradient Gels
In its apoprotein state, S6803 rHb has limited stability (4). Preliminary measurements indicated no difference due to the A69S replacement. This was confirmed by analyzing denaturation profiles obtained with urea gradient gels of wild-type and A69S apoproteins. The transitions observed for the apoglobins run individually lacked a well-defined range of urea concentration in which the native state was fully populated. For all practical purposes, the transition was complete at 2 M urea at pH 7.5. Electrophoresis of mixtures of the two apoproteins (so that they were subjected to identical gradients) yielded indistinguishable traces. Such a gel is shown in Figure 7. It is noteworthy that, under the chosen conditions, the unfolding and refolding rates were sufficiently fast to yield a detectable, continuous band of protein. This indicated that the half-times of the species co-existing at any urea concentration were shorter than 0.1 times the duration of the electrophoretic separation (49).
Figure 7.

Urea-gradient gel electrophoresis of wild-type and A69S apoproteins stained with Coomassie brilliant blue. The scale represents approximate urea concentrations. “0” and “8” indicate regions of the gel with constant urea concentration. The proteins begin to unfold immediately upon exposure to urea; by 2 M urea, the protein is completely unfolded, and the horizontal line corresponds to protein having an electrophoretic mobility consistent with the unfolded state. The two traces, which were observed individually, overlay perfectly in the mixture indicating that the substitution did not affect the apoprotein's resistance to urea denaturation.
Urea gradient gel eletrophoresis was also performed on the holoproteins (Figure 8) in an effort to obtain equilibrium and kinetic information simultaneously. Both Coomassie staining and heme staining were applied to reveal protein and dissociated heme, respectively. The gels were loaded either with native protein (0 M urea; Figure 8) or unfolded protein (8 M urea; data not shown) and returned identical results in a demonstration that the process was fully reversible. In the wild-type S6803 rHb-R gels, a native band with a constant mobility was observed up to ∼ 2 M urea (Figure 8A). At this concentration, a faint band was also detected with mobility consistent with unfolded protein; this band became intense above 2.5 M. Between 2 and 2.5 M urea, there was no detectable amount of protein with intermediate mobility. As the concentration of urea increased, the rate at which the heme was lost from the holoprotein also increased. The interrupted pattern of the stained gel indicated that, when the urea concentration neared 2 M, heme loss became irreversible because released heme migrated through the gel matrix faster than the protein could refold and recapture it. Under those conditions, the urea concentrations were sufficient to denature the apoprotein completely, resulting in the population of the unfolded rather than the folded state. Thus, the profile obtained for the holoprotein was not expected to mimic the transition observed for the urea titration, which takes place between 3.2 and 6 M urea (Figure 5). Heme staining of gels run for a shorter time confirmed that at ∼2.2 M urea, the heme had dissociated irreversibly from the protein (Figure 8B). The sensitivity of these experiments was often insufficient to detect the heme when bound to the protein.
Figure 8.
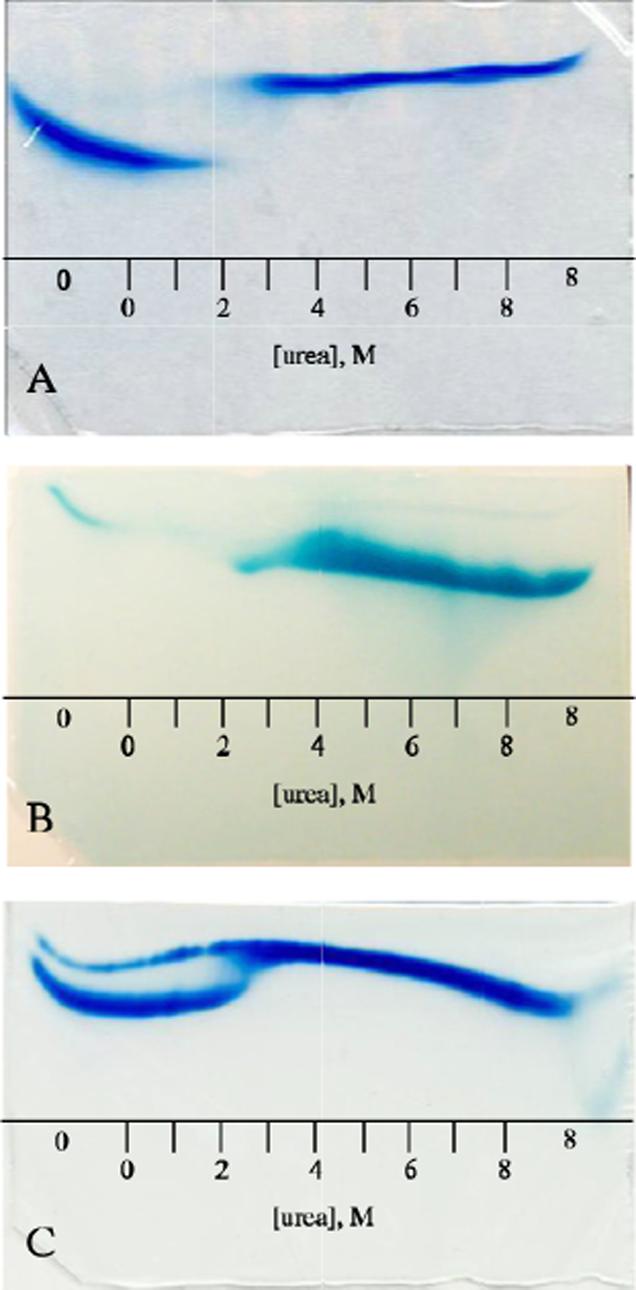
Holoprotein urea-gradient gels: A) Four-hour electrophoresis of wild-type S6803 ferric rHb-R visualized by Coomassie blue. The two species observed are the natively folded holoprotein (0−2 M) and the unfolded apoprotein (∼2.5−8 M). B) 40-min electrophoresis of wild-type and A69S S6803 ferric rHb-R stained for heme. The thin band in the 0-M buffer region of the gel corresponds to heme that is specifically bound to the natively folded protein; the thick band seen from 2−8 M urea arises from heme that has escaped irreversibly from the protein. C) Four-hour electrophoresis of A69S ferric S6803 rHb-R visualized by Coomassie blue. The sample contained both the apo and holo forms of the protein. The apoprotein (thin band) mirrors the transition seen in Figure 7. The A69S holoprotein (thick band) follows the pattern of the wild-type holoprotein (Figure 8A) except that evidence of transition is seen at 2−2.5 M urea. In all cases the proteins were loaded on the gel in the native state. The scale represents approximate urea concentrations. “0” and “8” indicate regions of the gel with constant urea concentration.
When the same experiments were performed on A69S rHb-R, the profile was similar, but not identical (Figure 8C). Although a discontinuity was observed at approximately the same urea concentration as for the wild-type rHb-R, detectable amounts of protein had intermediate mobility at the edges of the transition.
Cyanide Binding by Ferric A69S S6803 rHb-R
In the process of binding cyanide, the distal ligand to the heme iron (His46) is ultimately replaced by CN−, the dissociation product of HCN. Hence, multiple equilibria must be considered, including:
| (6) |
| (7) |
| (8) |
| (9) |
Wild-type S6803 rHb-R binds cyanide slowly at neutral pH (37): a 10-μM solution of protein is half-converted to the cyanomet form in the presence of 5 mM total cyanide (HCN and CN−) in approximately 10 min. The apparent bimolecular rate constant for the major kinetic phase is ∼ 3 × 10−1 M−1 s−1, three orders of magnitude slower than in vertebrate globins and pentacoordinate 2-on-2 globins (55).
The H117A replacement results in a protein that binds cyanide 8 times more slowly than wild type (37). This effect is noteworthy as His117 is located on the proximal side of the protein and is not positioned in the ligand access tunnel described in 2-on-2 globins (30). To probe a distinct proximal influence, the binding experiment was repeated with A69S S6803 rHb-R. Upon addition of a 200-, 500-, or 1000-fold excess of cyanide to the protein solution, the absorption at 423 nm was measured as a function of time. This manual mixing procedure yielded the relative apparent rates of association. To test the hypothesis that HCN dissociation played a significant role, the experiments were repeated in D2O.
Apparent pseudo-first-order rate constants obtained for the wild-type and A69S ferric rHb-R and rHb-A are shown in Figure 9 as a function of total cyanide concentration. These constants are associated with the major of four kinetic phases required to approach randomly distributed residuals in the fit of the time courses. The major phase accounted for approximately 80% (rHb-R) or 70% (rHb-A) of the total protein population. As shown by the slope of the lines, binding to the A69S variant was slower than to the wild-type protein. The ratio of the apparent second-order rate constants for the two proteins was approximately 2. Thus, the A69S replacement, as the H117A replacement, decelerated cyanide binding. In D2O, the number of kinetic phases remained the same in the A69S and wild-type proteins; the major phase was decelerated by a factor of 1.5 − 2 in both proteins (data not shown). At pH 8.2, the pseudo-first-order rate constant obtained for a solution containing a 200-fold excess of total cyanide was accelerated by a factor of 5 relative to the same solution at pH 7.2 (data not shown). This increase in pH corresponded to a 9-fold increase in free cyanide concentration; the acceleration did not scale according to the apparent bimolecular rate constant obtained at pH 7.2. In addition, an acceleration by a factor of 3 (wild-type) or 5 (A69S) was observed in the presence of the cross-link (Figure 9).
Figure 9.
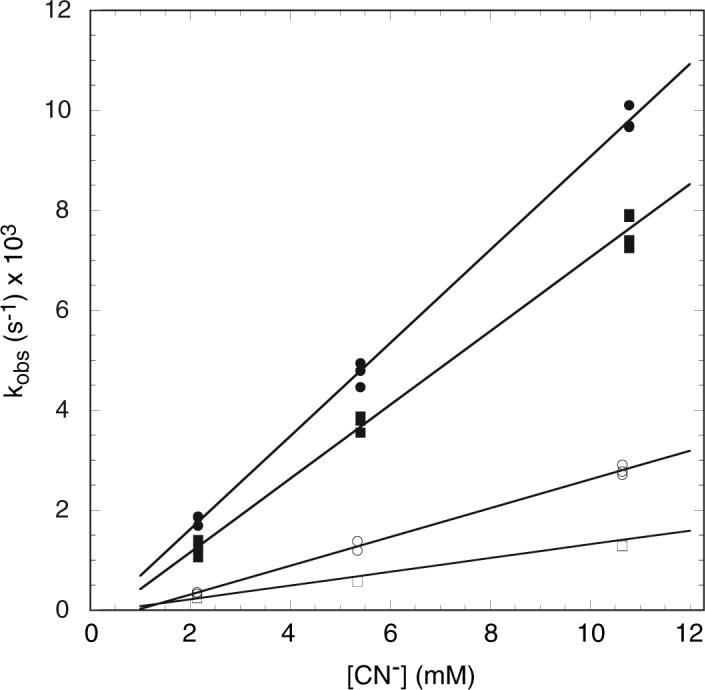
Apparent pseudo-first-order rate constants for association of cyanide with (○) wild-type ferric S6803 rHb-R, (□) A69S ferric S6803 rHb-R, (●) wild-type ferric S6803 rHb-A, and (■) A69S ferric S6803 rHb-A. The protein concentrations were 10.7 to 10.8 μM and the rate constants are for the major kinetic phase. The slopes of the solid lines represent apparent bimolecular rate constants.
Thermal denaturation of the cyanomet complex of wild-type (data not shown) and A69S rHb-R (Figure 4) in the presence of a 1000-fold excess of cyanide showed that heme release occurred at a higher temperature and with a steeper slope than in the absence of the exogenous ligand. This is also observed with H117A rHb-R (37).
DISCUSSION
The results obtained on A69S S6803 rHb-R are significant when compared to Mb and Lb, proteins for which the role of residue F7 has been analyzed in detail by several methods. In pig Mb (Ser92 at F7), disruption of the F7-F8 and F7−7-propionate H-bonding interactions causes an increase in affinity for small ligands and an enhanced propensity to lose heme. Interactions between the 7-propionate and His97 (FG3) are also affected, and the new geometry increases permeability to solvent (26). In human Mb, ligand binding is unaffected by F7 replacement, and the orientation of the proximal histidine rotates from the eclipsed position by a few degrees (27). Slight rotation of the proximal histidine is also reported for the S92D variant of horse heart Mb in the cyanomet state (28). The role of proximal interactions was reinspected by Peterson and coworkers, who studied variants containing multiple replacements and 8-residue F-helix swaps (56). These authors conclude that the hydrogen bond network involving Ser92 serves principally to stabilize the proximal pocket and retain the heme within the protein. The reverse, stabilization upon introduction of a hydrogen-bonding residue at F7, was tested with the V91S variant of Lb; in fact, heme loss is accelerated by the replacement (21). In this protein, the orientation of the proximal histidine is staggered with respect to the pyrrole nitrogens, a property achieved via steric interactions in the absence of hydrogen bonds (21). Common effects of F7 replacement are, therefore, small structural changes and holoprotein destabilization.
Structure
In S6803 rHb-R, His70 adopts a staggered conformation that presumably stabilizes distal ligand binding (20). The A69S replacement affected the chemical shift of the axial histidines moderately (Table 1), with larger proximal than distal deviations. Minor deviations were also observed for the heme group. Peripheral substituents (protons in α to PPIX ring) experienced an upfield shift on pyrrole A and C and a downfield shift on pyrrole B and D. In previous work, the paramagnetic shift of the methyl groups was used to assess the orientation of the axial histidines in the wild-type protein (34). The same heuristic approach (57) applied to the A69S variant showed that the shifts were consistent with a rotation of a few degrees of the bisector of the two histidine planes and a slight decrease in the angle between the two imidazole planes. The method is approximate and does not provide information on individual ligands; comparison of distal and proximal shifts, however, supported that a change in the orientation of the proximal histidine was most likely responsible for the effect. The shift pattern suggested that His70 rotated to align the projection of its plane closer to the x-axis (Figure 2). Such a move could accommodate the Ser69 hydroxyl group and, perhaps, reorient the NδH vector to favor hydrogen bonding. However, no direct evidence for formation of this hydrogen bond was found. The 7-propionate, which could potentially form a hydrogen-bond with Ser69 OH as in Mb, interacts with Lys42 in the wild-type protein. NOE perturbations did not suggest rearrangement near the propionates. Other perturbations were minor as well and suggested conserved geometric and electronic properties in the variant.
Stability
Urea-induced denaturation of the A69S and wild-type apoproteins, as observed by urea-gradient gel electrophoresis, indicated that the substitution had no effect on the resistance to denaturant. In both cases, the unfolding transition began at the lowest urea concentration and was complete by 2 M. The observation of a single, narrow protein band in the gel on which a mixture of wild-type and A69S proteins was run suggested that alteration of the free energy of holoprotein folding could be attributed to changes in heme-protein interactions.
The temperature- and urea-induced denaturation results indicated that the substitution had only slight effects on the relative free energies of the folded and unfolded states of the protein in the presence of the heme group. The most notable feature of the denaturation curves was the non-coincidence of the CD and absorption data. Through the thermal experiments, the A69S substitution revealed a decoupling of heme-iron decoordination and loss of secondary structure. This was consistent with the lower helical propensity of serine compared to alanine and with an intrinsically less stable turn of helix encompassing that position. In the urea experiments, neither protein followed a two-state mechanism. In all non-coincident cases, the CD data displayed a lower Cm than the absorbance data; therefore, backbone unraveling began at lower denaturant concentration than heme loss. A possible interpretation is that the serine side chain introduced strain into the F helix, made it more susceptible to denaturation, and facilitated decoordination of His70 once this helix started to fray. In this view, an intermediate state would be populated that contains heme specifically bound to a protein with a partially folded F helix. An extreme case of decoupling has been reported in Drosophila cytochrome c, where replacement of Pro30 is thought to disrupt a hydrogen-bond network involving the axial histidine (58).
The urea-induced denaturation of the holoproteins in solution yielded dramatically different results than the same experiment performed within the confines of an acrylamide gel. In solution, the system is allowed to come to equilibrium, and the folding and unfolding rates (as well as the rates of specific and non-specific heme association and dissociation) must only be considered in the sense that these quantities determine the time required to reach equilibrium and the equilibrium constant at each urea concentration. In the urea-gradient gel, however, an additional factor must be considered. Because the molecules are migrating, the ability of the protein to remain associated with the heme prosthetic group depends critically on the relative rates of protein refolding, heme association, and protein and heme migration. The urea-gradient gel method offers kinetic insights that are otherwise experimentally difficult to obtain.
The overall gel pattern obtained for the wild-type and A69S holoproteins suggested that, at ∼ 2 M urea, the rate of heme migration exceeded the rate of holoprotein refolding, and the two species observed in the gel were the folded holoprotein and the unfolded apoprotein. There was, however, a reproducible difference between the profiles obtained for the wild-type and the variant proteins. Whereas the wild-type gel showed only native and unfolded baselines, the A69S gel displayed hints of a transition. This could be explained in one of two ways: either the variant protein had a higher non-specific heme affinity (slower kdiss) or it refolded slightly faster than the wild-type protein. The former effect was not expected because non-specific heme affinity arises from low solubility of the cofactor, a property counteracted by urea. In addition, the two proteins differed by an OH group with limited ability to affect non-specific interactions. Hence, the dissimilarity between the wild-type and variant likely reflected a change in the refolding rate. The slight decrease in the free energy of folding for the variant relative to wild type (Table 3) indicated that the unfolding rate must have been increased also, arguing for a lowering of the transition state.
Kinetic studies performed on cytochrome b562 have indicated that holoprotein folding from a partially-folded apo state with which the heme group is associated proceeds significantly faster than refolding from the totally denatured state (59). This scenario is expected to apply only to hemoproteins that have high-stability apo forms remaining in the folded state at denaturant concentrations sufficient to disrupt the specific heme-protein contacts formed in the holoprotein. The S6803 rHb-R apoproteins began unfolding immediately upon addition of urea. In addition, no evidence for the population of the folded apoprotein was detected in either the holoprotein urea-gradient gels or the titration experiments performed in solution. Thus, the change in the staining pattern observed in the urea-gradient gel upon the A69S substitution likely reflected an alteration of the kinetics of folding to the holoprotein state from the fully unfolded state.
The acid-induced denaturation of the two proteins provided another measure of relative heme affinities. Two of the three protonation events linked with heme release are expected to involve the axial ligands. The 0.2-unit increase in the pH midpoint for the decoordination of the heme iron in the A69S protein was consistent with a moderate decrease in the strength of the association with the cofactor. Assuming once more that non-specific heme affinity remained unchanged, these results indicated a modest ΔΔG° (∼ 1 kJ mol−1) relative to the total free energy of the process and were consistent with those for the urea-induced denaturation experiments.
Cyanide Binding
Fitting the time course of cyanide binding by S6803 rHb-R under pseudo-first-order conditions requires several kinetic phases. This is unlike other globins, which exhibit monophasic behavior (55). The major phase extracted from wild-type S6803 rHb-R data has an apparent bimolecular rate constant comparable to that observed in Glycera dibranchiata Hb component III (60). In general, cyanide binding occurs minimally through two processes, one involving HCN (with bimolecular rate constant kHCN) and another CN− (with bimolecular rate constant kCN-). The observed rate constant contains a contribution from each process, weighted by the HCN and CN− fractional populations at the given pH. In G. dibranchiata Hb, the slow observed rate constant is attributed to the absence of a functional group (e.g, His or H2O) capable of catalyzing HCN dissociation in the heme cavity. The preference for CN− binding over HCN binding (kCN- > kHCN) is manifested in the pH dependence of the reaction kinetics. It is noteworthy that the 5-fold acceleration caused by a change of pH from 7.0 to 8.0 (60) is also comparable to the effect noted in S6803 rHb-R. Distinct isoforms of G. dibranchiata Hb display rate constants that vary within an order of magnitude and are likely to reflect the influence of minor structural perturbations on the balance of HCN and CN− binding.
The similarity of S6803 rHb-R and G. dibranchiata Hb with respect to cyanide binding is intriguing. In S6803 rHb, not only can HCN dissociation contribute to slow binding, but also decoordination of His46. The rate constants for His46 ligation (kH) and deligation (k−H) in the ferrous and presumably cross-linked state have been reported (38); they are rapid (kH = 4200 s−1 and k−H = 930 s−1, resulting in an equilibrium constant KH of 4.5) and suggest that the dissociation of HCN may dominate the reaction. The k−H value, however, depends on whether exogenous ligand binding is initiated by flash photolysis (38) or rapid mixing (39); decoordination occurs almost two orders of magnitude more slowly in the rapid mixing case (k−H = 14 s−1, dictating an equilibrium constant KH of 300). Rapid mixing conditions resemble more closely our experimental method, though the oxidation and heme cross-link states of the protein still differ. NMR data on the ferric S6803 rHb-R do not reveal any spectral contribution from a pentacoordinate species at room temperature, in support of an equilibrium favoring the hexacoordinate state (KH >> 5).
Previous studies on the H117A variant of S6803 rHb indicate that the presence of His117 increases the rate of cyanide binding (37) relative to an alanine at this position. The solution structure of the wild-type globin (34) suggests that His117, the residue that cross-links the heme, samples rapidly multiple conformations, both in and out of the heme pocket. A role for His117 in the dissociation of HCN is therefore conceivable. A similar role for Ala69 is not as readily envisioned. The side chain is located at the edge of the heme pocket, and polarity is increased by the serine substitution. An alternative interpretation is that the residues at position 69 and 117 affect the association and dissociation kinetics of His46. In its dissociated state, His46 can serve as a proton acceptor (as HisE7 does in vertebrate globins) and is a good candidate for catalyzing HCN dissociation. In this view, proton transfer efficiency would depend on the residence time of His46 in the decoordinated state. It is possible that the A69S replacement consolidates the His46-Fe bond, thereby slowing down HCN dissociation. The observation of larger-than-average chemical shift deviations for His46 in the variant may reflect this subtle alteration. A similar effect accompanied by reseating of the heme group can be invoked for the deceleration caused by the H117A replacement. A confirmation of the role of His117 and connection to His46 kinetics appeared in the behavior of rHb-A, which bound cyanide faster than rHb-R. The small solvent isotope effect (kH2O/kD2O = 1.5 to 2) was inconclusive as it could have arisen from a HCN bending mode or betray indirect influences associated with the replacement of labile hydrogens on the protein. As already discussed, the multiphasic character of the binding reaction complicated data analysis and interpretation in all cases.
Assuming that the deceleration in A69S rHb-R was related to a reinforcement of the distal coordination bond, the difference in rate constants compared to wild type indicated a trans effect being exerted by the proximal substitution. Because the acid-induced heme release and the denaturation monitored in the visible region supported a lowering of the heme affinity via axial bond weakening, the stabilization of the His46-Fe bond would appear smaller than the destabilization of the His70-Fe bond, with a net effect of holoprotein destabilization.
A recent study of neuroglobin and cytoglobin, both bis-histidine globins, relates the hyperthermostability of these proteins to their high KH in the ferrous state (13). In addition, exogenous ligand binding and distal histidine substitution cause a reduction of this stability. S6803 rHb-R, with a Tm of 75 °C and KH of 300, does not cluster with the neuroglobin/cytoglobin set. In fact, the thermal denaturation of wild-type, A69S (current study), and H117A (37) proteins show that the cyanide-bound state is more stable than the hemichrome state. In wild-type S6803 rHb-R, cyanide binding results in helical rearrangement and formation of a distal hydrogen-bonding network between the ligand and the protein (36, 37). These conformational changes are more extensive than observed in neuroglobin upon ligand binding (61) and lead to a repacked S6803 rHb-R interior with additional favorable interactions in the native state. Thus, the outcomes of hexacoordination and endogenous-exogenous ligand substitution differ in the cyanobacterial 2-on-2 globins and other types of globins.
CONCLUSION
The 2-on-2 globin fold has structural features that distinguish it from the canonical globin fold, including unique pathways to the heme pocket and a ligand stabilization mechanism. Additional data will be necessary to achieve a full description of these traits and their effects. Available information, however, already indicates that the residues near the heme group are essential for controlling interactions with heme and small ligands, as in other globins. The A69S replacement in S6803 rHb revealed that structural stability and ligand binding properties could be finely adjusted by proximal influences.
The amino acid preceding the proximal histidine in globins has been recognized as a partial determinant of properties such as the azimuthal angle of the proximal histidine and heme affinity. The introduction of a residue capable of hydrogen bonding at F7 in the hexacoordinate S6803 rHb-R resulted in modest decreases in resistance to temperature-, urea-, and acid-induced denaturation as well as changes in exogenous ligand-binding kinetics. A decrease in helical propensity triggering trans effects can explain the observations. A behavior common to globins emerges by which alteration of F7 alone, regardless of the nature of the change, deteriorates heme affinity. Proximal networks therefore appear optimized for each specific combination of residues. In contrast to cytochrome b5, which remains endogenously hexacoordinate even in the presence of high concentrations of potential ligands, and other bis-histidine complexes that have the ability to bind ligands but become destabilized in the process, the S6803 rHb-R protein seems to maintain relative proximal and distal bond strengths that allow for increased stability in the exogenous-ligand-bound state. This could speak to a physiological role, particularly if covalent attachment of the heme to His117 is not a permanent characteristic of the protein in vivo.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Nancy Scott for assistance throughout the project, Nicole Love for protein preparation, Dr. Daniel Jones for mass spectrometry data, and Dr. Christopher Falzone for discussion and careful reading of the manuscript.
Footnotes
This study was supported by National Science Foundation grants MCB-091182 and MCB-0349409, National Institutes of Health grant GM-054217, and NASA grant NNG04GN33H (DAV).
Abbreviations: APS, ammonium persulfate; DQF-COSY, double-quantum-filtered correlated spectroscopy; Hb, hemoglobin; HMQC, heteronuclear multiple quantum coherence; Lb, leghemoglobin; Mb, myoglobin; metMb, ferric myoglobin; metMbCN, cyanomet myoglobin; NOE, nuclear Overhauser effect; NOESY, two-dimensional nuclear Overhauser effect spectroscopy; PPIX, protoporphyrin IX; rHb, recombinant hemoglobin; rHb-A, recombinant hemoglobin with covalently linked heme; rHb-R, recombinant heme-reconstituted hemoglobin; S6803, Synechocystis sp. PCC 6803; TEMED, N,N,N′,N′-tetramethylethylenediamine; TOCSY, totally correlated spectroscopy; trHb, truncated hemoglobin.
SUPPORTING INFORMATION AVAILABLE
Four figures (portions of the NOESY spectrum used to assign proton resonances of A69S S6803 rHb-R in the ferric, bis-histidine state). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Walker FA. Models of the bis-histidine-ligated electron-transferring cytochromes. Comparative geometric and electronic structure of low-spin ferro- and ferrihemes. Chem. Rev. 2004;104:589–615. doi: 10.1021/cr020634j. [DOI] [PubMed] [Google Scholar]
- 2.de Sanctis D, Dewilde S, Vonrhein C, Pesce A, Moens L, Ascenzi P, Hankeln T, Burmester T, Ponassi M, Nardini M, Bolognesi M. Bishistidyl heme hexacoordination, a key structural property in Drosophila melanogaster hemoglobin. J. Biol. Chem. 2005;280:27222–9. doi: 10.1074/jbc.M503814200. [DOI] [PubMed] [Google Scholar]
- 3.Couture M, Das TK, Savard PY, Ouellet Y, Wittenberg JB, Wittenberg BA, Rousseau DL, Guertin M. Structural investigations of the hemoglobin of the cyanobacterium Synechocystis PCC 6803 reveal a unique distal heme pocket. Eur. J. Biochem. 2000;267:4770–4780. doi: 10.1046/j.1432-1327.2000.01531.x. [DOI] [PubMed] [Google Scholar]
- 4.Lecomte JTJ, Scott NL, Vu BC, Falzone CJ. Binding of ferric heme by the recombinant globin from the cyanobacterium Synechocystis sp. PCC 6803. Biochemistry. 2001;40:6541–6552. doi: 10.1021/bi010226u. [DOI] [PubMed] [Google Scholar]
- 5.Scott NL, Falzone CJ, Vuletich DA, Zhao J, Bryant DA, Lecomte JTJ. The hemoglobin of the cyanobacterium Synechococcus sp. PCC 7002: evidence for hexacoordination and covalent adduct formation in the ferric recombinant protein. Biochemistry. 2002;41:6902–6910. doi: 10.1021/bi025609m. [DOI] [PubMed] [Google Scholar]
- 6.Dewilde S, Kiger L, Burmester T, Hankeln T, Baudin-Creuza V, Aerts T, Marden MC, Caubergs R, Moens L. Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J. Biol. Chem. 2001;276:38949–38955. doi: 10.1074/jbc.M106438200. [DOI] [PubMed] [Google Scholar]
- 7.Trent JT, 3rd, Hargrove MS. A ubiquitously expressed human hexacoordinate hemoglobin. J. Biol. Chem. 2002;277:19538–19545. doi: 10.1074/jbc.M201934200. [DOI] [PubMed] [Google Scholar]
- 8.Burmester T, Ebner B, Weich B, Hankeln T. Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Mol. Biol. Evol. 2002;19:416–421. doi: 10.1093/oxfordjournals.molbev.a004096. [DOI] [PubMed] [Google Scholar]
- 9.Sawai H, Kawada N, Yoshizato K, Nakajima H, Aono S, Shiro Y. Characterization of the heme environmental structure of cytoglobin, a fourth globin in humans. Biochemistry. 2003;42:5133–42. doi: 10.1021/bi027067e. [DOI] [PubMed] [Google Scholar]
- 10.Feng L, Gell DA, Zhou S, Gu L, Kong Y, Li J, Hu M, Yan N, Lee C, Rich AM, Armstrong RS, Lay PA, Gow AJ, Weiss MJ, Mackay JP, Shi Y. Molecular mechanism of AHSP-mediated stabilization of α-hemoglobin. Cell. 2004;119:629–640. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Feng L, Zhou S, Gu L, Gell DA, Mackay JP, Weiss MJ, Gow AJ, Shi Y. Structure of oxidized α-haemoglobin bound to AHSP reveals a protective mechanism for haem. Nature. 2005;435:697–701. doi: 10.1038/nature03609. [DOI] [PubMed] [Google Scholar]
- 12.Weiland TR, Kundu S, Trent JT, 3rd, Hoy JA, Hargrove MS. Bis-histidyl hexacoordination in hemoglobins facilitates heme reduction kinetics. J. Am. Chem. Soc. 2004;126:11930–11935. doi: 10.1021/ja046990w. [DOI] [PubMed] [Google Scholar]
- 13.Hamdane D, Kiger L, Dewilde S, Uzan J, Burmester T, Hankeln T, Moens L, Marden MC. Hyperthermal stability of neuroglobin and cytoglobin. FEBS J. 2005;272:2076–2084. doi: 10.1111/j.1742-4658.2005.04635.x. [DOI] [PubMed] [Google Scholar]
- 14.Ptitsyn OB, Ting KL. Non-functional conserved residues in globins and their possible role as a folding nucleus. J. Mol. Biol. 1999;291:671–682. doi: 10.1006/jmbi.1999.2920. [DOI] [PubMed] [Google Scholar]
- 15.Knapp JE, Bonham MA, Gibson QH, Nichols JC, Royer WE., Jr. Residue F4 plays a key role in modulating oxygen affinity and cooperativity in Scapharca dimeric hemoglobin. Biochemistry. 2005;44:14419–14430. doi: 10.1021/bi051052+. [DOI] [PubMed] [Google Scholar]
- 16.Cheng XD, Schoenborn BP. Neutron diffraction study of carbonmonoxymyoglobin. J. Mol. Biol. 1991;220:381–399. doi: 10.1016/0022-2836(91)90020-7. [DOI] [PubMed] [Google Scholar]
- 17.Sinclair R, Hallam S, Chen M, Chance B, Powers L. Active site structure in cytochrome c peroxidase and myoglobin mutants: effects of altered hydrogen bonding to the proximal histidine. Biochemistry. 1996;35:15120–15128. doi: 10.1021/bi961064i. [DOI] [PubMed] [Google Scholar]
- 18.Roncone R, Monzani E, Murtas M, Battaini G, Pennati A, Sanangelantoni AM, Zuccotti S, Bolognesi M, Casella L. Engineering peroxidase activity in myoglobin: the haem cavity structure and peroxide activation in the T67R/S92D mutant and its derivative reconstituted with protohaemin-l-histidine. Biochem. J. 2004;377:717–724. doi: 10.1042/BJ20030863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liong EC, Dou Y, Scott EE, Olson JS, Phillips GN., Jr. Waterproofing the heme pocket: Role of proximal amino acid side chains in preventing hemin loss from myoglobin. J. Biol. Chem. 2001;276:9093–9100. doi: 10.1074/jbc.M008593200. [DOI] [PubMed] [Google Scholar]
- 20.Samuni U, Ouellet Y, Guertin M, Friedman JM, Yeh SR. The absence of proximal strain in the truncated hemoglobins from Mycobacterium tuberculosis. J. Am. Chem. Soc. 2004;126:2682–2683. doi: 10.1021/ja038093i. [DOI] [PubMed] [Google Scholar]
- 21.Kundu S, Snyder B, Das K, Chowdhury P, Park J, Petrich JW, Hargrove MS. The leghemoglobin proximal heme pocket directs oxygen dissociation and stabilizes bound heme. Proteins. 2002;46:268–277. doi: 10.1002/prot.10048. [DOI] [PubMed] [Google Scholar]
- 22.Hargrove MS, Olson JS. The stability of holomyoglobin is determined by heme affinity. Biochemistry. 1996;35:11310–11318. doi: 10.1021/bi9603736. [DOI] [PubMed] [Google Scholar]
- 23.Olson JS, Phillips GN., Jr. Kinetic pathways and barriers for ligand binding to myoglobin. J. Biol. Chem. 1996;271:17593–17596. doi: 10.1074/jbc.271.30.17593. [DOI] [PubMed] [Google Scholar]
- 24.Inaba K, Ishimori K, Imai K, Morishima I. Substitution of the heme binding module in hemoglobin alpha- and beta-subunits. Implication for different regulation mechanisms of the heme proximal structure between hemoglobin and myoglobin. J. Biol. Chem. 2000;275:12438–12445. doi: 10.1074/jbc.275.17.12438. [DOI] [PubMed] [Google Scholar]
- 25.Inaba K, Ishimori K, Morishima I. Structural and functional roles of heme binding module in globin proteins: identification of the segment regulating the heme binding structure. J. Mol. Biol. 1998;283:311–327. doi: 10.1006/jmbi.1998.2073. [DOI] [PubMed] [Google Scholar]
- 26.Smerdon SJ, Krzywda S, Wilkinson AJ, Brantley RE, Jr., Carver TE, Hargrove MS, Olson JS. Serine92 (F7) contributes to the control of heme reactivity and stability in myoglobin. Biochemistry. 1993;32:5132–5138. doi: 10.1021/bi00070a023. [DOI] [PubMed] [Google Scholar]
- 27.Shiro Y, Iizuka T, Marubayashi K, Ogura T, Kitagawa T, Balasubramanian S, Boxer SG. Spectroscopic study of Ser92 mutants of human myoglobin: hydrogen bonding effect of Ser92 to proximal His93 on structure and property of myoglobin. Biochemistry. 1994;33:14986–14992. doi: 10.1021/bi00254a006. [DOI] [PubMed] [Google Scholar]
- 28.Lloyd E, Burk DL, Ferrer JC, Maurus R, Doran J, Carey PR, Brayer GD, Mauk AG. Electrostatic modification of the active site of myoglobin: characterization of the proximal Ser92Asp variant. Biochemistry. 1996;35:11901–11912. doi: 10.1021/bi9608976. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Chien EY, Sligar SG, La Mar GN. Influence of proximal side mutations on the molecular and electronic structure of cyanomet myoglobin: an 1H NMR study. Biochemistry. 1998;37:6979–6990. doi: 10.1021/bi9728295. [DOI] [PubMed] [Google Scholar]
- 30.Wittenberg JB, Bolognesi M, Wittenberg BA, Guertin M. Truncated hemoglobins: A new family of hemoglobins widely distributed in bacteria, unicellular eukaryotes and plants. J. Biol. Chem. 2002;277:871–874. doi: 10.1074/jbc.R100058200. [DOI] [PubMed] [Google Scholar]
- 31.Vuletich DA, Lecomte JTJ. A phylogenetic and structural analysis of truncated hemoglobins. J. Mol. Evol. 2006;62:196–210. doi: 10.1007/s00239-005-0077-4. [DOI] [PubMed] [Google Scholar]
- 32.Milani M, Pesce A, Nardini M, Ouellet H, Ouellet Y, Dewilde S, Bocedi A, Ascenzi P, Guertin M, Moens L, Friedman JM, Wittenberg JB, Bolognesi M. Structural bases for heme binding and diatomic ligand recognition in truncated hemoglobins. J. Inorg. Biochem. 2005;99:97–109. doi: 10.1016/j.jinorgbio.2004.10.035. [DOI] [PubMed] [Google Scholar]
- 33.Dantsker D, Samuni U, Ouellet Y, Wittenberg BA, Wittenberg JB, Milani M, Bolognesi M, Guertin M, Friedman JM. Viscosity-dependent relaxation significantly modulates the kinetics of CO recombination in the truncated hemoglobin TrHbN from Mycobacterium tuberculosis. J. Biol. Chem. 2004;279:38844–38853. doi: 10.1074/jbc.M401513200. [DOI] [PubMed] [Google Scholar]
- 34.Falzone CJ, Vu BC, Scott NL, Lecomte JTJ. The solution structure of the recombinant hemoglobin from the cyanobacterium Synechocystis sp. PCC 6803 in its hemichrome state. J. Mol. Biol. 2002;324:1015–1029. doi: 10.1016/s0022-2836(02)01093-8. [DOI] [PubMed] [Google Scholar]
- 35.Hoy JA, Kundu S, Trent JT, 3rd, Ramaswamy S, Hargrove MS. The crystal structure of Synechocystis hemoglobin with a covalent heme linkage. J. Biol. Chem. 2004;279:16535–16542. doi: 10.1074/jbc.M313707200. [DOI] [PubMed] [Google Scholar]
- 36.Trent JT, 3rd, Kundu S, Hoy JA, Hargrove MS. Crystallographic analysis of synechocystis cyanoglobin reveals the structural changes accompanying ligand binding in a hexacoordinate hemoglobin. J. Mol. Biol. 2004;341:1097–1108. doi: 10.1016/j.jmb.2004.05.070. [DOI] [PubMed] [Google Scholar]
- 37.Vu BC, Nothnagel HJ, Vuletich DA, Falzone CJ, Lecomte JTJ. Cyanide binding to hexacoordinate cyanobacterial hemoglobins: Hydrogen bonding network and heme pocket rearrangement in ferric H117A Synechocystis Hb. Biochemistry. 2004;43:12622–12633. doi: 10.1021/bi048726l. [DOI] [PubMed] [Google Scholar]
- 38.Hvitved AN, Trent JT, 3rd, Premer SA, Hargrove MS. Ligand binding and hexacoordination in Synechocystis hemoglobin. J. Biol. Chem. 2001;276:34714–34721. doi: 10.1074/jbc.M105175200. [DOI] [PubMed] [Google Scholar]
- 39.Smagghe BJ, Sarath G, Ross E, Hilbert JL, Hargrove MS. Slow ligand binding kinetics dominate ferrous hexacoordinate hemoglobin reactivities and reveal differences between plants and other species. Biochemistry. 2006;45:561–570. doi: 10.1021/bi051902l. [DOI] [PubMed] [Google Scholar]
- 40.Vu BC, Jones AD, Lecomte JTJ. Novel histidine-heme covalent linkage in a hemoglobin. J. Am. Chem. Soc. 2002;124:8544–8545. doi: 10.1021/ja026569c. [DOI] [PubMed] [Google Scholar]
- 41.Scott NL, Lecomte JTJ. Cloning, expression, purification, and preliminary characterization of a putative hemoglobin from the cyanobacterium Synechocystis sp. PCC 6803. Protein Sci. 2000;9:587–597. doi: 10.1110/ps.9.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vu BC, Vuletich DA, Kuriakose SA, Falzone CJ, Lecomte JTJ. Characterization of the heme-histidine cross-link in cyanobacterial hemoglobins from Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002. J. Biol. Inorg. Chem. 2004;9:183–194. doi: 10.1007/s00775-003-0512-1. [DOI] [PubMed] [Google Scholar]
- 43.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 44.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 45.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 2006. [Google Scholar]
- 46.Bilsel O, Zitzewitz JA, Bowers KE, Matthews CR. Folding mechanism of the α-subunit of tryptophan synthase, an α/β barrel protein: global analysis highlights the interconversion of multiple native, intermediate, and unfolded forms through parallel channels. Biochemistry. 1999;38:1018–1029. doi: 10.1021/bi982365q. [DOI] [PubMed] [Google Scholar]
- 47.Pace CN, Shirley BA, Thomson JA. Protein structure: a practical approach. IRL Press; Oxford; New York: 1989. [Google Scholar]
- 48.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Creighton TE, Shortle D. Electrophoretic characterization of the denatured states of staphylococcal nuclease. J. Mol. Biol. 1994;242:670–682. doi: 10.1006/jmbi.1994.1616. [DOI] [PubMed] [Google Scholar]
- 50.Thomas PE, Ryan D, Levin W. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal. Biochem. 1976;75:168–176. doi: 10.1016/0003-2697(76)90067-1. [DOI] [PubMed] [Google Scholar]
- 51.Goodhew CF, Brown KR, Pettigrew GW. Haem staining in gels, a useful tool in the study of bacterial c-type cytochromes. Biochim. Biophys. Acta. 1986;852:288–294. [Google Scholar]
- 52.La Mar GN, Satterlee JD, de Ropp JS. Nuclear magnetic resonance of hemoproteins. In: Smith KM, Kadish K, Guilard R, editors. The Porphyrin Handbook. Academic Press; Burlington, MA: 1999. pp. 185–298. [Google Scholar]
- 53.Shiro Y, Iizuka T, Makino R, Ishimura Y, Morishima I. 15N NMR study of cyanide (C15N) complex of cytochrome P-450cam. Effects of d-camphor and putidaredoxin on iron-ligand structure. J. Am. Chem. Soc. 1989;111:7707–7711. [Google Scholar]
- 54.Mukhopadhyay K, Lecomte JTJ. A relationship between heme binding and protein stability in cytochrome b5. Biochemistry. 2004;43:12227–12236. doi: 10.1021/bi0488956. [DOI] [PubMed] [Google Scholar]
- 55.Milani M, Ouellet Y, Ouellet H, Guertin M, Boffi A, Antonini G, Bocedi A, Mattu M, Bolognesi M, Ascenzi P. Cyanide binding to truncated hemoglobins: a crystallographic and kinetic study. Biochemistry. 2004;43:5213–5221. doi: 10.1021/bi049870+. [DOI] [PubMed] [Google Scholar]
- 56.Peterson ES, Friedman JM, Chien EY, Sligar SG. Functional implications of the proximal hydrogen-bonding network in myoglobin: a resonance Raman and kinetic study of Leu89, Ser92, His97, and F-helix swap mutants. Biochemistry. 1998;37:12301–12319. doi: 10.1021/bi980752u. [DOI] [PubMed] [Google Scholar]
- 57.Bertini I, Luchinat C, Parigi G, Walker FA. Heme methyl 1H chemical shifts as structural parameters in some low-spin ferriheme proteins. J. Biol. Inorg. Chem. 1999;4:515–519. doi: 10.1007/s007750050337. [DOI] [PubMed] [Google Scholar]
- 58.Koshy TI, Luntz TL, Schejter A, Margoliash E. Changing the invariant proline-30 of rat and Drosophila melanogaster cytochromes c to alanine or valine destabilizes the heme crevice more than the overall conformation. Proc. Natl. Acad. Sci. U. S. A. 1990;87:8697–8701. doi: 10.1073/pnas.87.22.8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garcia P, Bruix M, Rico M, Ciofi-Baffoni S, Banci L, Ramachandra Shastry MC, Roder H, de Lumley Woodyear T, Johnson CM, Fersht AR, Barker PD. Effects of heme on the structure of the denatured state and folding kinetics of cytochrome b562. J. Mol. Biol. 2005;346:331–344. doi: 10.1016/j.jmb.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 60.Mintorovitch J, van Pelt D, Satterlee JD. Kinetic study of the slow cyanide binding to Glycera dibranchiata monomer hemoglobin components III and IV. Biochemistry. 1989;28:6099–6104. doi: 10.1021/bi00440a055. [DOI] [PubMed] [Google Scholar]
- 61.Vallone B, Nienhaus K, Matthes A, Brunori M, Nienhaus GU. The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand affinity. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17351–17356. doi: 10.1073/pnas.0407633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.