

Abstract
Lysophosphatidic acid bearing a benzophenone group in either the sn-1 or sn-2 chain of an oleoyl-type ester or oleyl-type ether chain and 32P in the phosphate group were synthesized. The benzophenone moiety was introduced by selective hydroboration of the double bond of enyne 11 at low temperature, followed by Suzuki reaction with 4-bromobenzophenone. The key intermediates for the preparation of ester-linked LPA 1 and 3 were obtained in one pot by a modified DIBAL-H reduction of orthoformate intermediate 22. These probes were shown to covalently modify a single protein target in rat plasma containing albumin and several protein targets in rat plasma containing a low level of albumin.
Introduction
Lysophosphatidic acid (LPA) has the simplest structure of natural phospholipids, yet has attracted significant attention because of the diversity and importance of its biological effects.1-5 LPA is not a single compound; rather, it comprises a family of related mediators that differ in the length and degree of unsaturation of a hydrocarbon chain linked to a glycerol backbone via ester,6 ether,7 or vinyl ether8 bonds at either the sn-1 or sn-2 position. Different LPA family members have distinct biological activities.9 LPA plays a key role in a myriad of physiological processes, including Ca2+ flux, vascular and neuronal function, cell growth/death, and cell migration.2,10 It is now appreciated that LPA has numerous intracellular, extracellular, and cell surface targets. LPA exerts its activities mainly through several cell-surface G protein-coupled receptors (GPCR), including the widely reported LPA1, LPA2, and LPA3 receptor subtypes. Recently, the LPA4/GPR23/P2Y9 receptor11 and the nuclear transcription factor PPARγ12 were identified as LPA receptors. LPA also interacts with numerous binding proteins, including albumin13 and gelsolin.14 It has been speculated that interactions with binding proteins may modulate the biological activity of LPA.
Despite recent progress elucidating many of the biological properties of LPA, a detailed understanding of the key roles of LPA in many disease processes has not yet been achieved. To gain insights into the protein targets of LPA, we have prepared photoreactive analogues of LPA. Photoactivatable lipid analogues have been widely employed to study hydrophobic binding sites of lipid targets.15 Recently, we reported the first synthesis and characterization of two photoactivable analogues of sphingosine 1-phosphate (S1P).16 Likewise, photoactivatable analogues of bioactive glycerophospholipids17 and lysosphingophospholipids18 have been reported recently. One previous study reported the synthesis and characterization of a photoreactive LPA analogue containing a trifluoromethylphenyldiazirine group for labeling of fetal bovine serum and LPA responsive cells.19 In this paper, we report the syntheses of additional photoactivable analogues of acyl- and O-alkyl-linked LPAs in which the long chain resembles an oleoyl (ester) or oleyl (ether) chain, respectively. The rationale for this approach is that the oleoyl chain has been shown to represent the maximally effective fatty acyl chain of natural LPAs in previous structure-activity studies.20 We have prepared both the 2-lyso (1 and 2) and 1-lyso (3 and 4) regioisomeric analogues of [32P]-LPA, as well as O-methoxymethyl (MOM)-protected photoreactive analogues of [32P]-LPA (denoted as 20P and 27P). In the current analogues, benzophenone was employed as the photoprobe for labeling of proteins because it is readily activated with long-wavelength UV light, is stable in most of the chemical reactions used during the synthesis of the probe, and generally yields covalent products modified at a single site.21 In the four probes reported herein, the benzophenone moiety is linked to the end of a Δ9 cis hydrocarbon chain. These analogues were characterized via the covalent modification of rat plasma proteins.

Results and Discussion
Retrosynthetic Analysis
Our retrosynthetic strategy for the synthesis of 1 and 2 is outlined in Scheme 1. Analogues 1 and 2 were envisioned as being produced via enzymatic phosphorylation and deprotection of 2-MOM-protected 3-hydroxy-glyceryl ester 5 (X = O) or ether (X = H2), respectively. Compound 5 can be generated by the selective removal of the sn-3 protecting group (PG) from 6, whereas intermediate 6 is accessible from carboxylic acid 7 (X = O) or from the corresponding alcohol (X = H2). The introduction of the benzophenone probe into 7 may be achieved by the regioselective addition of 9-BBN to the double bond in 8, provided that no reaction occurs at the internal triple bond, followed by Suzuki coupling with 4-bromobenzophenone. Compounds 3 and 4 can be prepared from the regioisomers of 5−8 using similar schemes.
Scheme 1.

Retrosynthetic Strategy
Conversion of 2-Decyn-1-ol to Alcohol 14
Scheme 2 outlines the synthesis of Δ9-alcohol 14. The acetylene zipper reaction of commercially available 2-decyn-1-ol22 gave terminal acetylide 9. After the hydroxy group was protected as a THP ether to give 10 in nearly quantitative yield, addition of allyl bromide to the lithium anion of 9 provided enyne 11 in 79% yield. The benzophenone moiety was introduced by selective hydroboration of the double bond of 11 at low temperature, without reaction of the organoborane with the triple bond, followed by Suzuki reaction of the terminal boronate with 4-bromobenzophenone. After the THP group of 12 was removed, Lindlar-catalyzed hydrogenation of the triple bond in 13 afforded Z alcohol 14 in very high yield.
Scheme 2. Synthesis of Long-Chain Alcohol 14.
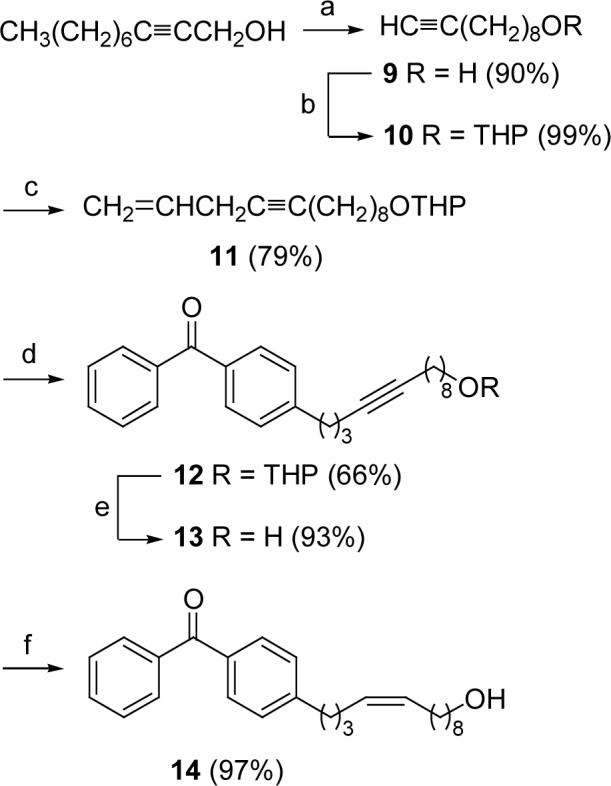
(a) LiNH(CH2)3NH2, t-BuOK/H2N(CH2)3NH2; (b) DHP, PPTS, CH2Cl2; (c) i. n-BuLi, THF, −78 °C to rt; ii. allyl bromide, CuI, THF, −78 °C; (d) i. 9-BBN, THF, −15 °C to rt; ii. 4-bromobenzophenone, Pd(PPh3)4, K3PO4, THF/DMF, reflux, 2 h; (e) PPTS (cat.), MeOH; (f) H2, Lindlar, MeOH.
Synthesis of Ester-linked LPA Probes 1 and 3. Conversion of Alcohol 14 to Glyceride 20
Oxidation of alcohol 14 with PDC gave acid 15, which was converted to ester 16 by reaction with (R)-isopropylidene glycerol (Scheme 3). After removal of the acetonide protecting group in 16 afforded diol 17, monoacylation of the resultant primary hydroxy group was accomplished according to a reported procedure23 with 1 equiv. of FMOC-chloroformate, providing secondary alcohol 18 in 28% yield based on diol 17. This yield is comparable to that reported with a similar diol.23 Treatment of 18 with MOMCl provided glyceride 19, and removal of the FMOC group (10% piperidine in CH2Cl2) afforded 20. Although this multistep synthesis is straightforward, the overall yield of ester 20 is low and the method does not allow for the preparation of the regioisomer of 20. To overcome these deficiencies, an alternative synthetic strategy was designed (see Scheme 4).
Scheme 3. Synthesis of Glyceride 20 via FMOC-ester 18.
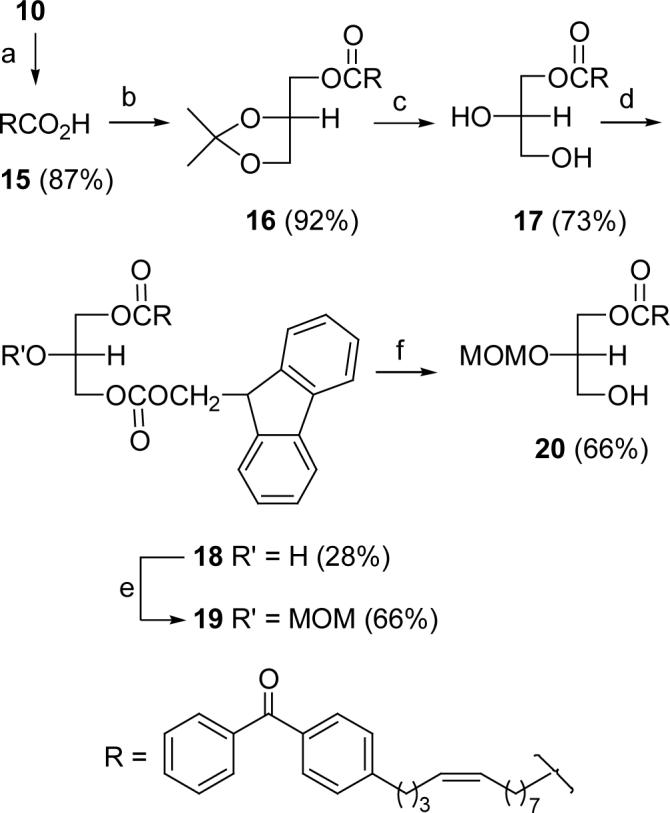
(a) PDC/DMF, rt , 2 d; (b) (R)-(-)-2,2-dimethyl-1,3-dioxolane-4-methanol, DCC, DMAP, CH2Cl2, rt; (c) 0.4 N HCl/90% dioxane, rt; (d) FMOC-chloroformate, DMAP, CH2Cl2, −10 °C; (e) MOMCl, i-Pr2NEt, CH2Cl2; (f) piperidine, CH2Cl2.
Scheme 4. Synthesis of 1 and 3.
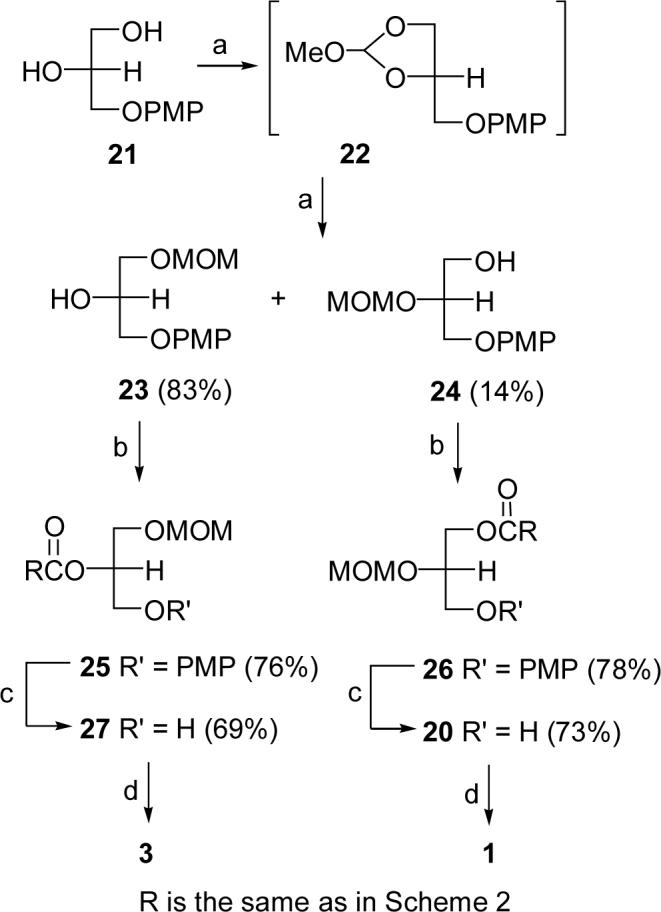
(a) (i). HC(OMe)3, CSA (cat.), CH2Cl2, rt; ii. DIBAL-H, 0 °C; (b) 15, PPh3, DIAD, THF, 0 °C-rt; (c) CAN, CH3CN/H2O (6:1); (d) i. DGK, [32P]ATP, pH 6.6, 37 °C, 2 h; ii. TMSBr, CH2Cl2, 0 °C.
Syntheses of 1 and 3 via 3-O-PMP-sn-glycerol (21)
Our revised synthetic scheme begins with the conversion of 3-O-PMP-sn-glycerol (21)24 to glyceride 20 and its regioisomer 27 (Scheme 4). The MOM protecting group was introduced into diol 21 prior to ester formation by transesterification with trimethyl orthoformate using a modification of a previously reported procedure.25 The key step in this scheme is the protection of one of the hydroxy groups of diol 21 as a MOM ether via orthoformate intermediate 22, followed by reduction with DIBAL-H in toluene. When the reduction was conducted at −78 °C, regioisomers 23 and 24 were obtained in a ratio of 23:1 and an overall yield of 96% after chromatography. Thus, this methodology is a highly efficient route to the precursor of 1-lyso-LPA product 3, but not to the 2-lyso-LPA product 1. We altered the reduction conditions to obtain an adequate amount of primary alcohol 24 for conversion to 1-acyl-LPA product 1, which is needed in the photolabeling experiments. When the DIBAL-H reduction was carried out at 0 °C, compound 24 was obtained in 14% yield, which sufficed for the preparation of 1. Esterification of 23 and 24 with acid 15 using Mitsunobu reaction conditions, followed by removal of the PMP group in 25 and 26 with CAN, provided 27 and 20. Compounds 1 and 3 were obtained by phosphorylation of 27 and 20 with diacylglycerol kinase (DGK) and [32P]ATP.26 The final step, deprotection of the MOM group of 20P and 27P, was accomplished in quantitative yield with trimethylsilyl bromide in dry CH2Cl2 at 0 °C.
Synthesis of Ether-linked LPA Probes 2 and 4
Scheme 5 depicts the strategy we used to introduce an O-alkyl chain bearing a benzophenone probe into the sn-1 or sn-2 positions of glycerol. Reaction of diol 21 with dibutyltin oxide afforded cyclic tin intermediate 29,27 which was used without purification. Ethers 30 and 31 were obtained in 80% and 10% yields, respectively, by nucleophilic substitution of bromide 28 with intermediate 29. After the hydroxy group in 30 or 31 was protected as a MOM ether, the PMP group was removed with CAN to give precursor 34 or 35 in high yield. Ether-linked probes 2 and 4 were obtained from 34 and 35 using the same phosphorylation and deprotection methodology described for the preparation of 1 and 3.
Scheme 5. Synthesis of Ether-linked Probes 2 and 4.
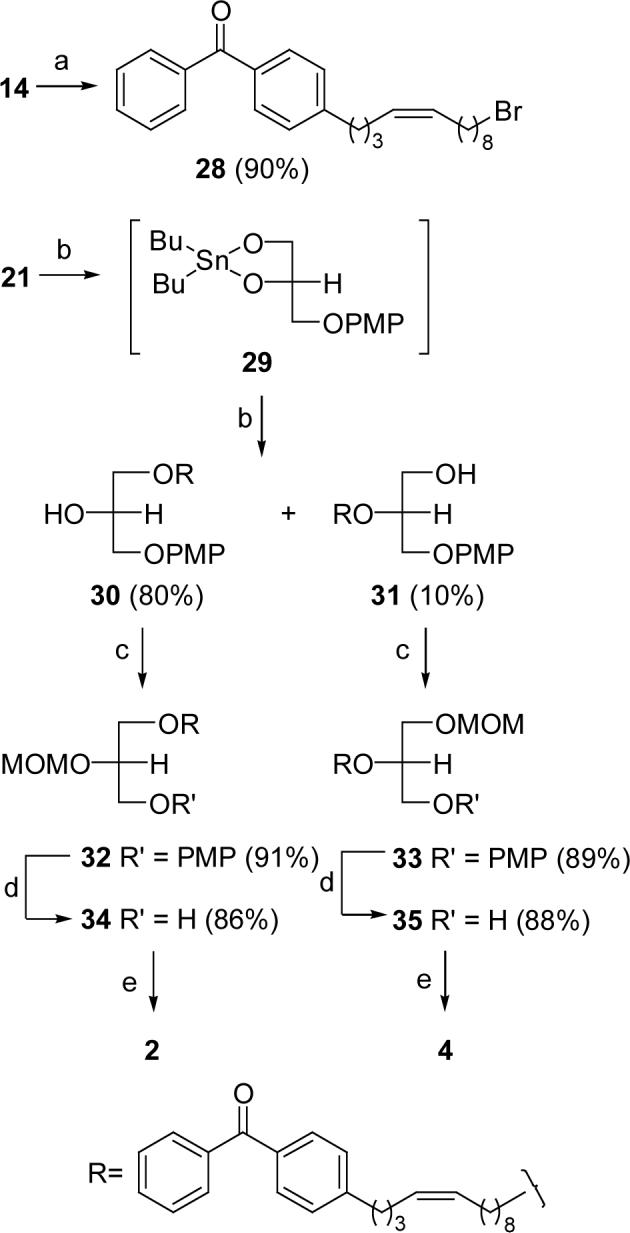
(a) (i) CH3SO2Cl, Et3N, 0 °C-rt; (ii) LiBr, THF, reflux; (b) (i) (n-Bu)2SnO, CHCl3/MeOH (10:1), reflux; (ii) 28, CsF, 18-crown-6, DMF, rt, 1 d; (c) MOMCl, i-Pr2NEt, CH2Cl2; (d) CAN, CH3CN/H2O (6:1); (e) (i) DGK, [32P]ATP; (ii) TMSBr, CH2Cl2, 0 °C.
Plasma Protein Photolabeling
The goal of this research was to generate tools that can be used to distinguish targets of individual LPA family members, i.e., acyl vs. O-alkyl and sn-1 vs. sn-2 linked aliphatic benzophenones. To that end, we tested five of the synthesized target compounds and intermediates for the ability to photolabel plasma proteins. Probes 1 and 2 allow for a comparison of the sn-1 acyl- vs. O-alkyl analogues whereas 2 and 4 allow for a comparison of the sn-1 vs. sn-2 regioisomers. Comparisons between compounds 1 and 3 were not made because of facile acyl migration inherent in probe 3.28 However, the sn-3 phosphorylation products (20P and 27P) of synthetic intermediates 20 and 27 afford additional comparison between the acyl regioisomers of LPA without the risk of acyl migration. Plasma from the Sprague-Dawley rat (“normal rat plasma,” NRP) contains 19.4 mg of albumin per mL,29 and was used as a surrogate for normal human plasma. Conversely, plasma from the Nagase analbuminemic rat (“analbuminemic rat plasma,” ARP) contains only 0.4 mg of albumin per mL.29 This material was used as a tool to examine binding to plasma proteins other than albumin.
Benzophenone-containing LPA probes were characterized by UV-dependent labeling of plasma proteins (Figure 1). Probes 1, 2, 4, 20P, or 27P all showed time-(Figure 1A) and concentration- (data not shown) dependent labeling of predominantly one protein band in NRP. Based on the fact that LPA was initially purified from serum as biological activity associated with albumin13 and the size of the labeled band in this study (∼70 kDa), we conclude that this protein is likely albumin. In contrast, 4 or 5 targets were identified on photolabeling of plasma with a low content of albumin (ARP) with the same probes (Figure 1B). Once again, labeling for all probes was time (Figure 1B) and dose (data not shown) dependent. The intensity of the individual labeled bands varied among the probes but all of the probes modify similar targets in both NRP and ARP. This was expected since these targets are presumed to be transport proteins in which the interactions are based more on hydrophobicity than on stereochemistry or regiochemistry. Further identification and characterization of plasma protein targets of LPA-containing benzophenones, as well as examination of the pharmacology of these reagents at individual LPA GPCR and cellular targets, is currently underway in our laboratories.
Figure 1.


Covalent modification of plasma proteins by benzophenone-containing LPA analogues. Plasma (5% in PBS) from Sprague-Dawley (19.4 mg albumin/mL) A or Nagase rats (0.4 mg albumin/mL) B was incubated with compounds 1, 2, 4, 20P, or 27P (50 nM, DMSO 10% final concentration) in the dark for 30 min prior to UV irradiation for up to 10 min on ice. Samples were diluted with 2 × sample loading buffer and separated by SDS-PAGE. Gels were subsequently dried and radioactive bands were visualized by autoradiography.
Experimental Section
Tetrahydro-2-(tridec-12-en-9-ynyloxy)-2H-pyran (11)
To a solution of alkyne 10 (2.00 g, 8.40 mmol) in 20 mL of dry THF, n-BuLi (3.5 mL, 10.1 mmol, a 2.89 M solution in hexane) was added at −78 °C under N2. After being stirred for 1 h at −78 °C, the reaction mixture was warmed to room temperature and stirred for an additional 1.5 h. The mixture was then slowly added to a solution of allyl bromide (1.22 g, 10.1 mmol) and CuI (100 mg, 0.50 mmol) in 20 mL of THF at −78 °C. After the reaction mixture was stirred overnight at room temperature, the mixture was filtered through a short silica gel column. The solvent was removed, and the residue was purified by chromatography (hexane/EtOAc 20:1) to afford 11 (1.85 g, 79%) as a colorless oil. 1H NMR δ 1.24−1.90 (m, 18H), 2.14−2.22 (m, 2H), 2.90−2.97 (m, 2H), 3.34−3.41 (m, 1H), 3.48−3.52 (m, 1H), 3.69−3.76 (m, 1H), 3.84−3.89 (m, 1H), 4.55−4.59 (m, 1H), 5.06−5.11 (m, 1H), 5.27−5.36 (m, 1H), 5.77−5.84 (m, 1H); 13C NMR δ 18.7, 19.6, 23.1, 25.4, 26.1, 28.8, 29.0, 29.0, 29.3, 29.7, 30.7, 62.2, 67.6, 76.4, 82.8, 98.8, 115.5, 133.3; HR-MS [MNa+] m/z calcd C18H30O2Na 301.2138, found 301.2107.
4-(13-(Tetrahydro-2H-pyran-2-yloxy)tridec-4-ynyl)-benzophenone (12)
A flask charged with 11 (1.11 g, 4.00 mmol) and 15 mL of THF was cooled to −15 °C in a salt-ice bath, and 9-BBN (12 mL, 6.0 mmol, a 0.5 M solution in THF) was added dropwise under N2. After the addition, the reaction mixture was warmed to room temperature and stirred for 5 h. 4-Bromobenzophenone (1.04 g, 4.00 mmol), Pd(PPh3)4 (230 mg, 0.20 mmol), DMF (15 mL), and anhydrous K3PO4 (1.57 g, 8.00 mmol) were added, and the resulting mixture was heated at reflux for 2 h. The reaction mixture was poured into 100 mL of water and extracted with CH2Cl2 (4 × 40 mL). The combined organic layers were washed with 100 mL of water and dried (Na2SO4). The solvent was removed under vacuum and the residue was purified by chromatography (hexane/EtOAc 8:1) to provide 12 (1.21 g, 66%) as a colorless oil. 1H NMR δ 1.28−1.90 (m, 20H), 2.12−2.22 (m, 4H), 2.80 (t, 2H, J = 7.6 Hz), 3.32−3.40 (m, 1H), 3.43−3.50 (m, 1H), 3.68−3.74 (m, 1H), 3.82−3.87 (m, 1H), 4.53−4.58 (m, 1H), 7.28−7.80 (m, 9H). 13C NMR δ 18.0, 18.5, 19.5, 25.3, 26.0, 28.6, 28.9, 29.2, 29.5, 30.2, 30.6, 34.6, 62.1, 67.4, 77.2, 79.1, 80.9, 98.6, 128.0, 128.3, 129.7, 130.1, 132.0, 135.1, 137.7, 146.9, 196.1; HR-MS [MNa+] m/z calcd for C31H40O3Na 483.2870, found 483.2880.
4-(13-Hydroxytridec-4-ynyl)-benzophenone (13)
A solution of 12 (370 mg, 0.80 mmol) and a catalytic amount of PPTS in MeOH was heated at reflux for 3 h. Concentration, followed by purification of the residue by chromatography (hexane/EtOAc 4:1), provided 13 (282 mg, 93%). 1H NMR δ 1.25−1.60 (m, 12H), 1.80−1.85 (m, 2H), 2.14−2.21 (m, 4H), 2.75 (s, 1H), 2.80 (t, 2H, J = 7.6 Hz), 3.58 (t, 2H, J = 6.8 Hz), 7.27−7.79 (m, 9H); 13C NMR δ 17.9, 18.5, 25.5, 28.5, 28.8, 28.9, 29.1, 30.0, 32.4, 34.5, 62.3, 79.1, 80.9, 127.9, 128.2, 129.6, 130.1, 132.0, 134.9, 137.5, 146.9, 196.2; HR-MS [MNa+] m/z calcd for C26H32O2Na 399.2294, found 399.2285.
4-((Z)-13-Hydroxytridec-4-enyl)-benzophenone (14)
Lindlar catalyst (15 mg, 5.5% by weight) was added to a solution of 13 (270 mg, 0.72 mmol) in 10 mL of MeOH. The mixture was stirred under a hydrogen balloon until the starting alkyne was consumed (monitored by TLC, hexane/EtOAc 2:1). The solvent was removed under vacuum, and the residue was purified by chromatography (hexane/EtOAc 4:1) to give 14 (262 mg, 97%) as an oil. 1H NMR δ 1.25−1.48 (m, 10H). 1.50−1.56 (m, 2H), 1.68−1.74 (m, 2H), 1.96−2.13 (m, 4H), 2.45 (s, 1H), 2.69 (t, 2H, J = 7.6 Hz), 3.59 (t, 2H, J = 6.8 Hz), 5.33−5.40 (m, 2H), 7.25−7.77 (m, 9H); 13C NMR δ 25.6, 26.5, 27.1, 29.0, 29.2, 29.3, 29.5, 30.9, 32.5, 35.2, 62.5, 128.0, 128.2, 128.7, 129.7, 130.1, 130.5, 132.0, 134.9, 137.6, 147.7, 196.4; HR-MS [MNa+] m/z calcd for C26H34O2Na 401.2451, found 401.2454.
(Z)-13-(4-Benzoylphenyl)-9-tridecenoic acid (15)
A solution of 14 (730 mg, 1.93 mmol) and PDC (7.3 g, 19.3 mmol) in DMF (20 mL) was stirred at room temperature for 2 days. Water (100 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with water (100 mL) and dried (Na2SO4). The solvent was removed under vacuum and the residue was purified by chromatography (hexane/EtOAc 2:1) to give 15 (657 mg, 87%) as a viscous oil. 1H NMR δ 1.24−1.40 (m, 8H), 1.58−1.75 (m, 4H), 1.96−2.12 (m, 4H), 2.33 (t, 2H, J = 7.2 Hz), 2.70 (t, 2H, J = 7.6 Hz), 5.33−5.44 (m, 2H), 7.26−7.79 (m, 9H); 13C NMR δ 24.6, 26.7, 27.2, 28.9, 29.0, 29.1, 29.5, 31.0, 34.0, 35.4, 128.1, 128.3, 128.9, 129.9, 130.3, 130.6, 132.1, 135.0, 137.8, 147.8, 179.9, 196.5; HR-MS [MNa+] m/z calcd for C26H32O3Na 415.2244, found 415.2255.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenoyl)-3-O-FMOC-sn-glycerol (18)
DCC (6.3 mg, 0.031 mmol) and DMAP (1.0 mg, 8.1 μmol) were added to a solution of 15 (10.0 mg, 0.025 mmol) and (R)-(-)-2,2-dimethyl-1,3-dioxolane-4-methanol (4.0 mg, 0.031 mmol) in 1 mL of CH2Cl2. After the mixture was stirred at room temperature for 40 h, the solvent was removed under vacuum, and the residue was purified by chromatography (hexane/EtOAc 4:1) to provide (Z)-13-benzoylphenyl-9-tridecenoic acid (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl ester (16) (11.9 mg, 92%). 1H NMR δ 1.23−1.37 (m, 8H), 1.37 (s, 3H), 1.43 (s, 3H), 1.60−1.75 (m, 4H), 1.96−2.12 (m, 4H), 2.34 (t, 2H, J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz), 3.71−3.75 (m, 1H), 4.05−4.18 (m, 3H), 4.28−4.33 (m, 1H), 5.33−5.42 (m, 2H), 7.26−7.80 (m, 9H); 13C NMR δ 24.8, 25.3, 26.6, 26.7, 27.1, 29.0, 19.0, 29.0, 29.5, 31.0, 34.0, 35.4, 64.4, 66.2, 73.5, 109.7, 128.1, 128.4, 128.9, 129.8, 130.2, 130.5, 132.1, 135.0, 137.8, 147.7, 173.5, 196.3.
One drop of concentrated HCl was added to a solution of 16 (90 mg, 0.18 mmol) in 90% dioxane (3 mL). After the mixture was stirred at room temperature for 4 h, the solvent was removed under vacuum and the residue was purified by chromatography (hexane/EtOAc 1:1) to give 1-((Z)-13-(4-benzoylphenyl)-9-tridecenoyl))-sn-glycerol (17) (60 mg, 73%). 1H NMR δ 1.23−1.38 (m, 8H), 1.55−1.68 (m, 4H), 1.95−2.12 (m, 4H), 2.33 (t, 2H, J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz), 2.75 (br s, 2H), 3.55−3.68 (m, 2H), 3.88−3.94 (m, 1H), 4.15 (t, 2H, J = 4.8 Hz), 5.34−5.43 (m, 2H), 7.25−7.79 (m, 9H); 13C NMR δ 24.8, 26.6, 27.1, 29.0, 29.0, 29.0, 29.5, 31.0, 34.0, 35.4, 63.3, 65.0, 70.1, 128.2, 128.3, 128.9, 129.9, 130.3, 130.6, 132.2, 135.0, 137.8, 147.9, 174.2, 196.7.
A solution of 17 (58 mg, 0.12 mmol) and DMAP (15 mg, 0.12 mmol) in 1 mL of dry CH2Cl2 was cooled to −10 °C in a salt-ice bath. A solution of FMOC-chloroformate (32 mg, 0.12 mmol) in 2 mL of dry CH2Cl2 was added dropwise. After the reaction mixture was stirred for 30 min, the solution was washed with water (5 mL) and dried (Na2SO4). A small amount (12 mg) of starting material 17 was recovered after chromatography, and 18 (19 mg, 28%) was also obtained after chromatography (hexane/EtOAc 1:3). 1H NMR δ 1.22−1.76 (m, 12H), 1.95−2.21 (m, 5H), 2.35 (t, 2H), J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz), 4.10−4.28 (m, 5H), 4.41 (d, 2H, J = 7.2 Hz), 5.33−5.42 (m, 2H), 7.24−7.80 (m, 17H); 13C NMR δ 24.8, 26.7, 27.2, 29.1, 29.1, 29.6, 30.9, 31.1, 34.1, 35.5, 46.7, 64.8, 68.1, 68.5, 70.1, 120.1, 125.1, 127.2, 127.9, 128.2, 129.0, 129.9, 130.3, 130.6, 132.2, 135.1, 137.9, 141.3, 143.2, 147.8, 155.2, 173.8, 196.6, 207.0; HR-MS [MNa+] m/z calcd for C44H48O7Na 711.3292, found 711.3302.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenoyl)-2-O-methoxymethyl-3-O-FMOC-sn-glycerol (19)
Chloromethyl methyl ether (7 mg, 87 μmol) was added to a stirred solution of 18 (6 mg, 8.7 μmol) and i-Pr2NEt (11 mg, 87 μmol) in dry CH2Cl2 (1 mL) under N2 at 0 °C. After the mixture was stirred at 0 °C for 2 h and at room temperature for 2 days, water (5 mL) and CH2Cl2 (5 mL) were added. The organic layer was washed with brine (5 mL), water (5 mL), and dried (Na2SO4). Product 19 (4.2 mg, 66% yield) was obtained after removal of the solvent and purification by chromatography (hexane/EtOAc 4:1). 1H NMR δ 1.21−1.39 (m, 12H), 1.57−1.75 (m, 4H), 1.95−2.12 (m, 4H), 2.34 (t, 2H, J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz), 3.40 (s, 3H), 4.03−4.33 (m, 5H), 4.41 (d, 2H, J = 7.1 Hz), 4.73 (s, 2H), 5.33−5.44 (m, 2H), 7.24−7.83 (m, 17H); 13C NMR δ 24.9, 26.8, 27.3, 29.1, 29.6, 31.1, 34.1, 35.5, 46.7, 55.7, 63.0, 67.0, 70.0, 72.4, 96.0, 100.0, 120.1, 125.1, 127.2, 127.9, 128.2, 128.4, 129.0, 130.0, 130.3, 130.7, 132.2, 135.1, 137.9, 141.3, 143.3, 147.8, 155.0, 173.4, 196.5; HR-MS [MNa+] m/z calcd for C46H52O8Na 755.3560, found 755.3598.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenoyl)-2-O-methoxymethyl-sn-glycerol (20) from 19
Piperidine (0.2 mL) was added to a solution of 19 (1.3 mg, 1.7 μmol) in dry CH2Cl2 (2 mL) under nitrogen. The mixture was stirred at room temperature until the starting material was consumed (TLC, hexane/EtOAc 4:1). Concentration and purification by chromatography (hexane/EtOAc 2/1) afforded 20 (0.6 mg, 66%). The Rf and 1H NMR spectrum were identical with data obtained for the same compound prepared from 26 (see below).
1-(4-Methoxyphenoxy)-3-(methoxymethoxy)propan-(2R)-2-ol (23) and 3-(4-Methoxyphenoxy)-(2R)-2-(methoxymethoxy)propan-1-ol (24)
Trimethyl orthoformate (0.66 mL, 6.0 mmol) was added to a stirred suspension of diol 21 (594 mg, 3.0 mmol) and d-10-camphorsulfonic acid (12 mg, 50 μmol) in 36 mL of dry CH2Cl2 at at room temperature under nitrogen. The reaction mixture was stirred at room temperature until diol 21 was fully consumed (TLC, hexane/EtOAc 1:1). The mixture was cooled to 0 °C, and 12 mL of DIBAL-H (18 mmol, a 1.5 M solution in toluene) was added slowly. Stirring was continued at 0 °C until the ortho ester intermediate disappeared (TLC, hexane/EtOAc 4:1). A solution of potassium sodium tartrate (10.2 g, 36.0 mmol) in water (40 mL) was added cautiously to quench the reaction, and the resulting mixture was stirred at room temperature overnight. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts were washed with water (50 mL) and dried (Na2SO4 and a small amount of K2CO3). The solvent was removed under vacuum, and the residue was purified by chromatography (a gradient of hexane/EtOAc from 10:1 to 1:1) to provide 603 mg (83%) of 23 and 99 mg (14%) of 24. Data for compound 23: 1H NMR δ 3.31 (s, 1H), 3.35 (s, 3H), 3.65−3.77 (m, 5H), 3.96 (d, 2H, J = 5.8 Hz), 4.08−4.15 (m, 1H), 4.65 (s, 2H), 6.77−6.86 (m, 4H); 13C NMR δ 55.0, 55.3, 68.9, 69.2, 69.4, 96.6, 114.4, 115.3, 152.5, 153.8; HR-MS [MNa+] m/z calcd for C12H18O5Na 265.1046, found 265.1049. Data for compound 24: 1H NMR δ 2.69 (s, 1H), 3.43 (s, 3H), 3.71−3.86 (m, 5H), 3.93−4.04 (m, 3H), 4.79 (s, 2H), 6.78−6.87 (m, 4H); 13C NMR δ 55.6, 62.9, 68.3, 78.0, 96.7, 114.6, 115.4, 152.6, 154.0; HR-MS [MNa+] m/z calcd for C26H34O2Na 265.1046, found 265.1037.
1-O-Methoxymethyl-2-O-(13-(4-benzoylphenyl)-9(Z)-tridecenoyl)-3-O-(4-methoxyphenyl)-sn-glycerol (25)
DIAD (10 mg, 49.5 μmol) was added to a solution of acid 15 (16 mg, 41.3 μmol), alcohol 23 (10 mg, 41.3 μmol), and PPh3(13 mg, 49.5 μmol), in dry THF (2 mL) at 0 °C. After the mixture was warmed to room temperature and stirred for 24 h, water (5 mL) and CH2Cl2 (5 mL) were added. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic layers were washed twice with water (5 mL) and dried (Na2SO4). After the solvents were removed under vacuum, the residue was purified by chromatography (hexane/EtOAc 4:1) to give 25 (19 mg, 76%). 1H NMR δ 1.24−1.36 (m, 8H), 1.58−1.76 (m, 4H), 1.95−2.12 (m, 4H), 2.35 (t, 2H, J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz) 3.34 (s, 3H), 3.76 (s, 3H), 3.79 (d, 2H), 4.06−4.13 (m, 2H), 4.64 (s, 2H), 5.29−5.35 (m, 1H), 5.36−5.44 (m, 2H), 6.80−6.87 (m, 4H), 7.27−7.82 (m, 9H); 13C NMR δ 24.9, 26.8, 27.3, 29.0, 29.1, 29.2, 29.6, 31.1, 34.3, 35.5, 55.3, 55.7, 65.8, 67.0, 70.8, 96.5, 114.6, 115.6, 128.2, 128.4, 129.0, 130.0, 130.4, 130.7, 132.2, 135.1, 137.9, 147.8, 152.7, 154.1, 173.3, 196.5; HR-MS [MNa+] m/z calcd for C38H48O7Na 639.3292, found 639.3298.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenoyl)-2-O-methoxymethyl-3-O-(4-methoxyphenyl)-sn-glycerol (26)
Compound 26 was prepared in 78% yield according to the procedure used to prepare compound 25. 1H NMR δ 1.24−1.35 (m, 8H), 1.58−1.78 (m, 4H), 1.95−2.13 (m, 4H), 2.30−2.35 (m, 2H), 2.68−2.73 (m, 2H), 3.41 (s, 3H), 3.76 (s, 3H), 4.02 (d, 2H, J = 5.3 Hz), 4.11−4.17 (m, 1H), 4.22−4.28 (m, 1H), 4.33−4.38 (m, 1H), 4.77 (s, 2H), 5.36−5.42 (m, 2H), 6.80−6.86 (m, 4H), 7.26−7.82 (m, 9H); 13C NMR δ 24.9, 26.8, 27.2, 29.1, 29.1, 29.2, 29.6, 31.1, 34.1, 35.5, 55.6, 55.7, 63.6, 68.2, 73.3, 96.1, 114.6, 115.5, 128.2, 128.3, 129.0, 129.9, 130.3, 130.6, 132.1, 135.1, 137.9, 147.8, 152.7, 154.1, 173.5, 196.5; HR-MS [MNa+] m/z calcd for C38H48O7Na 639.3292, found 639.3297.
1-O-Methoxymethyl-2-O-(13-(4-benzoylphenyl)-9(Z)-tridecenoyl)-sn-glycerol (27)
CAN (181 mg, 0.33 mmol) was added to a solution of 25 (85 mg, 0.14 mmol) in 7 mL of CH3CN/water (6:1) at room temperature. The mixture was stirred until compound 25 was consumed (∼1 h, TLC, hexane/EtOAc 1:1), and was then diluted with water (10 mL) and CH2Cl2 (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 6 mL), and the combined organic extracts were washed with brine (10 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified by chromatography (hexane/EtOAc 2:1) to furnish 27 (49 mg, 69%). 1H NMR δ 1.23−1.38 (m, 8H), 1.57−1.77 (m, 4H), 1.96−2.13 (m, 4H), 2.35 (t, 2H, J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz), 3.36 (s, 3H), 3.70−3.83 (m, 4H), 4.63 (s, 2H), 5.01−5.07 (m, 1H), 5.34−5.45 (m, 2H), 7.27−7.81 (m, 9H); 13C NMR δ 24.9, 26.7, 27.2, 29.1, 29.1, 29.2, 29.6, 31.1, 34.3, 35.5, 55.4, 62.3, 66.3, 73.1, 96.6, 128.2, 128.4, 129.0, 130.0, 130.4, 130.6, 132.2, 135.1, 137.9, 147.9, 173.6, 196.6; HR-MS [MNa+] m/z calcd for C31H42O6Na 533.2874, found 533.2865.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenoyl)-2-O-methoxymethyl-sn-glycerol (20) from Compound 26
Compound 20 was prepared in 73% yield by the same procedure used to prepare compound 27. 1H NMR δ 1.24−1.38 (m, 8H), 1.56−1.76 (m, 4H), 1.95−2.17 (m, 4H), 2.33 (t, 2H, J = 7.6 Hz), 2.71 (t, 2H, J =7.6 Hz), 3.42 (s, 3H), 3.58−3.71 (m, 2H), 3.80−3.85 (m, 1H), 4.14−4.24 (m, 2H), 4.74 (s, 2H), 5.35−5.44 (m, 2H), 7.24−7.82 (m, 9H); 13C NMR δ 24.9, 26.7, 27.2, 29.1, 29.1, 29.6, 31.1, 34.1, 35.5, 55.7, 62.6, 63.2, 77.7, 96.6, 128.2, 128.3, 129.0, 129.9, 130.3, 130.6, 132.2, 135.1, 137.9, 147.8, 173.7, 196.5; HR-MS [MNa+] m/z calcd for C31H42O6Na 533.2874, found 533.2863.
4-((Z)-Bromotridec-4-enyl)-benzophenone (28)
To 3 mL of CH2Cl2 under N2 were added alcohol 14 (108 mg, 0.29 mmol) and Et3N (43 mg, 0.43 mmol) at 0 °C. After the mixture was stirred for 5 min, methanesulfonyl chloride (49 mg, 0.43 mmol) was added, and the resulting reaction mixture was stirred at room temperature overnight, quenched with 5 mL of water, and extracted with CH2Cl2 (3 × 5 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated. The crude mesylate was dissolved in dry THF (5 mL), and lithium bromide (50 mg, 0.57 mmol) was added. The reaction mixture was heated at reflux overnight, and then cooled to room temperature and poured into water (5 mL) and CH2Cl2 (5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL). The combined organic phases were washed with water (10 mL), dried (Na2SO4), and concentrated. The resulting yellow oil was purified by chromatography (hexane/EtOAc 10:1) to provide 28 (113 mg, 90%) as a colorless oil. 1H NMR δ 1.22−1.45 (m, 10H), 1.68−1.86 (m, 4H), 1.96−2.12 (m, 4H), 2.70 (t, 2H, J = 7.8 Hz), 3.37 (t, 2H, J = 6.8 Hz), 5.35−5.44 (m, 2H), 7.25−7.82 (m, 9H); 13C NMR δ 26.6, 27.1, 28.0, 28.6, 29.1, 29.2, 29.5, 31.0, 32.7, 33.9, 35.4, 128.1, 128.2, 128.9, 129.8, 130.2, 130.5, 132.0, 135.0, 137.8, 147.7, 196.2; HR-MS [MNa+] m/z calcd for C26H33BrONa 463.1607, found 463.1600.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenyl)-3-O-(4-methoxyphenyl)-sn-glycerol (30) and 2-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenyl)-3-O-(4-methoxyphenyl)-sn-glycerol (31)
Di-n-butyltin oxide (64 mg, 0.26 mmol) was added to a solution of diol 21 (51 mg, 0.26 mmol) in 3 mL of CHCl3/MeOH (10:1). After the resulting suspension was heated at reflux for 3 h to give a clear solution, the solvents were evaporated to give cyclic stannoxane intermediate 29 as a white solid. Cesium fluoride (78 mg, 0.51 mmol) and 18-crown-6 (8 mg, 26 μmol) were added, and the solid mixture was dried overnight under high vacuum. After DMF (3 mL) and bromide 28 (71 mg, 0.16 mmol) were added, the reaction mixture was stirred at room temperature for 1 day. Water (0.5 mL) and EtOAc (10 mL) were added, and the mixture was stirred for an additional 1 h and was subsequently filtered through a pad of silica gel to remove an insoluble by-product. The filtrate was washed with water (5 mL), dried (Na2SO4), and concentrated. The residue was purified by chromatography (hexane/EtOAc 2:1) to provide 30 (72 mg, 80%) and 31 (8.7 mg, 10%). Data for compound 30: 1H NMR δ 1.21−1.38 (m, 8H), 1.52−1.76 (m, 4H), 1.94−2.12 (m, 4H), 2.54 (br s, 1H), 2.70 (t, 2H, J = 7.6 Hz), 3.43−3.64 (m, 4H), 3.75 (s, 3H), 3.93−4.01 (m, 2H), 4.09−4.16 (m, 1H), 5.33−5.45 (m, 2H), 6.79−6.88 (m, 4H), 7.25−7.82 (m, 9H); 13C NMR δ 26.0, 26.7, 27.2, 29.2, 29.4, 29.4, 29.5, 29.6, 31.0, 35.4, 55.6, 69.1, 69.7, 71.5, 71.7, 114.6, 115.5, 128.1, 128.3, 128.9, 129.9, 130.3, 130.7, 132.1, 135.1, 137.9, 147.8, 152.7, 154.0, 196.4; HR-MS [MNa+] m/z calcd for C36H46O5Na 581.3237, found 581.3249. Data for compound 31: 1H NMR δ 1.22−1.38 (m, 8H), 1.52−1.77 (m, 4H), 1.95−2.14 (m, 4H), 2.68 (t, 2H, J = 7.8 Hz), 3.52−3.86 (m, 5H), 3.76 (s, 3H), 3.94−4.04 (m, 2H), 5.34−5.45 (m, 2H), 6.79−6.91 (m, 4H), 7.25−7.81 (m, 9H); 13C NMR δ 26.1, 26.8, 27.3, 29.3, 29.5, 29.5, 29.7, 30.1, 31.1, 35.5, 55.7, 62.5, 68.1, 70.7, 78.2, 114.7, 115.5, 128.2, 128.4, 129.0, 130.0, 130.4, 130.8, 132.2, 135.1, 138.0, 147.9, 152.8, 154.0, 196.6; HR-MS [MNa+] m/z calcd for C36H46O5Na 581.3237, found 581.3252.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenyl)-2-O-methoxymethyl-3-O-(4-methoxyphenyl)-sn-glycerol (32)
Chloromethyl methyl ether (40 mg, 497 μmol) was added under N2 to a stirred solution of 30 (56 mg, 99 μmol) and i-Pr2NEt (64 mg, 497 μmol) in dry CH2Cl2 (3 mL) at 0 °C. The reaction mixture was stirred at room temperature for 2 days. The solvent was removed and the residue was purified by chromatography (hexane/EtOAc 4:1) to afford unreacted 30 (10 mg) and 32 (45 mg, 91%). 1H NMR δ 1.22−1.38 (m, 8H), 1.52−1.76 (m, 4H), 1.96−2.15 (m, 4H), 2.70 (t, 2H, J = 7.8 Hz), 3.41 (s, 3H), 3.41−3.50 (m, 2H), 3.56−3.65 (m, 2H), 3.75 (s, 3H), 3.99−4.11 (m, 3H), 4.79 (s, 3H), 5.34−5.45 (m, 2H), 6.79−6.87 (m, 4H), 7.28 (d, 2H, J = 8.3 Hz), 7.47 (t, 2H, J = 7.8 Hz), 7.57 (t, 1H, J = 7.3 Hz), 7.72−7.81 (m, 4H); 13C NMR δ 26.0, 26.7, 27.2, 29.2, 29.4, 29.4, 29.6, 29.6, 31.1, 35.4, 55.4, 55.6, 68.7, 70.4, 71.7, 84.5, 96.2, 114.5, 115.4, 128.1, 128.3, 128.9, 129.9, 130.3, 130.7, 132.1, 135.1, 137.9, 147.8, 152.9, 153.8, 196.4; HR-MS [MNa+] m/z calcd for C38H50O6Na 625.3500, found 625.3519.
1-O-Methoxymethyl-2-O-(13-(4-benzoylphenyl)-9(Z)-tridecenyl)-3-O-(4-methoxyphenyl)-sn-glycerol (33)
Compound 33 was prepared in 89% yield from 31 by the same procedure used to prepare 32. 1H NMR δ 1.22−1.39 (m, 8H), 1.55−1.62 (m, 2H), 1.68−1.76 (m, 2H), 1.97−2.04 (m, 2H), 2.07−2.13 (m, 2H), 2.70 (t, 2H, J = 7.8 Hz), 3.34 (s, 3H), 3.58−3.80 (m, 5H), 3.76 (s, 3H), 3.97−4.08 (m, 2H), 4.65 (s, 2H), 5.34−5.44 (m, 2H), 6.80−6.88 (m, 2H), 7.26−7.81 (m, 9H); 13C NMR δ 26.0, 26.7, 27.3, 29.3, 29.4, 29.5, 29.7, 30.0, 31.1, 35.5, 55.2, 55.7, 66.9, 68.2, 70.7, 77.1, 96.7, 114.6, 115.5, 128.2, 128.3, 128.9, 129.9, 130.3, 130.8, 132.1, 135.1, 137.9, 147.8, 153.0, 153.9, 196.5; HR-MS [MNa+] m/z calcd for C38H50O6Na 625.3500, found 625.3497.
1-O-(13-(4-Benzoylphenyl)-9(Z)-tridecenyl)-2-O-methoxymethyl-sn-glycerol (34)
Compound 34 was prepared in 86% yield using the procedure described to prepare compound 27. 1H NMR δ 1.24−1.39 (m, 8H), 1.52−1.60 (m, 2H), 1.68−1.77 (m, 2H), 1.97−2.04 (m, 2H), 2.06−2.13 (m, 2H), 2.70 (t, 2H, J = 7.6 Hz), 3.42 (s, 3H), 3.40−3.80 (m, 7H), 4.75 (s, 2H), 5.34−5.45 (m, 2H), 7.27−7.82 (m, 9H); 13C NMR δ 26.0, 26.7, 27.3, 29.2, 29.4, 29.4, 29.6, 29.7, 31.1, 35.5, 55.6, 63.7, 71.1, 71.8, 78.5, 96.6, 128.2, 128.3, 128.9, 129.9, 130.3, 130.7, 132.1, 135.1, 137.9, 147.8, 196.5; HR-MS [MNa+] m/z calcd for C31H44O5Na 519.3081, found 519.3079.
1-O-Methoxymethyl-2-O-(13-(4-benzoylphenyl)-9(Z)-tridecenyl)-sn-glycerol (35)
The same method used to prepare 34 was used to prepare 35 in 88% yield. 1H NMR δ 1.22−1.40 (m, 8H), 1.52−1.77 (m, 4H), 1.95−2.13 (m, 4H), 2.70 (t, 2H, J = 7.6 Hz), 3.37 (s, 3H), 3.46−3.77 (m, 7H), 4.63 (s, 2H), 5.34−5.45 (m, 2H), 7.27−7.82 (m, 9H); 13C NMR δ 26.1, 26.7, 27.3, 29.2, 29.4, 29.5, 29.7, 30.0, 31.1, 35.5, 55.3, 62.6, 67.0, 70.3, 78.5, 96.7, 128.2, 128.3, 128.9, 129.9, 130.3, 130.7, 132.2, 135.1, 137.9, 147.8, 196.5; HR-MS [MNa+] m/z calcd for C31H44O5Na 519.3081, found 519.3095.
Supplementary Material
Acknowledgments
We thank Dr. George Kaysen, University of California at Davis, for providing analbuminemic rat plasma. Financial support from National Institutes of Health Grant CA92160 is gratefully acknowledged.
Footnotes
Supporting Information Available: Procedures for enzymatic incorporation of [32P]-phosphate into the benzophenone-containing analogues, for covalent modification of plasma proteins, and for the preparation of compounds 9 and 10, and 1H and 13C NMR spectra for all new compounds and intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bittman R. Chem. Phys. Lipids. 2004;129:111–131. doi: 10.1016/j.chemphyslip.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Tigyi G, Parrill AL. Prog. Lipid Res. 2003;42:498–526. doi: 10.1016/s0163-7827(03)00035-3. [DOI] [PubMed] [Google Scholar]
- 3.Mills GB, Moolenaar WH. Nat. Rev. Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 4.Luquain C, Sciorra VA, Morris AJ. Trends Biochem. Sci. 2003;28:377–383. doi: 10.1016/S0968-0004(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 5.Ye X, Ishii I, Kingsbury MA, Chun J. Biochim. Biophys. Acta. 2002;1585:108–113. doi: 10.1016/s1388-1981(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 6.Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi G. J. Anal. Biochem. 2001;292:287–295. doi: 10.1006/abio.2001.5063. [DOI] [PubMed] [Google Scholar]
- 7.Tokumura A, Sinomiya J, Kishimoto S, Tanaka T, Kogure K, Sugiura T, Satouchi K, Waku K, Fukuzawa K. Biochem. J. 2002;365:617–628. doi: 10.1042/BJ20020348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liliom K, Fischer DJ, Virag T, Sun G, Miller DD, Tseng JL, Desiderio DM, Seidel MC, Erickson JR, Tigyi G. J. Biol. Chem. 1998;273:13461–13468. doi: 10.1074/jbc.273.22.13461. [DOI] [PubMed] [Google Scholar]
- 9.Bandoh K, Aoki J, Taira A, Tsujimoto M, Arai H, Inoue K. FEBS Lett. 2000;478:159–165. doi: 10.1016/s0014-5793(00)01827-5. [DOI] [PubMed] [Google Scholar]
- 10.Ishii I, Fukushima N, Ye X, Chun J. Annu. Rev. Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 11.Noguchi K, Ishii S, Shimizu T. J. Biol. Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 12.McIntyre TM, Pontsler AV, Silva AR, Hilaire A. St., Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Proc. Nat. Acad. Sci. USA. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Tigyi G, Henschen A, Miledi R. J. Biol. Chem. 1991;266:20602–20609. [PubMed] [Google Scholar]; b Tigyi G, Miledi R. J. Biol. Chem. 1992;267:21360–21367. [PubMed] [Google Scholar]
- 14.a Meerschaert K, De Corte V, De Ville Y, Vandekerckhove J, Gettemans J. EMBO J. 1998;17:5923–5930. doi: 10.1093/emboj/17.20.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goetzl EJ, Lee H, Azuma T, Stossel TP, Turck CW, Karliner JS. J. Biol. Chem. 2000;275:14573–14578. doi: 10.1074/jbc.275.19.14573. [DOI] [PubMed] [Google Scholar]
- 15.For a recent review, see: Lala AK. Chem. Phys. Lipids. 2002;116:177–188. doi: 10.1016/s0009-3084(02)00026-9..
- 16.Lu X, Cseh S, Byun H-S, Tigyi G, Bittman R. J. Org. Chem. 2003;68:7046–7050. doi: 10.1021/jo034828q. [DOI] [PubMed] [Google Scholar]
- 17.a Li G, Samadder P, Arthur G, Bittman R. Tetrahedron. 2001;57:8925–8932. [Google Scholar]; b Wang P, Blank DH, Spencer TA. J. Org. Chem. 2004;69:2693–2702. doi: 10.1021/jo030329d. [DOI] [PubMed] [Google Scholar]
- 18.Lu X, Bittman R. J. Org. Chem. 2005;70:4746–4750. doi: 10.1021/jo050513u. [DOI] [PubMed] [Google Scholar]
- 19.van der Bend RL, Brunner J, Jalink K, van Corven EJ, Moolenaar WH, van Blitterswijk WJ. EMBO J. 1992;11:2495–2501. doi: 10.1002/j.1460-2075.1992.tb05314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujiwara Y, Sardar V, Tokumura A, Baker DL, Murakami-Murofushi K, Parrill AL, Tigyi G. J. Biol. Chem. 2005;280:35038–35050. doi: 10.1074/jbc.M504351200. [DOI] [PubMed] [Google Scholar]
- 21.Dormán G, Prestwich GD. Trends Biotechnol. 2000;18:64–77. doi: 10.1016/s0167-7799(99)01402-x. For a review, see. [DOI] [PubMed] [Google Scholar]
- 22.Abrams SR, Shaw AC. Org. Synth. 8:146–148. Coll. [Google Scholar]
- 23.Bibak N, Hajdu J. Tetrahedron Lett. 2003;44:5875–5877. [Google Scholar]
- 24.a Wang ZM, Zhang XL, Sharpless KB. Tetrahedron Lett. 1993;34:2267–2270. [Google Scholar]; b Vilchèze C, Bittman R. J. Lipid Res. 1994;35:734–738. [PubMed] [Google Scholar]; c Byun H-S, Kumar ER, Bittman R. J. Org. Chem. 1994;59:2630–2633. [Google Scholar]; d Marino-Albernas J, Bittman R, Peters A, Mayhew E. J. Med. Chem. 1996;39:3241–3247. doi: 10.1021/jm960164j. [DOI] [PubMed] [Google Scholar]
- 25.He L, Byun H-S, Bittman R. J. Org. Chem. 2000;65:7618–7626. doi: 10.1021/jo001225v. [DOI] [PubMed] [Google Scholar]
- 26.Waggoner DW, Gómez-Muñoz A, Dewald J, Brindley DN. J. Biol. Chem. 1996;271:16506–16509. doi: 10.1074/jbc.271.28.16506. See Supporting Information for experimental details. [DOI] [PubMed] [Google Scholar]
- 27.Yokoyama K, Baker DL, Virag T, Liliom K, Byun H-S, Tigyi G, Bittman R. Biochim. Biophys. Acta. 2002;1582:295–308. doi: 10.1016/s1388-1981(02)00184-1. [DOI] [PubMed] [Google Scholar]
- 28. For the occurrence of intramolecular acyl migration in a related lysophospholipid, see. [Google Scholar]; a Plückthun A, Dennis EA. Biochemistry. 1982;21:1743–1750. doi: 10.1021/bi00537a007. [DOI] [PubMed] [Google Scholar]; b Croset M, Brossard N, Polette A, Lagarde M. Biochem. J. 2000;345:61–67. [PMC free article] [PubMed] [Google Scholar]
- 29.Hama K, Bandoh K, Kakehi Y, Aoki J, Arai H. FEBS Lett. 2002;523:187–192. doi: 10.1016/s0014-5793(02)02976-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.