

Abstract
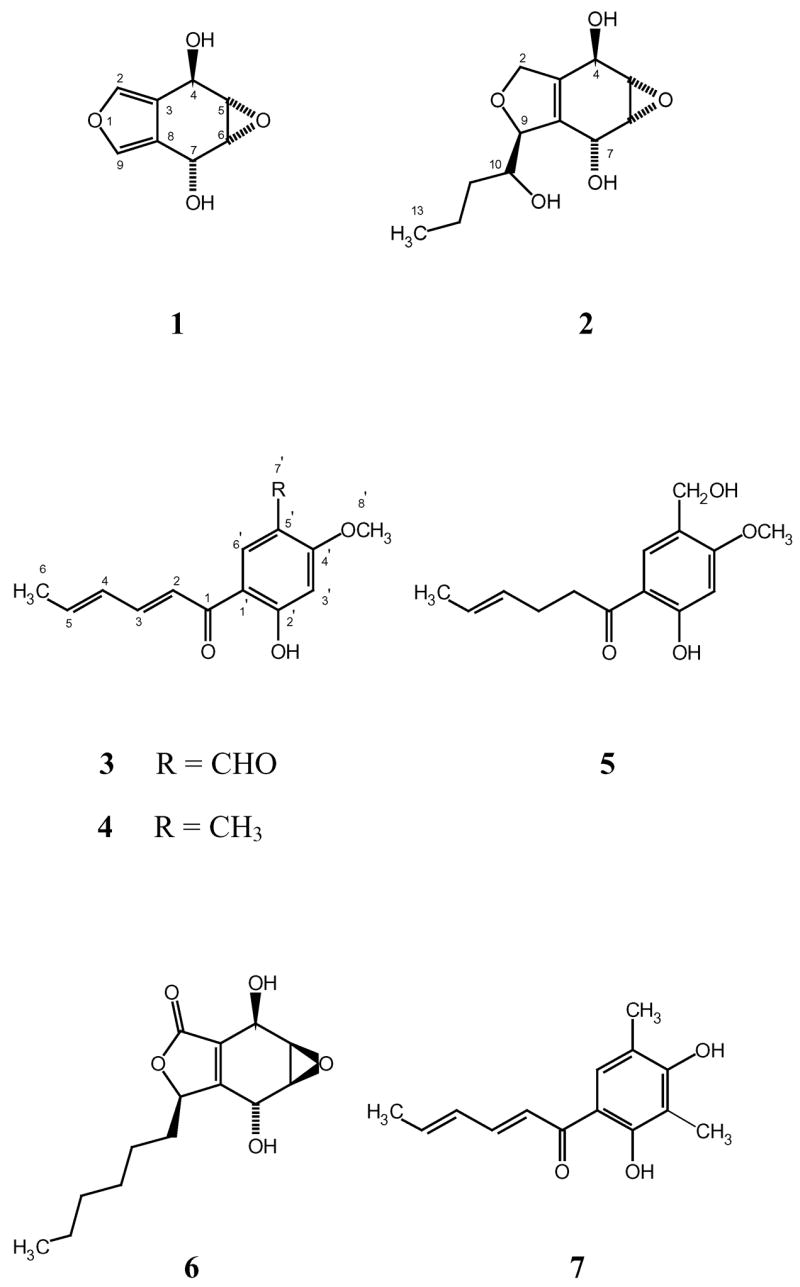
Two new benzofuran-derived metabolites of polyketide origin called phaeofurans A and B (1–2), along with three sorbicillin analogs (3–5) have been obtained from a fungicolous isolate of the genus Phaeoacremonium (NRRL 32148). The structures were determined by analysis of MS and 2D NMR data. The antifungal effects of the extract were ascribed to the sorbicillin analogs.
Previous chemical studies of mycoparasites and fungicolous fungi that attack the nutrient-rich stromata of wood-decay fungi have yielded novel metabolites with antifungal effects and other biological activities.1–6 As part of our ongoing studies of these types of fungi, chemical investigations of a culture identified as Phaeoacremonium sp. (Incertae sedis; NRRL 32148) isolated from field-collected stromata of Hypoxylon truncatum (Xylariaceae) produced two new polyketides named phaeofurans A and B (1–2) and three sorbicillin analogs (3–5), two of which have not been previously reported. Details of the isolation and structure determination of these compounds are presented here.
Results and discussion
Solid-substrate fermentation cultures of a Phaeoacremonium species grown on rice were extracted with ethyl acetate. The resulting extract showed antifungal activity against Aspergillus flavus and Fusarium verticillioides, and was partitioned between acetonitrile and hexane to yield an acetonitrile-soluble mixture that was subsequently fractionated over Sephadex LH-20. Active fractions were subjected to silica gel flash chromatography and/or reversed phase HPLC to yield compounds 1–5.
Phaeofuran A (1) was isolated as a white film that formed fine needles upon evaporation of an acetone solution. The formula of 1 was tentatively assigned as CgHgCX based on analysis of EIMS (apparent M+ at m/z 168) and NMR data (Table 1). The intensities of the EIMS M+ ion and the ESIMS M+H ion proved to be insufficient for obtaining high resolution MS data, but the NMR data suggested the presence of two exchangeable protons in the molecule, so a diacetate derivative of 1 was prepared. HRESIMS analysis of this derivative (obsd m/z 253.0712 [M+H]+, calcd for C12H13O6, 253.0712), together with analysis of its 1H and 13C NMR spectra confirmed the molecular formula of 1 as C8H8O4.
Table 1.
NMR Data for Phaeofuran A (1) in Acetone-d6
| Position | δH(mult; J in Hz)a | δCb | HMBCc H# → C# | NOESYc H# → H# |
|---|---|---|---|---|
| 2 | 7.41 (ddd; 1.6, 1.6, 0.5) | 140.3 | 3, 4, 7, 8, 9 | 4 |
| 3 | --- | 123.4 | --- | --- |
| 4 | 5.06 (dd; 1.8, 1.6) | 64.0 | 2, 3, 8 | 2, 5 |
| 4-OH | 4.46 (br s)c | --- | --- | --- |
| 5 | 3.38 (ddd; 3.9, 1.8, 0.5) | 55.5 | 2, 3, 4, 6, 7 | 4, 6 |
| 6 | 3.40 (dd; 3.9, 2.5) | 55.9 | 4, 5, 7, 8, 9 | 5, 7 |
| 7 | 5.05 (d; 2.5) | 60.5 | 3, 5, 8, 9 | 6, 9 |
| 7-OH | 4.46 (br s)c | --- | --- | --- |
| 8 | --- | 123.1 | --- | --- |
| 9 | 7.49 (d; 1.6) | 141.6 | 2, 3, 6, 7, 8 | 7 |
Recorded at 400 MHz.
Recorded at 100 MHz.
Variable; sometimes resolved, sometimes observed as a single broad signal that integrated as two protons, J -values to the OH signals were not observed in the 1H NMR spectra obtained for 1.
Several HMBC and NOESY correlations observed for 1 (and 2) in the acetone-d6 data were somewhat ambiguous due to signal overlap, but were confirmed by analysis of data recorded in CD3OD.
With five unsaturations, four olefinic or aromatic sp2 carbon atoms (two of them protonated), four oxygenated sp3 methines, and no carbonyl carbons, phaeofuran A (1) must contain three rings, two double bonds, and two OH groups. In addition, observation of four pairs of signals with very similar 13C NMR chemical shifts (Table 1) suggested a pseudo-symmetrical structure. The NMR shifts for two of the oxymethine units (δC 55.9 and 55.5/δH 3.40 and 3.38) were diagnostic for the presence of a 1,2-disubstituted epoxide. Each of the epoxide protons was coupled to one of the other two oxymethines with a small J-value, enabling construction of a substructure corresponding to the C4–C7 unit in 1. The same unit was evident in the 1H NMR spectrum of the diacetate, except that the signals for the oxymethine protons at C-4 and C-7 were downfield shifted from δ 5.05/5.06 to δ 6.12/6.18. The remaining two 1H NMR signals in the spectrum of 1 corresponded to olefinic/aromatic protons that showed long-range coupling (1.6 Hz) to each other. These data, together with the number of unsaturations and the Sc values for the sp2 carbons, limit the number of possible structures, with one containing fused furan and cyclohexane rings as most likely to accommodate these requirements. HMBC correlations (Table 2) of H-4 with C-2, C-3, and C-8, and of H-7 with C-3, C-8, and C-9, enabled closure of the six-membered ring, and established the locations of all of the sp2 carbons. Correlations of H-2 with C-9 and of H-9 with C-2 supported assignment of the furan ring, thereby completing the gross structure of phaeofuran A as shown in 1. NOESY correlations (Table 2) of olefinic/aromatic protons H-2 and H-9 to the oxymethine proton signals for H-4 (8 5.06) and H-7 (8 5.05), respectively, supported this assignment. The structure is consistent with additional HMBC correlations observed, and with the multiple small J-values observed for the signals in the 1H NMR spectrum. Residual one-bond correlations in the HMBC spectrum of 1 allowed assignment of protons to their respective carbon atoms, and the shift assignments and relevant J-values observed were consistent with the presence of the relatively rare 3,4-dialkylfuran moiety.7,8
Table 2.
NMR data for Phaeofuran B (2) in Acetone-d6
| Position | δH(mult; J in Hz)a | δCb | HMBC H# → C# | NOESY H# → H# |
|---|---|---|---|---|
| 2α | 4.42 (m) | 76.0 | 3, 8, 10 | 2β, 9 |
| 2β | 4.73 (ddd; 12, 5.9, 3.4) | --- | 3,4,7,8,10 | 2α, 4, 10, 11 |
| 3 | --- | 133.8 | --- | --- |
| 4 | 4.36 (br d; 6) | 61.7 | 3, 5, 6, 8 | 5 |
| 4-OH | 4.43 (br d; 6) | --- | 3, 4, 5 | --- |
| 5 | 3.33 (dd; 3.6, 1.2) | 56.8 | 2, 3, 4, 6 | 4, 6 |
| 6 | 3.42 (ddd; 3.6, 3.3, 0.5) | 55.3 | 5, 7, 8, 9 | 5, 7 |
| 7 | 4.64 (br m) | 64.5 | --- | 6, 10 |
| 7-OH | 4. 18 (d; 7.2) | --- | 6, 7, 8 | --- |
| 8 | --- | 134.3 | --- | --- |
| 9 | 4.74 (m) | 90.0 | 3, 4, 7, 8 10 | 2α, 10, 11 |
| 10 | 3.64 (m) | 74.3 | --- | 2β, 7, 9, 11 |
| 10-OH | 3.84 (d; 5.4) | --- | 9, 10, 11 | --- |
| 11 | 1.42 (m), 1.57 (m) | 35.7 | 9, 10, 12, 13 | 9, 10, 12, 13 |
| 12 | 1.37 (m), 1.56 (m) | 19.5 | 10, 11,13 | 11, 13 |
| 13 | 0.89 (t; 6.6) | 14.5 | 11, 12 | 10, 11,12 |
Recorded at 400 MHz.
Recorded at 100 MHz.
Interestingly, the relative stereochemistry of phaeofuran A (1) was readily assigned on the basis of symmetry considerations. Of the three possible diastereomeric structures of phaeofuran A, two of them have H-4 and H-7 cis to each other, leading to a plane of symmetry that would give rise to an achiral molecule showing only three 1H NMR methine signals and four 13C NMR signals. Therefore, H-4 and H-7 must be trans to each other in phaeofuran A, thereby establishing the relative stereochemistry as shown in 1. However, specific position assignments for the NMR signals proved to be somewhat problematic. Although NOESY data enabled association of each furan proton with its nearby hydroxymethine counterpart, they did not enable assignment of which hydroxymethine proton was cis to the epoxide oxygen (both hydroxymethine protons showed correlations of similar intensity with the corresponding vicinal epoxy CH proton). As expected, the JH4–H5 and JH6–H7 couplings were different, but only minimally so (1.8 and 2.5 Hz). Unfortunately, to our knowledge, no literature examples with a dihydroxyepoxycyclohexane ring fused to a furan moiety as in structure 1 are available, and a conclusive assignment based on coupling values for somewhat analogous compounds proved to be elusive. The corresponding trans-type relationships in several compounds containing somewhat similar l,4-dioxy-2,3-epoxycyclohexene units range from 1 to 4 Hz,9–13 while examples of J-values for cis-type relationships range from < 1 to 3 Hz,10,13–15 and in one instance, values of 3.0 and 1.2 Hz are reported for two different cis relationships in the same molecule.15 In the case of what is perhaps the closest literature analog (integrasone; 6), values of 3.0 and 1.5 Hz were assigned to cis- and trans-type relationships, respectively.16 In the latter instance, the J-values were cited as reasons for the assignment, as well as a notable difference in intensity for the NOESY correlations between the hydroxymethine protons and their vicinal epoxide neighbors (cis > trans). However, despite the close proximity of the H-5/H-6 and H4/H-7 signals in the spectrum of 1, it was possible at 600 MHz to observe distinct, strong NOESY correlations for both pairs (i.e., H-4 with H-5 and H-6 with H-7), with approximately equal intensity. Molecular modeling calculations for 1 (Spartan ’02; Wavefunction, Inc.) supported the concept that the value for the cis relationship should be larger, since the expected dihedral bond angle would be approximately 30°, while that for the trans relationship would be nearly 90°, but it has been noted that employment of Karplus-type calculations relating bond geometry to J-values in such instances can be equivocal.13 Ultimately, the larger J-value was assigned to the cis relationship as shown in Table 1 on the basis of the these considerations and by analogy to the assignment made for 2 (see below), though this is not considered to be definitive.
Phaeofuran B (2) was isolated as an oil, and its molecular formula was assigned as C12H18O5 on the basis of HRESIMS, 1H NMR (Table 1), 13C NMR (Table 1), and DEPT spectral data. Analysis of the ID NMR spectra revealed the presence of one methyl, three sp3 methylenes (one oxygenated), one tetrasubstituted olefin unit, and six oxygenated sp2 methines. With four unsaturations and only two sp2 carbons, phaeofuran B (2) must also contain three rings.
COSY correlations revealed the presence of two spin-systems corresponding to the C9–C13 and the C4–C7 structural units in 2. The 1H NMR data for the latter spin-system displayed a coupling pattern and δ-values very similar to those in the corresponding segment of phaeofuran A (1), suggesting the presence of an analogous unit. HMQC data for 2 enabled assignment of protons to their respective carbons. HMBC correlations (Table 2) of H-5 to C-3, and of H-6 to C-8, connected the C4-C7 unit to the sp2 carbons, revealing the presence of a six-membered ring unit similar to that found in phaeofuran A (1). The presence of this unit was corroborated by additional correlations of OH-4 to C-3, C-4, and C-5, and of OH-7 to C-6, C-7, and C-8. Correlations of a third hydroxyl proton doublet at δ 3.84 to C-9, C-10, and C-l1 placed this OH group at C-10. This leaves only one oxygen atom which must connect to both C-2 and C-9 to form the remaining ring. Although no HMBC correlations between H2-2 and C-9 or between H-9 and C-2 were observed using acetone-d6 as the solvent, a weak correlation was observed between H-2α and C-9 when the data were recorded using CD3OD. Four-bond correlations of H-5 and H-6 to C-2 and C-9, respectively, were also observed, and are most likely if C-2 and C-9 are connected to C-3 and C-8, respectively. Based on these data, the gross structure of phaeofuran B was proposed as shown in 2. This structure was further supported by additional HMBC correlations.
Observations of a strong HMBC correlation of H-2α to C-10 and a weak correlation of H-2β to C-10 were initially unexpected. However, these long-range correlations could be rationalized by the occurrence of heteronuclear homoallylic coupling analogous to the particularly large homoallylic 1H-1H couplings anticipated17 and observed in this system. The coupling constants for H-2α and H-2β were measured in CD3OD to suppress the hydroxyl signals of 2 (and the coupling thereto), and the splitting patterns of H-2α (dddd, J = 12, 3.1, 2.3, 0.8 Hz) and H-2β (dddd, J = 12, 5.8, 3.3, 0.4 Hz) reflected the large homoallylic couplings of H-2α (7 = 3.1 Hz) and H-2β (7 = 5.8 Hz) with H-9. Dihydrofuran rings of this type are rare among natural products, and a suitable model compound was difficult to find in the literature. The closest known analog of phaeofuran B (2) appears to be integrasone (6),16 a recently reported HIV-integrase inhibitor. Comparison of the data for 2 and 6 (recorded using the same solvent; CD3OD) indicated close similarities in the relevant features of the data, particularly for the dihydroxyepoxycyclohexene unit.
Unlike the case for 1, the relative stereochemistry was not independently dictated by symmetry considerations. NOESY correlations (Table 2) observed for phaeofuran B (2) fully supported assignment of the regiochemistry shown. In addition, a key NOESY correlation was observed between H-7 and H-10 that allowed placement of H-7 and the hydroxybutyl side-chain on the same face of the molecule. The configurations at C-4, C-5, C-6, and C-7 were assigned by analysis of J-values in comparison to those of phaeofuran A (1) and integrasone (6). The presence of 3.3- and 1.2-Hz couplings for JH4–H5 and JH6–H7 again supported the assignment of a trans relationship between H-4 and H-7, as these values were very similar to those reported for 6 (3.0 and 1.5 Hz),16 and described above for 1. As was reported for 6 (but unlike the situation in 1), the epoxide proton signal displaying the larger (3.3-Hz) vicinal coupling showed a significantly more intense NOESY correlation to its hydroxymethine neighbor, supporting assignment of the 3.3-Hz coupling to a cis relationship. However, a subtle difference in the data argued for a slight stereochemical variation between 2 and 6. In the data for 6, the oxymethine signal displaying the 1.5-Hz (trans) coupling to its vicinal epoxide neighbor showed a NOESY correlation with a side-chain signal (H2-10).16 However, in the data for 2, the oxymethine signal that showed this key correlation was the one that displays a 3.3-Hz (cis) coupling with its vicinal epoxide proton neighbor. This minor distinction led to assignment of a different overall diastereoisomeric configuration in 2 relative to that assigned for 6, and resulted in proposal of the relative stereochemistry of phaeofuran B (with the exception of position 10) as shown in 2.
Known metabolite 318 and two new analogs (4 and 5) were also obtained in this investigation. Compounds 3–5 are close analogs of the known antibiotic sorbicillin (7),19 and the structures of 4 and 5 were determined by comparison of NMR data with those for related known metabolites, including 3, which was identified by inspection of its NMR spectra and comparison of these data to literature values.18 Compound 4 was obtained as a yellow solid, and its formula was assigned as C14H16O3 by analysis of EIMS (M+ at m/z 232), 1H NMR, 13C NMR, and DEPT data (Table 3). The chemical shifts of the olefinic protons of the side chain are very similar to those of 3, and their coupling constants are consistent with an E, E configuration. Additional features of 4 include signals for a 1,2,4,5-tetrasubstituted benzene ring with two singlet aromatic proton signals indicating placement para to each other, an O-methyl group, and a hydrogen-bonded hydroxyl group (δ 13.4) at positions analogous to those in 3 (based on δC values). The chemical shift of the ketone carbon carbonyl (δ 192.2) is consistent with its connection to olefinic and aryl groups. Replacement of the aldehyde NMR resonances observed for 3 with signals characteristic of an extra aryl methyl group (δH 2.13/δC 18.9) allowed assignment of the structure as l-(2′-hydroxy-4′-methoxy-5′-methylphenyl)-2,4-E,E-hexadiene-l-one, as shown in 4.
Table 3.
1H and 13C NMR Data for Compounds 4 and 5 (CDCl3)
| 4 | 5 | |||
|---|---|---|---|---|
| Position | δh (mult; J in Hz) | δC | δh (mult; J in Hz) | δC |
| 1 | --- | 192.2 | -- | 204.3 |
| 2 | 6.92 (d; 15) | 121.8 | 2.96 (t; 7.4) | 37.9 |
| 3 | 7.45 (dd; 15, 10) | 144.5 | 2.39 (m) | 27.3 |
| 4 | 6.31 (m) | 130.6a | 5.49 (dtq; 15,5.4, 1.1) | 130.3 |
| 5 | 6.28 (dq; 15,6.1) | 141.2 | 5.47 (dqt; 15,5.9, 1.1) | 126.2 |
| 6 | 1.90 (d; 6.1) | 15.7 | 1.64 (ddt; 5.9, 1.1, 1.1) | 17.9 |
| 2′(OH) | 13.40 (s) | 161.2 | 12.91 (s) | 163.7 |
| 3′ | 6.39 (s) | 99.1 | 6.42 (s) | 99.6 |
| 4′ | -- | 165.2 | -- | 165.3 |
| 5′ | -- | 113.0 | -- | 120.8 |
| 6′ | 7.49 (br s) | 130.5a | 7.64 (s) | 129.3 |
| 7′ | 2. 13 (d; 0.5) | 18.9 | 4.61 (br s);2.02 (br s; OH) | 61.1 |
| 8′ | 3.84 (s) | 55.7 | 3.87 (s) | 55.8 |
These assignments may be interchanged.
Compound 5 was obtained as a white powder, and its formula was assigned as C14H18O4 by analysis of EIMS (M+ at m/z 250), 1H NMR, 13C NMR, and DEPT data (Table 3), and by comparison with data for another close analog which differs from 5 only by replacement of the hydroxymethylene unit with an aldehyde group.18 Inspection of the 1H NMR spectrum of 5 revealed the presence of a downfield oxymethylene proton signal at δ 4.61, a broad OH singlet at δ 2.02, and a side-chain containing only one olefin unit (at C-4 and C-5) instead of two as in 3, 4, and 7. The chemical shift of the ketone carbonyl (δ 204.3) is consistent with the absence of conjugation with an olefin unit. Comparison of the remaining chemical shifts with the corresponding values for other analogs18,19 again revealed a 1,2,4,5-tetrasubstituted benzene ring bearing a chelated hydroxyl group (δ 12.91) and a methoxy group arranged meta to each other. These data enabled assignment of the structure of 5 as l-(2′-hydroxy-4′-methoxy-5′-hydroxymethylphenyl)-E-4-hexene-1 -one.
Phaeofurans A and B (1–2) appear to be of polyketide origin. They did not show antifungal activity against Fusarium verticillioides (NRRL 25457) or Aspergillus flavus (NRRL 6541), nor did they show activity against Staphylococcus aureus (ATCC 29213), Escherichia coli (ATCC 25922), or Candida albicans (ATCC 14053) in standard disk diffusion bioassays (at 200 μg/disk). However, they are chemically distinctive. While isobenzofuranoids occur occasionally as fungal metabolites, most such examples retain the aromaticity of the benzene ring, rather than that of the furan moiety. Representatives that incorporate reduced benzene ring units are precedented,16,20 but are more unusual, with the recently reported integrasone (6) being the closest analog to our knowledge. Compounds 3–5 are also presumably of polyketide (hexaketide) origin, with the extra carbon atom at C-5′ derived from a C1-donor such as S-adenosylmethionine, and with 3 and 5 being further oxidized at this position relative to 4. The known compound 3 displayed potent activity in disk assays21 against A. flavus (40-mm diameter clear zone at 48 h) and moderate activity against F. verticillioides (18-mm zone of partial clearing at 48 h; 200 μg/disk in each case). New analog 5 showed similar activity against F. verticillioides (18-mm zone of partial clearing at 48 h), but somewhat weaker activity against A. flavus (18-mm zone of reduced sporulation at 48 h). Compound 4 showed no activity in these assays when tested at the same level. Compounds 3 and 5 were obtained in limited quantities, but the amounts actually isolated are not representative of their concentration in the extract, and their potency suggests that they account for a significant amount of the antifungal activity originally observed.
Experimental Section
General Experimental Procedures
Procedures and instrumentation used for measurement of melting points, optical rotations, IR, UV, and MS data have been described elsewhere.22 1H NMR spectra were recorded with Bruker AMX-600 or DRX-400 instruments, and 13C NMR and DEPT spectra were recorded with the DRX-400. All 13C NMR multiplicities were verified by DEPT data and are consistent with the assignments provided in the Tables. HMBC and HMQC spectra were recorded at 400 or 600 MHz (1H dimension, Bruker DRX-400 or Bruker AMX-600). All NMR spectra were referenced to the residual solvent signals for acetone-d6 (δH 2.04/δC 29.8) or CD3OD (δH 3.30/δC 49.0). HPLC isolation was performed using a Beckman 110B solvent delivery module and a Beckman 168 diode array detector.
Fungal material
Phaeoacremonium sp. was isolated by D.T.W. from the surface of stromata of Hypoxylon truncatum (Schw.:Fr.) J.H. Miller that had formed on a dead hardwood branch collected at Chehaw Park, Albany, Georgia, by Dr. Bruce W. Horn on April 28, 2000. A subculture has been deposited in the Agricultural Research Service (ARS) collection at the NCAUR (accession number NRRL 32148). The isolate was recognized as Phaeoacremonium by Dr. Richard Summerbell of the Centraalbureau voor Schimmelcultures (CBS), Utrecht, The Netherlands. However, the characteristic features of this species did not match those of any previously described species of Phaeoacremonium, none of which are reported to be fungicolous.23,24
Fermentation, extraction, and isolation
The culture was grown on several slants of potato dextrose agar (PDA) for 14 days (25°C). A hyphal fragment-spore suspension (propagule density, 103/mL of sterile distilled water) prepared from the potato dextrose agar slants served as the inoculum. Fermentations were carried out in two 500-mL Erlenmeyer flasks, each containing 50 g of rice (Botan Brand; J.F.C. International). Distilled water (50 mL) was added to each flask, and the contents were soaked overnight before being autoclaved at 15 lb/in2 for 30 min. After the flasks had cooled to room temperature, they were inoculated with 1.0 mL of the hyphal fragment-spore suspension and incubated for 30 days at 25°C. Following incubation, the fermented rice substrate in each flask was first fragmented with a spatula and then extracted three times with EtOAc (50 mL each time). The combined EtOAc extracts were filtered and evaporated to yield an oil (542 mg) was stored at −20°C until processing. This extract was partitioned between CH3CN (2 mL) and hexane (3 × 2 mL) to obtain an CH3CN-soluble mixture (421 mg) that was fractionated through a Sephadex LH-20 column [60 g swelled in hexane-CH2Cl2 (1:4) and loaded in a 2-cm i.d. glass column] using a step gradient of 180 mL hexane-CH2Cl2 (1:4), 190 mL CH2Cl2-acetone (3:2), 220 mL CH2Cl2-acetone (1:4), 195 mL acetone, and 200 mL MeOH, collecting a total of 14 fractions.
A 104-mg fraction eluted with CH2Cl2-acetone (1:4) was processed by reversed-phase HPLC (C18-Dynamax 60A, 250 × 21.4 mm, 8-μm) using CH3CN-H2O gradient elution (10% – 27% CH3CN over 40 min, 27% – 69% CH3CN over 20 min, 69% – 100% CH3CN over 1 min, and 100% for 5 min) at 10 mL/min and UV detection at 215 nm to afford phaeofuran A (1; 4 mg, tR 7.0 min) and phaeofuran B (2; 6 mg, tR 11.6 min).
An 8-mg fraction eluted with hexane-CH2Cl2 (1:4) was processed by reversed-phase HPLC (C18-Alltech HS Hyperprep, 250 × 10 mm, 8-μm) using a MeOH-H2O gradient (70% – 100% MeOH over 30 min, 100% MeOH for 5 min) at 2 mL/min with UV detection at 230 nm to afford 1-(5′-formyl-2′-hydroxy-4′-methoxy)-2,4-hexadiene-1-one (3; 1 mg, tR 38.1 min) and 1-(2-hydroxy-4-methoxy-5-methyl)-2,4-hexadiene-1-one (4; 2 mg, tR 42.0 min). Compound 3 was identified by comparison of its MS and NMR data to literature data.18,19 A 13-mg fraction eluted with hexane-CH2Cl2 (1:4) was processed by reversed-phase HPLC (C18-Alltech HS Hyperprep, 250 × 10 mm, 8-μm) using a CH3CN-H2O gradient (30% – 65% over 20 min, 65% for 10 min, 65% – 100% over 5 min, and 100% for 5 min) at 2 mL/min with UV detection at 230 nm to afford 1-(2-hydroxy-4-methoxy-5-hydroxymethyl)-4-hexene-1-one (5; 3 mg, tR 27.2 min)
Phaeofuran A (1): white crystals, mp 127–129 °C; [α]D +76° (c 0.1, CH3OH); UV (CH3OH) λmax 212 (ε 5000) nm; IR (KBr) νmax 3429, 3263, 3144, 3127, 1554, 1432, 1349, 1126, 1049, 1034, 1020; 1H and 13C NMR data, see Table 1; HMBC and NOESY data, see Table 2; EIMS (70 eV) m/z 168 ([M]+; rel int 15), 121 (39), 94 (21), 74 (20), 65 (100), 50 (96).
Phaeofuran B (2): oil; [α]D −3° (c 0.2, CH3OH); UV (CH3OH) λmax 207 (ε 7000) nm; IR (KBr) νmax 3370, 1638, 1459, 1420, 1383, 1223, 1115, 1035; 1H and 13C NMR data in acetone-d6, see Table 1; HMBC and NOESY data, see Table 2; 1H NMR (600 MHz, CD3OD) δ 4.88 (m, H-9), 4.79 (dddd, 12, 5.8, 3.3, 0.4, H-2β, 4.58 (m, H-7), 4.47 (dddd, 12, 3.1, 2.3, 0.8, H-2α), 4.36 (br s, H-4), 3.72 (dt, 9.2, 3.6, H-10), 3.47 (ddd, 3.7, 3.0, 0.6, H-6), 3.39 (dd, 3.7, 1.4, H-5), 1.57 (m, H-12a), 1.52 (m, H-11a), 1.47 (m, H-11b), 1.38 (H-12b, m), 0.94 (t, 7.2, H3-13); 13C NMR (125 MHz, CD3OD) delta; 134.7 (C-3), 133.5 (C-8), 90.9 (C-9), 76.8 (C-2), 74.3 (C-10), 64.6 (C-7), 62.0 (C-4), 57.4 (C-5), 55.9 (C-6), 34.8 (C-11), 20.0 (C-12), 14.3 (C-13); EIMS (70 eV) m/z 169 ([M-C4H9O]+; rel int 1), 151 (2), 123 (13), 105 (5), 95 (22), 73 (24), 65 (16), 55 (100); HRESIMS m/z 265.1063 [M+Na]+, calcd. for C12H18O5Na 265.1052.
Acetylation of Phaeofuran A (1)
Approximately 0.5 mg of phaeofuran A (1) was mixed with 500 μL of acetic anhydride and two drops of pyridine. The mixture was stirred at room temperature for 17 hours and the reaction was quenched by adding 1 mL of distilled H2O. The mixture was partitioned with CHC13 (3 × 1 mL) and the organic phase was washed with 0.1 N NaOH (3 × 1 mL), and then with distilled H2O (7 × 1 mL) until the pH of the aqueous phase was approximately 7 as checked with litmus paper. The organic phase was dried over Na2SO4 and then filtered through glass wool and evaporated under a stream of air to afford 0.5 mg of the diacetyl derivative of 1: 1H NMR (400 MHz, CDC13) delta; 7.52 (H-2, d, J = 1.6), 7.31 (H-9, dd, J = 1.6, 1.6), 6.22 (H-7, dd, J = 1.6, 1.6), 6.16 (H-4, d, J = 2.5), 3.62 (H-6, ddd J = 3.6, 1.6, 0.4), 3.55 (H-5, dd, J = 3.6, 2.5), 2.20 (CH3, s), 2.04 (CH3, s); 1H NMR (400 MHz, acetone-d6) δ 7.60 (H-2, d, J = 1.6), 7.45 (H-9, ddd, J = 1.6, 1.6, 0.4), 6.18 (H-7, dd, J = 1.6, 1.6 Hz), 6.12 (H-4, d, J = 2.4 Hz), 3.62 (H-6, ddd J = 3.6, 1.6, 0.4 Hz), 3.61 (H-5, dd, J = 3.6, 2.4), 2.15 (CH3, s), 2.01 (CH3, s); HRESIMS obsd m/z 253.0712 [M+H]+, calcd for C12H12O6+H, 253.0712.
1-(2′-Hydroxy-4′-methoxy-5′-methylphenyl)-E,E′-2,4-hexadiene-1-one (4): yellow powder, mp 68–70°C; UV (CH3OH) λmax 307 (ε 4000), 359 (ε 2000) nm; IR (film on NaCl plate) νmax 3308, 1618, 1567, 1382, 1365, 1261, 1148cm−1; 1H and 13C NMR data, see Table 3; EIMS (70 eV) m/z 232 (M+; rel int 51), 217 (84), 191 (29), 165 (37), 77 (43), 71 (57), 69 (64), 57 (100), 55 (91).
1-(2′-Hydroxy-4′-methoxy-5′-hydroxymethylphenyl)-E-4-hexene-1-one (5): white powder, mp 59–61°C; UV (CH3OH) λmax 216 (ε 9000), 230 (ε 7000), 275 (ε 6000), 320 (ε 3000) nm; IR. (film on NaCl plate) νmax 3307, 1635, 1498, 1446, 1378, 1273, 1209, 1141, 1029 cm−1; 1H and 13C NMR data, see Table 3; EIMS (70 eV) m/z 250 (M+; rel int 6), 232 (19), 217 (4), 181 (100), 77 (12), 69 (13), 65 (18), 55 (50).
Supplementary Material
1H and 13C NMR spectra for phaeofurans A and B (1–2) and sorbicillin analogs 4 and 5 are available free of charge via the Internet at http://pubs.acs.org.
Figure 1.
Acknowledgments
We thank the National Science Foundation (CHE-0315591) and the National Institutes of Health (R01 GM 60600) for financial support, and the University of Iowa NMR and MS Central Facility staff for technical assistance. We also thank Dr. Richard Summerbell of the Centraalbureau voor Schimmelcultures for examining the fungal culture.
References
- 1.Angawi RF, Swenson DC, Gloer JB, Wicklow DT. Tetrahedron Lett. 2003;44:7593–7596. [Google Scholar]
- 2.Che Y, Gloer JB, Wicklows DT. J Nat Prod. 2002;65:399–402. doi: 10.1021/np010519o. [DOI] [PubMed] [Google Scholar]
- 3.Angawi RF, Swenson DC, Gloer JB, Wicklow DT. J Nat Prod. 2003;66:1259–1262. doi: 10.1021/np0301285. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Li C, Swenson DC, Gloer JB, Wicklow DT, Dowd PF. Org Lett. 2003;5:773–776. doi: 10.1021/ol0340686. [DOI] [PubMed] [Google Scholar]
- 5.Höller U, Gloer JB, Wicklow DT. J Nat Prod. 2002;65:876–882. doi: 10.1021/np020097y. [DOI] [PubMed] [Google Scholar]
- 6.Soman AG, Gloer JB, Wicklow DT. J Nat Prod. 1999;62:386–388. doi: 10.1021/np980411h. [DOI] [PubMed] [Google Scholar]
- 7.Harrison LJ, Becker H. Phytochemistry. 1989;28:1261–1262. [Google Scholar]
- 8.Freeman-Cook KD, Halcomb RL. J Org Chem. 2000;65:6153–6159. doi: 10.1021/jo000665j. [DOI] [PubMed] [Google Scholar]
- 9.Duke RK, Rickards RW. J Org Chem. 1984;49:1898–1904. [Google Scholar]
- 10.Soman AG, Gloer JB, Koster B, Malloch D. J Nat Prod. 1999;62:659–661. doi: 10.1021/np980563c. [DOI] [PubMed] [Google Scholar]
- 11.Garlaschelli L, Vidari G, Vita-Finzi P. Tetrahedron Lett. 1996;37:6223–6226. [Google Scholar]
- 12.De Silva LB, Herath WHMW, Gunawardena DSS, Wijesundera RLC, Medis SA, Choudhary MI, Clardy J. Tetrahedron Lett. 1995;36:1997–2000. [Google Scholar]
- 13.Rashid A, Read G. J Chem Soc. 1969:2053–2058. [Google Scholar]
- 14.Betts RL, Murphy ST, Johnson CR. Tetrahedron: Asymmetry. 2004;15:2853–2860. [Google Scholar]
- 15.Mühlenfeld A, Achenbach H. Phytochemistry. 1988;27:3853–3855. [Google Scholar]
- 16.Herath KB, Jayasuriya H, Bills GF, Polishook JD, Dombrowski AW, Guan Z, Felock PJ, Hazuda DJ, Singh SB. J Nat Prod. 2004;67:872–874. doi: 10.1021/np0340504. [DOI] [PubMed] [Google Scholar]
- 17.Pretsch E, Bühlmann P, Affolter C. Tables of Spectral Data. 3. Springer-Verlag; Berlin: 2000. Structure Determination of Organic Compounds; p. 205. [Google Scholar]
- 18.Geigert J, Stermitz FR, Schroeder HA. Tetrahedron. 1973;29:2343–2345. [Google Scholar]
- 19.Trifonov LS, Bieri JH, Prewo R, Dreiding AS, Hoesch L, Rast DM. Tetrahedron. 1983;39:4243–4256. [Google Scholar]
- 20.Oh H, Swenson DC, Gloer JB, Shearer CA. J Nat Prod. 2003;66:73–79. doi: 10.1021/np020342d. [DOI] [PubMed] [Google Scholar]
- 21.Wicklow DT, Joshi BK, Gamble WR, Gloer JB, Dowd PF. Appl Environ Microbiol. 1998;64:4482–4484. doi: 10.1128/aem.64.11.4482-4484.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reátegui RF, Gloer JB, Campbell J, Shearer CA. J Nat Prod. 2005;68:701–705. doi: 10.1021/np050035i. [DOI] [PubMed] [Google Scholar]
- 23.Crous PW, Gams W, Wingfield MJ, van Wyk PS. Mycologia. 1996;88:786–796. [Google Scholar]
- 24.Mostert L, Crous PW, Groenwald JZ, Gams W, Summerbell RC. Mycologia. 2003;95:654–659. doi: 10.1080/15572536.2004.11833069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H and 13C NMR spectra for phaeofurans A and B (1–2) and sorbicillin analogs 4 and 5 are available free of charge via the Internet at http://pubs.acs.org.