

Abstract
A coordinatively unsaturated ruthenium complex catalyzed the formation of a carbon-carbon bond between two judiciously chosen alkene and alkyne partners in good yield, and in a chemo- and regioselective fashion, in spite of the significant degree of unsaturation of the substrates. The resulting 1,4-diene forms the backbone of the cytotoxic marine natural product amphidinolide P. The alkene partner was rapidly assembled from (R)-glycidyl tosylate, which served as a linchpin in a one-flask, sequential three-components coupling process using vinyllithium and a vinyl cyanocuprate. The synthesis of the alkyne partner made use of an unusual anti-selective addition under chelation control conditions of an allyltin reagent derived from tiglic acid. In addition, a remarkably E-selective E2 process using the azodicarboxylate-triphenylphosphine system is featured. Also featured is the first example of the use of a β-lactone as a thermodynamic spring to effect macrolactonization. The oxetanone ring was thus used as a productive protecting group that increased the overall efficiency of this total synthesis. This work was also an opportunity to further probe the scope of the ruthenium-catalyzed alkene-alkyne coupling, in particular using enynes, and studies using various functionalized substrates are described.
Introduction
Within the last decade, marine microorganisms have become an important source of biologically active substances. Unicellular eukaryotes known as dinoflagellates produce some of the most structurally complex and most toxic substances known to man such as brevetoxin, ciguatoxin, okadaic acid and saxitoxin, all of which are increasingly the source of human intoxication.1 Although ninety percent of these organisms are planktons, a number of photosynthetic dinoflagellates take up residence within other organisms as symbiotic partners. In 1986, the group of Kobayashi isolated a novel macrolide, named amphidinolide A, from a strain of laboratory-cultured symbiotic dinoflagellates of the genus Amphidinium sp., which are found inside the cells of the Okinawan flatworm Amphiscolops sp.2 New members of this structurally varied class of compounds have been continually discovered by the group of Kobayashi ever since, and close to 40 amphidinolides have been isolated.3 These macrolides have all demonstrated antineoplastic activity against murine lymphoma L1210 and human epidermoid carcinoma KB cells in vitro. Although most of them have an IC50 in the low micromolar range, amphidinolide N displays subpicomolar activity against these two cell lines.3c The biological activity of these compounds, along with their very limited availability and challenging structures, have made them popular targets for total synthesis. Numerous strategies have been disclosed4 and several amphidinolides have succumbed to total synthesis.5 A common feature to the vast majority of amphidinolides is the presence of one, or more commonly, several exo-methylene units. We envisioned that the ruthenium-catalyzed alkene-alkyne coupling reaction developed in our laboratories6 would provide a tool to develop convergent syntheses of these compounds (the proposed mechanism is shown in Figure 1). Reciprocally, total synthesis of judiciously chosen members of this family would provide a stringent test for the chemoselectivity of this reaction and an opportunity for further development. We have successfully applied it, both inter- and intramolecularly, to the synthesis of amphidinolide A.5e,f We now report in full details our efforts which led to the completion of the synthesis of amphidinolide P.5c Amphidinolide P (1), which was isolated by Kobayashi in a yield of 0.0002%, exhibits cytotoxicity against murine lymphoma L1210 and human epidermoid carcinoma KB cells in vitro (IC50 = 4.0 and 14.6 μM, respectively).7 The structure and relative configuration of amphidinolide P was determined by extensive 1H NMR and 13C NMR studies and molecular mechanics calculations. These studies revealed a backbone consisting of a 15-membered macrolactone with three exo-methylene units, one hemiketal forming a tetrahydropyran moiety, an epoxide moiety and seven chiral centers. The proposed structure and relative configuration of 1 was confirmed by total synthesis.5d
Figure 1.

Proposed catalytic cycle for the ruthenium-catalyzed alkene-alkyne coupling reaction.
Our initially envisioned retrosynthetic analysis is depicted in Scheme 1. Amphidinolide P (1) was anticipated to derive from precursor 2 via a thermal macrocyclisation.8 Although β-ketoesters also undergo thermal macrocyclization (via the same acylketene intermediate),5d,9 the dioxenone can be conveniently carried through multiple synthetic steps. We therefore initially envisioned 6 as the desired alkene addition partner. An intriguing feature of 1, and of 2 by extension, is the presence of an exo-methylene unit in conjugation with an olefin, forming a 1,3-diene moiety. Synthesis of this moiety by a ruthenium-catalyzed alkene-alkyne coupling reaction would therefore require enyne 5. This type of substrate had never been investigated before and it was unclear at the onset of this project what the outcome would be. As shown in Figure 1, the alkyne partner can adopt two orientations in the cationic ruthenium(+2) complex, leading to either a linear or a branched 1,4-diene product (although the alternative orientation of the alkene may also lead to a ruthenacycle, syn-β-hydrogen elimination would in this case most likely be precluded for geometrical reasons). Our results have shown that as the size of R increases, the branched to linear ratio decreases, indicating that steric interaction between the alkene and alkyne is an important factor in determining the regioselectivity of the reaction. Based on steric factors, the enyne was therefore expected to largely favor the formation of the desired branched product. However, on electronic grounds, one might expect that attack of the ruthenium at the terminal carbon of the alkyne would be less favorable since the conjugated olefin reduces the polarization of the triple bond. Conversely, we have shown that increasing the polarization of the triple bond, by appending a trimethylsilyl group at the terminal carbon, improved the branched to linear product ratio.6b Based on these considerations, TMS-alkyne 5 was envisioned to be the desired addition partner. Herein we disclose a detailed account of our studies, leading to a synthesis of amphidinolide P.
Scheme 1.

Initial retrosynthetic analysis.
Results and Discussion
Synthesis of the alkyne coupling partner
Several routes toward alkyne 5 were investigated in the course of this project. We envisioned that 5 could be the product of the allylation of the corresponding aldehyde, as depicted in Scheme 2. The required aldehyde 10 was prepared by an unusual partial reduction of known ester 9.10 The reaction of allyltin reagent 12 and 13 (obtained in two steps from commercially available angelic acid methyl ester and tiglic acid, respectively)11 with aldehyde 10 in the presence of a stoichiometric amount of BF3.Et2O provided syn-11 in quantitative yield, as a 2.6:1 and 6.5:1 mixture of diastereomers, respectively. Given literature precedents, the major diastereomer was assumed to be the syn isomer. This was confirmed by a selective synthesis of anti-11.12 Various enantioselective versions of the allylation reactions shown in Scheme 2 have been reported. Addition of allyltin reagents to aldehydes, which proceed through open transition states, are usually syn-selective.13 An exception to this trend was discovered by Yamamoto, who showed that methallyl- and crotyltrialkyltin reagents react with aldehydes in the presence of AgOTf-BINAP to give the anti adduct, irrespective of the geometry of the starting material.14 However, to the best of our knowledge, the use of trialkyl-(β-methylcrotyl)stannane has not been reported in this process. The reaction of 10 and 12 in the presence of 20 mol% AgOTf-(R)-Binap at −20 °C, according to Yamamoto’s procedure,14b was attempted. Unfortunately, the reaction was prohibitively slow, and only minute traces of product could be detected. Warming the mixture to room temperature did not afford any further conversion. Yamamoto reported good yields with both crotyltributyltin and methallyltributyltin,14b and it appears that substitution at both the β- and γ-position is detrimental to the reactivity of the allylmetal reagent. We found however that 13 reacted with aldehyde 10 in the presence Yamamoto’s CAB catalyst15 to afford scalemic syn-11. Without optimization, the reaction proceeded in 80% yield and 5:1 syn/anti ratio. Conversion of the mixture into the O-methyl mandelate esters,16 and 500 MHz 1H NMR spectroscopy analysis indicated a 6.5:1 e.r. for the syn isomer and 2:1 e.r. for the anti isomer. This was not a viable route however, since, as one would anticipate, inversion of stereochemistry at the alcohol carbon using Mitsunobu conditions resulted in intractable mixtures of SN2′ and elimination products, as well as the desired product.
Scheme 2.
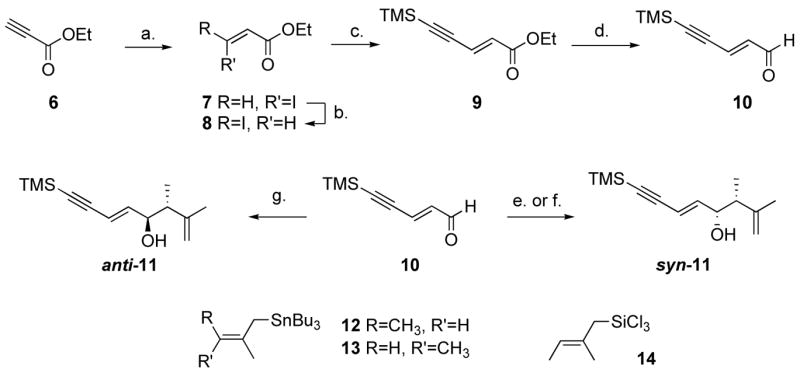
Synthesis of racemic enyne systems.a
a Reagents and conditions: (a) 1.5 equiv. NaI, AcOH, 70 °C, 13 h, Z/E>49:1. (b) 0.01 equiv. aq. HI, benzene, 1.7 M, 80 °C, 8 h, E/Z 16:1. (c) 1.1 equiv. trimethylsilylacetylene, 0.005 equiv. CuI, 0.01 equiv. Pd(PPh3)2Cl2, Et3N, 50 °C, 13 h, 81% (3 steps). (d) 1.1 equiv. DIBAL-H, toluene, −95 °C, 1 h, 70%. (e) 1.1 equiv. BF3.Et2O, 1.3 equiv. 12, CH2Cl2, −78 °C, 5 min, quant., syn/anti 2.6:1. (f) 1.1 equiv. BF3.Et2O, 1.3 equiv. 13, CH2Cl2, −78 °C, 5 min, quant., syn/anti 6.5:1. (g) 1.0 equiv. HMPA, 2.0 equiv. 14, CH2Cl2, −78 °C, 12 h, 23%, syn/anti 1:19.
The stereochemistry of the allylation product can usually be dictated by the geometry of the starting allylmetal reagent when the reaction goes through closed transition states, and axial-axial interactions in a Zimmermann-Traxler transition state become the controlling factor. This has been shown to be the mode of reaction of allyltrichlorosilanes in the presence of nucleophilic catalysts.17 Again, to the best of our knowledge, the use of trichloro-(β-methylcrotyl)silane (14) has not been reported in this process. Although trichlorosilanes, including β-substituted crotylsilane, 17c,e are known to be relatively stable, off-the-shelf compounds, 14 appeared to be an exception. The isolation of 14 proved to be problematic, and it showed poor intrinsic stability, as decomposition was noted after overnight storage at −15 °C under argon. Given the difficulties we encountered with the preparation and handling of this compound, we did not pursue the asymmetric synthesis of anti-11 using this reagent. Instead, we decided to investigate a substrate-controlled approach to the allylmetal addition problem, as depicted in Scheme 3. This idea was based on previous results disclosed by Nakai and co-workers, who found that the addition of trimethyl-(β-methylcrotyl)silane to scalemic α-benzyloxypropionaldehyde under chelation-control conditions afforded the unusual anti product in excellent selectivities, regardless of the geometry of the starting silane reagent.18
Scheme 3.

A substrate-controlled approach to alkyne 5.
The optimized synthesis of 16 is described in Scheme 4. Use of the aluminum ate-complex derived from lithium trimethylsilylacetylide19 resulted in a quantitative yield for the addition reaction to commercially available (S)-glycidyl butyrate (19) in the presence of BF3.Et2O. The benzyl protection of alcohol 20 using benzyl-2,2,2-trichloroacetimidate in mixtures of CH2Cl2-hexane20 was quite sluggish and we found that dioxane was an excellent solvent for this reaction, giving clean and complete conversion within 15 min, in the presence of 20 mol% of trifluoromethanesulfonic acid and using crude, freshly prepared acetimidate.21 DIBAL-H deprotection of the crude ether 21 gave alcohol 22, which was essentially clean. No purification of the intermediates was found to be necessary, and after a Moffat-Swern oxidation, aldehyde 18 was isolated in 71% yield over the 4 steps. This aldehyde was stable to chromatography on silica gel. A Kumada coupling between a 7:3 isomeric mixture of 2-bromo-2-butene and trimethylsilylmagnesium chloride, using a modified literature procedure, gave the silane 17 in 52% yield as a 1:1 mixture of diastereomers.15a We initially conducted the reaction at −78 °C in neat CH2Cl2 and a 42% yield of product was obtained. As judged from 500 MHz 1H NMR spectroscopy analysis, only traces of non-chelation product was detected, and the product resulting from chelation control (16) was isolated as a 6:1 mixture, epimeric at C-6. The 4,5-syn-5,6-anti relationship for the major product was tentatively assigned on the basis of the coupling constants for H-6, H-5, H-4, (dq, J 9.0, 7.0), (dd, J 9.0, 2.0), (ddd, J 8.0, 6.0, 2.0), respectively. This assignment was later supported by nOe studies on a cyclopentane derivative (vide infra). The corresponding signals for the minor diastereomer were masked, but the two methyl doublets, as well as one benzylic hydrogen doublet were resolved and could be integrated. Using a CH2Cl2-pentane mixture, the temperature could be lowered to −110 °C, and we found that using 2 equiv. of silane and 1 equiv. of SnCl4 in a 1:1 mixture of CH2Cl2-pentane, the product could be isolated in 77% yield and 9:1 d.r.
Scheme 4.

Synthesis of alcohol 16.a
a Reagents and conditions: (a) added to 1.3 equiv. lithium acetylide, 1.3 equiv. AlMe3, then 1.3 equiv. BF3.Et2O added, ether, −78 °C, 0.5 h. (b) 2.0 equiv. benzyl-2,2,2-trichloroacetimidate, 0.2 equiv. TfOH, dioxane, 24 °C, 0.5 h. (c) 1.3 equiv. DIBAL-H, CH2Cl2, −78 °C, 15 min. (d) 2.0 equiv. oxalyl chloride, 4.0 equiv. DMSO, 5.0 equiv. Et3N, CH2Cl2, −78 °C to 0 °C, 71% (4 steps). (e) 1.0 equiv. SnCl4, 2.0 equiv. 17, CH2Cl2-pentane 1:1, −110 °C, 15 min, 77%, 9:1 d.r at C-6.
The alcohol was protected as the TIPS ether to give 23 in good yield, and the two diastereomers were separated at this stage. Cleavage of the benzyl group with lithium di-tert-butylbiphenylide resulted in the partial migration of the TIPS group. Both BCl3 and transfer hydrogenation gave complex mixtures. Various Lewis acids were tested, and they all promoted rapid cyclization to give the tetrahydrofuran derivative 24 (Scheme 5).
Scheme 5.

Debenzylation of alkyne 16.a
a Reagents and conditions: (a) 3.0 equiv. TIPSOTf, 4.0 equiv. 2,6-lutidine, CH2Cl2, 24 °C, 6 h, 82%. (b) 2.0 equiv. DDQ, dichloroethane-buffer (pH 7) 9:1 v/v, reflux, 45 min, 82%. (c) 1.3 equiv. 9-Br-9-BBN, CH2Cl2, −78 °C, 5 min, 59%. (d) 1.3 equiv. 9-I-9-BBN, CH2Cl2, −78 °C, 5 min, 75 %. (e) 1.3 equiv. FeCl3, CH2Cl2, 0 °C to 24 °C, 30 min, 39%. (f) 2.0 equiv. SnCl4, CH2Cl2, 0 °C, 30 min, complete conversion.
This facile process is precedented,22 and could be due to the presence of traces of water in the solvent, or of protic acid in the commercial solution of Lewis acid. Hydrochloric acid has been shown to promote this reaction,23 and although it has been found that the CeCl3.7H2O/NaI system was an efficient cyclization promoter,24 this might also be due to the presence of Brønsted acid. In the event, although analysis of the 1H NMR spectrum of tetrahydrofuran derivative 24 was ambiguous (H-3, qd, J 7.5, 4.5; H-4, dd, J 4.5, 4.0; H-5, ddd, J 8.0, 5.5, 4.0) with regard to the relative stereochemistry, nOe’s of 5.0% (H-3 irradiation) and 4.1% (H-5 irradiation) were measured between H-3 and H-5 (Scheme 5). Although nOe’s between H-4 and H-3, and H-4 and H-5 are less diagnostic in a 5-membered ring, the large values observed (7.2 and 8.8%, respectively) also pointed to an all-syn arrangement in 24, consistent with a (chelation-controlled) anti-selective silane addition, and this was in agreement with Nakai’s precedent.18 Eventually we found that the use of an excess of DDQ in a boiling mixture of dichloroethane and aqueous buffer (pH 7) rapidly cleaved the benzyl ether to give alcohol 15 in excellent yields (82–86%) (Scheme 5). This easy oxidation might be facilitated by the inductive effect of the neighboring silyl ether.
Initial elimination attempts focused on converting alcohol 15 into the sulfonate derivative, followed by base-promoted elimination. DBU-promoted elimination of the mesylate derivative afforded the alkene 5 in 60% yield, albeit in an unacceptable 1.6:1 E/Z ratio. Attempt to improve this ratio by making the triisopropylbenzenesulfonyl derivative failed, as the alcohol was too unreactive toward trisyl chloride. We turned our attention to the use of the azodicarboxylate-triphenylphosphine system. We were pleased to find that DIAD-PPh3 (3 equiv.) in toluene at 80 °C gave a clean reaction to afford 5 in 83% yield (Scheme 6) and a very satisfying 9:1 E/Z ratio (E isomer: 2 d, δ 5.70 and 6.09, J 16.0, 5.0 and 16.0, 2.0; Z isomer: 1 d, δ 5.49, J 11.0 and 1 dd, δ 5.89, J 11.0, 9.0). The two isomers were inseparable, and traces of starting material remained. Extended reaction time afforded no further conversion. Neither higher temperatures nor the use of tert-butyl azodicarboxylate had any effect on the selectivity and conversion. On scale-up, those conditions reliably afforded 5 in 75–83% yield and 8-9:1 E/Z ratios. We therefore had access to alkyne 5 in 8 steps and 32% overall yield from commercially available (S)-glycidyl butyrate (19). The TMS group could be removed using standard conditions in 96% yield, to give alkyne 25.
Scheme 6.
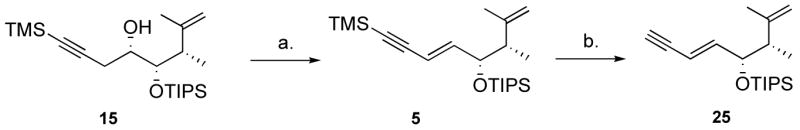
Conversion of alcohol 15 to alkene 5.a
a Reagents and conditions: (a) 3.0 equiv. PPh3, 3.0 equiv. diisopropyl azodicarboxylate, toluene, 80 °C, 20 min, 75%, E/Z 8:1. (b) 1.0 equiv. K2CO3, MeOH, 24 °C, 2 h, 96%.
Synthesis of the alkene coupling partner
We initially envisioned that alkene 4 could be prepared using the sequence outlined in Scheme 7. The chirality in this fragment could be introduced using an asymmetric allylation reaction, and this chiral center could be used to induce additional asymmetry. Alkyloxy-directed aldol reactions between propionate-derived silylketene acetals and β-alkoxyaldehydes have been described and shown to proceed with good simple diastereoselectivity, to give 1,2-syn products, and high levels of 1,3-induction to give predominantly the 2,4-anti-diastereomer.25 Although there has been no reported precedent for the use of silyl dienolates derived from ethyl dioxenone in this process, the substrate-controlled reaction of a silyl dienolate derived from methyl dioxenone with a β-alkoxyaldehyde was recently disclosed (it proceeded stereorandomly).26
Scheme 7.
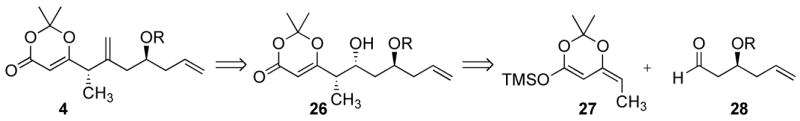
Retrosynthetic analysis for the preparation of 4.a
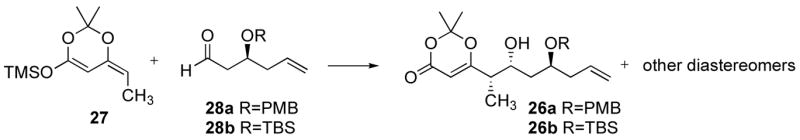
We studied this unprecedented reaction with racemic 28a27 and 28b.28 Silylketene acetal 27 was prepared following a known procedure,29 as a 1.6:1 mixture of isomers, starting from 6-ethyl-2,2-dimethyl-[1,3]-dioxin-4-one.30 Although Sato et al. reported that the Z isomer was the major product of the reaction,29 nOe studies established that the E isomer was the major product in our hands.31
Treatment of 27 and the TBS protected aldehyde 28b with TiCl4 as Lewis acid in dichloromethane at −78 °C resulted in decomposition of the starting material (Table 1, entry 1). Applying the same conditions to the reaction of the PMB protected aldehyde 28a resulted in cleavage of the benzyl group (entry 3), and the diols could be obtained in good yield in a 2.6:1 ratio for the 2,4-anti/syn diastereomers, which could be separated by column chromatography. The 2,4-anti product was found to be a 4:1 diastereomeric mixture, favoring the desired syn-isomer. The yield of the desired product was however unacceptably low and we sought to improve on this result. Use of BF3.Et2O resulted in very poor selectivities (entries 2 and 4). The switch to TiCl2(OiPr)2 gave cleaner reactions, with no PMB deprotection and improved 2,4-anti/syn ratios. Although the four diastereomers were inseparable, only two AB systems were observed for the methylene group of the PMB ether, which corresponded to each pair of 2,4-anti and 2,4-syn diastereomers, and these could be integrated. Likewise, the 1,2-anti/syn diastereomeric ratio was determined by integration of proton signals of the methyl group in the α position of the hydroxyl group, which gave only two doublets corresponding to each pair of 1,2-anti and 1,2-syn diastereomers.31 Replacing dichloromethane with toluene consistently improved the 2,4-anti selectivity (entries 6 vs. 5, 8 vs. 7, 10 vs. 9). However, 1,2-anti/syn ratios were poor and we therefore investigated whether modifying the isomeric ratio for 27 could lead to improved results. When a 10:1 mixture was used, the 1,2-anti/syn ratio increased (entries 7 and 8). It was possible to obtain a 1:2 E/Z solution of 27 from a 10:1 E/Z solution by treating 27 with iodine in dichloromethane. Unfortunately, no major improvement of the 1,2-anti/syn ratio could be observed using this 1:2 E/Z solution of 27 (entries 9 and 10). Using simple esters and thioesters instead of the dioxenone did not provide any satisfactory solution,32 nor did the use of chiral Lewis acids.29 Thus, we abandoned this route.
Table 1.
Addition of silylketene acetal 27 to aldehydes 28a and 28b.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R | E/Z-ratio 27 | lewis acid | solvent | 2,4-anti/syna | 1,2-anti/synb | yieldc |
| 1 | TBS | 1.6:1 | TiCl4 | CH2Cl2 | - | - | decomp. |
| 2 | TBS | 1.6:1 | BF3·OEt2 | CH2Cl2 | 2:1 | 1:1 | 78 % |
| 3d | PMB | 1.6:1 | TiCl4 | CH2Cl2 | 2.6:1 | 1:4e | 61 % |
| 4 | PMB | 1.6:1 | BF3·OEt2 | CH2Cl2 | 1.7:1 | 1:1 | 76 % |
| 5 | PMB | 1.6:1 | TiCl2(OiPr)2 | CH2Cl2 | 3.4:1 | 1:1.3 | 80 % |
| 6 | PMB | 1.6:1 | TiCl2(OiPr)2 | toluene | 7.5:1 | 1:1 | 89 % |
| 7 | PMB | 10:1 | TiCl2(OiPr)2 | CH2Cl2 | 3:1 | 2:1 | 75 % |
| 8 | PMB | 10:1 | TiCl2(OiPr)2 | toluene | 8:1 | 3:1 | 73 % |
| 9 | PMB | 1:2 | TiCl2(OiPr)2 | CH2Cl2 | 4:1 | 1:1.4 | 80 % |
| 10 | PMB | 1:2 | TiCl2(OiPr)2 | toluene | 5:1 | 1.1:1 | 72 % |
All four diastereomers were inseparable; the ratio was determined by integration of the PMB benzylic protons, which gave one AB system for each pair of 2,4-anti and 2,4-syn diastereomers.
Determined by integration of the protons of the methyl α to the hydroxyl, which gave one doublet for each pair of 1,2-anti and 1,2-syn diastereomers.
Combined yield of all diastereomers.
Loss of the PMB group was observed.
The 2,4-anti/syn diastereomers were separable; the 1,2-anti/syn ratio is given for the desired 2,4-anti product.
Capitalizing on a hydrosilylation reaction developed in our laboratories,33 a different approach to dioxenone 27 was envisioned using alkyne 31 (Scheme 8). Oxidation of the vinylsilane 29 to the corresponding ketone could lead to dioxenone 4. We hoped that a regioselective addition to epoxide 32 would afford alkyne 31.
Scheme 8.
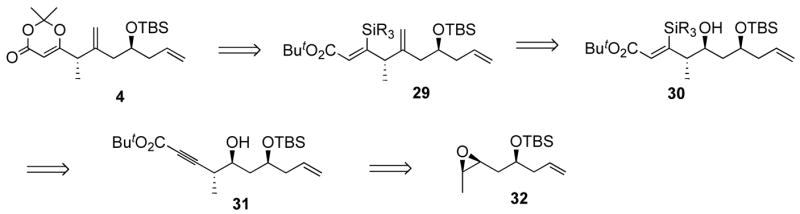
Retrosynthetic analysis for a hydrosylilation-based approach to dioxenone 4.
Epoxide 32 was prepared as depicted in Scheme 9. Sharpless et al. reported that Grignard reagents reacted chemoselectively with p-toluenesulfonic acid glycidyl ester (34) in the presence of Li2CuCl4, although they reported incomplete conversions for this reaction.34 Commercially available Z-1-bromoprop-1-ene (33) could be converted at r.t. without apparent loss of stereochemistry to the corresponding Grignard reagent,35 which reacted with 34 in the presence of Li2CuCl4 to give alcohol 35 in 97% yield. We found that simply using a slight excess of Grignard reagent did afford complete conversion in less than 5 min on a 20 g scale. Treating alcohol 35 with KH for 7–22 h gave the corresponding epoxide, which was not isolated, but rather was treated with the lithium salt of trimethylsilylacetylene in the presence of BF3.Et2O, to afford alkyne 36. Crude 36 was directly treated with catalytic VO(acac)2 and excess TBHP36 to afford epoxide 37 in an excellent 80% yield over the two steps. Pleasingly, 1H NMR spectroscopy analysis indicated a 19:1 diastereomeric ratio. O-Silylation (TBSCl, TMEDA), followed with C-desilylation (K2CO3 in methanol) and Lindlar reduction of the alkyne gave the desired epoxide 32 in 86% overall yield for the three steps.
Scheme 9.
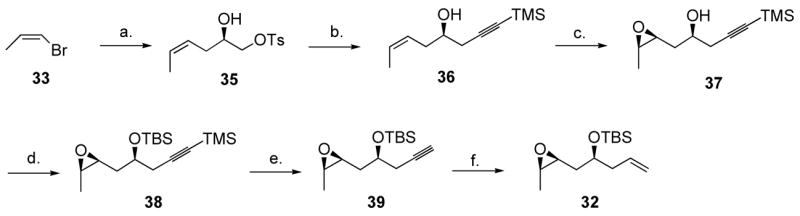
Synthesis of epoxide 32.
a Reagents and conditions: (a) 1.0 equiv. Mg, THF, 23 °C, 2 h then added to 0.05 equiv. Li2CuCl4, THF, −35 °C, 35 min, then 0.7 equiv. 34, −35 °C, 10 min, 97%, Z/E >49:1. (b) 1.2 equiv. KH, THF, 0 °C to 23 °C, 22 h then added to 2.0 equiv. lithium trimethylsilylacetylide (prepared from trimethylsilylacetylene and nBuLi, THF, −78 °C, 10 min), THF-hexane, −78 °C, 10 min then 1.1 equiv. BF3.Et2O. (c) 0.07 equiv. VO(acac)2, 2.2 equiv. TBHP, CH2Cl2-decane, 23 °C, 16 h, 71%, d.r. 19:1. (d) 3.2 equiv. TMEDA, 2.0 equiv. TBSCl, DMF, 23 °C, 13 h. (e) 1.1 equiv. K2CO3, MeOH, 23 °C, 6 h. (f) 1 atm H2, 0.02 equiv. Lindlar catalyst, 2.1 equiv. quinoline, hexane, 23 °C, 15 min, 86% (3 steps).
Unfortunately, regioselectivity for the epoxide opening using BF3.Et2O turned out to be very low (1.7:1 in favor of the desired isomer), giving the two separable isomers 41a and 42a in 85% combined yield (Table 2, entry 1). The two products were unambiguously identified by the splitting pattern of the hydrogen α to the alkyne, i.e. dq for 41a and dt for 42a. The use of Et2AlCl instead of BF3.Et2O gave only a mixture of epichlorhydrins (entry 2). We also tested the alane prepared from tert-butylpropiolate (n-Buli, AlMe3) in this reaction, but it was unreactive (entry 3). The use of the alane derived from the trimethylsilylacetylide also resulted in low selectivities, favoring the undesired isomer 42b (entry 4).
Table 2.
Opening of epoxide with alkynylmetal reagents.
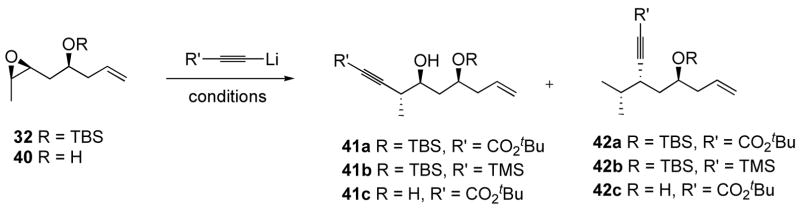 | ||||
|---|---|---|---|---|
| entry | R | R′ | conditions | result |
| 1 | TBS | CO2tBu | 1.0 equiv. of BF3.Et2O | 85 %, 41a/42a 1.7:1 |
| 2 | TBS | CO2tBu | 1.0 equiv. of Et2AlCl | mixtures of epichlorhydrin |
| 3 | TBS | CO2tBu | 1.0 equiv. of AlMe3, then 1.0 equiv. of BF3.Et2O | no conversion |
| 4 | TBS | TMS | 1.0 equiv. of AlMe3, then 1.0 equiv. of BF3.Et2O | 62%, 41b/42b 1:1.3 |
| 5 | H | CO2tBu | i. Ti(OiPr)4 ii. BF3.Et2O | 39%, 41c/42c 0:1 |
| 6 | H | CO2tBu | 1.0 equiv. of BF3.Et2O | 74%, 41c/42c 0:1 |
| 7 | H | CO2tBu | i. aluminum tris(2,6-diphenylphenoxide) ii. BF3.Et2O | 37%, 41c/42c 1.3:1 |
| 8 | H | CO2tBu | Sc(OTf)3 | 50%
43 
|
| 9 | H | CO2tBu | SnCl4, Et3N | no reaction |
| 10 | H | CO2tBu | Mg(OTf)2 | no reaction |
We sought to increase the steric bulk on the alkoxy side of the epoxide by preparing a TIPS analog of epoxide 32. However this alcohol was unreactive toward TIPSCl, even under forcing conditions, whereas TIPSOTf caused decomposition of the epoxide and TIPSH under rhodium catalysis gave no reaction. A trityl analog of 32 could be prepared (1.5 equiv. TrCl, 2.0 equiv. DBU, CH2Cl2, 22 °C, 21 h), but the ratio of products under the conditions of entry 1 was still only 2:1, favoring the desired product (not shown). We then decided to test the unprotected alcohol (40) in the presence of bidentate Lewis acids, in the hope that a five-membered chelate should favor alkylation at the desired position. To the best of our knowledge there is no precedent for this reaction with β-(1,2-disubstituted)-epoxy alcohols. Strong Lewis acids are required to activate the oxirane toward attack by carbon nucleophiles, and BF3.OEt2 has been used extensively,37 with Et2AlCl being the other metal complex of choice. The use of catalytic AlMe3 in conjunction with alkynyllithium reagents and β- or γ-epoxy ethers results in an equilibrium between the aluminum ate-complex and the chelate complex with the epoxide, to give good yields of product.38 Crucially however, this has only been demonstrated with monosubstituted epoxides. First treating 40 with Ti(OiPr)4, and adding it to the lithiated propiolate and BF3.OEt2 resulted in the exclusive formation of the undesired isomer 42c in 39% yield (entry 5). Using the same conditions, but in the absence of Ti(OiPr)4, gave only the undesired isomer in 74% yield (entry 6). Pre-complexation with a very bulky Lewis acid39 gave the desired isomer 41c in low selectivity and low yield (entry 7). Use of Sc(OTf)3 gave a product whose structure was tentatively assigned as the tetrahydrofuran derivative 43 (entry 8). We also tested a variety of Lewis acids with alcohol 40 and trimethylacetylide, but were not able to find conditions that afforded the desired product. Although the BF3.Et2O catalyzed reaction with tert-butylpropiolate (Table 2, entry 1) represented an improvement (85% yield, 1.7:1 ratio of separable isomers) over the results obtained with the aldol route (Table 1, entry 6), the remaining difficulties associated with this route made it a dicey bet for a rapid access to the long-awaited dioxenone 4. We therefore decided to settle for a safer, less ambitious but nonetheless concise route, which we expected would afford a straightforward access to 4.
Our third approach is depicted in Scheme 10, with commercially available (R)-glycidyl tosylate (34) and (R)-hydroxyisobutyric acid methyl ester (Roche ester, 48) envisioned as starting material. We planned to prepare vinyl bromide 47 from alcohol 48. We envisioned that epoxide 34 would serve as a linchpin to connect metallated 47 and vinyl lithium, thus exploiting the difference of reactivity between the two electrophilic sites of 34. Alcohol 46 thus obtained would then be converted in five steps to dioxenone 4, via 45 and 44.
Scheme 10.

Retrosynthetic analysis for the preparation of dioxenone 4.
The Roche ester (48) was protected with TBDPSCl in quantitative yield (Scheme 11). Initially, the crude product 49 was reduced to the corresponding aldehyde with DIBAL-H, which was converted to alkyne 50 using the Seyferth-Ohira-Bestmann reagent.40 Bestmann’s conditions, using K2CO3 in methanol at 0 °C to effect deacetylation of the reagent, induced significant elimination and 50 was isolated in a modest 40% yield. We found that the homogeneous conditions optimized by Nicolaou et al. (1 equiv. NaOMe/phosphonate, THF, −78 °C to r.t)41 were very efficient, allowing isolation of alkyne 50 in a very reproducible 76% yield over the three steps {[α]D26 −5.3, c 4.1, CHCl3}. Only on an 80 mmol scale, did we observe a drop in the yield (59%), and this was largely due to the formation of a larger amount of alcohol in the DIBAL-H reduction step. We surmised that aluminum salts should not prevent the alkynylation reaction and that it should be possible to prepare 50 without isolating the intermediate aldehyde. After stirring 49 with 1.15 equivalents of DIBAL-H in CH2Cl2 at −78 °C for 1 h, 1.35 equivalents of MeOH was added and the mixture was warmed to r.t., and then added to 2.5 equivalents of Seyferth-Ohira-Bestmann reagent which had been premixed with 2.5 equivalents of NaOMe in THF at −78 °C. After warming to 0 °C over 20 min and standard work-up, alkyne 50 was isolated in an improved 83% yield from 48 (Scheme 11). The drawback of this procedure is the excess of Seyferth-Ohira-Bestmann reagent needed, as 2.2 equivalents gave a 62% yield and 1.5 equivalents afforded 50 in ca. 40% yield. Alkyne 50 could then be converted into 47 in excellent yields, using 9-Br-9-BBN, followed by an acetic acid quench. The standard hydrogen peroxide-sodium hydroxide work-up led to lower yields of product and was omitted. Although this meant that the crude product was contaminated with large amounts of material of very low solubility, it did not prove to be detrimental to the purification of 47 by flash silica gel chromatography. The coupling of 47 with (R)-glycidyl tosylate 34 required extensive optimization. We initially focused on forming the Grignard reagent and found that it could only form at the reflux of THF, with 1,2-dibromoethane-mediated activation of the magnesium, and this reaction was always accompanied with the formation of unacceptable amounts of debrominated alkene. We were able to effect clean bromine-lithium exchange, providing that this reaction was carried out in ether, using a fresh solution of t-BuLi. Formation of Lipshutz’ mixed cyanocuprate42 afforded epoxide 51, which upon treatment with vinyllithium in the presence of BF3.Et2O afforded alcohol 46 in good yields. The two operations could be done in one flask, without isolation of 51, with no detrimental effect on the yield. The stereochemistry of 46 was confirmed by preparing the corresponding (R) and (S)-O-methyl mandelate esters derivatives.16 500 MHz 1H NMR spectroscopy analysis showed a single diastereomer for each compound, and analysis of the chemical shifts unambiguously confirmed the S configuration of the alcohol (Figure 2).
Scheme 11.

Synthesis of the alkene coupling partner.a
Reagents and conditions: (a) 1.0 equiv. TBDPSCl, 1.3 equiv. imidazole, CH2Cl2, 23 °C, 0.5 h. (b) 1.15 equiv. DIBAL-H, CH2Cl2, −78 °C, 60 min, then 1.35 equiv. MeOH, −78 °C to 24 °C then added to 2.5 equiv. CH3(CO)CHN2P(O)(OMe)2, 2.5 equiv. NaOMe, THF, −78 °C to 0 °C, 20 min, 83% (2 steps). (c) 2.0 equiv. 9-Br-9-BBN, CH2Cl2-hexane, 0 °C, 6 h then 14 equiv. AcOH, 0 °C, 1 h, 96%. (d) 2.0 equiv. t-BuLi, ether, −78 °C, 1 h, then 1.3 equiv. ThCu(CN)Li, THF, −78 °C to −45 °C, −45 °C, 1 h, then 2.0 equiv. 34, THF, −45 to 0 °C, 0 °C, 5 h, then 2.0 equiv. vinyllithium, 2.0 equiv. BF3.Et2O, THF, −78 °C, 20 min, 71%. (e) 1.8 equiv. TBSOTf, 4.0 equiv. 2,6-lutidine, CH2Cl2, 0 °C, 5 min. (f) 1.2 equiv. TBAF.3H2O, 1.2 equiv. AcOH, DMF, 23 °C, 13 h, 77% (2 steps). (g) 2.0 equiv. (COCl)2, 4.0 equiv. DMSO, 4.6 equiv. Et3N, CH2Cl2, −78 °C to −20 °C, 20 min. (h) 4.0 equiv. t-BuOAc, 4.0 equiv. LDA, THF-hexanes, −78 °C, 1 h, then 45, THF, −78 °C, 10 min, 78% (2 steps). (i) 1.5 equiv. TBAF, THF, 24 °C, 4 h, 89%.
Figure 2.

500MHz 1H NMR spectroscopy analysis of the esters derived from 46 and (R)- and (S)-methoxyphenyl acetic acid (MPA) confirmed the absolute stereochemistry of alcohol 46.
TBS protection of 46 afforded compound 52 which was used in the next step without purification. Selective hydrolysis of the primary silyl ether using TBAF in the presence of acetic acid in DMF,43 gave alcohol 53 (Scheme 11). Moffat-Swern oxidation, followed by addition of the lithium enolate of tert-butyl acetate gave ester 54 in 78% yield, and as a 2.8:1 mixture of diastereoisomers (the presumably major Felkin-Anh product is shown). As the formation of the dioxenone proved problematic and the study of the alkene-alkyne coupling progressed (vide infra), the desilylated substrate 55 became attractive and could be obtained from 54 in 89% yield using TBAF in THF. We were unable to find conditions that would allow us to prepare 55 without resorting to intermediate TBS protection of the secondary alcohol.
Conditions for the formation of the dioxenone were initially examined on a model system. Precedents for this reaction stem from studies by Eastman chemists, Clemens, Witzeman and Hyatt, who studied the formation and mechanism thereof of acylketene from β-ketoesters and dioxenones.9 In particular, they established that formation of acylketene was most favorable with tert-butyl acetoacetate compared with methyl, ethyl, isopropyl and isobutyl,9a and also that isopropenyl acetoacetate forms 2,2,6-trimethyl-1,3-dioxen-4-one upon heating with excess acetone.9b We prepared isopropenyl ester 57 from commercially available methylpropionaldehyde (56) and submitted it to Clemens and Witzeman’s conditions (Scheme 12).9b Upon heating with 100 equiv. of acetone in toluene in a stoppered flask, 58 afforded dioxenone 59 in 77% yield, although the purity of the product was modest as judged by 1H NMR spectroscopy.
Scheme 12.

Model studies for dioxenone formation.a
a Reagents and conditions: (a) 3 equiv. isopropenyl acetate, 3 equiv. LDA, −78 °C, 5 min. (b) 4 equiv. PCC, 1 equiv. NaOAC, 4 Å MS, CH2Cl2, 18 h, 24% (2 steps). (c) toluene-acetone (100 equiv.) 2:1 v/v, 90 °C, 40 min, 77%.
We then prepared the isopropenyl ester (61) derived from aldehyde 45 (Scheme 13), but when heated in the presence of acetone, none of the desired dioxenone was formed. Instead, two products were isolated (conditions A), methylketone 62 and dioxenone 63, presumably via the mechanism depicted in Scheme 13. In neat acetone (conditions B), the reaction still proceeded, although at a lower rate and only the dioxenone 63 was observed by TLC. This was unanticipated as Williams used a similar β-ketoester, going through a similar acylketene to accomplish the macrocyclization.5d In the complete amphidinolide P system, the acylketene got smoothly trapped by the alcohol 12 carbons away to form the 15-membered ring (starting from the methyl ester, 90 min, toluene, reflux). With alkenes 54 and 55 in hand, we could certainly envisage completing the synthesis, and we did not do any further studies on dioxenone synthesis.
Scheme 13.

Attempted dioxenone formation.a
and conditions: (a) 5 equiv. isopropenyl acetate, 5 equiv. LDA, −78 °C, 5 min, 45%. (b) 4 equiv. PCC, 1 equiv. NaOAC, 4 Å MS, CH2Cl2, 4 h, 50%. (c) Conditions A: toluene-acetone (100 equiv.) 2:1 v/v, 90 °C, 90 min; conditions B: acetone, 56 °C, 3.5 h.
Studies of the ruthenium catalyzed enyne-alkene coupling
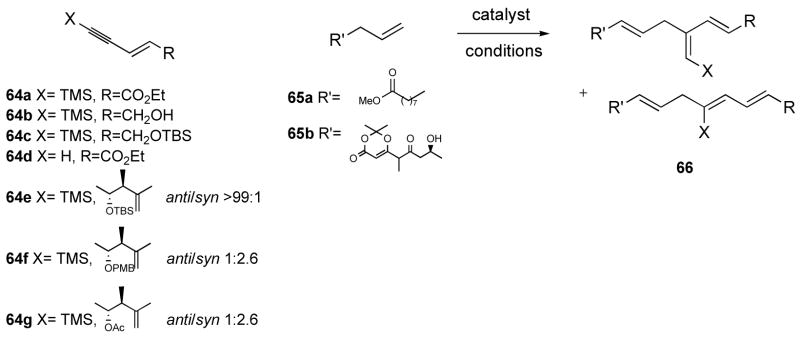
As mentioned in the introduction, enynes had never been tested as substrates for the alkene-alkyne coupling, and we therefore carried out some model studies. Enynes 64a–g and alkenes 65a and 65b were prepared31 (we were not able to identify conditions to convert ketone 65b into the desired olefin) and coupled under various conditions using [CpRu(CH3CN)3]PF6 (67) or CpRu(COD)Cl (68) and the results are compiled in Table 3.
Table 3.
Addition of alkene 65a–b to enynes 64a–g.
 | |||||||
|---|---|---|---|---|---|---|---|
| entrya | alkyne | alkene | catalyst (mol%) | solvent | temp. °C | product % (brsm)b | branched to linear ratio |
| 1 | 64a | 65a | 67 (10) | acetone | 24 | 66aa 31 (70) | 1:0 |
| 2 | 64a | 65a | 67 (10) | DMF | 24 | 66aa 5 (73) | n.d. |
| 3 | 64a | 65a | 67 (10) | DMF | 55 | 66aa 5 (65) | n.d. |
| 4 | 64b | 65a | 67 (10) | acetone | 24 | no reaction | - |
| 5 | 64c | 65a | 67 (10) | acetone | 24 | 66ca 45 (75) | 1:0 |
| 6 | 64d | 65a | 67 (10) | acetone | 24 | 66da 26 (75) | 1:2 |
| 7 | 64d | 65a | 67 (10) | DMF | 24 | 66da 36 (78) | 1.8:1 |
| 8 | 64d | 65a | 67 (10) | DMF | 55 | 66da 50 (76) | 1.8:1 |
| 9 | 64d | 65a | 67 (10) | DMF | 70 | 66da 56 (68) | 2.7:1 |
| 10 | 64d | 65a | 68 (5) | MeOH | 65 | 66da 13 (53) | 2.3:1 |
| 11 | 64e | 65a | 67 (10) | acetone | 24 | 66ea 46 (65) | 1:0 |
| 12 | 64f | 65b | 67 (10) | acetone | 24 | 66fb 27 (100) | n.d. |
| 13 | 64f | 65b | 67 (100) | acetone | 24 | 66fb 10 (25) | n.d. |
| 14 | 64f | 65b | 67 (10) | DMF | 60 | 66fb no reaction | - |
| 15 | 64g | 65b | 67 (10) | acetone | 24 | 66gb 10 (40) | n.d. |
| 16c | 64g | 65b | 67 (10) | acetone | 24 | 66gb traces | - |
| 17 | 64g | 65b | 67 (10) | DMF | 65 | 66gb no reaction | - |
All reactions were run at 0.1M for 1–4 h using a 1:1 ratio of alkene to alkyne.
brsm indicates the yield based on recovered alkene.
1 equivalent of malonic acid was added.
We first studied the reaction with methyl 10-undecenoate (65a) in the presence of 10 mol% of catalyst 67. With TMS-alkynes 64a–c fast conversions (<10 min) and low turn-overs were observed (Table 3, entries 1–5), although as expected, only the branched product was detected by 1H NMR spectroscopic analysis. Removing the TMS group resulted in increased turn-over (entry 6 vs. entry 1) although the linear product was now the major product. Switching to DMF and increasing the temperature improved the yield further and favored the branched product (entries 7–9). The yield could be increased up to 56% by heating the reaction mixture at 70 °C in DMF (pre-heated oil bath), and an improved branched to linear ratio of 2.7:1 was observed. The CpRu(COD)Cl (68) catalyst fared poorly in this reaction (entry 10). Next we studied racemic alkynes 64e–g. The result obtained with 64c (entry 5) was nicely reproduced with the desired, more functionalized analog 64e since the coupling reaction with olefin 65a in a 1:1 ratio in acetone at room temperature in the presence of 67 (10 mol%) yielded the desired compound 66ea in 46% yield (brsm 65%) as well as several unidentified by-products (entry 11). Treatment of 66ea in acetone in the presence of 20 mol% 67 led to a 95 % recovery, which pointed to the stability of the product. Using dioxenone 65b, the reaction was carried out in acetone at r.t., and again rapid conversion and low yield of product was observed (entry 12). Poor reactivity of alkene 65b might be inferred from the facts that the alkyne was fully consumed and that alkene recovery was excellent. Unlike what has been occasionally observed,6c adding another portion of catalyst resulted in no further conversion. Using 1 equivalent of catalyst 67 gave worse conversion (entry 13) which might indicate the formation of catalytically inactive aggregates, or self-catalyzed decomposition, although poor mass recovery points to a possible different reaction manifold. Curiously, when DMF was used, no reaction was observed (entry 14). The reaction of 64g also proceeded poorly (entry 16), while the addition of a bidentate acid to the medium was detrimental to the conversion (entry 17). Again DMF was not a suitable solvent, affording no product (entry 17).
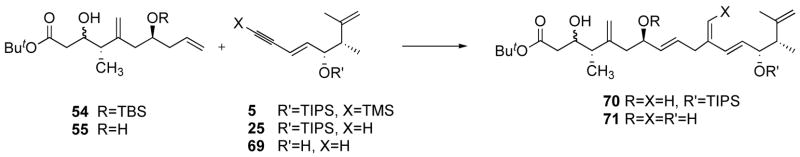
At this point in time we had alkenes 54 and 55 in hand, and the alkene-enyne coupling was then tested with those substrates, as shown in Table 4. Alkene 54 was unstable in the presence of the catalyst 67 in acetone (entry 1), and no reaction occurred in DMF (entry 2), except under forcing conditions (100 °C), where the silyl ether was hydrolyzed, demonstrating the Lewis acid character of the ruthenium(II) species. Surmising that steric hindrance might preclude coordination of both coupling partners to the ruthenium center, we removed the TMS group, and tested the reaction with alkyne 25, to no avail (entry 4). Pushing the idea further, we carried out the reaction with diol 55 and alkyne 25, and were pleased to isolate the product 70 in 28% yield (entry 5). Using an excess of alkene was essential in order to obtain good conversion. Importantly, no linear isomer was detected by 500 MHz 1H NMR spectroscopy, and unreacted alkene recovery was good. In order to try and obtain full conversion, we tested the CpRu(COD)Cl (68) catalyst.6c,d At the reflux of methanol in the presence of ammonium ion and using a three-fold excess of alkene, full conversion was obtained and product 70 was obtained in 57% yield (entry 6). Again, no linear isomer was detected by 500 MHz 1H NMR spectroscopy, and unreacted alkene recovery was good.
Table 4.
Studies of the coupling reaction between alkynes 5, 25 and 69 and alkenes 54 and 55.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | alkene | alkyne | alkene:alkyne ratio | catalyst (mol%) | solvent (alkyne concentration) | Temp. °C | reaction time | product %a (recovered alkene, recovered alkyne) |
| 1 | 54 | 5 | 1:1 | 67 (10) | acetone (0.15) | 24 | 2 h | complex mixture |
| 2 | 54 | 5 | 1:1 | 67 (10) | DMF (0.15) | 24 to 100 | 2 h | - (60,i 100) |
| 3 | 54 | 5 | 2:1 | 67 (10) | DMF-acetone 3:1 (0.20) | 24 | 16 h | - (100, 100) |
| 4 | 54 | 25 | 1.1:1 | 67 (10) | acetone (0.25) | 24 | 1.5 h | - (57, n.d.) |
| 5 | 55 | 25 | 2:1 | 67 (10) | acetone (0.15) | 24 | 2 h | 28 (80, 45) |
| 6 | 55 | 25 | 2.7:1 | 68 (10)b | methanol (0.10) | 67 | 20 min | 57c (80, -d) |
| 7 | 55 | 25 | 4.5:1 | 68 (5)b | methanol (0.06) | 67 | 75 min | 61c (86, -d) |
| 8 | 55 | 25 | 4.5:1 | 68 (2)b | methanol (0.06) | 67 | 2 h | 43c (77, 35) |
| 9e | 55 | 25 | 4.5:1 | 67 (10) | acetone (0.06) | 24 | 15 h | 72 (95, -d) |
| 10f | 55 | 25 | 4.5:1 | 67 (10) | acetone (0.06) | 24 | 13 h | 50 (42, -d) |
| 11 | 55 | 25 | 4.5:1 | 67 (10)g | acetone (0.06) | 24 | 3 h | - (100, 100) |
| 12 | 55 | 25 | 4.5:1 | 67 (10)h | methanol (0.10) | 67 | 3 h | traces (n.d., n.d.) |
| 13 | 55 | 69 | 1.3:1 | 68 (5)b | methanol (0.10) | 67 | 48 h | 17c (96, -d) |
| 14 | 55 | 69 | 5.5:1 | 68 (5)b | methanol (0.06) | 67 | 16 h | 20c (84, -d) |
| 15 | 55 | 69 | 3:1 | 68 (10)b | methanol (0.05) | 67 | 3 h | 30c (70, -d) |
| 16 | 55 | 69 | 2.5:1 | 67 (5)b | acetone (0.05) | 24 | 3 h | traces (n.d., n.d.) |
In all the cases where the product was isolated, the branched to linear ratio was found to be >49:1 as judged by 500MHz 1H NMR analysis.
2 equiv./Ru of NH4PF6 was added.
yield adjusted for the amount of alkyne consumed in the [2+2+2] reaction with the COD ligand.
Full alkyne conversion was observed.
0.04 mmol scale.
1 mmol scale.
10 mol% tetra-n-butylammonium chloride was added.
10 mol% tetra-n-butylammonium chloride and 10 mol% NH4PF6 were added.
Desilylation was observed.
Since the reaction was almost quantitative in alkene 55, and significant decomposition of the alkyne occurs, the reaction was attempted at lower catalyst loading and lower alkyne concentration (entries 7 and 8). Only a marginal improvement was observed with 5 mol% of 68 using a 4.5:1 ratio of 55/25, whereas using 2 mol% resulted in incomplete conversion, although the yield based on recovered starting material was 66%. The quality of the solvent was crucial in this process, since the use of methanol purified using a column solvent purification apparatus,44 which was most likely contaminated with basic alumina, led to no conversion. We returned to catalyst 67 (10 mol%) using a 4.5:1 ratio of 55/25 at 0.06M, and found that the reaction proceeded slowly but cleanly in dry acetone at r.t. to give 70 in 72% yield (entry 9). However, on scale-up, a lot of decomposition was observed (entry 10). This difference of catalyst activity might be due to a difference in water concentration between the small scale and large scale reactions, and we hypothesized that water might be a ligand for the active catalytic species. Similar results as those of entry 9 were obtained on a small scale when acetone from a wash bottle was used, in which case, at 0.06M, the molar ratio of water to ruthenium was at least fifteen. More work will need to be done to understand the effect of water in the alkene-alkyne coupling using catalyst 67. To this day, it remains unclear what the structure of the active catalyst is. Using the optimized conditions (entry 9), adding 10 mol% of TBAC totally shut down the reaction (entry 11, TBAC and 67 were mixed under argon, acetone was added, followed with 55 and 25, which were both recovered quantitatively after several hours). Using the conditions of entry 7, but replacing 68 with 10 mol% 67 and 10 mol% of TBAC, only traces of 70 were observed (entry 12). These results would seem to indicate that the active catalyst is different in acetone and methanol (notwithstanding the role of the solvent as a ligand), with the chloride remaining bound to the ruthenium when methanol is used as solvent. Adding chloride to 67 in acetone shut down the reaction (entry 11), and conversely, it could be that no active chloride-bound ruthenium catalyst was formed when 67 and TBAC were mixed in methanol (entry 12). Alkene-alkyne couplings do proceed with catalyst 68 in methanol in the absence of NH4PF6 (where presumably the active catalyst is a Cp-ruthenium chloride species), and in fact NH4PF6 provides only modest improvements.6d We did not however run this experiment (entry 6 conditions) without NH4PF6.
In view of the subsequent macrocyclization step, it was interesting to find out whether the reaction could be carried out with desilylated 69, which was obtained from 25 (3 equiv. of TBAF, THF, r.t., 15 min, 50 % unoptimized). It turned out 69 afforded very low rates compared to 25, presumably because 69 is a better ligand than 25 and is not displaced easily by the alkene (entries 13–16). None of it was recovered and only low yields of 71 were observed.
Completion of the synthesis
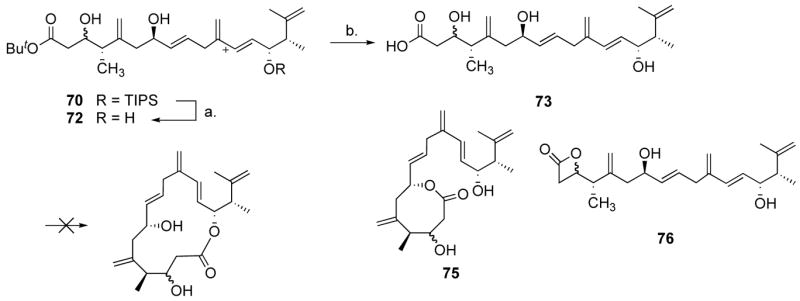
With the full backbone of amphidinolide P in hand (70), we could now focus on the final steps of the synthesis. Without the dioxenone functionality, and with the C-3 and C-7 alcohols both deprotected, macrocyclization through acylketene formation seemed precluded. Even if we could selectively oxidize the C-3 alcohol, we thought that formation of a stable hemi-acetal might considerably slow down or even shut down the formation of the acylketene. We thus decided to test more standard macrocyclization techniques, through acyl activation of the corresponding acid. Compound 70 was therefore treated with excess TBAF to afford alcohol 72 in excellent yield (Scheme 14), and 72 was subjected to a variety of conditions to convert it to the acid, all leading to extensive decomposition. In spectacular contrast, we found that TMSOTf was an excellent Lewis acid for this transformation,45 and after an aqueous HCl work-up, acid 73 was obtained in quantitative yield and did not require additional purification. Reversing the order of steps also gave acid 73 in good yield, but purification was then required. We found that acid 73 was a very unstable compound, which decomposed in a few days upon standing, even at −20 °C. It was nonetheless submitted to a variety of macrocyclization conditions. The macrolactonization methods reported by Yamaguchi,46 Trost,47 Mukaiyama,48 Keck,49 and Mitsunobu50 all gave complex mixtures. Using Mukaiyama’s or Keck’s systems, some residue could be isolated that displayed IR stretching frequencies of 1720 and 1830 cm−1, indicative of a mixture of medium-size lactone and β-lactone, respectively. When the Corey-Nicolaou methodology51 was employed, 8-membered ring 75 was isolated in 20–30% yields. Intrigued by the possibility that β-lactone 76 was an intermediate in the formation of 75, and rather than trying to optimize the reaction with this unstable seco-acid, we wondered whether we could not use the β-lactone functionality52 as an activated acyl system, stable enough to undergo several synthetic steps, albeit reactive enough to undergo transesterification to some larger, more stable ring-systems. This novel strategy for macrolactonization would not require a redesign of our synthetic route since, in theory, aldehyde 45 could undergo a [2+2] cycloaddition reaction to form a β-lactone (Scheme 15), which would provide an interesting substrate for our alkene-alkyne coupling reaction. A potentially big advantage of intermediate 78 over 70 was the presence of only one free hydroxyl, which could reduce chemoselectivity problems in the end-game. Indeed, studies of the hydroxyl-directed epoxidation of ester 70 led to complex mixtures, partly due to lack of chemoselectivity. In this respect, the β-lactone would act as a “productive protecting group”.
Scheme 14.
Preparation of seco-acid 73 and attempted macrolactonization.a
a Reagents and conditions: (a) 4.0 equiv. TBAF, THF, 24 °C, 2 h, 94%. (b) 7.5 equiv. TMSOTf, 11.5 equiv. 2,6-lutidine, 0 °C, 3 h, 24 °C, 30 min, quant.
Scheme 15.

Novel end-game for the synthesis of 1, using a β-lactone precursor.
We investigated conditions to form β-lactone 79 from aldehyde 45. The Lewis acid catalyzed cycloaddition of ketene and an aldehyde has been known for some time.53 However, the generation of ketene requires burdensome equipment. Alternatively, a stable ketene equivalent such as trimethylsilyketene (80)54 or dichloroketene could be used, where the stabilizing substituents could be removed in the product; another alternative is to generate ketene in situ, by dehydrohalogenation of acetyl halides with an amine base.55 We initially focused on the latter, inspired by the work of Nelson et al.,56 who generated ketene from Hünig’s base and acetyl chloride, and used Al(SbF6)3 (generated in situ from AgSbF6 and AlCl3) as a Lewis acid to promote the cycloaddition. Although we had some degree of success with this protocol, in our hands it was a very capricious reaction that led to unreproducible results, and none of the alternative Lewis acids tested gave satisfactory results (replacing AlCl3 with GaCl3, InCl3, Al(OTf)3 or Me2AlCl). AcBr offered no improvement, and various sulfonamide/trimethylaluminum systems57 offered only modest amounts of β-lactone. The LiClO 4 methodology reported by Cossio58 was also inneffective. We briefly studied the tandem aldol-lactonization reaction,59 using ketene triethylsilylthioacetal and 45 in the presence of ZnCl2, but again only low yields of β-lactone were obtained. We next turned our attention to the use of trimethylsilylketene (80).54 This compound can be prepared very conveniently by silylation of ethyl ethynylether to give ethyl trimethylsilylethynylether, which upon heating to 120 °C, undergoes a 1,5-hydrogen shift to give off ethylene and 80 (b.p. 81–82 °C) in 70% yield. Ketene 80 was stored in the freezer and no decomposition was observed after 6 weeks. The cycloaddition of 80 and 45 did not proceed when catalyzed by MgBr2.Et2O,60 whereas BF3.Et2O gave the lactone (81) in 49% yield. Me2AlCl however afforded 81 as an inconsequential 1.6:1 mixture of diastereomers in a very reproducible 90% yield, using just 1.1 equivalent of 80 (Scheme 16).61 This was consistent with literature results that show that Al(III) is predominantly the metal catalyst of choice for [2+2] reactions between aldehydes and ketenes.56,57,61,62 Next, we looked for conditions that would cleave both the O-Si bond and the C-Si bond in one pot. TBAF gave the fully desilylated product 79 (υC=O 1827 cm−1) in only 26% yield, and aqueous HF did not cleave the C-Si bond. It is known that KF.2H2O desilylates β-lactones,60 so 81 was first treated with KF.2H2O until TLC analysis indicated complete conversion, whereupon the mixture was cooled to 0 °C, and aqueous HF was added. Using this procedure, 79 was very reliably obtained in 69% yield over the three steps. As an added bonus, the two diasteromers were separable, and although this epimeric center would eventually be destroyed, working with a single diastereomer simplified the studies of the remaining steps.
Scheme 16.

Synthesis of β-lactone 79.a
a Reagents and conditions: (a) 2.0 equiv. (COCl)2, 4.0 equiv. DMSO, 4.6 equiv. Et3N, CH2Cl2, −78 °C to 0 °C. (b) 1.0 equiv. Me2AlCl, 1.1 equiv. 80, CH2Cl2, −78 °C, 0.5 h. (c) 1.5 equiv. KF.2H2O, CH3CN, 25 °C, 1 h, then 40% aq. HF, 0 °C, 0.5 h, 69% (3 steps), d.r. 1.6:1.
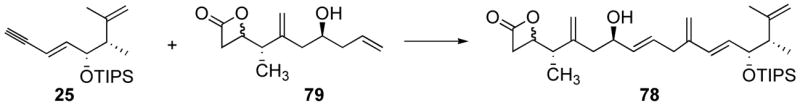
Despite slight concerns about the compatibility of the somewhat Lewis acidic (see for example Table 4, entry 2) catalyst 67 and the β-lactone functionality, coupling between alkene 79 and alkyne 25 proceed well (Table 5, entry 1). However, a steady decrease of the yield was observed as the scale of the reaction was increased (entry 1 vs. 2 vs. 3 vs. 4). This was accompanied with a higher recovery of the excess alkene 79, indicating a greater propensity for the alkyne 25 to decompose. The use of 3.5 equivalents of alkene seemed optimal, since a slight decrease in yield was observed when only 2.8 equiv. were used (68% vs. 64%, entry 3 vs. entry 5). It is worth noting that a similar result was observed when acetone from a wash bottle (entry 6) was used instead of distilled acetone (entry 1).
Table 5.
Alkene-alkyne coupling of 79 and 25.
 | ||||
|---|---|---|---|---|
| entry | conditions | Scale (mmol 25) | yield 78 (%) | recovered 79 (%) |
| 1a | 10 mol% 67, acetone, 0.05 M, r.t., 13 h, 25/79 1:3.5 | 0.14 | 75 | 87 |
| 2a | 10 mol% 67, acetone, 0.05 M, r.t., 10 h, 25/79 1:3.5 | 0.20 | 69 | 86 |
| 3a | 10 mol% 67, acetone, 0.05 M, r.t., 12 h, 25/79 1:3.3 | 0.44 | 68 | 87 |
| 4a | 10 mol% 67, acetone, 0.05 M, r.t., 12 h, 25/79 1:3.5 | 0.80 | 56 | 91 |
| 5a | 10 mol% 67, acetone, 0.05 M, r.t., 10 h, 25/79 1:2.8 | 0.36 | 64 | 90 |
| 6b | 10 mol% 67, acetone, 0.05 M, r.t., 22 h, 25/79 1:3.4 | 0.04 | 72 | 75 |
acetone was distilled from CaCl2.
acetone was taken from a wash bottle.
We then submitted 78 to TBAF and obtained 82 in 71% yield (Scheme 17). When we heated oxetanone 82 at the reflux of hexane in the presence of 10 mol% of Otera’s catalyst63 (85) at 0.001 M for 20 min, we only isolated 8-membered lactone 83 in quantitative yield. Lowering the catalyst loading to 1 mol% gave 83 in 88% yield after 45 min. We did not observe any conversion to the 15-membered macrolide after 3 h using 10 mol% catalyst, which would suggest, somewhat counterintuitively, that 83 is in fact more stable than the corresponding 15-membered ring (84). This somewhat unanticipated result suggested, at the cost of one extra step, an excellent strategy to differentiate between the three secondary alcohols. While the two alcohols at C-3 and C-14 were protected as a β-lactone and a TIPS ether (78, Scheme 17), respectively, the alcohol at C-7 would be used to direct the epoxidation. Leaving the TIPS group on, and after isomerization from the 4- to the 8-membered lactone, we could anticipate oxidizing the newly unmasked alcohol at C-3. Removing the TIPS would then reveal the C-14 allylic alcohol. We expected that with this substrate, the 8-to 15-membered ring isomerization would be favored, driven by concomitant hemi-acetal formation and giving the natural product amphidinolide P (1). We briefly investigated the substrate-directed epoxidation of alkene 78. It is well established in the literature that E-1,2-disubstituted olefins are poor substrates for hydroxyl-directed epoxidation with allylic alcohols, since they sustain minimal A-1,2 and A-1,3 interactions in the transition state, usually giving the syn product with very poor selectivities.64 Only the VO(acac)2/TBHP system is known to be anti-selective for this particular class of allylic alcohols. We tested this system with syn-78 in various solvents (dichloromethane, hexane, toluene, benzene, chlorobenzene), and although mass recovery was good, close to 1:1 ratios were obtained in all cases. The major product in the toluene, hexane, CH2Cl2 experiment was assigned the anti configuration based on literature precedent (the coupling constants are not diagnostic in these systems), and later, on the result of the reagent-controlled epoxidation (vide infra). A reversal of selectivity was observed in chlorobenzene and benzene (which gave the highest selectivity, 1:2). We then resorted to the Katsuki-Sharpless tartrate/Ti(OiPr)4 system.65 Based on multiple literature precedents,66 the use of (−)-tartrate was expected to be a matched case. Indeed the reaction with anti-78 gave 3,4-anti-77 in 87% yield and a single diastereomer using (−)-diethyl tartrate (diisopropyl tartrate gave a similar result, but was inseparable from the product). Reaction with the mixture of diastereomers anti-78 and syn-78 gave a partially separable mixture of 3,4-syn-77 and 3,4-anti-77 in 83% yield (Scheme 18). As anticipated, when we submitted 77 to catalyst 85, 8-membered lactone 86 (υC=O 1732 cm−1) was obtained in 93% yield using 5 mol% catalyst at 0.002 M in hexane. The C-3 alcohol could then be oxidized using Dess-Martin periodinane to give ketone 87 in 83% yield (υC=O 1756 and 1715 cm−1). Desilylation using excess TBAF in THF at r.t. gave alcohol 88 in near quantitative yield. This was a very clean reaction, and no double-bond isomerization or epimerization were observed. No enol was detected in CDCl3, as judged from the 1H NMR spectroscopy spectrum. Finally, when 88 was submitted to 20 mol% 85 for 8 h at 0.001 M in hexane at reflux, amphidinolide P (1) was isolated in an excellent 84% yield.
Scheme 17.
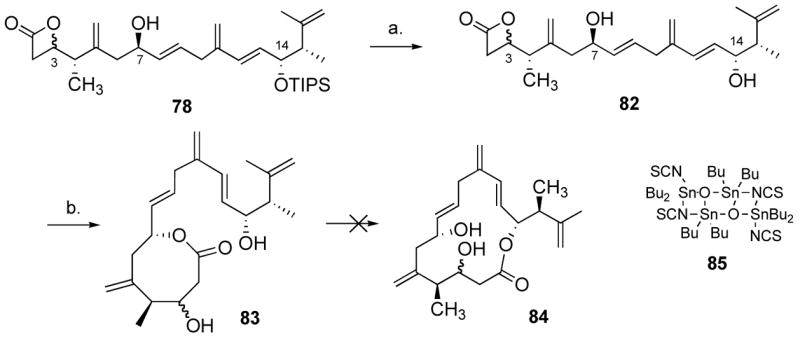
Attempted formation of the amphidinolide 15-membered ring system.a
a Reagents and conditions: (a) 4.0 equiv. TBAF, THF, 0 °C, 5 h, 71%. (b) 0.1 equiv. 85, hexane, 0.001M, reflux, 20 min, quant.
Scheme 18.
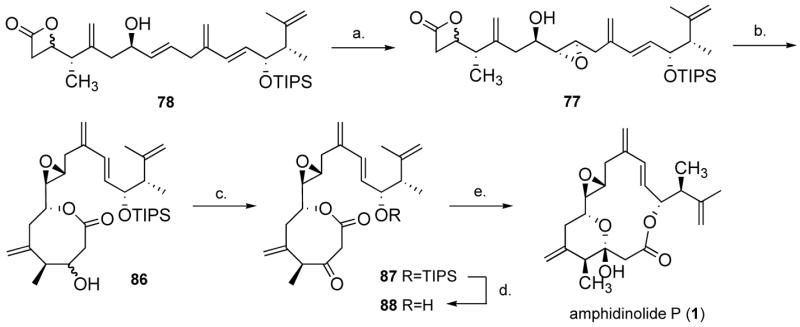
Final steps.
a Reagents and conditions: (a) 1.0 equiv. Ti(OiPr)4, 1.2 equiv. (−)-DET, 2.0 equiv. TBHP, 4Å MS, CH2Cl2, −20 °C, 2 h, 83%. (b) 0.05 equiv. 85, hexane, 0.002 M, reflux, 1 h, 93%. (c) 3.0 equiv. Dess-Martin periodinane, CH2Cl2, 23 °C, 1 h, 82%. (d) 5.0 equiv. TBAF, THF, 0 °C to 23 °C, 23 °C, 1 h, 95 %. (e) 0.20 equiv. 85, hexane, 0.001 M, reflux, 8 h, 84%.
Data for synthetic 1 was identical to the data reported for the natural product, except for the optical rotation: [α]D23 −27.4 (c 0.17, MeOH), lit.7 [α ]D20 +31 (c 0.098, MeOH). Four optical rotation measurements in absolute methanol at slightly different concentrations gave consistent values. Concentrations of 0.09, 0.17, 0.19, 0.23 gave [α]D23 values of −27.2, −27.4, −31.7 and −28.3, respectively. No change of optical rotation was observed after 5 h of storage in methanol, and the 1H NMR spectra of 1 in C6D6 and CD3OD were also unchanged. Williams et al. reported a synthesis of 1 which relied on two Sharpless asymmetric epoxidations to introduce the chirality, both of them using the (+)-diethyl tartrate ligand, and which should give synthetic 1 of opposite absolute configuration to the one reported herein.5d Yet they also reported a negative optical rotation, [α]D23 −30 (c 0.09, MeOH). Unfortunately, Professor Williams was not able to provide us with a sample of synthetic 1, and no direct comparative measurement could be done.
Conclusion
The synthesis of amphidinolide P demonstrated that β-lactones could be used as a handle for the construction of medium-sized rings, and as an alternative macrolactonization strategy. The use of a β-lactone in this work also allowed for the differenciation of three secondary alcohols, thereby minimizing the use of protecting groups in the end-game and increasing the efficiency of the synthesis. This work also highlighted the chemo- and regioselectivity of the ruthenium-catalyzed addition of alkene to alkynes. In the course of these studies, we showed that this reaction was compatible with silyl ethers, esters, β-lactones, allylic alcohols, and disubstituted alkenes, and that enynes gave perfect regioselectivity for the branched product to give 2-allylated-1,3-dienes. As a result, a novel highly convergent synthetic strategy emerged for the synthesis of amphidinolide P. Indeed the required alkene was prepared in 9 steps and 30% yield, and the alkyne also in 9 steps and 26% yield, both from readily available and inexpensive chiral building blocks.
Experimental Section
(E)-(5S,6S)-6,7-Dimethyl-5-triisopropylsilanyloxy-1-trimethylsilanyl-octa-3,7-dien-1-yne (5)
To a solution of 15 (1.73 g, 4.36 mmol) and triphenylphosphine (3.46 g, 13.19 mmol) in dry toluene (20 mL) was added diisopropyl azodicarboxylate (2.67 g, 13.20 mmol) and the flask was lowered into a preheated oil bath (80 °C). After stirring at this temperature for 20 min, the volatiles were removed in vacuo and the residue was purified by flash silica gel column chromatography (petroleum ether) to give alkyne 5 (1.37 g, 3.61 mmol, 83%) as a colorless oil and an 8:1 inseparable E/Z mixture (Found: C, 69.59; H, 11.14. C22H42OSi2 requires C, 69.77; H, 11.18%); [α]D23 +1.7 (c 3.41, CHCl3); Rf 0.40 (petroleum ether); νmax/cm−1 2945, 2868, 2361, 2134, 1464, 1250, 1059, 958, 883, 843, 760, 679, 654; E isomer: δH (500 MHz, CDCl3) 0.18 (9 H, s), 0.97 (3 H, d, J 7.0), 1.07 (21 H, s), 1.76 (3 H, s), 2.40 (1 H, br. quin., J 6.0), 4.46 (1 H, td, J 5.0, 2.0), 4.75 (1 H, s), 4.85 (1 H, s), 5.70 (1 H, dd, J 16.0, 2.0), 6.09 (1 H, dd, J 16.0, 5.0); δC (125 MHz, CDCl3) 0.0, 12.3, 12.5, 18.1, 22.2, 47.1, 74.1, 94.1, 103.8, 110.0, 111.9, 144.5, 146.0; Z isomer: δH (500 MHz, CDCl3) 0.17 (9 H, s), 0.97 (3 H, d, J 7.0), 1.06 (21 H, s), 1.80 (3 H, s), 2.40 (1 H, masked), 4.46 (1 H, masked), 4.75 (1 H, s), 4.85 (1 H, s), 5.49 (1 H, d, J 11.5), 5.89 (1 H, dd, J 11.5, 9.0).
(4S,7S)-8-(tert-Butyl-diphenyl-silanyloxy)-7-methyl-6-methylene-oct-1-en-4-ol (46)
To a solution of thiophene (0.76 g, 9.03 mmol) in THF (8 mL) at −30 °C was added n-BuLi (2.58 M, 3.50 mL, 9.03 mmol) dropwise. The mixture was stirred for 30 min, whereupon it was cannulated into a slurry of CuCN (99.99 %, 809 mg, 9.03 mmol) in THF (8 mL) at −78 °C. The cooling bath was removed and upon reaching r.t., a clear brown solution was obtained. This solution was kept at ca. −20 °C until the the vinyl lithium reagent was ready (vide infra).
To a solution of vinyl bromide 47 (2.79 g, 6.91 mmol) in ether (28 mL) was added t-BuLi (1.44 M, 10 mL, 14.4 mmol) at −78 °C over 10 min. After another 45 min, the freshly prepared solution of 2-thienyl lithiumcyanocuprate was cannulated into it. The pale brown heterogeneous mixture was warmed to −45 °C (chlorobenzene/dry ice bath), and stirred at this temperature for 1 h. A solution of (R)-glycidyl tosylate (34) (3.1 g, 13.58 mmol) in THF (11 mL) was then cannulated into the mixture, and the resulting slurry was warmed to 0 °C over 10 min. After an additional 5 h at 0 °C, the mixture was recooled to −78 °C and a vinyl lithium solution (13.93 mmol, prepared from nBuLi and tetravinyltin at −78 °C, 45 min then warming to 24 °C) in THF (14 mL) was added, followed after 5 min, with BF3.Et2O (1.97 g, 13.93 mmol). The resulting mixture was stirred for 20 min, then quenched with a 9:1 solution of saturated aqueous NH4Cl solution/NH4OH and diluted with ether. After 20 min of vigorous stirring followed by filtration through Celite, the organic phase was washed with brine. The combined aqueous phase was back-extracted twice with ether. After drying the combined organic phase over MgSO4, the volatiles were removed in vacuo to give a residue that was purified by silica gel flash chromatography (petroleum ether-ethyl acetate, 19:1 to 9:1) to afford the alcohol 46 (2.01 g, 4.92 mmol, 71%) as a colorless oil (Found: C, 76.43; H, 9.02. C26H36O2Si requires C, 76.42; H, 8.88%); [α]D22 −13.1 (c 3.22, CHCl3); Rf 0.30 (petroleum ether-ethyl acetate, 9:1); νmax/cm−1 3448, 2960, 2931, 2858, 1472, 1428, 1121, 1080, 823, 740, 702, 614; δH (400 MHz, CDCl3) 1.05 (9 H, s), 1.07 (3 H, d, J 7.0), 2.04 (1 H, dd, J 14.0, 9.5), 2.19–2.23 (3 H, m), 2.35 (1 H, broad sex, J 7.0), 3.49 (1 H, dd, J 10.0, 7.0), 3.62 (1 H, dd, J 10.0, 6.0), 3.71 (1 H, dddd, J 9.5, 6.0, 6.0, 4.5), 4.93 (1 H, s), 4.94 (1 H, s), 5.09–5.14 (2 H, m), 5.83 (1 H, ddt, J 17.0, 10.5, 7.0), 7.35–7.43 (6 H, m), 7.64–7.68 (4 H, m); δC (100 MHz, CDCl3) 16.7, 19.2, 26.8, 41.4, 43.6, 68.2, 68.5, 112.7, 117.5, 127.6, 129.6, 133.6, 133.7, 134.9, 135.6, 135.6, 148.8.
(8E,12E)-(4S,7R,14R,15S)-3,7-Dihydroxy-4,15,16-trimethyl-5,11-dimethylene-14-triisopropylsilanyloxy-heptadeca-8,12,16-trienoic acid tert-butyl ester (70)
Conditions of Table 4, Entry 7: A dry flask was charged with alkene 55 (2.8:1 d.r., 83 mg, 0.292 mmol) and alkyne 25 (20 mg, 0.065 mmol) and flushed with argon. Methanol (1.1 mL) was added, followed with CpRu(COD)Cl (1.0 mg, 0.003 mmol) and NH4PF6 (1.0 mg, 0.006 mmol) and the mixture was heated to reflux over 10 min. After 75 min, the mixture was allowed to cool and concentrated in vacuo. Purification by flash silica gel column chromatography (petroleum ether-ethyl acetate, 4:1 to 7:3) afforded some recovered alkene 55 (72 mg, 0.252 mmol) and the ester 70 (24 mg, 0.040 mmol, 61 %) as a yellow oil and an inseparable 2.8:1 mixture of C-3 epimers (Found: C, 71.01; H, 10.77. C35H62O5Si requires C, 71.14; H, 10.57 %); [α]D22 −6.0 (c 4.06, CHCl3); Rf 0.39 (petroleum ether-ethyl acetate, 7:3); νmax/cm−1 3427, 2966, 2942, 2867, 1729, 1462, 1368, 1255, 1154, 1059, 970, 884; δH (500 MHz, CDCl3, minor diastereomer in brackets) 0.97 (3 H, d, J 7.0), 1.05 (21 H, s), 1.10 (3 H, d, J 7.0), 1.45 (1.46) (9 H, s), 1.75 (3 H, s), 2.21 (1 H, dd, J 14.5, 9.0), 2.18–2.40 (3 H, m), 2.36 (1 H, dd, J 16.0, 9.0), 2.42 (2.49) (1 H, dd, J 16.0, 3.5 (2.5)), 2.90 (2 H, d, J 6.5), 3.97 (1 H, ddd, J 9.0, 5.5, 3.5), 4.21–4.30 (1 H, m), 4.36 (1 H, broad t, J 5.5), 4.70 (1 H, s), 4.78 (1 H, s), 4.93 (1 H, s), 4.98 (1 H, s), 4.99 (1 H, s), 5.04 (5.02) (1 H, s), 5.53–5.58 (1 H, m), 5.62 (1 H, dd, J 16.0, 7.0), 5.73–5.79 (1 H, m), 6.16 (1 H, d, J 16.0); δC (125 MHz, CDCl3, minor diastereomer in brackets) 12.5, 13.3, 14.9 (15.6), 18.1 (18.1), 21.8, 28.1(29.7), 35.0, 39.9 (39.5), 44.0 (43.9), 44.2 (45.3), 47.8, 70.3 (71.2), 70.4 (70.9), 75.5, 81.2, 111.5, 113.8 (114.1), 115.7, 129.2 (128.8), 130.5, 131.9, 133.8 (133.9), 143.9, 146.9, 148.2 (148.3), 172.5.
Conditions of Table 4, Entry 9: To a solution of alkyne 25 (13 mg, 0.042 mmol) and alkene 55 (2.8:1 d.r., 54 mg, 0.190 mmol) in dry acetone (0.7 mL) at 0 °C was added [CpRu(CH3CN)3]PF6 (1.8 mg, 0.004 mmol). The mixture was warmed to r.t. and stirred for 15 h, whereupon it was concentrated in vacuo. The residue was purified by silica gel flash chromatography (petroleum ether-ethyl acetate, 30 %) to afford some recovered alkene 55 (43 mg, 0.151 mmol) and the ester 70 (18 mg, 0.030 mmol, 72 %) as a yellow oil and an inseparable 2.8:1 mixture of C-3 epimers.
4-((1S,4S)-4-Hydroxy-1-methyl-2-methylene-hept-6-enyl)-oxetan-2-one (79)
To a solution of DMSO (1.68 g, 21.56 mmol) in CH2Cl2 (75 mL) at −78 °C was added oxalyl chloride (1.36 g, 10.77 mmol) and the mixture was stirred for 20 min, whereupon a solution of alcohol 53 (1.54 g, 5.41 mmol) was added dropwise. After another 20 min at −78 °C, triethylamine (3.26 g, 32.29 mmol) was added and the cooling bath was removed. Upon reaching 0 °C, the mixture was partitioned between ether and saturated aqueous NH4Cl. The organic phase was washed with saturated aqueous NH4Cl, brine, dried over MgSO4 and concentrated in vacuo. The crude aldehyde (45), which was obtained as a yellow oil (1.55 g), was immediately redissolved in CH2Cl2 (50 mL) and cooled to −78 °C. Me2AlCl (1.0M in hexanes, 5.4 mL, 5.4 mmol) was added over 5 min. The bright yellow mixture was stirred for 3 min, whereupon neat trimethylsilylketene (0.65 g, 5.72 mmol) was added dropwise. After another 30 min, 0.5M aqueous NaHSO4 (20 mL) and ether (100 mL) were added and the mixture was allowed to warm to r.t. with vigorous stirring. Additional 0.5M aqueous NaHSO4 (150 mL) and ether (100 mL) were added and the two clear phases were separated. The organic phase was washed with brine (100 mL) and the combined organic phase was back-extracted with ether (2 × 50 mL), dried over MgSO4 and concentrated in vacuo. The yellow residue (81, 2.15 g) was taken up in acetonitrile (60 mL) and KF.2H2O (0.76 g, 8.06 mmol) was added. The mixture was vigorously stirred for 1 h, whereupon it was cooled to 0 °C. Aqueous 49 % HF (13 mL, 364 mmol) was added dropwise and the mixture was stirred at 0 °C for 30 min. After dilution with ether (100 mL), solid NaHCO3 (30 g) was added portionwise over 5 min. After stirring for another 5 min, the mixture was filtered through a sintered funnel packed with MgSO4. The solids were well rinsed with ether and the combined filtrate was concentrated in vacuo. The residue was purified by silica gel flash chromatography (petroleum ether-ethyl acetate, 7:3 to 3:2) to afford the lactone 79 (0.78 g, 3.71 mmol, 69 %) as a yellow oil and a 1.6:1 mixture of separable diastereomers (Found: M+, 210.1254. C12H18O3 requires M 210.1256, 0.7 ppm, EIMS);
one C-3 epimer
[α]D 26 +20.8 (c 1.73, CHCl3); Rf 0.19 (petroleum ether-ethyl acetate, 7:3); νmax/cm−1 3417, 2924, 1827, 1642, 1412, 1278, 1127, 914, 867; δH (400 MHz, CDCl3) 1.22 (3 H, d, J 7.0), 2.14–2.35 (4 H, m), 2.50 (1 H, br. quin, J 7.0), 3.15 (1 H, dd, J 16.5, 4.5), 3.45 (1 H, dd, J 16.5, 6.5), 3.77–3.83 (1 H, m), 4.45 (1 H, ddd, J 8.5, 6.5, 4.5), 4.94 (1 H, s), 5.04 (1 H, s), 5.16 (1 H, d, J 18.0), 5.17 (1 H, d, J 11.0), 5.33 (1 H, dddd, J 18.0, 11.0, 7.5, 7.0); δC (100 MHz, CDCl3) 16.1, 41.6, 41.8, 43.1, 43.7, 68.9, 73.8, 113.9, 118.6, 134.2, 146.5, 168.1.
other C-3 epimer
[α]D 26 −14.4 (c 1.4, CHCl3); Rf 0.13 (petroleum ether-ethyl acetate, 7:3); νmax/cm−1 3417, 2933, 1827, 1642, 1412, 1278, 1127, 913, 869; δH (500 MHz, CDCl3) 1.10 (3 H, d, J 7.0), 2.16–2.26 (3 H, m), 2.29–2.34 (2 H, m), 2.52–2.58 (1 H, m), 3.13 (1 H, dd, J 16.5, 4.5), 3.48 (1 H, dd, J 16.5, 6.0), 3.78–3.83 (1 H, m), 4.46 (1 H, ddd, J 8.0, 6.0, 4.5), 5.05 (1 H, s), 5.06 (1 H, s), 5.12–5.17 (2 H, m), 5.80–5.89 (1 H, m); δC (125 MHz, CDCl3) 14.7, 41.4, 41.6, 43.0, 43.1, 68.6, 73.4, 114.0, 118.2, 134.4, 146.3, 167.8.
4-((5E,9E)-(1S,4R,11R,12S)-4-Hydroxy-1,12,13-trimethyl-2,8-dimethylene-11-triisopropylsilanyloxy-tetradeca-5,9,13-trienyl)-oxetan-2-one (78)
To a solution of alkyne 25 (42 mg, 0.137 mmol) and alkene 79 (1.6:1 d.r., 100 mg, 0.475 mmol) in dry acetone (2.5 mL) at 0 °C was added [CpRu(CH3CN)3]PF6 (6.0 mg, 0.0138 mmol). The mixture was warmed to r.t. and stirred for 13 h, whereupon it was concentrated in vacuo. The residue was purified by silica gel flash chromatography (petroleum ether-ethyl acetate, 20 to 40%) to afford some recovered 79 (62 mg, 0.295 mmol, 87%) and the lactone 78 (52 mg, 0.100 mmol, 75%) as a yellow oil and a 1.6:1 mixture of C-3 epimers (Found: [M+Na]+, 539.3517. C31H52O4Si requires M+Na 539.3533, 2.9 ppm, ESIMS); [α]D26 −0.2 (c 0.85, CHCl3); Rf 0.40 (petroleum ether-ethyl acetate, 7:3); νmax/cm−1 3441, 2943, 2866, 1831, 1645, 1462, 1374, 1125, 1059, 970, 882; δH (500 MHz, CDCl3, minor diastereomer in brackets) 0.97 (3 H, d, J 7.0), 1.05 (21 H, s), 1.20 (1.09) (3 H, d, J 7.0), 1.75 (3 H, s), 2.18–2.33 (2 H, m), 2.38 (1 H, br quin., J 7.0), 2.90 (2 H, d, J 6.5), 3.12 (3.13) (1 H, dd, J 16.5, 4.5), 3.42 (3.45) (1 H, dd, J 16.5, 5.5), 4.20–4.25 (1 H, m), 4.35–4.38 (1 H, m), 4.43 (4.46) (1 H, ddd, J 7.0, 5.5, 4.5), 4.69 (1 H, s), 4.78 (1 H, s), 4.91 (2 H, s), 4.98 (5.03) (1 H, s), 5.02 (5.06) (1 H, s), 5.53 (5.55) (1 H, dd, J 15.0, 7.0), 5.61 (5.62) (1 H, dd, J 16.0, 6.5), 5.76 (5.76) (1 H, dt, J 15.0, 7.0), 6.16 (1 H, d, J 16.0); δC (125 MHz, CDCl3, minor diastereomer in brackets) 12.4, 13.2 (13.3), 16.0, 18.1, 21.8, 35.0, 41.6 (41.2), 43.4 (42.9), 43.8 (43.6), 47.7, 71.1 (70.6), 73.8 (73.3), 75.3 (75.4), 111.4, 113.9 (114.1), 115.7, 129.6 (129.3), 130.5 (130.4), 131.8 (131.9), 133.7 (133.6), 143.7 (143.8), 146.0 (145.7), 146.9, 168.1 (167.8).
(5S,8R)-8-[(2S,3R)-3-((E)-(5R,6S)-6,7-Dimethyl-2-methylene-5-triisopropylsilanyloxy-octa-3,7-dienyl)-oxiranyl]-4-hydroxy-5-methyl-6-methylene-oxocan-2-one (86)
Lactone 77 (1:1 mixture of C-3 epimers, 128 mg, 0.240 mmol) and distannoxane 85 (14 mg, 0.011 mmol) were placed in a dry flask, and dry hexane (120 mL) was added. The mixture was stirred at reflux for 1 h, cooled and concentrated in vacuo. The residue was purified by silica gel flash chromatography (petroleum ether-ethyl acetate, 17:3) to afford the lactone 86 (119 mg, 0.223 mmol, 93%) as a pale yellow oil and an inseparable 1:1 mixture of C-3 epimers (Found: M+, 532.3567. C31H52O5Si requires M 532.3584, 3.2 ppm, EIMS); [α]D25 +26.0 (c 1.93, CHCl3); Rf 0.21 (petroleum ether-ethyl acetate, 7:3); νmax/cm−1 3448, 2943, 2886, 1732, 1644, 1462, 1373, 1251, 1162, 1127, 1102, 1059, 1014, 992, 987, 884; EIMS m/z 532 (M+, 3), 463 [(M–C5H9]+, 100);
one C-3 epimer
δH (500 MHz, CDCl3) 0.98 (3 H, d, J 7.0), 1.05 (21 H, s), 1.18 (3 H, d, J 7.0), 1.76 (3 H, s), 2.10 (1 H, dq, J 9.5, 7.0), 2.37–2.54 (4 H, m), 2.52 (1 H, dd, J 11.5, 7.0), 2.55 (1 H, dd, J 13.5, 3.0), 2.71 (1 H, dd, J 11.5, 5.0), 2.89 (1 H, dd, J 5.5, 2.5), 3.11 (1 H, td, J 5.5, 2.0), 3.66 (1 H, m), 4.25 (1 H, ddd, J 11.0, 5.5, 2.0), 4.40 (1 H, br. t, J 6.0), 4.68 (1 H, br. s), 4.78 (1 H, br. s), 5.03 (1 H, br. s), 5.06 (1 H, br. s), 5.07 (1 H, br. s), 5.12 (1 H, br. s), 5.60 (1 H, dd, J 16.0, 6.5), 6.20 (1 H, d, J 16.0); δC (125 MHz, CDCl3) 12.4, 13.0, 18.1, 21.9, 34.17, 37.9, 41.2, 42.7, 45.0, 47.6, 56.4, 58.4, 73.3, 75.1, 81.1, 111.5, 116.7, 118.9, 130.64, 131.8, 140.8, 144.9, 146.8, 171.7.
other C-3 epimer
δH (500 MHz, CDCl3) 0.98 (3 H, d, J 7.0), 1.05 (21 H, s), 1.23 (3 H, d, J 7.0), 1.76 (3 H, s), 2.01 (1 H, dq, J 9.5, 7.0), 2.37–2.54 (4 H, m), 2.49 (1 H, dd, J 12.5, 5.0), 2.61 (1 H, dd, J 14.0, 1.5), 2.91 (1 H, dd, J 5.0, 2.0), 2.97 (1 H, dd, J 12.5, 4.0), 3.14 (1 H, ddd, J 6.0, 5.0, 2.0), 4.10 (1 H, m), 4.40 (1 H, br. t, J 6.0), 4.59 (1 H, ddd, J 11.0, 5.0, 2.5), 4.68 (1 H, br. s), 4.78 (1 H, br. s), 5.07 (2 H, br. s), 5.12 (1 H, br. s), 5.21 (1 H, br. s), 5.60 (1 H, dd, J 16.0, 6.5), 6.20 (1 H, d, J 16.0); δC (125 MHz, CDCl3) 12.4, 13.0, 18.1, 21.9, 34.21, 37.9, 41.2, 42.8, 43.7, 47.6, 55.8, 58.5, 73.3, 75.1, 79.3, 111.5, 116.1, 118.9, 130.68, 131.8, 140.8, 144.9, 147.4, 172.2.
Amphidinolide P (1)
Lactone 88 (14.0 mg, 0.037 mmol) and distannoxane 85 (9 mg, 0.007 mmol) were placed in a dry flask, and dry hexane (37 mL) was added. The mixture was stirred at reflux for 8 h, cooled and concentrated in vacuo. The residue was purified by silica gel flash chromatography (petroleum ether-ether, 17:3) to afford amphidinolide P (1) (11.7 mg, 0.031 mmol, 84%) as a colorless oil; [α]D23 −27.4 (c 0.17, MeOH); Rf 0.35 (petroleum ether-ethyl acetate, 17:3); νmax/cm−1 3482, 3084, 2971, 2942, 1712, 1650, 1433, 1376, 1361, 1291, 1243, 1189, 1111, 988, 967, 896; δH (500 MHz, C6D6) 0.91 (3 H, d, J 7.0), 0.92 (3 H, d, J 7.0), 1.67 (3 H, br. s), 1.93–1.96 (1 H, m), 2.10 (1 H, dd, J 12.7, 11.5), 2.17 (1 H, br. dd, J 13.5, 9.5), 2.27 (1 H, d, J 12.0), 2.36 (1 H, d, J 12.0), 2.43 (1 H, dq, J 9.5, 7.0), 2.48 (1 H, dt, J 9.5, 1.5), 2.52 (1 H, dd, J 12.7, 2.7), 2.62 (1 H, dd, J 8.5, 1.5), 2.68 (1 H, br. d, J 13.5), 3.47 (1 H, ddd, J 11.5, 8.5, 2.7), 4.27 (1 H, d, J 2.0), 4.77 (1 H, m), 4.81 (1 H, br. s), 4.81–4.82 (1 H, m), 4.87–4.89 (1 H, m), 4.89–4.90 (1 H, m), 4.94 (1 H, m), 5.29 (1 H, br. t, J 8.5), 5.60 (1 H, dd, J 16.2, 7.5), 6.20 (1 H, d, J 16.2); δC (125 MHz, C6D6) 11.8, 16.1, 19.5, 36.3, 39.4, 45.0 (_ 2), 45.2, 58.2, 62.7, 73.5, 78.5, 99.2, 110.0, 112.3, 118.2, 129.1, 133.6, 142.2, 143.7, 146.5, 172.4.
Supplementary Material
Experimental procedures and characterization data for compounds 6-11, 14-28, 32, 35-42, 44-47, 49, 50, 52-55, 64-66, 70, 77-79, 82, 83, 86-88 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health (GM-33049) and the National Science Foundation for their generous support of our programs. Mass spectra were provided by the Mass Spectrometry Regional Center of the University of California – San Francisco supported by the NIH Division of Research Resources.
References
- 1.a) Stommel EW, Watters MR. Curr Treat Options Neurol. 2004;6:105. doi: 10.1007/s11940-004-0020-9. [DOI] [PubMed] [Google Scholar]; (b) Van Dolah FM. Environ Health Perspect. 2000;108:133. doi: 10.1289/ehp.00108s1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobayashi J, Ishibashi M, Nakamura H, Ohizumi Y, Yamasu T, Sasaki T, Hirata Y. Tetrahedron Lett. 1986;27:5755. [Google Scholar]
- 3.(a) Tsuda M, Kariya Y, Iwamoto R, Fukushi E, Kawabata J, Kobayashi J. Marine Drugs. 2005;3:1. [Google Scholar]; (b) Kubota T, Sakuma Y, Tsuda M, Kobayashi J. Marine Drugs. 2004;2:83. [Google Scholar]; (c) Kobayashi J, Tsuda M. Nat Prod Rep. 2004;21:77. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]; (d) Chakraborty TK, Das S. Curr Med Chem: Anti-Cancer Agents. 2001;1:131. doi: 10.2174/1568011013354660. [DOI] [PubMed] [Google Scholar]; (e) Kobayashi J. In: Comprehensive Natural Products Chemistry. Mori K, editor. Vol. 8. Elsevier; New York: 1999. p. 619. [Google Scholar]; (f) Ishibashi M, Kobayashi J. Heterocycles. 1997;44:543. [Google Scholar]; (g) Ishibashi M, Kobayashi J. Chem Rev. 1993;93:1753. [Google Scholar]
- 4.For the most recent studies, see: Amphidinolide F: Shotwell JB, Roush WR. Org Lett. 2004;6:3865. doi: 10.1021/ol048381z. Amphidinolides B1, B2, and B3: Zhang W, Carter RG, Yokochi AFT. J Org Chem. 2004;69:2569. doi: 10.1021/jo035829l. Amphidinolide O: Pang JH, Ham YJ, Lee DH. Bull Kor Chem Soc. 2003;24:891.. Amphidinolide C: Kubota T, Tsuda M, Kobayashi J. Tetrahedron. 2003;59:1613.
- 5.Amphidinolides T1 and T4: Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2005;127:4297. doi: 10.1021/ja042733f. Amphidinolide X: Lepage O, Kattnig E, Fürstner A. J Am Chem Soc. 2004;126:15970. doi: 10.1021/ja044130+. Amphidinolide P: Trost BM, Papillon JPN. J Am Chem Soc. 2004;126:13618. doi: 10.1021/ja045449x.Williams DR, Myers BJ, Mi L. Org Lett. 2000;2:945. doi: 10.1021/ol0000197. Amphidinolide A, revised structure: Trost BM, Wrobleski ST, Chisholm JD, Harrington PE, Jung M. J Am Chem Soc. 2005;127:13589. doi: 10.1021/ja0533646.Trost BM, Harrington PE, Chisholm JD, Wrobleski ST. J Am Chem Soc. 2005;127:13598. doi: 10.1021/ja053365y.Trost BM, Chisholm JD, Wrobleski ST, Jung M. J Am Chem Soc. 2002;124:12420. doi: 10.1021/ja027883+.Maleczka RE, Jr, Terrell LR, Geng F, Ward JS., III Org Lett. 2002;4:2841. doi: 10.1021/ol0262284.Lam HW, Pattenden G. Angew Chem, Int Ed. 2002;41:508. doi: 10.1002/1521-3773(20020201)41:3<508::aid-anie508>3.0.co;2-7. Amphidinolide W: Ghosh AK, Gong G. J Am Chem Soc. 2004;126:3704. doi: 10.1021/ja049754u.Amphidinolide T1: Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2004;126:998. doi: 10.1021/ja039716v.Ghosh AK, Liu C. Strat Tact Org Synth. 2004;5:255.Ghosh AK, Liu C. J Am Chem Soc. 2003;125:2374. doi: 10.1021/ja021385j.Amphidinolide T1, T3, T4, and T5: Aiessa C, Riveiros R, Ragot J, Fürstner A. J Am Chem Soc. 2003;125:15512. doi: 10.1021/ja038216z.Amphidinolide T4: Fürstner A, Aissa C, Riveiros R, Ragot J. Angew Chem, Int Ed. 2002;41:4763. doi: 10.1002/anie.200290042.Amphidinolide K: Williams DR, Meyer KG. J Am Chem Soc. 2001;123:765. doi: 10.1021/ja005644l.Amphidinolide J: Williams DR, Kissel WS. J Am Chem Soc. 1998;120:11198.
- 6.Trost BM, Shen HC, Pinkerton AB. Chem Eur J. 2002;8:2341. doi: 10.1002/1521-3765(20020517)8:10<2341::AID-CHEM2341>3.0.CO;2-A.Trost BM, Machacek M, Schanderbeck MJ. Org Lett. 2000;2:1761. doi: 10.1021/ol0059504.Trost BM, Indolese AF, Müller TJJ, Treptow B. J Am Chem Soc. 1995;117:615.Trost BM, Probst GD, Schoop A. J Am Chem Soc. 1998;120:9228.Trost BM, Pinkerton AB, Toste FD, Sperrle M. J Am Chem Soc. 2001;123:12504. doi: 10.1021/ja012009m. For use in natural product syntheses, see: Trost BM, Yang H, Probst GD. J Am Chem Soc. 2004;126:48. doi: 10.1021/ja038787r.Trost BM, Gunzner JL. J Am Chem Soc. 2001;123:9449. doi: 10.1021/ja011424b.(h) ref. 5c, 5e, 5f. See also: Trost BM. Acc Chem Res. 2002;35:695. doi: 10.1021/ar010068z.Trost BM, Toste DF, Pinkerton AB. Chem Rev. 2001;101:2067. doi: 10.1021/cr000666b.
- 7.Ishibashi M, Takahashi J, Kobayashi J. J Org Chem. 1995;60:6062. [Google Scholar]
- 8.Boeckman RK, Pruitt JR. J Am Chem Soc. 1989;111:8286.Quinn EK, Olmstead MM, Kurth MJ. J Org Chem. 1993;58:5011. (c) ref. 6g.
- 9.(a) Witzeman JS, Nottingham WD. J Org Chem. 1991;56:1713. [Google Scholar]; (b) Clemens RJ, Witzeman JS. J Am Chem Soc. 1989;111:2186. [Google Scholar]; (c) Clemens RJ, Hyatt JA. J Org Chem. 1985;50:2431. [Google Scholar]; (d) Hyatt JA. J Org Chem. 1984;49:5102. [Google Scholar]; (e) Hyatt JA, Feldman PE, Clemens RJ. J Org Chem. 1984;49:5105. [Google Scholar]
- 10.Iijima T, Endo Y, Tsuji M, Kawachi E, Kagechika H, Shudo K. Chem Pharm Bull. 1999;47:398. doi: 10.1248/cpb.47.398. [DOI] [PubMed] [Google Scholar]
- 11.Weigand S, Brückner R. Synthesis. 1996:475. [Google Scholar]
- 12.Kobayashi S, Nishio K. J Org Chem. 1994;59:6620.Denmark SE, Coe DM, Pratt NE, Griedel BD. J Org Chem. 1994;59:6161. doi: 10.1021/jo052202p.For the preparation of 14, see Supporting Information and: Aoki S, Mikami K, Terada M, Nakai T. Tetrahedron. 1993;49:1783.Haynes RK, Katsifis AG, Vonwiller SC, Hambleyf TW. J Am Chem Soc. 1988;110:5423.
- 13.(a) Yanagisawa A. ch 27 In: Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Vol. 2. Springer-Verlag; Heidelberg: 1999. [Google Scholar]; (b) Yamamoto Y, Asao N. Chem Rev. 1993;93:2207. [Google Scholar]; (c) Keck GE, Savin KA, Cressman ENK, Abbott DE. J Org Chem. 1994;59:7889. [Google Scholar]
- 14.(a) Yanagisawa A, Nakashima H, Ishiba A, Yamamoto H. J Am Chem Soc. 1996;118:4723. [Google Scholar]; (b) Yanagisawa A, Ishiba A, Nakashima H, Yamamoto H. Synlett. 1997:88. [Google Scholar]
- 15.(a) Ishiara K, Mouri M, Gao Q, Maruyama T, Furuta K, Yamamoto H. J Am Chem Soc. 1993;115:11490. [Google Scholar]; (b) Marshall JA, Tang Y. Synlett. 1992:653. [Google Scholar]
- 16.Trost BM, Belletire JL, Godleski S, McDougal PG, Balkovec JM, Baldwin JL, Christy ME, Ponticello GS, Varga SL, Springer JP. J Org Chem. 1986;51:2370. [Google Scholar]
- 17.(a) Denmark SE, Fu J. J Am Chem Soc. 2000;122:12021. [Google Scholar]; (b) Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Fu J. Org Lett. 2002;4:1951. doi: 10.1021/ol025971t. [DOI] [PubMed] [Google Scholar]; (d) Iseki K, Mizuno S, Kuroki Y, Kobayashi Y. Tetrahedron Lett. 1998;53:2767. [Google Scholar]; (e) Marshall JA, Liao J. J Org Chem. 1998;63:5962. doi: 10.1021/jo980603h. [DOI] [PubMed] [Google Scholar]
- 18.Mikami K, Kawamoto K, Loh TP, Nakai T. J Chem Soc, Chem Commun. 1990:1161. [Google Scholar]
- 19.(a) Skrydstrup T, Bénéchie M, Khuong-Huu F. Tetrahedron Lett. 1990;31:7145. [Google Scholar]; (b) Watanabe H, Watanabe H, Bando M, Kido M, Kitchara T. Tetrahedron. 1999;55:9755. [Google Scholar]
- 20.Wessel HP, Iversen T, Bundle DR. J Chem Soc, Perkin Trans 1. 1985:2247. [Google Scholar]
- 21.Clizbe LA, Overman LE. Org Synth. 1978;58:4. [Google Scholar]
- 22.Xu Z, Johannes CW, Houri AF, La DS, Cogan DA, Hofilena GE, Hoveyda AH. J Am Chem Soc. 1997;119:10302. [Google Scholar]
- 23.Paquette LA, Balogh D, Engel P. Heterocycles. 1981;15:271. [Google Scholar]
- 24.Marrota E, Foresti E, Marcelli T, Peri F, Righi P, Scardovi N, Rosini G. Org Lett. 2002;4:4451. doi: 10.1021/ol026955z. [DOI] [PubMed] [Google Scholar]
- 25.(a) Reetz M, Kessler K, Jung A. Tetrahedron. 1984;40:4327. [Google Scholar]; (b) Mahrwald R. Chem Rev. 1999;99:1095. doi: 10.1021/cr980415r. [DOI] [PubMed] [Google Scholar]
- 26.Munakata R, Katakai H, Ueki T, Kurosaka J, Takao K, Tadano K. J Am Chem Soc. 2003;125:14722. doi: 10.1021/ja038732p. [DOI] [PubMed] [Google Scholar]
- 27.Racemic aldehyde 28a was prepared by dihydroxylation, followed by oxidative cleavage of known PMB-protected hepta-1,6-dien-4-ol: Shepherd JN, Na J, Myles DC. J Org Chem. 1997;62:4558.
- 28.Racemic aldehyde 28b was prepared in four standard steps (allylation, TBS protection, DDQ-mediated PMB cleavage and Moffat-Swern oxidation) from known 3-(4-methoxy-benzyloxy)-propionaldehyde: Oka T, Marai A. Tetrahedron. 1998;54:1.
- 29.Sato M, Sunami S, Sugita Y, Kaneko C. Heterocycles. 1995;41:1435. [Google Scholar]
- 30.(a) Oikawa Y, Sugano K, Yonemitsu O. J Org Chem. 1978;43:2087. [Google Scholar]; (b) Sato M, Ogasawara H, Komatsu S, Kato T. Chem Pharm Bull. 1984;32:3848. [Google Scholar]
- 31.See supporting information for details.
- 32.Use of a 1:5 E/Z mixture of silylketene acetal derived from ethyl propionate in the presence of TiCl2(OiPr)2 in toluene gave a 2.5:1 1,2-anti/syn ratio and a better than 10:1 2,4-anti/syn ratio. Use of a 1:6 E/Z mixture of silylketene acetal derived from tert-butyl thiopropionate in the presence of TiCl2(OiPr)2 in dichloromethane gave a 1:1 1,2-anti/syn ratio and an 8:1 2,4-anti/syn ratio. In both cases, alternative conditions gave higher 1,2-anti/syn ratio.
- 33.(a) Trost BM, Ball ZT. J Am Chem Soc. 2004;126:13942. doi: 10.1021/ja045971j. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Ball ZT. J Am Chem Soc. 2001;123:12726. doi: 10.1021/ja0121033. [DOI] [PubMed] [Google Scholar]
- 34.Klunder JM, Onami T, Sharpless KB. J Org Chem. 1989;54:1295. [Google Scholar]
- 35.Kant J. J Org Chem. 1993;58:2296. and references cited therein. [Google Scholar]
- 36.Sharpless KB, Verhoeven TR. Aldrichimica Acta. 1979;12:63. [Google Scholar]
- 37.(a) Yamaguchi M, Hirao I. Tetrahedron Lett. 1983;24:391. [Google Scholar]; (b) Eis MJ, Wrobel JE, Ganem B. J Am Chem Soc. 1994;106:3693. [Google Scholar]
- 38.Ooi T, Kagoshima N, Ichikawa H, Maruoka K. J Am Chem Soc. 1999;121:3328. [Google Scholar]
- 39.(a) Maruoka K, Imoto H, Saito S, Yamamoto H. J Am Chem Soc. 1994;116:4131. [Google Scholar]; (b) Saito S, Yamamoto H. Chem Commun. 1997:1585. [Google Scholar]
- 40.(a) Müller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521. [Google Scholar]; (b) Ohira S. Synth Commun. 1989;19:561. [Google Scholar]
- 41.Nicolaou KC, Li Y, Fylaktadikou KC, Mitchell HJ, Wei HX, Weyershausen B. Angew Chem Int Ed. 2001;40:3849. [PubMed] [Google Scholar]
- 42.(a) Lipshutz BH, Kozlowski JA, Parker DA, Nguyen SL, McCarthy KE. J Organomet Chem. 1985;285:437. [Google Scholar]; (b) Taylor RJK, editor. Organocopper Reagents: A practical Approach. Oxford University Press; Oxford: 1994. [Google Scholar]
- 43.Higashibayashi S, Shinko K, Ishizu T, Hashimoto K, Shirahama H, Nakata M. Synlett. 2000:1306. [Google Scholar]
- 44.Pangborn AB, Giardello MA, Grubbs RH. Organometallics. 1996;15:1518.Alaimo PJ, Petrs DW, Arnold J, Bergman RG. J Chem Ed. 2001;78:64. (c) http://www.glasscontour.com/.
- 45.Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J Am Chem Soc. 1999;121:7540. [Google Scholar]
- 46.(a) Inanaga J, Hirata H, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]; (b) Hikato M, Sakurai Y, Horita K, Yonemitsu O. Tetrahedron Lett. 1990;31:6367. [Google Scholar]; (c) Evans DA, Kim AS. J Am Chem Soc. 1996;118:11323. [Google Scholar]
- 47.Trost BM, Chisholm JD. Org Lett. 2002;4:3743. doi: 10.1021/ol026726c. [DOI] [PubMed] [Google Scholar]
- 48.Mukaiyama T, Usui M, Saigo K. Chem Lett. 1976:49. [Google Scholar]
- 49.Keck GE, Boden EP. J Org Chem. 1985;50:2394. [Google Scholar]
- 50.Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 51.Corey EJ, Nicolaou KC. J Am Chem Soc. 1974;96:5614. [Google Scholar]
- 52.(a) Yang HW, Romo D. Tetrahedron. 1999;55:6403. [Google Scholar]; (b) Pommier A, Pons JM. Synthesis. 1995:729. [Google Scholar]; (c) Pommier A, Pons JM. Synthesis. 1993:441. [Google Scholar]; (d) Lowe C, Vederas JC. Org Prep Proc Int. 1995;27:305. [Google Scholar]
- 53.(a) Zaitseva GS, Vinokurova NG, Baukov YI. Zh Obshch Khim. 1975;45:1398. [Google Scholar]; (b) Brady WT, Saidi K. J Org Chem. 1979;44:733. [Google Scholar]
- 54.(a) Ruden RA. J Org Chem. 1974;39:3607. [Google Scholar]; (b) Shchukovskaya LL, Pal’chik RI, Lazarev AN. Dokl Akad Nauk SSSR. 1965;164:357. [Google Scholar]
- 55.Hyatt JA, Raynolds PW. Org React. 1994;45:159. [Google Scholar]
- 56.Nelson SG, Wan Z, Peelen TJ, Spencer KL. Tetrahedron Lett. 1999;40:6535. [Google Scholar]
- 57.(a) Dymock BW, Kocienski PJ, Pons JM. Synthesis. 1998:1655. [Google Scholar]; (b) Dymock BW, Kocienski PJ, Pons JM. Chem Comm. 1996:1053. [Google Scholar]; (c) Tamai T, Yoshiwara H, Someya M, Fukumoto J, Miyano S. J Chem Soc, Chem Comm. 1994:2281. [Google Scholar]
- 58.Lecea B, Arrieta A, Arrastia I, Cossio FP. J Org Chem. 1998;63:5216. doi: 10.1021/jo9804745. [DOI] [PubMed] [Google Scholar]
- 59.(a) Yang HW, Zhao C, Romo D. Tetrahedron. 1997;53:16471. [Google Scholar]; (b) Yang HW, Romo D. J Org Chem. 1998;63:1344. [Google Scholar]
- 60.Zemribo R, Romo D. Tetrahedron Lett. 1995;36:4159. [Google Scholar]
- 61.For the use of Et2AlCl and other aluminium-based lewis acids, see: Concepcion AB, Maruoka K, Yamamoto H. Tetrahedron. 1995;51:4011.
- 62.(a) Tamai T, Someya M, Fukumoto J, Miyano S. J Chem Soc, Perkin Trans I. 1994:1549. [Google Scholar]; (b) Romo D, Harrison PHM, Jenkins SI, Riddoch RW, Park K, Yang HW, Zhao C, Wright GD. Bioorg Med Chem. 1998;6:1255. doi: 10.1016/s0968-0896(98)00114-x. [DOI] [PubMed] [Google Scholar]; (c) Nelson SG, Cheung WS, Kassick AJ, Hilfiker MA. J Am Chem Soc. 2002;124:13654. doi: 10.1021/ja028019k. [DOI] [PubMed] [Google Scholar]
- 63.(a) Otera J, Ioka S, Nozaki H. J Org Chem. 1989;54:4013. [Google Scholar]; (b) Otera J, Dan-oh N, Nozaki H. J Org Chem. 1991;56:5307. [Google Scholar]; (c) Orita A, Sakamoto K, Hamada Y, Mitsutome A, Otera J. Tetrahedron. 1999;55:2899. [Google Scholar]
- 64.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307. [Google Scholar]
- 65.Rossiter BE, Katsuki T, Sharpless KB. J Am Chem Soc. 1981;103:464. [Google Scholar]
- 66.For an excellent example demonstrating the exquisite selectivity of the Katsuki-Sharpless reagent, see: Ahmed A, Hoegenauer EK, Enev VS, Hanbauer M, Kaehlig H, Öhler E, Mulzer J. J Org Chem. 2003;68:3016. doi: 10.1021/jo026743f.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for compounds 6-11, 14-28, 32, 35-42, 44-47, 49, 50, 52-55, 64-66, 70, 77-79, 82, 83, 86-88 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.