

Abstract
UV/VIS absorbance measurements and associated studies of cytochrome P450 BM-3 in complex with N-palmitoylglycine (NPG) indicate that a conformational change occurs in the active site of the complex where the terminal atoms of the ligand move from a site distant from the heme iron, as seen in the low temperature crystal structure to a site proximal to the heme iron at biological temperatures. We employ replica exchange molecular dynamics simulations to study this conformational change. The population of the proximal state is found to increase with temperature in agreement with UV/VIS absorbance and NMR measurements. In addition to the conformations characterized by x-ray crystallography and computer modeling, this study shows that a new conformational state is significantly populated at room temperature. The observed increase in the population of conformations where the terminal atoms of NPG is proximal to the heme iron with increasing temperature indicates that the proximal state is stabilized by conformational entropy. A proposal for the origin of this entropic stabilization is provided based on the structure of the newly identified state. We use the temperature weighted histogram (T-WHAM) method to characterize the transition state regions of the conformational ensemble and propose a mechanism of interconversion between these low free energy conformational states.
Introduction
The cytochrome P450 enzymes catalyze the oxidation of a wide variety of hydrophobic substrates.1 P450 enzymes are ubiquitous; they are found in bacteria, yeast, fungi, worms, plants, fish and mammals. In humans it is found in the liver and is important in cellular “housekeeping” processes including the metabolism of pharmaceutical agents and detoxification.1 P450 enzymes are thus important in the study of drug metabolism and toxicity.
The mechanism of catalysis by P450 is centered on the iron of the heme group.1,2 The iron is hexa-coordinated, one of the co-ordination sites being occupied by a water molecule. On binding to the substrate, the substrate displaces the water molecule bound to the heme iron,3 changing the coordination of the iron and the spin state from +1/2 to +5/2. The spin state change is accompanied by a shift in the UV/VIS spectral band from 418 nm to 390 nm.1,4 The change in spin state modulates the redox potential of the heme allowing reduction of the heme iron from ferric to ferrous.1 Oxygen binding then proceeds and the steps towards the oxidation of the substrate are initiated. Crystal structures of P450 enzymes including mammalian P450 2C5,5,6 2C97 and bacterial P450cam8–11 and P450eryF12,13 bound to different substrates show that the respective substrates bind proximal to the iron of the heme,, understandably considering the above mechanism. However the crystal structures of many P450 enzyme-substrate complexes such as, P450 BM-3 bound to N-palmitoylglycine (NPG),14 mammalian CYP2C9 bound to warfarin,15 CYP3A4 with progesterone,16 and CYP2C8 with palmitic acid17 show the substrate bound distant to the iron in a position that, based on the above mechanism, is evidently unproductive for chemistry. The low temperature x-ray crystal structure of P450 BM-3 bound to N-palmitoylglycine (NPG) shows the substrate atoms where hydroxylation occurs, to be 7.5 Å away from the iron14 and a water molecule is present at the sixth coordination site of the iron. Jovanovic et al.18,19 have recently proposed that the structure of the P450 BM-3/NPG complex depends on temperature and that at biologically relevant temperatures the ligand moves from a position distant from the heme iron, as seen in the low temperature x-ray crystal structure, into a position proximal to the iron, leading to the displacement of the iron coordinated water molecule and the initiation of the enzymatic mechanism described above.
Jovanovic et al. based their conclusion on the following observations18,19 : 1) at low temperatures UV/VIS absorbance and NMR measurements indicate that the enzyme is largely in the low spin state, consistent with the distant siting of the ligand and the presence of the water molecule bound to the iron, as in the low temperature x-ray crystal structure14 and 2) at biologically relevant temperature. UV/VIS absorbance and NMR measurements indicate that the enzyme is almost completely in the high spin state, which would occur if the ligand has moved proximal to the heme iron and has displaced the coordinated water molecule.18,19 Solid state NMR and magic angle spinning experiments18 also show a change in conformation localized in the active site at temperatures between −30 °C and room temperature. Using a novel induced fit docking protocol20 based on the PLOP (protein local optimization program) methodology for high resolution protein structure prediction,21 a low energy conformation with the ligand proximal to the iron has been proposed.18 The shift of ligand position is made possible by a conformational change involving mainly the side chain of Phe87, which is interposed between the ligand and the heme group in the low temperature x-ray crystal structure (see Figure 1a),14 whereas it assumes a different rotameric state that allows the hydrocarbon tail of the substrate to move closer to the heme iron (see Figure 1b) at ambient temperature. The potential energy of the proposed structure with ligand proximal to the heme was predicted to be only 5.8 kcal/mol above that of the x-ray structure.18
Figure 1.
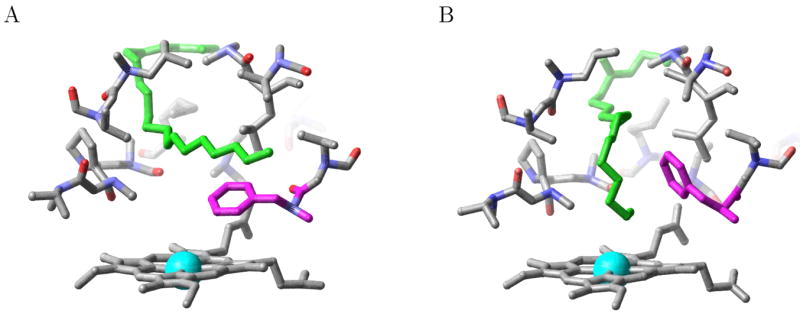
The active site of the P450 BM-3/NPG complex in (a) the low temperature x-ray conformation (PDB id 1jpz) representative of distal state where the NPG (shown in green) is distant from the heme iron, with Phe87 (shown in magenta) interposed between NPG and heme iron (shown in blue) (b) the alternative active site of the conformation predicted by Jovanovic et al. representative of the proximal state where Phe87 has changed its rotameric state to allow NPG to approach the heme iron.
In this structure the ω–1 carbon atom, one of the hydroxylation sites of NPG (shown in Figure 2), is within 3.6 Å of the iron as compared to 7.6 Å in the low temperature crystal structure. It was thus concluded that the proposed structure is a likely model for a conformation of the P450 BM-3/NPG complex at room temperature.
Figure 2.
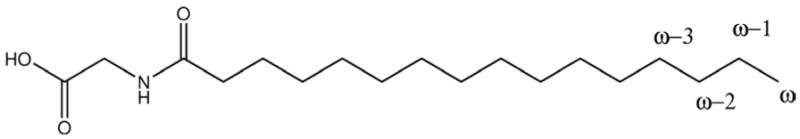
The chemical structure of N-palmitoylglycine. The terminal carbon atom that is close to the heme iron is labeled by ω. The sites which undergo hydroxylation are labeled as ω–1, ω–2 and ω–3.
In this study we use replica exchange molecular dynamics (REMD)22,23 to study the equilibrium between, the conformations of the P450 BM-3/NPG complex in which the terminal carbon atoms of NPG is distant from the heme iron as in the low temperature x-ray crystal (henceforth addressed as distal state) and conformations with the terminal carbon atoms of NPG proximal to the heme iron as in the conformation proposed by Jovanovic et al. (henceforth addressed as proximal state),18 of the P450 BM-3/NPG complex. Replica exchange is an advanced sampling algorithm that allows the efficient sampling of the rough free energy landscape of biomolecular systems. REMD involves running parallel simulations of several replicas of the system each at a different temperature and occasionally exchanging conformations according to a modified Metropolis transition probability. These conformational exchanges allow rapid interconversion between stable conformations through intermediates sufficiently populated only at higher temperatures. REMD is ideally suited for this problem not only because it improves conformational sampling but also because it yields the distribution of conformations as a function of temperature. REMD simulations have been used in the study of peptide folding,23–25 NMR structure refinement,26 and loop modeling.27 We use replica exchange molecular dynamics to calculate the relative population of the proximal and distal states of the P450 BM-3/NPG complex at room temperature. The population of the proximal state is also studied as a function of temperature. We characterize the ensemble of conformations generated by the replica exchange simulation at room temperature to identify additional conformational states. A proposal for the origin of the entropic stabilization of the proximal state over distal states at room temperature is made. To obtain information about relevant high energy transition states regions which are scarcely resolved at room temperature we use the temperature weighted histogram (T-WHAM)28 method which combines data from simulations at several temperatures and we postulate a mechanism for the conformational interconversion between the different states.
Methodology
We employ the replica exchange molecular dynamics method (REMD)22,23 to sample conformational states of the P450 BM-3/NPG complex. The replica exchange method consists of running a series of simulations at different specified temperatures. Each simulation is called a replica of the system. An exchange of conformations between the replicas at temperatures Tm and Tn is attempted periodically and it is accepted according to the following Metropolis transition probability22
| (1) |
where βm =1/kTm and Em is the current potential energy of the replica m-th replica. The REMD method gives a canonical distribution of conformations at each of these temperatures. The method has been implemented in the molecular simulations package IMPACT.29
The crystal structure of the P450 BM-3/NPG complex14 was obtained from the protein data bank30 (PDB id 1jpz) and used for the REMD simulation. To prevent unfolding of the protein at high temperatures, but to nevertheless allow enough flexibility of the protein to observe the conformational change at the active site, we employed the restraining scheme described below. The positions of the Cα atoms belonging to the 120 residues of the protein that are within 5 Å from any atom of the ligand in the x-ray structure are harmonically restrained with a force constant of 0.3 kcal/mol/Å2, which allows a 4 Å range of motion well above thermal motion at room temperature (see Supporting Information). Side chains and other backbone atoms in this region are left completely free. Atoms outside this region are fixed. By restraining only the Cα atoms of these residues we achieve conservation of secondary structure while still allowing considerable motion within these secondary structure elements. The magnitude of the force constant was chosen so as to prevent unfolding at high temperatures without significantly affecting conformational distributions at room temperature. All the atoms of Phe87, a residue known to undergo a large conformational change, as well as well as those belonging to the same loop (residues Leu86 to Asn95) are left free. The carbon atom of the carboxylate group of NPG was harmonically constrained to prevent the ligand from dissociating from the protein at high temperatures. All the other atoms of the ligand are left free. This restraining scheme is similar to that employed by Jovanovic et al.18
Before carrying out the final REMD simulation, the system was carefully annealed and equilibrated using the following scheme. Single processor molecular dynamics runs at various temperatures revealed that the conformational transition whereby the Phe87 sidechain undergoes a rotameric state change and the ligand moves proximal to the heme occurs frequently at 700 K. Keeping 700 K as the upper limit of the temperature range, and given the chosen number of replicas, we carried out a 1 ns replica exchange simulation with 24 replicas ranging from 400 K to 700 K. The structures obtained at the end of this simulation were then used to seed a replica exchange simulation of 1 ns duration with temperatures ranging from 310 K to 555 K. These two simulations at higher temperatures ensured proper equilibration and removal of correlation from the starting structures and provided seeding structures for the final production replica exchange simulation. The final production REMD simulation was carried out for 1 ns over a temperature range from 260 K to 463 K with 24 replicas, at temperatures 260, 266, 273, 280, 287, 294, 302, 310, 318, 326, 334, 342, 351, 360, 369, 379, 388, 398, 409, 419, 430, 441, 452 and 463 K. The temperature range was chosen to study the system at biologically relevant temperatures and at the same time 1) to connect with low temperature experimental information and 2) to enhance sampling at low temperature.
The REMD simulations of the P450 BM-3/NPG complex employed the OPLS-AA fixed charge all-atom force field31 using the same charge model as previously employed18 and the AGBNP32 implicit solvent model to mimic the water environment. Simulations using implicit solvent models are not as computationally intensive as those using explicit solvent models; this is particularly true for REMD simulations because many fewer replicas are needed to achieve reasonable replica exchange rates due to the smaller system size. In recent years accurate and computationally efficient implicit solvent models have been applied successfully to the study of biomolecules.33–35 The mechanism of spin state change18 involves the displacement by the substrate of a water molecule from the sixth coordination site of the iron. We have analyzed the solvent accessible surface area of representative conformations of the proximal and distal states. In agreement with the generally recognized activation mechanism, the heme iron is water accessible in the distal conformations, in which the ligand is distant from the heme iron and water inaccessible in the proximal state, where the ligand is in contact with the heme iron. The implicit solvent model we adopted, because it includes desolvation free energy terms which qualitatively take into account the loss of favorable iron-water interactions, is able to capture some of the essential energetic features that determine the equilibrium between the water-bound and substrate-bound forms of the heme iron. The molecular dynamics time step of the simulation is 1 fs. Exchange of temperatures was attempted every 100 fs. Velocity rescaling was used with a temperature relaxation time of 0.4 ps. The replica exchange acceptance ratio was 25% on average. The total simulation time, including equilibration, was 3 ns for 24 replicas for a total of 72 ns.
Population distributions (see below), were obtained by collecting the ω–1-Fe distance, the side chain torsional angles, and the potential energy of conformations from 10 replicas in the temperature range from 260 K to 357 K. These quantities are binned into histograms which are then used as the input for the temperature weighted histogram method (T-WHAM)28 to finally give population distributions at room temperature. T-WHAM28 makes it possible to resolve the population distributions corresponding to conformations of relatively high free energy, which are rarely sampled at room temperature, but are needed to determine the mechanism of interconversion between stable conformations. T-WHAM accomplishes this by exploiting information contained in the high temperature replicas where high free energy conformations are generated.
Results and Discussion
The Population of the proximal state as a function of temperature
The population distributions as a function of ω–1-Fe distance at 260 K (solid line), at 302 K (dashed line) and 318 K (dotted line) are shown in Figure 3. The ω–1-Fe distance in the low temperature x-ray crystal structure,14 which corresponds to the distal state, is 8.5 Å and in the conformation proposed by Jovanovic et al.,18 which corresponds to the proximal state, is 4.5 Å. These distances are indicated by arrows in Figure 3. As temperature increases, the population corresponding to the proximal state increases and the population of distal state decreases. At 260 K most of the population is concentrated in a peak corresponding to the distal state (see Figure 3). As the temperature is increased to 302 K (see Figure 3), a new population peak corresponding to the proximal state emerges.18,19 At 302 K the proximal state constitutes 70% of the total population and the distal state the remaining 30% population (see below). Thus at biologically relevant temperatures the P450 BM-3/NPG complex exists in an equilibrium between proximal and distal states. A free energy barrier of roughly 1.2 kcal/mol separates the two states (not shown here), which is small enough to allow relatively rapid interconversion at room temperature. As temperature is increased beyond 318 K most of the population of the complex shifts to the proximal state (see Figure 3).
Figure 3.
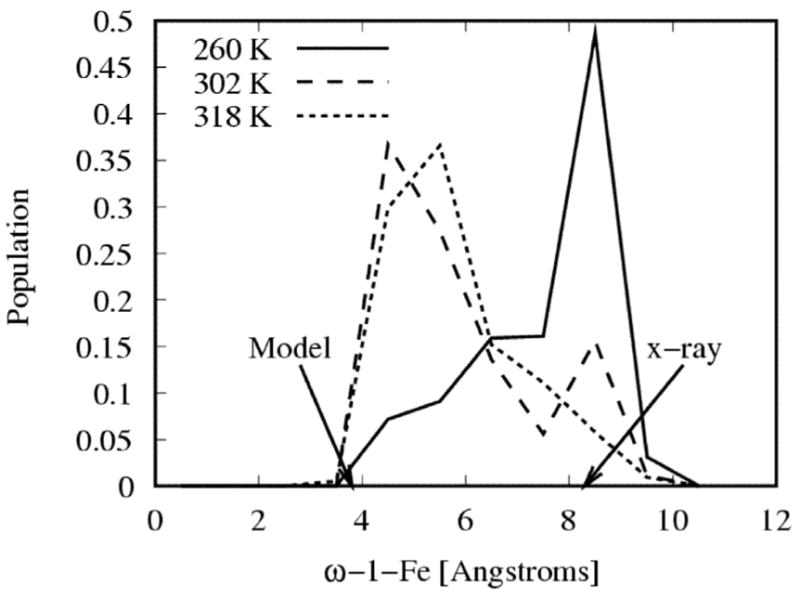
The population distribution of conformations of P450 BM-3/NPG complex as a function of ω–1-Fe distance at temperatures 260 K (solid line), 302 K (dashed line) and 318 K (dotted line). The arrows indicate the values of the ω–1-Fe distance in the x-ray crystal structure (1jpz) and the conformation modeled by Jovanovic et al. At 260 K most of the population is concentrated in the peak corresponding to the distal state. At 302 K population peaks corresponding to both proximal and distal states are seen. At 318 K the proximal state is the most populated.
By defining the proximal state to be all conformations with ω–1-Fe distances less than 6.5 Å we obtain the population of the proximal state as a function of temperature, by integration of the population distribution in Figure 3 over the ω–1-Fe distance at each temperature of the REMD simulation (see Figure 4). The population of the proximal state is 32% at 260 K, increases with temperature (see Figure 3) and finally plateaus at 318 K with 90% of the population in the proximal state. Both proximal and distal states exist at all temperatures. Rather than a sharp conformational transition from distal to proximal state at a specific transition temperature, a shift in population from distal to proximal state occurs gradually with increasing temperature. The increase in population of the proximal state with increasing temperature indicates (see Figure 4) that the proximal state is stabilized by conformational entropy. Our simulation results are in agreement with reported observations. The low temperature x-ray crystal structure14 shows the ligand is distal to the heme. UV/VIS absorbance and solid state NMR measurements of cytochrome P450 BM-318,19 show that the low spin species (corresponding to distal state) is predominant at low temperatures and the high spin species (corresponding to proximal state) predominates at room temperature. Our predicted midpoint of the transition from the distal to the proximal state (see Figure 4) is 268 K, ~20 degrees higher than the experimentally observed transition temperature from the low spin to the high spin species.19
Figure 4.
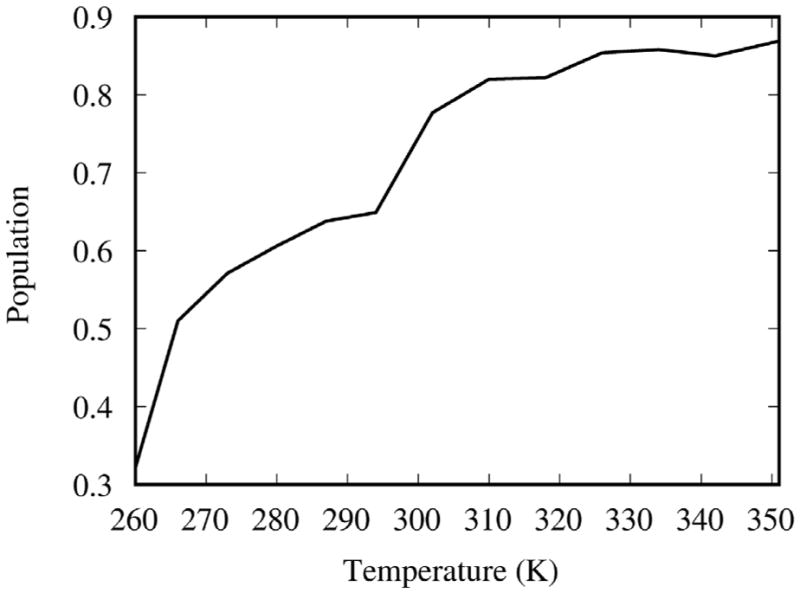
The population as a function of temperature p(T) corresponding to the conformations in which ligand is proximal to the heme iron (obtained by integration of the population distribution from Figure 2 along the ω–1-Fe distance coordinate between 0 Å to 6.5 Å ). The proximal state population increases monotonically with temperature indicating that the proximal state is stabilized by conformational entropy at temperatures greater than at least 268 K. This is borne out by the expression for the conformational entropy difference between the proximal and the distal states, ΔS = k ln[p/(1−p)]+{kT/[p(1−p)]}∂p/∂p, where the second term is positive and the first term is positive for T >268 K (p(T) >1/2).
Structural analysis of the ensemble of conformations at ambient temperature
The conformational change in the active site of the P450 BM-3/NPG complex involves primarily NPG and the active site residue Phe87. We show in Figure 5a the population distribution at 302 K as a function of the ω–1-Fe distance and the χ1 side chain torsional angle of Phe87 and in Figure 5b, the population distribution as a function of the ω–1-Fe distance and the χ2 side chain torsional angle of Phe87. In these plots the coordinates corresponding to the x-ray crystal conformation14 is marked by a cross and the coordinates corresponding to the high temperature conformation proposed by Jovanovic et al.18 is marked by a circle. Three distinct states can be identified in Figures 5a and 5b. The first state (20% population) identified by the population peak centered at (ω–1-Fe =8.0 Å, χ1 =190°) in Figure 5a and at (ω–1-Fe = 8.0 Å, χ2 = −60°) in Figure 5b, corresponds to the distal state, which also includes the x-ray crystal conformation. The second state (4% population) identified by the population peak centered at (ω–1-Fe=5.0 Å, χ1 =170°) in Figure 5a and at (ω–1-Fe=5.0 Å, χ2 =30°) in Figure 4b, contributes to the proximal state and includes the previously proposed proximal conformation.18
Figure 5.
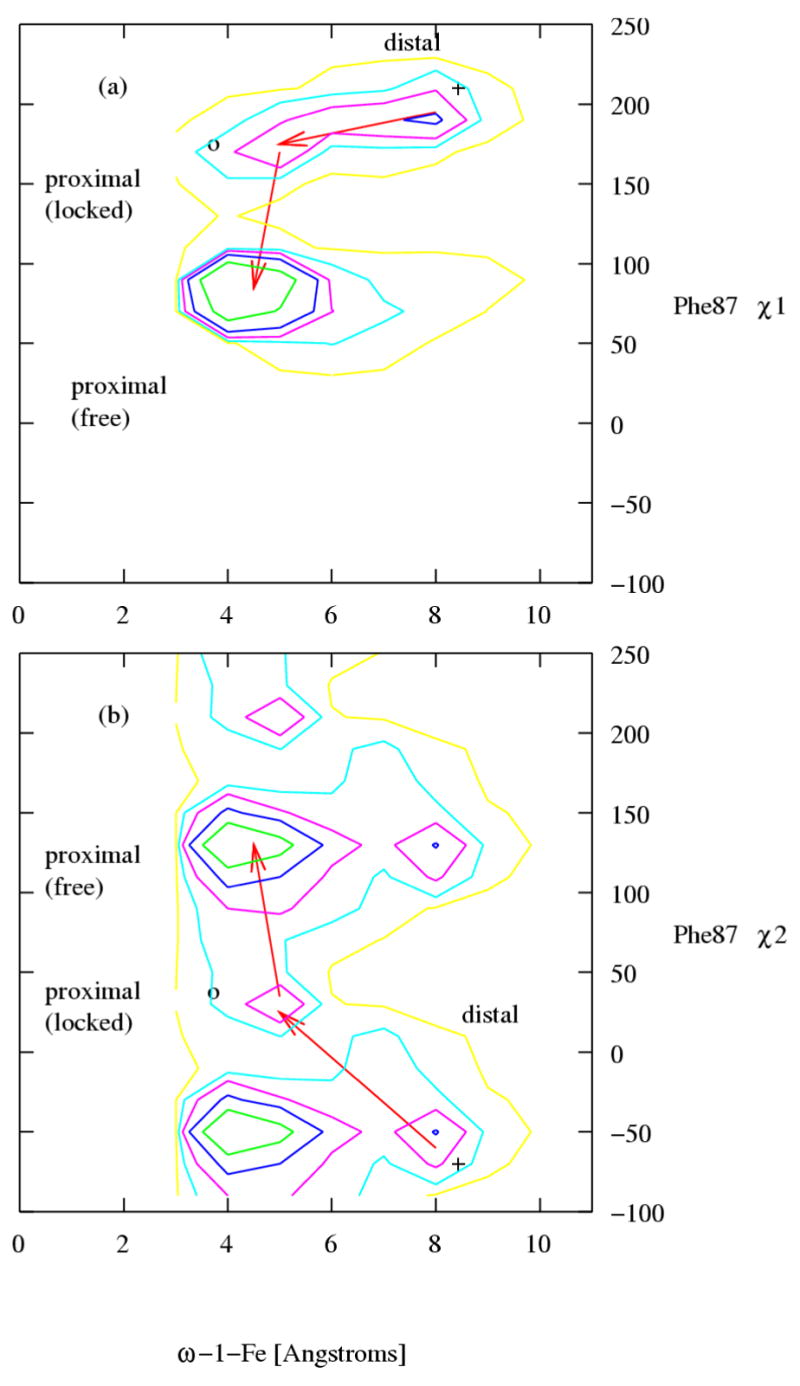
The population distributions at 302 K of P450 BM-3/NPG conformations as a function of (a) the ω–1-Fe distance and the χ1 angle of Phe87 and (b) the ω–1-Fe distance and the χ2 angle of Phe87. The coordinates corresponding to the x-ray crystal conformation and the proximal state conformation predicted by Jovanovic et al. are indicated by a cross and a circle respectively. The distal state, the proximal locked ligand and proximal free ligand states are indicated. Contours are drawn at 1×10−3, 1×10−2, 2.5×10−2, 5×10−2 and 1×10−1 fractional population.
Besides these two states corresponding to previously identified conformations, a new conformational state is observed. This conformational state, which is the most populated of the three (74 % population), is centred at (ω–1-Fe=4.5 Å, χ1=85°) in Figure 5a and at (ω–1-Fe=4.5 Å, χ2=135°) in Figure 5b. The proximal state primarily consists of conformations from this newly identified state.
A representative conformation of this newly identified state is shown in Figure 6. The Phe87 side chain (in magenta) assumes a rotameric state that, as in the conformation predicted by Jovanovic et al. does not block the terminal carbon atoms of the ligand from approaching the heme iron14 (see Figure 1a). However, unlike the conformation proposed by Jovanovic et al. (see Figure 1b), in this conformation the Phe87 sidechain does not form close contacts with the terminal carbon atoms of the ligand (compare to Figure 1b). Instead, Phe87 assumes a rotameric state (see Figure 6) so as to allow unhindered motion of the terminal Carbon atoms of the ligand towards and away from the heme, unlike in the structure modeled by Jovanovic et al. (see Figure 1b), in which the close contacts Phe87 forms with the terminal atoms of the ligand prevents the latter from moving away from the heme. The state corresponding to the conformation proposed by Jovanovic et al.18 is henceforth referred to as “locked ligand” proximal state and the newly identified state is instead referred to as the “free ligand” proximal state. The larger conformational flexibility in the free ligand proximal state relative to the locked ligand proximal and distal states is evidenced by the widths of the population distributions shown in figures 5a and 5b. The range of ω–1-Fe distances measured by the extent of the region with fractional population greater than 2.5 ×10−3 (contained in the red contours in figures 5a and 5b ), in the free ligand state (4 Å) is approximately twice that, corresponding to the ligand locked state (1.5 Å) and corresponding to the distal state (2 Å). Similarly, the range of motion of the Phe87 side chain, measured by the distribution of χ1 and χ2 angles, is significantly broader for the free ligand proximal state than for the locked ligand proximal or the distal states. We conclude that the conformational entropy associated with the newly identified free ligand conformational state is the origin of the observed entropic stabilization of the proximal state over the distal state, as the temperature is increased.
Figure 6.
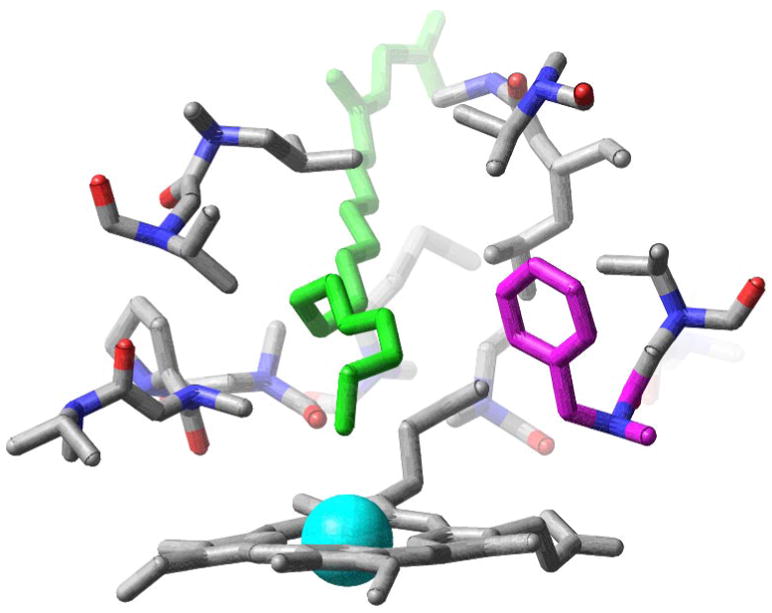
Active site of the P450 BM-3/NPG complex in a representative proximal free ligand conformation. Note that in this conformation not only has the side chain Phe87 (shown in magenta) changed its rotameric state to allow NPG (shown in green) to approach the heme iron (shown in blue), but also Phe87 is oriented so as to give NPG the ability to move more freely (as compared to the the proximal state conformation predicted by Jovanovic et. al.) to and away from the heme.
Mechanism of interconversion between states
T-WHAM is used in this study to calculate the population distribution as a function of the ω–1-Fe distance and χ1 or χ2 at room temperature (Figure 5). The T-WHAM method by combining and reweighting the data from replicas at different temperatures in a replica exchange simulation, makes it possible to obtain the population distribution at room temperature in regions rarely sampled at room temperature. In particular, transition state regions that connect the stable states cannot be resolved by simply binning the data from the walker at room temperature.
The arrows in Figures 5a and 5b trace the transition pathways between the proximal, free ligand and locked ligand states. These pathways have been identified by searching for the low free energy transition states as well as by inspection of the molecular dynamics trajectories. As indicated by the first of the two arrows (in Figure 5a and Figure 5b), the protein first goes from the distal state (see Figure 1a) to the proximal locked ligand state (see Figure 1b). The direct interconversion between distal and free ligand proximal states is not possible because of the lack of population density between distal and proximal free ligand states (see Figure 5a). The change from distal to locked ligand proximal state is achieved by a change largely, in χ2 of Phe87 of 90° (see Figure 5b) and a simultaneous decrease of the ω–1–Fe distance. During this change χ1, of Phe87 readjusts by 20° (see Figure 5a) to accommodate the change in ligand position. A free energy barrier separates these two states as seen in Figure 5b. As indicated by the second of the two arrows (in Figure 5a and Figure 5b), from the locked-ligand proximal state (see Figure 1b) the aromatic ring of Phe87 repositions, χ2 changing by 90° (see Figure 5b) and χ1 by 85° (see Figure 5a) resulting in the free ligand proximal state (see Figure 6).
In going from the locked ligand proximal state to the free ligand proximal state the system encounters a second free energy barrier. The locked ligand proximal state appears to be an intermediate in the conversion from the distal to free ligand proximal state.
Conclusions
Replica exchange molecular dynamics22 simulations were carried out using a fixed charge all-atom force field in implict solvent to study the conformational equilibrium in the active site of the cytochrome P450 BM-3/NPG complex. The enzymatic mechanism of cytochrome P450 BM-3 hydroxylation involves a spin state change of the heme iron connected to a conformational change18 in the active site of the complex, where by the terminal atoms of the ligand move from a site distant from the heme iron, as seen in the low temperature crystal structure (the distal state),14 to a site proximal to the heme iron, as in the conformation modeled by Jovanovic et al. (the proximal state).18 In agreement with recent UV/VIS and solid state NMR measurements,18,19 our simulations show that while the distal state is most populated at low temperatures, the population of the proximal state increases with temperature. At room temperature the majority of the population is in the proximal state and a minor population corresponds to the distal state. The increase in population of the proximal state with increasing temperature suggests an entropic origin for the stabilization of the proximal state.
In addition to the conformational states corresponding to previously identified conformations,18 we have characterized a new conformational substate (the free ligand proximal state), that helps in rationalizing the more favorable entropy of the proximal state. The high spin character of P450 at room temperature is primarily attributed to this state since it is the most highly populated state in our simulation. In this state the terminal carbon atoms of the ligand are positioned close to the heme iron and Phe87 (a side chain known to undergo substantial conformational change) is oriented so as to allow considerable motion of the ligand. We have quantified the conformational flexibility in the active site by calculating the distribution of conformations as a function of the ω–1-Fe distance, and the side chain torsional angles of Phe87. We find that the newly identified state exhibits significantly greater conformational flexibility than the other two states, as indicated by the spread of the ω–1-Fe distance and the torsional angles of Phe87. The greater range of motion in this state provides the basis for the observed entropic stabilization of the proximal state. The fact that this state is primarily stabilized by conformational entropy rather than potential energy suggests why it was not located in the conformational search methods employed in ref. 18. The free energy model employed to rank order structures in the induced fit protocol used in ref. 18 does not take account of protein or ligand entropy (solvent entropy is approximately included in the implicit solvation model), and hence the free ligand proximal state was ranked less favorably in these calculations than it should have been. Based on the present work, incorporation of a better estimation of conformational entropy into binding mode and binding affinity calculations of protein-ligand complexes appears to be an important research direction to pursue.
In conclusion, the cytochrome P450 BM-3/NPG complex is predicted to exist in an ensemble of conformations distributed among three distinct conformational states. Detectable populations of the complex exist in these states at all temperatures. Varying the temperature results in redistribution of the populations among these different states. T-WHAM analysis of transition states reveals that interconversion between the different states occurs relatively rapidly at room temperature and that the locked ligand proximal state is an intermediate between the free ligand proximal and distal states. Finally, the results are consistent with the experimental data of Jovanovic et. al.,18,19 with regard to the temperature at which the conformational transition takes place, the predicted entropic stabilization of the high temperature state, and the identification of the key protein residues involved in the transition.
This is the first time to our knowledge that REMD22 has been used to probe possible reaction paths for the migration of ligands within a protein using temperature dependent population distributions. The results obtained here suggest that the methodology can be used to study conformational equilibria of other P450 systems, such as P450-cam which is known to exhibit spin equilibria at low temperatures,8–11 and others such as mammalian CYP2C9 bound to warfarin,15 CYP3A4 with progesterone,16 and CYP2C8 bound to palmitic acid17 which show the substrate bound distant to the heme iron and are therefore expected to show substrate mobility and active site plasticity similar to the P450 BM-3/NPG complex studied here.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the NIH to RAF (GM40526) and RML (GM30580).
References
- 1.Montellano PRO. Cytochrome P450: Structure, Mechanism and Biochemistry. 2. Plenum Press; New York and London: 1995. [Google Scholar]
- 2.Guallar V, Friesner RA. J Am Chem Soc. 2004;126:8501. doi: 10.1021/ja036123b. [DOI] [PubMed] [Google Scholar]
- 3.Poulos TL, Raag R. FASEB J. 1992;6:674. doi: 10.1096/fasebj.6.2.1537455. [DOI] [PubMed] [Google Scholar]
- 4.Sligar SG. Biochemistry. 1976;15:5399. doi: 10.1021/bi00669a029. [DOI] [PubMed] [Google Scholar]
- 5.Wester MR, Johnson EF, Marques-Soares C, Dijols S, Dansette PM, Mansuy D, Stout CD. Biochemistry. 2003;42:9335. doi: 10.1021/bi034556l. [DOI] [PubMed] [Google Scholar]
- 6.Wester MR, Johnson EF, Marques-Soares C, Dansette PM, Mansuy D, Stout CD. Biochemistry. 2003;42:6370. doi: 10.1021/bi0273922. [DOI] [PubMed] [Google Scholar]
- 7.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:35630. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 8.Poulos TL, Finzel BC, Howard AJ. J Mol Biol. 1987;195:687. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 9.Schlichting I, Jung C, Schulze H. FEBS Letters. 1997;415:253. doi: 10.1016/s0014-5793(97)01135-6. [DOI] [PubMed] [Google Scholar]
- 10.Bell SG, Chen XH, Sowden RJ, Xu F, Williams JN, Wong LL, Rao ZH. J Am Chem Soc. 2003;125:705. doi: 10.1021/ja028460a. [DOI] [PubMed] [Google Scholar]
- 11.Raag R, Poulos TL. Biochemistry. 1991;30:2674. doi: 10.1021/bi00224a016. [DOI] [PubMed] [Google Scholar]
- 12.Cupp-Vickery JR, Poulos TL. Nature Struct Biol. 1995;2:144. doi: 10.1038/nsb0295-144. [DOI] [PubMed] [Google Scholar]
- 13.Cupp-Vickery J, Anderson R, Hatziris Z. Proc Natl Acad Sci USA. 2000;97:3050. doi: 10.1073/pnas.050406897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haines DC, Tomchick DR, Machius M, Peterson JA. Biochemistry. 2001;40:13456. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- 15.Williams PA, Cosme J, Ward A, Angova HC, Vinkovic DM, Jhoti H. Nature. 2003;424:464. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 16.Williams PA, Cosme JMVD, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Science. 2004;305(19):683. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 17.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:9497. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 18.Jovanovic T, Farid R, Friesner RA, McDermott AE. J Am Chem Soc. 2005;127:13548. doi: 10.1021/ja0524604. [DOI] [PubMed] [Google Scholar]
- 19.Jovanovic T, McDermott AE. Biochemistry. 2006 submitted. [Google Scholar]
- 20.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. Proteins: Struct Funct Bioinf. 2004;55:351. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 22.Sugita Y, Okamoto Y. Chem Phys Lett. 1999;314:141. [Google Scholar]
- 23.Felts AK, Harano Y, Gallicchio E, Levy RM. Proteins: Struct Funct Bioinf. 2004;56:310. doi: 10.1002/prot.20104. [DOI] [PubMed] [Google Scholar]
- 24.Nymeyer H, Gnanakaran S, Garcia AE. Methods Enzym. 2004;383:119. doi: 10.1016/S0076-6879(04)83006-4. [DOI] [PubMed] [Google Scholar]
- 25.Rhee YM, Pande VS. Biophys J. 2003;84:775. doi: 10.1016/S0006-3495(03)74897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Won HS, Im W, Dyson HJ, Brooks CL., III J Biomol NMR. 2005;31(1):59. doi: 10.1007/s10858-004-6056-z. [DOI] [PubMed] [Google Scholar]
- 27.Fenwick MK, Escobedo FA. Biopolymers. 2003;68:160. doi: 10.1002/bip.10291. [DOI] [PubMed] [Google Scholar]
- 28.Gallicchio E, Andrec M, Felts AK, Levy RM. J Phys Chem B. 2005;109:6722. doi: 10.1021/jp045294f. [DOI] [PubMed] [Google Scholar]
- 29.Banks JL, Beard SB, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, et al. J Comp Chem. 2005;26:1752. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucl Acid Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118(45):11225. [Google Scholar]
- 32.Gallicchio E, Levy RM. J Comp Chem. 2004;25:479. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 33.Ravindranathan KP, Gallicchio E, Levy RM. J Mol Biol. 2005;353:196. doi: 10.1016/j.jmb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Trylska J, McCammon JA, Brooks CL., III J Am Chem Soc. 2005;127:11125. doi: 10.1021/ja052639e. [DOI] [PubMed] [Google Scholar]
- 35.Vendruscolo M, Paci E, Karplus M, Dobson CM. Proc Natl Acad Sci USA. 2003;100:14817. doi: 10.1073/pnas.2036516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.