

Abstract
Specific metal-binding sites have been found in not only proteins, but also DNA and RNA molecules. Together, these metalloenzymes consist of a major portion of the enzyme family and can catalyze some of the most difficult biological functions. Designing these metalloenzymes can be both challenging and rewarding, as it can provide deeper insights into the structure and function of proteins and provide cheaper and more stable alternatives for biochemical and biotechnological applications. Towards this goal, both rational and combinatorial approaches have been used. The rational approach is good for designing metalloenzymes that are well characterized, such as heme proteins, while the combinatorial approach is better at designing those whose structures are poorly understood, such as metallo-DNA/RNAzymes. Among the rational approaches, de novo design is at its best when metal-binding sites reside in a scaffold whose structure has been designed de novo (e.g., α-helical bundles). Otherwise, design using native scaffolds can be equally effective, allowing more choices of scaffolds whose structural stability is often more resistant to multiple mutations. In addition, computational and empirical designs have both enjoyed successes. Due to the limitation in defining structural parameters for metal-binding sites, a computational approach is restricted to mostly metal-binding sites that are well defined, such as mononuclear or homonuclear centers. An empirical approach, even though it is less restrictive in the metal-binding sites to be designed, depends heavily on one’s knowledge and choice of templates and targets. An emerging approach is a combination of both computational and empirical approaches. The success of these approaches can be measured not only by threedimensional structural comparison between the designed and target enzymes, but also by the total amount of insight obtained from the design process and studies of the designed enzymes. One of the biggest advantages of designed metalloenzymes is the potential of placing two different metal-binding sites in the same protein framework for comparison. A final measure of success is how one can utilize the insight gained from the intellectual exercise to design new metalloenzymes, including those with unprecedented structures and functions. Future challenges include designing more complex metalloenzymes such as heteronuclear metal centers with strong nanomolar or lower, affinities. A key to meeting this challenge is to focus on the design of not only primary but also secondary coordination spheres using a combination of improved computer programs, experimental design and high-resolution crystallography.
Introduction
It is hard to imagine a biomolecular world without metal ions; they impart not only new colors and magnetic properties to biomolecules, but also highly tunable redox and catalytic activities that can carry out the most difficult biological functions. It is no wonder that metalloproteins consist of ~1/3 of structurally characterized proteins and ~1/2 of all proteins.1 In addition, advances in biology in the past 20 years has shown that DNA and RNA molecules are capable of catalyzing important biological reactions,2,3 including protein synthesis.4 With fewer numbers of building blocks (four bases instead of 20 natural amino acids), these catalytic DNA/RNA molecules (also called deoxyribozymes/ribozymes or DNA/RNAzymes) often require metal ions to expand and enhance their activities.5 Therefore, the study of metallo-DNA/RNAzymes have become a new frontier for bioinorganic chemists.
Fascinated by the complex, yet beautiful structures of these metalloenzymes, inorganic chemists and biochemists alike have been trying to design metalloenzymes with predicted structures and functions.6–36 In comparison to non-metalloenzyme design, the design of metalloenzymes has been recognized to be much more difficult because extra consideration is required for the different metal ions, their oxidation states, and their preferred geometry and ligand donor set. Furthermore, methods that are highly successful in designing non-metalloenzymes are often not as successful in metalloenzyme design. Despite these challenges, a number of approaches have been successfully developed. To beginners of this exciting field, such as the graduate students to whom this Forum is targeted to, it is difficult to decide which approach to use, how to recognize a successful design, and how to identify potential challenges. This article attempts to provide recent examples, mostly from the author’s own group, on how to choose a particular approach to tackle a particular project goal, to distinguish the pros and cons of each approach, and to address the issues important to advancing the field. By illustrating a number of unique new insights gained from the process, this article also conveys how rewarding metalloenzyme design can be, even though it is still a much underdeveloped field compared to other areas of inorganic chemistry and biology, such as synthetic modeling of metalloproteins using small organic molecules.
How to Design Metalloenzymes?
Rational Design or not?
In theory, rational design is always preferred. In practice, the choice may not be as clear, as it depends on how much in-depth knowledge a designer has on the structure and function of the target biomolecule. A large number of successes in rational design have occurred in the design of heme proteins,17,20–22,26,28–30,32,34–36 partly due to the fact that heme proteins are among the most well-understood metalloproteins.37 On the other hand, little is known about metal-binding sites in DNA/RNAzymes. Without knowledge, it is difficult to begin the rational design process. To complement the rational design approach, a “non-rational” or combinatorial approach called in vitro selection38–43 has been used to obtain, from a large random DNA or RNA library of up to 1015 different molecules, small groups of DNA or RNA that can bind specific metal ions (e.g., Pb(II),44–46 Cu(II),47–49 Zn(II),50,51 and Co(II)52,53), or metal-containing prosthetic groups (e.g., heme54) for different biological functions (e.g., phosphodiester transfer or porphyrin metallation). The sequence and proposed secondary structure of some of the selected metallo-DNAzymes (Figure 1) strongly suggest that it is possible to use combinatorial selection to obtain metal-specific DNA/RNAzymes.5
Figure 1.

Examples of metal-specific DNAzymes. (A) Mg2+-dependent “10–23” DNAzyme with RNA nuclease activity; B) Pb2+-dependent “8–17” DNAzyme with RNA nuclease activity; C) Cu2+-dependent DNAzyme with ligase activity; and D) DNAzyme that catalyzes porphyrin metallation. The letter “N” in the sequence represents any nucleotides capable for forming Watson-Crick base pairs. R=A or G, Y=C or T.
This combinatorial approach may also answer some of the questions that are very difficult to answer with rational design. For example, it is well known that Zn(II) and high spin Co(II) show a number of similarities, including identical charge and ionic radii. Zn(II) in proteins can often be replaced by Co(II) with few changes in metal-binding site structure and enzymatic activity. It has been very difficult to rationally design a metalloenzyme that has strong preference for Zn(II) over Co(II), or vice versa. By employing a combination of in vitro selection and a negative selection strategy to remove DNA sequences of low selectivity, two DNAzymes have been selected, one with selectivity for Zn(II) over Co(II) and the other with opposite selectivity.53 Interestingly, both of these DNAzymes share similar sequences and secondary structures, with only a four base difference (Figure 2). Further spectroscopic and structural studies of these and other metallo-DNAzymes selected with the non-rational approach may establish a new paradigm for bioinorganic chemistry in the nucleic acid world.5
Figure 2.
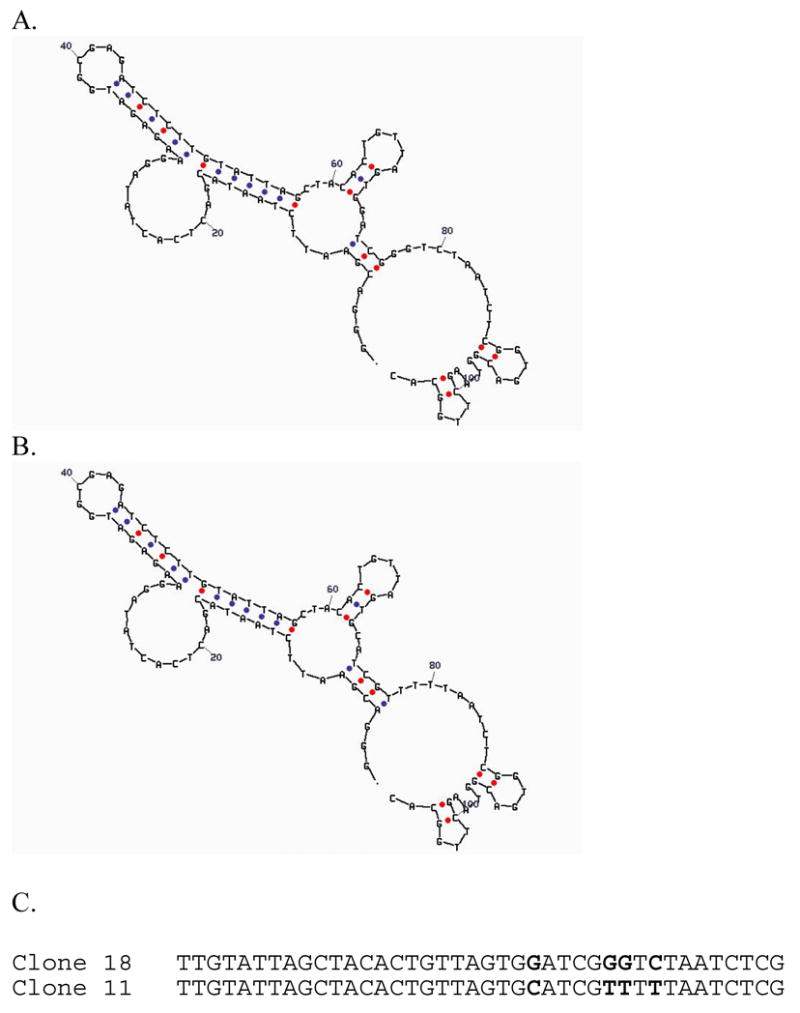
Two DNAzymes with different selectivity: Zn2+ selective (clone 18) and Co2+ selective (Clone 11). Predicted secondary structures of Clone 18 (A) and Clone 11 (B), and detailed sequence alignment in the sequence where it was randomized during the selection process (C).
While a combinatorial approach is quite useful in obtaining novel metalloenzymes, it is usually difficult to sample enough sequence variety (i.e., selection can sample up to only ~1015 different sequences out of a possible 440 or 1024 sequences for a typical DNA library with 40 randomized nucleotides). In addition, it is often tough to control the outcome of the selection, as it is sensitive to selection conditions. An effective strategy is to combine rational design with combinatorial design by using rational design to focus on a particular region of a metalloenzyme and using combinatorial design to optimize the metal-binding affinity and reactivity.
De Novo Design or Not?
If one chooses the rational design approach, he or she may face a question of whether or not to design de novo, i.e., from scratch. Metalloprotein design can be divided conceptually into two steps, design of overall structures or scaffolds, and design of metal-binding sites. De novo design encompasses both steps, represents the ultimate design goal, and is considered metalloprotein design in its purest and most challenging form. Tremendous progress has been made in the de novo design of metalloproteins and this progress has been summarized in articles in this Forum issue and elsewhere.20–22,26,32
An alternative and equally effective approach is the design of metal-binding sites into native scaffolds that are stable, easy to obtain in high yield, and well characterized.28 This approach is based on a closer look at protein sequence and structure databases. As of October 25, 2005, 196,277 protein sequences have been annotated in the UniProtKB/Swiss-Prot data bank.55 As of November 1, 2005, 31,830 solved protein structures have been deposited in the RCSB Protein Data Bank.56 Despite the large number of sequences and structures, only ~1,000 unique folds have been discovered. From these statistics, we can conclude that, rather than choosing a unique scaffold for each protein, Nature exploits only a limited number of thermodynamically stable scaffolds and uses them repeatedly to design many proteins with diverse active site structures and functions. For example, the Greek key β-barrel fold has been used by ~600 different types of proteins comprising of thousands of structures in the PDB, with diverse functions such as immunoglobins, oxidases, reductases, amylases and dismutases (Figure 3). Therefore, learning this “trick” from Nature is an important part of metalloprotein design. For instance, type 1 blue copper proteins and CuA-containing proteins such as cytochrome c oxidase and nitrous oxide reductase share a similar Greek key β-barrel scaffold.57,58 This structural homology has led to designs of the CuA site into the type 1 blue copper proteins amicyanin59 and azurin60 using a technique called loop-directed mutagenesis (vide infra).
Figure 3.
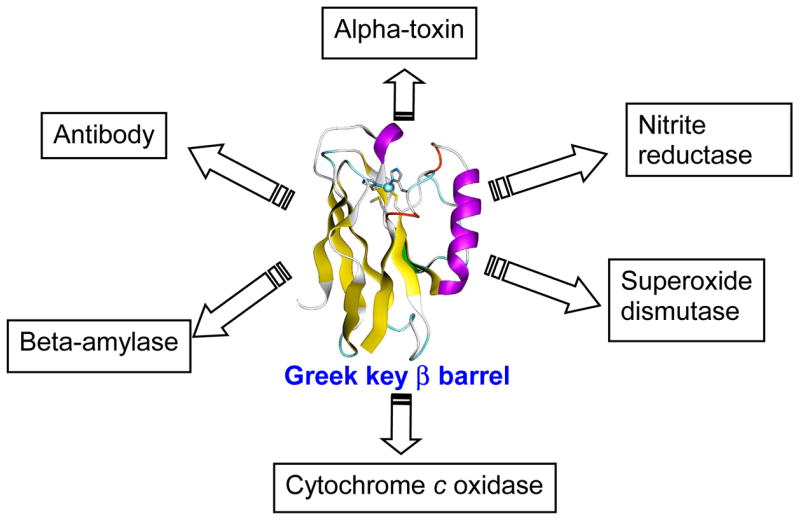
The Greek key β-barrel scaffold has been utilized by at least 600 different types of proteins with diverse active site structures and functions.
Another reason for utilizing a native protein scaffold to design metal binding sites is the beneficial properties of native scaffolds. Since protein folding remains a challenging problem, only a few scaffolds have been designed; the most well characterized scaffold is the α-helical bundle.61,62 If one is interested in designing metalloproteins with an α-helical bundle fold, the de novo design approach is extremely effective, as evidenced by much success in the design of heme proteins and dinuclear iron proteins.20–22,26,32,63 However, if one is interested in designing metalloproteins with folds that have not been designed de novo, such as the Greek key β-barrel fold mentioned above, choosing a protein with a similar scaffold is an excellent alternative. Furthermore, due to many years of evolution, native scaffolds are generally more stable or less vulnerable to amino acid residue changes and thus, are more tolerant to dramatic changes in its structure in order to create new metal-binding sites. For example, in designing a manganese peroxidase using yeast cytochrome c peroxidase, a stable and active enzyme was obtained even after 11 amino acid mutations at different locations in the protein.64
Computational or Empirical Design?
To rationally design metalloproteins, computer programs such as Dezymer65,66 and Metal Search,67,68 are available to aid in the design of metal-binding sites into proteins and exciting results have been obtained.66,69–74 However, since parameters or force fields used in defining a specific metal-binding site with a particular oxidation state, ligand and geometric preference, are highly variable and often ill-defined in general, current computer programs deal mostly with mononuclear metalbinding sites only, without much metal ion distinction, such as between Cu(II) and Zn(II). Few programs can design metal-binding sites with unusual geometry, such as the type 1 blue copper site,57,58 or complex metalloenzymes with heteronuclear metal assemblies such as the heme-manganese site in manganese peroxidase (MnP), the heme-copper site in heme-copper oxidases (HCOs), and the heme-non-heme iron site in nitric oxide reductase (NOR).37 While these programs will undoubtedly improve, a more empirical approach is needed in the meantime that relies on sequence or structural homology of proteins to guide the design efforts. The principles gained from the empirical approach can be used to improve these programs. Progress in both approaches is required for advancement of the metalloenzyme design field.
A representative example of empirical design based on structural homology is the design of manganese peroxidase (MnP) using cytochrome c peroxidase (CcP) as the scaffold.75 Both MnP and CcP are in the peroxidase family and share the same overall structure, despite low sequence homology of <15% (Figure 4A).76 They also share very similar metal-binding site structures, including the heme and proximal/distal histidines (Figure 4B). Even some of the hydrogen-bonding patterns are conserved between the two peroxidases. The major difference is the presence of a Mn(II)-binding site in MnP and Phe instead of Trp around the heme binding site (Figure 4C). While CcP oxidizes cytochrome c, MnP oxidizes Mn(II), which is then used to oxidatively degrade both lignin, the second most abundant biopolymer on earth, and aromatic pollutants. Biodegradation of lignin is a key step in the petroleum making process from trees while bioremediation of aromatic pollutants under physiological conditions can play an important role in environmental engineering.77 Therefore, it is interesting to design a Mn(II)-binding site with MnP activity into CcP, an abundant enzyme from baker’s yeast. To achieve the goal, a structural-based comparison revealed that all the ligands to the Mn(II)-binding site in MnP is either missing or occupied by another residue in CcP (Figure 4D). Those ligands have been introduced into CcP, resulting in a new protein with a Mn(II)-binding site and new MnP activity in CcP.78–80 Preliminary crystal structure characterization of a Co(II) derivative of a CcP variant containing the designed Mn(II)- site suggests that the designed site is similar to that of MnP (Figure 5).
Figure 4.
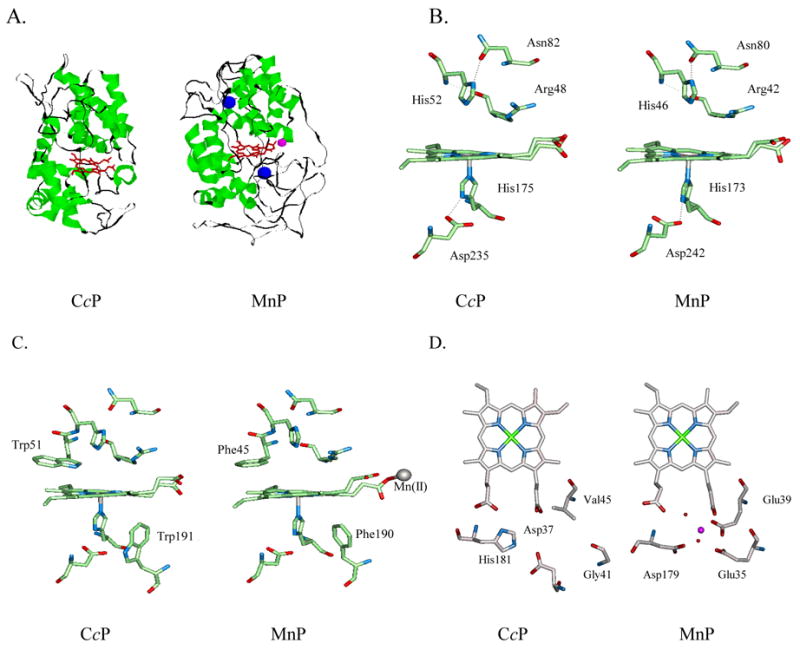
The similarities between CcP and MnP in the overall structure (A) and in the active site structure (B); the difference between CcP and MnP active site structure (C) and Mn(II)-binding site (D). The pink ball in (A) represents Mn(II) while the blue ball in (A) represents Ca(II).
Figure 5.

Structural comparison of the Co(II) derivative of a designed metal-binding site in CcP (blue) and Mn(II) center in MnP (red).
Further analysis also indicated that CcP contains two tryptophans (Trp 191 and Trp51) while MnP has phenylalanines at the corresponding positions (Figure 4C). Additional mutations of the two Trp residues to the corresponding Phe in MnP conferred even greater MnP activity.64,81 However, the two mutations do not contribute equally to increased activity. The Trp51Phe mutation resulted in a much larger increase because, in comparison to Trp191, Tpr51 is more capable of stabilizing compound II, an active species in the catalytic reaction mechanism of MnP.
The above example shows a relatively clear one-to-one correspondence between the starting template (CcP) and the target (MnP) protein. Sometimes such a correspondence may not be obvious. Designing a CuA center into type 1 blue copper proteins is a good example. Type 1 blue copper centers are classic electron transfer (ET) centers whose structure and function have been well established.57,58 CuA centers, found in cytochrome c oxidase (CcO) and nitrous oxide reductase (N2OR), are a new type of copper center and the first to contain a metal-metal bond in biology.57,58 A structure-based sequence alignment indicates that both proteins share a similar Greek β-barrel scaffold and the major difference between the two proteins lies in a loop between two β-strands in the Cterminus (Figure 6). However, there is no clear one-to-one correspondence between the residues in the loop of the two proteins that could serve as ligands for the two copper centers. For instance, the number of residues in the two loops (10 and 12 for the blue copper and CuA loop, respectively) and the number of metal ions (mononuclear and dinuclear for the blue copper and CuA center, respectively) are different for the two proteins (Figure 6). If one were to make mutations one at a time, it would be very difficult to determine which mutations and what combination of mutations to make. Instead, replacement of the entire loop sequence (i.e., loop-directed mutagenesis) of the blue copper center with the corresponding loop sequence of the CuA center was carried out, resulting in new a CuA center in amicyanin59 and azurin60,82 that closely mimics those in native CcO and N2OR.
Figure 6.

Schematic illustration of engineering the CuA center into azurin through loopdirected mutagenesis.
Metalloprotein design based on structural homology is a powerful approach and has produced a number of new designed metalloproteins. Sometimes, however, such a homology may not be apparent or non-existent. These cases may demand even deeper knowledge of the metal-binding sites in both the starting template protein and the final target protein. Type 1 blue copper centers contain a copper-thiolate center in a distorted tetrahedral geometry not normally seen in copper complexes (Figure 7A). To learn how to design a type 1 copper protein, copper-zinc superoxide dismutase (CuZnSOD), a normal type 2 copper protein, was used as a template protein to covert it into a type 1 copper protein. Even though CuZnSOD has a similar Greek key β-barrel fold as type 1 copper proteins, the metal-binding sites are located in entirely different positions in the proteins; no one-to-one residue or loop correspondence can be found. Since the type 2 copper center in CuZnSOD lacks the conserved cysteine in type 1 copper proteins, a cysteine was introduced into the metal-binding site of CuZnSOD (Figure 7B) with very different results, depending on the mutation.83 When a cysteine was introduced into the copper site of CuZnSOD by replacing one of the histidine ligands in a tetragonal geometry, a normal type 2 copper-thiolate center was obtained.84,85 On the other hand, replacing one of the histidines in the distorted tetrahedral zinc site of CuZnSOD with cysteine and replacing zinc with copper resulted in a type 1 copper-thiolate center.84,86 This result indicated that both the cysteine ligand and the distorted tetrahedral geometry are required to form the type 1 copper center.
Figure 7.

(A) Type 1 blue copper and replacement of cysteine and methionine with unnatural amino acids. (B). Type 2 copper protein CuZnSOD.
Interestingly, the type 1 copper center was also designed using a computational approach. 65,66 The program Dezymer was used to search for sites in thioredoxin, where a type 1 copper center can be created with similar geometry and ligand donor set. Based on the program design, a series of four primary designs and 32 variants were carried out.65,87,88 The most successful variant of the designed thioredoxin was able to mimic the type 1.5 copper center after a strong, exogenous ligand, azide, was introduced axially into the designed center.
The above example showed that the same protein can be designed by either a computational or an empirical approach, and each approach has its own strengths and weaknesses. While a computational approach can be used broadly to search almost any protein to design target metal sites, the empirical approach is restricted to a limited number of proteins that contain metal-binding sites with some relationship to the target site (e.g., ligand or geometry). On the other hand, because parameters or force fields in defining the type 1 copper center are not complete, designing a type 1 copper center using a computer program still remains an elusive goal, as no design of copper-containing exogenous ligand-free type 1 copper proteins has been successfully demonstrated despite a number of computational and experimental studies.65,87,88 In the meantime, design of a type 1 copper center using an empirical approach remains the only successful example.84,86 Further study of both native and empirically designed type 1 copper proteins may shed new light on how to design type 1 copper proteins and help improve a program’s chance for success.
Since both computational and empirical approaches have their own strengths and weaknesses, it is desirable to combine both approaches. An example of such a combination is the design of a CuB center into either CcP89 or myoglobin (Mb)90 to mimic the heme-copper center in CcO. CcP and Mb contain a heme center as in CcO, but lack the CuB center present in CcO. To design a CuB center into Mb at a similar location as in CcO, the heme centers of the Mb and CcO crystal structures were first overlaid on top of each other and the CuB center in CcO was used as a guide to locate sites to introduce the ligands (three histidines) necessary to create a CuB center in Mb. After careful rotation and movement of the structures on the computer, the distal histidine of Mb was identified as one of the His ligands that overlay closely with one of the His ligands in CcO (Figure 8A). From there, candidate residues for the two remaining His ligands for the new CuB center were evaluated first by visual inspection, and Leu29 and Phe43 were chosen based on how closely they occupy a similar space as the corresponding His ligands in CcO. After replacing the two residues with histidines and inserting a copper ion on the computer, the structure was minimized using a computer program. The final minimized metal-binding site structure (Mb(Leu29His/Phe43His), called CuBMb) resembles that of the CuB center in CcO (Figure 8B). This design, which involved an empirical comparison of two heme proteins based on knowledge and then computational modeling, was later confirmed experimentally by a variety of spectroscopic techniques. The same strategy was also successfully demonstrated in designing a CuB center into CcP.89
Figure 8.
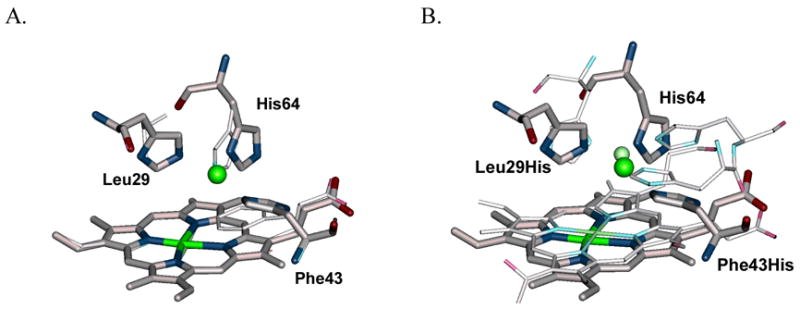
(A) Overlay of the crystal structure of WTMb (thin) and a structural model of CuBMb based on computer modeling and energy minimization (thick); (B) overlay of the crystal structure of heme-cooper center in CcO (thin) and the same structural model of CuBMb (thick) as in (A).
How to measure success?
One clear measure of success is how closely the designed metalloenzymes resembles the target native enzymes. For example, a high-resolution crystal structure of the designed CuA center in azurin showed that the active site structure of the designed CuA center is almost identical to that of the CuA center in native CcO, including not only the Cu2(SCys)2 core structure, but also the secondary coordination sphere such as the interactions with methionine and the peptide bond carbonyl oxygens that are perpendicular to the Cu2(SCys)2 plane (Figure 9). This measure, however, is not the only one, and not necessarily the best one, as it depends on the starting point. A design with the same final structure from a totally de novo peptide, although not yet achieved, is arguably more impressive than a design with a protein with a similar overall structure.
Figure 9.
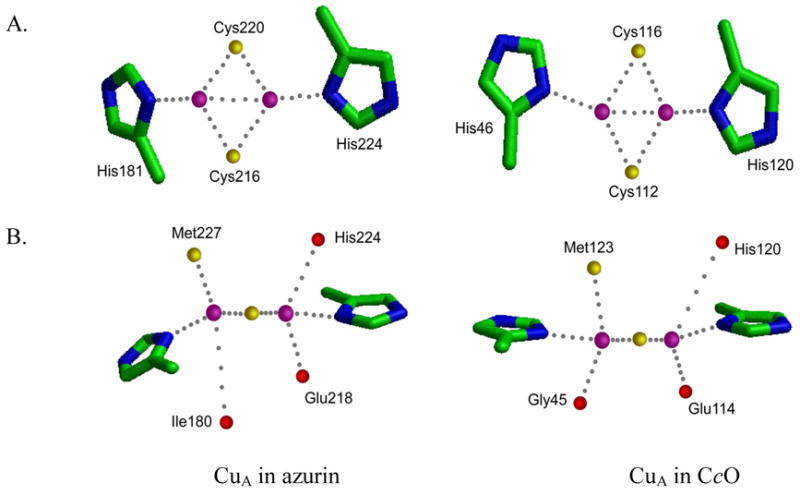
Crystal structures of CuA site in azurin and CcO as viewed from the Cu2(Scys)2 plane (A), and perpendicular to the plane (B).
Another and perhaps a more important measure of success is how much the design process or study of the designed metalloenzymes can offer new or deeper insights that are otherwise difficult to obtain by studying native enzymes. Because the designed proteins are often smaller, more stable, less complex (e.g., free from interfering chromophores as in CcO), or more amenable to spectroscopic and X-ray crystallographic studies, they offer considerable opportunities to provide newer or deeper insights. For example, a kinetic study of Cu(II) incorporation into the metal-free apo-protein of the designed CuA azurin showed that formation of the mixed-valence form [Cu(+1.5)..Cu(+1.5)] is fast and spontaneous and goes through a tetragonal intermediate preferred by the Cu(II) ion. In addition, the process requires an external reductant, without which sacrificial oxidation of free cysteines in the apo-protein may occur, reducing the yield.91 Furthermore, metal ion substitutions of the native copper ions with other metal ions suggest that the CuA center has a strong preference for an M(II)M(I) state.92,93 Finally a pH-dependent study of CuA azurin provided a rare example of a reversible pH-dependent transition between delocalized and trapped mixed valence states of a dinuclear copper center in inorganic chemistry.94 Further studies identified the C-terminal His as the site of protonation and demonstrated a dramatic increase in reduction potential when the CuA center was transformed from a delocalized to a trapped valance state through protonation. The CuA center is known to be the electron entry site in CcO. However, its role in proton-coupled electron transfer has not been clearly defined. Since the corresponding C-terminal His in CcO is located along a major electron transfer pathway from CuA to heme a, and since protonation can result in an increased reduction potential that could prevent electron transfer flow from CuA to heme a, the above results strongly indicate that CuA and the Cterminal histidine may play an important role in proton-coupled electron transfer in CcO.
Similarly, a study of the designed CuBMb clearly showed that CuB,90,95,96 heme type97 and proton or hydrogen bonding networks98 all play significant roles in CcO activity. For example, spectro-electrochemical titrations of CuBMb demonstrated that the presence of a metal ion in the designed CuB center has a significant effect on the reduction potential of heme Fe only when the two metal centers are coupled, indicating that spin coupling plays an important role in reduction potential regulation.95 In addition, the lower oxidation state of Cu(I) in the CuB center resulted in a much smaller increase (16 mV) in heme reduction potential than the higher oxidation state metal ion (e.g., Zn(II), 118 mV). Therefore, the heme reduction potential must be lowered after the first electron transfer to reduce heme Fe3+-CuB 2+ to Fe3+-CuB +. In order to raise the heme reduction potential to make the second electron transfer (i.e., reduction of Fe3+-CuB + to Fe2+-CuB +) favorable, most likely a proton or decoupling of the heme-copper center is needed in the heme copper site. These findings provide a strong argument for a thermodynamic driving force basis for redox-regulated proton transfer in heme copper oxidases.
One of the biggest advantages of designed metalloenzymes is the potential of placing two different metal-binding sites in the same protein framework for comparison. For example, both blue copper and CuA are electron transfer centers. It has been difficult to evaluate which one is a more efficient ET center, as they reside in different protein matrices and ET rate comparison is often complicated by difficulties in defining the coupling factor between the donor and acceptor. By placing the two centers in the same protein framework of azurin, the difference in coupling factor can be minimized, if not eliminated (Figure 10A), and a direct comparison can be carried out, which showed that the CuA center is a more efficient ET center, most likely due to a smaller reorganization energy.99 In addition, a study also found that the same set of mutations on the conserved methionine resulted in much less influence on the reduction potential in a CuA center than in a blue copper center (Figure 10B).100 These findings have shed new light onto the structure and function of the two ET centers. Blue copper centers have been used widely in biology to perform a variety of functions, ranging from photosynthesis to denitrification, with each redox partner having a very different reduction potential. In order to couple the ET function with those redox partners, the reduction potentials of blue copper centers should be widely varied to match that of their redox partners. A convenient way to tune such a potential is through variation in the axial ligand, from methionine in azurin (304 mV) to Gln in stellacyanin (180 mV) or Phe in laccase (790 mV vs. NHE). On the other hand, CuA centers are part of terminal oxidases such as CcO and the reduction potential difference between its redox partners is very small (~50 mV). Large variations in reduction potentials can result in reverse electron flow and loss of function. Therefore, the Cu2(SCys)2 diamond core structure is made to be more resistant to axial ligand variation. Despite this low reduction potential difference in CuA and its partners, CuA still needs to transfer electrons fast, and is built to be a more efficient ET center. Therefore, by placing two different metal-binding sites into the same protein, metalloprotein design can provide unique insights that are otherwise difficult to obtain by studying the native proteins or synthetic model compounds using organic molecules as ligands.
Figure 10.

(A) Overlay of backbone structures of type 1 blue copper azurin and designed CuA azurin; (B) effects of methionine mutations on the reduction potential of the metal site in CuA azurin and type 1 blue copper azurin.
A final measure of success is how one can utilize the insight gained from the intellectual exercise to design new metalloenzymes, including those with unprecedented structures and functions.34,101,102 Even though native metalloenzymes have the most diverse structures and functions among biomolecules, a close look indicates that they use only a limited number of natural amino acids or nucleotides (as ligands), and physiologically available metal ions and metal-containing cofactors. Introducing unnatural amino acids into metalloproteins such as azurin has allowed fine-tuning of their functional properties, such as reduction potential, linearly at almost an atomic level (Figure 7A).103–105 Such changes allow control of a single factor such as hydrophobicity, without changing other factors such as sterics. On the other hand, introducing non-native metal-containing cofactors such as MnSalen106–108 and metallocene (e.g., ferrocene109) into proteins have resulted in new biocatalysts and ET proteins that are soluble and stable in water, with controllable functional properties. For example, by placing ferrocene into the protein scaffold of azurin through bioconjugation with the conserved cysteine (Figure 11A), the reduction potential of ferrocene increased from 402 mV outside the protein, to 579 mV inside protein. This dramatic increase in the reduction potential strongly suggests that the hydrophobic encapsulation of the ferrocene induces destabilization of the higher oxidation state of the copper ion, resulting in a higher reduction potential. Further support of this conclusion comes from a study of the Phe15Cys/Cys112Gly/Met121Gly variant of azurin in which ferrocene was incorporated in the same way. This variant was constructed to place ferrocene even deeper into the protein pocket by replacing the original Cys112 to Gly, creating a new Cys bioconjugation point through the Phe15Cys mutation, and creating more room through the Met121Gly mutation (Figure 11B). Under identical conditions, the reduction potential of this new organometalloprotein increased even further to 610 mV, demonstrating the power of using the protein itself to control the functional properties of metal centers. These artificial metalloenzymes are welcome additions to the metalloenzyme family and may find a number of potential biochemical and biotechnological applications.
Figure 11.
Designed artificial organometalloproteins. (A) A blue copper azurin containing a covalently attached ferrocene; (B) a Phe15Cys/Cys112Gly/Met121Gly variant of the blue copper azurin containing a covalently attached ferrocene.
In almost all the cases mentioned above, knowledge gained from the study and design of other metalloenzymes have helped the design process. For example, to design heme enzymes, Nature has utilized different strategies such as non-covalent (Mb), single covalent-attachment (selected cytochrome c) and dual covalent-attachment (majority of cytochrome c’s). Similar strategies have been implemented in the successful design of MnSalen into Mb.106–108
The above examples showed that rational design based on native scaffolds can be powerful in designing new metalloenzymes. However, it has its own limitations. Since this rational design strategy starts with a native protein rather than a de novo designed one, structural features embedded in the starting template protein that help formation of the new target metal-binding site may not be obvious to the designer. For example, in designing a CuA center into a type 1 copper protein azurin using loop-directed mutagenesis, the work highlights the importance of the ligand loop in defining the CuA center.60,82 However, additional structural features in azurin may have helped CuA formation, such as electrostatic or hydrogen bonding interactions110 that cannot be identified with confidence. To reveal these features, one may need to use a template protein that is different from type 1 copper proteins.
What challenges are ahead?
The examples given in this article and in other articles from this Forum have clearly demonstrated that metalloenzyme design is quite fruitful and extremely rewarding. Like any young and emerging field, however, it has its own challenges. Most successes are in the design of relatively straightforward mononuclear or homonuclear metal-binding sites, with affinity often in the micromolar range for dissociation constants. To design more complex metalloenzymes with stronger affinities, one needs to focus on design of not only the primary coordination sphere (i.e., ligands that directly coordinate to the metal ions), but also the secondary coordination sphere (i.e., residues around the ligands). Without consideration of the secondary coordination sphere, the metal-binding sites may not even form, or may not have the desired functional properties. For example, in designing a cytochrome P450 using CcP as a template, replacing the proximal His in CcP with Cys as in P450, the key difference between the two proteins, was not enough, as cysteine was oxidized rapidly to cysteric acid.111 It was then recognized that a conserved Phe is present next to the Cys in P450 while a Glu is at the same position in CcP. Phe in P450 can help stabilize Cys, while the negatively charged Glu in CcP may have destabilized the Cys in the designed protein.112 To overcome this limitation, the Glu was replaced by Leu, an amino acid that is similar in structure to Glu, but can provide a similar hydrophobic environment to Cys. This change in the secondary coordination sphere resulted in a stable heme-thiolate, not only in the resting ferric state, but also in reduced ferrous state and in the presence of a strong trans ligand such as CN−.112,113
Another important element in the secondary coordination sphere is the presence of water and hydrogen bonding networks. A number of biochemical and biophysical studies have demonstrated that water and hydrogen bonding networks play a critical role in tuning the enzyme activity from one type, such as P450, to another type, such as heme oxygenase (HO).114 This factor has to be considered in metalloprotein design, such as designing a heme-copper center in myoglobin. Despite the fact that a CuB center was designed into the distal pocket of Mb that is similar to that in CcO,90 the designed protein (i.e., CuBMb) did not display oxidase activity when the oxy form of the enzyme was reduced by reductants.98 HO activity (conversion of heme to verdoheme) was shown instead. A number of control experiments carried out in the presence of catalase, ruled out poor protein stability or protein dynamics of the model protein as the main reason for the difference in reactivity, since the heme was stable and non-reactive when the reaction was carried out in the absence of copper, or in the presence of either redox inactive Zn(II) or Ag(I), or in the presence of copper and wild-type Mb.98 In addition, the use of H2O2, an oxidant with an equivalent number of electrons as O2 with two extra electrons and two extra protons, resulted in a ferryl species, a key intermediate in oxidase, but not HO reactivity.98 These results strongly suggest that the presence of extra protons or a hydrogen-bonding network is important for oxidase-like activity. It was proposed that the hydroxyl group of o-type and a-type hemes may play a role in providing the hydrogen bonding network.115,116 Replacing the b-type heme in Mb with a heme o mimic containing the extra hydroxyl group resulted in significant (19-fold) reduction of verdoheme formation.97 This work demonstrated the importance of the secondary coordination sphere to metalloprotein design. We will see many more such examples in further metalloprotein design and biomimetic modeling in general.
To meet these challenges, advances in both computational and empirical designs are required. It calls for better modeling programs with well-defined parameters/force fields for metal-binding sites that can delineate both the identity of the metal ions, not simply partial charges, and the secondary coordination sphere around the metal-binding site, including electrostatic, hydrophobicity and hydrogen bonding forces. It also demands clever empirical designs based on in-depth knowledge that can take all these elements into consideration. Critical to success is detailed structural characterizations of designed proteins, particularly high-resolution, three-dimensional crystal or NMR structures, as they will not only confirm how successful the design is, but they can also reveal surprises that can enrich our knowledge and give us information that can be applied to modeling programs for the next generation of design. Given the breath-taking advances in molecular, computational and structural biology, it should be possible in the near future to make “designer metalloenzymes” that can be custom made to bear desirable structures and functions.
Acknowledgments
I wish to thank all Lu group members whose work has been cited in the references for their dedication and hard work, and Ms. Natasha Yeung, Hee Jung Hwang, Thomas D. Pfister, and Dewain Garner for help with preparation of this paper. The Lu group work described in this paper has been generously supported by the NSF and NIH.
References
- 1.Thomson AJ, Gray HB. Curr Opin Chem Biol. 1998;2:155–158. doi: 10.1016/s1367-5931(98)80056-2. [DOI] [PubMed] [Google Scholar]
- 2.Doudna JA, Cech TR. Nature. 2002;418:222–228. doi: 10.1038/418222a. [DOI] [PubMed] [Google Scholar]
- 3.Joyce GF. Annu Rev Biochem. 2004;73:791–836. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- 4.Moore PB, Steitz TA. Nature. 2002;418:229–235. doi: 10.1038/418229a. [DOI] [PubMed] [Google Scholar]
- 5.Lu Y. Chem Eur J. 2002;8:4588–4596. doi: 10.1002/1521-3765(20021018)8:20<4588::AID-CHEM4588>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 6.Tainer JA, Roberts VA, Getzoff ED. Curr Opin Biotechnol. 1991;2:582–591. doi: 10.1016/0958-1669(91)90084-i. [DOI] [PubMed] [Google Scholar]
- 7.Tainer JA, Roberts VA, Getzoff ED. Curr Opin Biotechnol. 1992;3:378–387. doi: 10.1016/0958-1669(92)90166-g. [DOI] [PubMed] [Google Scholar]
- 8.Berg JM. Curr Opin Struct Biol. 1993;3:585–588. [Google Scholar]
- 9.Regan L. Annu Rev Biophys Biomol Struct. 1993;22:257–281. doi: 10.1146/annurev.bb.22.060193.001353. [DOI] [PubMed] [Google Scholar]
- 10.Regan L. Trends Biochem Sci. 1995;20:280–285. doi: 10.1016/s0968-0004(00)89044-1. [DOI] [PubMed] [Google Scholar]
- 11.Matthews DJ. Curr Opin Biotechnol. 1995;6:419–424. doi: 10.1016/0958-1669(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 12.Christianson DW, Fierke CA. Acc Chem Res. 1996;29:331–339. [Google Scholar]
- 13.Hellinga HW. In: Protein Eng. Cleland JL, Craik CS, editors. Wiley-Liss; New York, N. Y.: 1996. pp. 369–398. [Google Scholar]
- 14.Hellinga HW. Curr Opin Biotechnol. 1996;7:437–441. doi: 10.1016/s0958-1669(96)80121-2. [DOI] [PubMed] [Google Scholar]
- 15.Regan L. Adv Mol Cell Biol. 1997;22A:51–80. [Google Scholar]
- 16.Lu Y, Valentine JS. Curr Opin Struct Biol. 1997;7:495–500. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 17.Gibney BR, Rabanal F, Dutton PL. Curr Opin Chem Biol. 1997;1:537–542. doi: 10.1016/s1367-5931(97)80050-6. [DOI] [PubMed] [Google Scholar]
- 18.Hellinga HW. Folding Des. 1998;3:R1–R8. doi: 10.1016/S1359-0278(98)00001-7. [DOI] [PubMed] [Google Scholar]
- 19.Benson DE, Wisz MS, Hellinga HW. Curr Opin Biotechnol. 1998;9:370–376. doi: 10.1016/s0958-1669(98)80010-4. [DOI] [PubMed] [Google Scholar]
- 20.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 21.Gibney BR, Dutton PL. Adv Inorg Chem. 2001;51:409–455. [Google Scholar]
- 22.Kennedy ML, Gibney BR. Curr Opin Struct Biol. 2001;11:485–490. doi: 10.1016/s0959-440x(00)00237-2. [DOI] [PubMed] [Google Scholar]
- 23.Gilardi G, Fantuzzi A, Sadeghi SJ. Curr Opin Struct Biol. 2001;11:491–499. doi: 10.1016/s0959-440x(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 24.Baltzer L, Nilsson J. Curr Opin Biotechnol. 2001;12:355–360. doi: 10.1016/s0958-1669(00)00227-5. [DOI] [PubMed] [Google Scholar]
- 25.Xing G, DeRose VJ. Curr Opin Chem Biol. 2001;5:196–200. doi: 10.1016/s1367-5931(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 26.Lombardi A, Nastri F, Pavone V. Chem Rev. 2001;101:3165–3189. doi: 10.1021/cr000055j. [DOI] [PubMed] [Google Scholar]
- 27.Franklin SJ. Curr Opin Chem Biol. 2001;5:201–208. doi: 10.1016/s1367-5931(00)00191-5. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Berry SM, Pfister TD. Chem Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe Y. Curr Opin Chem Biol. 2002;6:208–216. doi: 10.1016/s1367-5931(02)00301-0. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi T, Hisaeda Y. Acc Chem Res. 2002;35:35–43. doi: 10.1021/ar000087t. [DOI] [PubMed] [Google Scholar]
- 31.Barker PD. Curr Opin Struct Biol. 2003;13:490–499. doi: 10.1016/s0959-440x(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 32.Reedy CJ, Gibney BR. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 33.Jantz D, Amann BT, Gatto GJ, Jr, Berg JM. Chem Rev. 2004;104:789–799. doi: 10.1021/cr020603o. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y. Curr Opin Chem Biol. 2005;9:118–126. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 35.Lu Y, Berry SM. In: Encyclopedia of Life Sciences (in press) Groups NP, editor. 2005. [Google Scholar]
- 36.Lu Y. In: Encyclopedia of Inorganic Chemistry. 2. Bruce King RE-i-C., editor. 2005. in press. [Google Scholar]
- 37.Turano P, Lu Y. In: Handbook on Metalloproteins. Bertini I, Sigel H, Sigel A, editors. Marcel Dekker, Inc; New York, NY: 2001. pp. 269–356. [Google Scholar]
- 38.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 39.Gold L, Polisky B, Uhlenbeck O, Yarus M. Annu Rev Biochem. 1995;64:763–797. doi: 10.1146/annurev.bi.64.070195.003555. [DOI] [PubMed] [Google Scholar]
- 40.Osborne SE, Ellington AD. Chem Rev. 1997;97:349–370. doi: 10.1021/cr960009c. [DOI] [PubMed] [Google Scholar]
- 41.Breaker RR. Chem Rev. 1997;97:371–390. doi: 10.1021/cr960008k. [DOI] [PubMed] [Google Scholar]
- 42.Wilson DS, Szostak JW. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 43.Joyce GF. In: The RNA World. 2. Gesteland RF, Cech TR, Atkins JF, editors. Vol. 37. Vol. 37. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1999. pp. 687–689. [Google Scholar]
- 44.Breaker RR, Joyce GF. Chem Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Lu Y. J Am Chem Soc. 2000;122:10466–10467. [Google Scholar]
- 46.Brown AK, Li J, Pavot CMB, Lu Y. Biochemistry. 2003;42:7152–7161. doi: 10.1021/bi027332w. [DOI] [PubMed] [Google Scholar]
- 47.Cuenoud B, Szostak JW. Nature. 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 48.Carmi N, Shultz LA, Breaker RR. Chem Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]
- 49.Wang W, Billen LP, Li Y. Chemistry & Biology. 2002;9:507–517. doi: 10.1016/s1074-5521(02)00127-8. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Zheng W, Kwon AH, Lu Y. Nucleic Acids Res. 2000;28:481–488. doi: 10.1093/nar/28.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santoro SW, Joyce GF, Sakthivel K, Gramatikova S, Barbas CF., III J Am Chem Soc. 2000;122:2433–2439. doi: 10.1021/ja993688s. [DOI] [PubMed] [Google Scholar]
- 52.Mei SHJ, Liu Z, Brennan JD, Li Y. J Am Chem Soc. 2003;125:412–420. doi: 10.1021/ja0281232. [DOI] [PubMed] [Google Scholar]
- 53.Bruesehoff PJ, Li J, Augustine AJ, Lu Y. Comb Chem High T Scr. 2002;5:327–335. doi: 10.2174/1386207023330264. [DOI] [PubMed] [Google Scholar]
- 54.Li Y, Sen D. Nat Struct Biol. 1996;3:743–747. doi: 10.1038/nsb0996-743. [DOI] [PubMed] [Google Scholar]
- 55.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LSL. Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vila AJ, Fernández CO. In: Handbook on Metalloproteins. Bertini I, Sigel A, Sigel H, editors. Marcel Dekker; New York, NY: 2001. pp. 813–856. [Google Scholar]
- 58.Lu Y. Biocoordination Chemistry. In: Que J, Lawrence Tolman WB, McCleverty JA, Meyer TJ, editors. Comprehensive Coordination Chemistry II: From Biology to Nanotechnology. Vol. 8. Elsevier; Oxford, UK: 2004. p. 8. [Google Scholar]
- 59.Dennison C, Vijgenboom E, de Vries S, van der Oost J, Canters GW. FEBS Lett. 1995;365:92–94. doi: 10.1016/0014-5793(95)00429-d. [DOI] [PubMed] [Google Scholar]
- 60.Hay M, Richards JH, Lu Y. Proc Natl Acad Sci U S A. 1996;93:461–464. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeGrado WF, Wasserman ZR, Lear JD. Science (Washington, D C, 1883-) 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 62.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Science (Washington, D C, 1883-) 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 63.Summa CM, Lombardi A, Lewis M, DeGrado WF. Curr Opin Struct Biol. 1999;9:500–508. doi: 10.1016/S0959-440X(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 64.Pfister TD, Gengenbach AJ, Syn S, Lu Y. Biochemistry. 2001;40:14942–14951. doi: 10.1021/bi011400h. [DOI] [PubMed] [Google Scholar]
- 65.Hellinga HW, Richards FM. J Mol Biol. 1991;222:763–785. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 66.Hellinga HW. Curr Opin Biotechnol. 1996;7:437–441. doi: 10.1016/s0958-1669(96)80121-2. [DOI] [PubMed] [Google Scholar]
- 67.Clarke ND, Yuan SM. Proteins: Struct, Funct, Genet. 1995;23:256–263. doi: 10.1002/prot.340230214. [DOI] [PubMed] [Google Scholar]
- 68.Desjarlais JR, Clarke ND. Curr Opin Struct Biol. 1998;8:471–475. doi: 10.1016/s0959-440x(98)80125-5. [DOI] [PubMed] [Google Scholar]
- 69.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, DeGrado WF, Dutton PL. Nature. 1994;368:425–432. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 70.Klemba M, Gardner KH, Marino S, Clarke ND, Regan L. Nat Struct Biol. 1995;2:368–373. doi: 10.1038/nsb0595-368. [DOI] [PubMed] [Google Scholar]
- 71.Rojas NRL, Kamtekar S, Simons CT, McLean JE, Vogel KM, Spiro TG, Farid RS, Hecht MH. Protein Sci. 1997;6:2512–2524. doi: 10.1002/pro.5560061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pinto AL, Hellinga HW, Caradonna JP. Proc Natl Acad Sci U S A. 1997;94:5562–5567. doi: 10.1073/pnas.94.11.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J Am Chem Soc. 1997;119:6195–6196. [Google Scholar]
- 74.Klemba MW, Munson M, Regan L. Proteins. 1998:313–353. [Google Scholar]
- 75.Gengenbach A, Wang X, Lu Y. Fundamentals and Catalysis of Oxidative Delignification Processes. In: Argyropoulos DS, editor. ACS Symposium Series, Vol 785. Vol. 785. American Chemical Society; Washington, D.C.: 2001. pp. 487–500. [Google Scholar]
- 76.Sundaramoorthy M, Kishi K, Gold MH, Poulos TL. J Biol Chem. 1994;269:32759–32767. [PubMed] [Google Scholar]
- 77.Gold MH, Youngs HL, Gelpke MDS. Met Ions Biol Syst. 2000;37:559–586. [PubMed] [Google Scholar]
- 78.Yeung BK, Wang X, Sigman JA, Petillo PA, Lu Y. Chem Biol. 1997;4:215–221. doi: 10.1016/s1074-5521(97)90291-x. [DOI] [PubMed] [Google Scholar]
- 79.Wilcox SK, Putnam CD, Sastry M, Blankenship J, Chazin WJ, McRee DE, Goodin DB. Biochemistry. 1998;37:16853–16862. doi: 10.1021/bi9815039. [DOI] [PubMed] [Google Scholar]
- 80.Wang X, Lu Y. Biochemistry. 1999;38:9146–9157. doi: 10.1021/bi990235r. [DOI] [PubMed] [Google Scholar]
- 81.Gengenbach A, Syn S, Wang X, Lu Y. Biochemistry. 1999;38:11425– 11432. doi: 10.1021/bi990666+. [DOI] [PubMed] [Google Scholar]
- 82.Hay MT, Ang MC, Gamelin DR, Solomon EI, Antholine WE, Ralle M, Blackburn NJ, Massey PD, Wang X, Kwon AH, Lu Y. Inorg Chem. 1998;37:191–198. [Google Scholar]
- 83.Lu Y, Roe JA, Gralla EB, Valentine JS. In: Bioinorganic Chemistry of Copper. Karlin KD, Tyeklar Z, editors. Chapman & Hall; New York: 1993. pp. 64–77. [Google Scholar]
- 84.Lu Y, Gralla EB, Roe JA, Valentine JS. J Am Chem Soc. 1992;114:3560–3562. [Google Scholar]
- 85.Lu Y, Roe JA, Bender CJ, Peisach J, Banci L, Bertini I, Gralla EB, Valentine JS. Inorg Chem. 1996;35:1692–1700. doi: 10.1021/ic9513189. [DOI] [PubMed] [Google Scholar]
- 86.Lu Y, LaCroix LB, Lowery MD, Solomon EI, Bender CJ, Peisach J, Roe JA, Gralla EB, Valentine JS. J Am Chem Soc. 1993;115:5907–5918. [Google Scholar]
- 87.Hellinga HW, Caradonna JP, Richards FM. J Mol Biol. 1991;222:787–803. doi: 10.1016/0022-2836(91)90511-4. [DOI] [PubMed] [Google Scholar]
- 88.Hellinga HW. J Am Chem Soc. 1998;120:10055–10066. [Google Scholar]
- 89.Sigman JA, Kwok BC, Gengenbach A, Lu Y. J Am Chem Soc. 1999;121:8949–8950. [Google Scholar]
- 90.Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 91.Wang X, Ang MC, Lu Y. J Am Chem Soc. 1999;121:2947–2948. [Google Scholar]
- 92.Hay MT, Milberg RM, Lu Y. J Am Chem Soc. 1996;118:11976–11977. [Google Scholar]
- 93.Hay MT, Lu Y. J Biol Inorg Chem. 2000;5:699–712. doi: 10.1007/s007750000158. [DOI] [PubMed] [Google Scholar]
- 94.Hwang HJ, Lu Y. Proc Natl Acad Sci U S A. 2004;101:12842–12847. doi: 10.1073/pnas.0403473101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao X, Yeung N, Wang Z, Guo Z, Lu Y. Biochemistry. 2005;44:1210–1214. doi: 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- 96.Zhao X, Nilges MJ, Lu Y. Biochemistry. 2005;44:6559–6564. doi: 10.1021/bi047465c. [DOI] [PubMed] [Google Scholar]
- 97.Wang N, Zhao X, Lu Y. J Am Chem Soc. 2005;127:16541–16547. doi: 10.1021/ja052659g. [DOI] [PubMed] [Google Scholar]
- 98.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Natl Acad Sci U S A. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Farver O, Lu Y, Ang MC, Pecht I. Proc Natl Acad Sci USA. 1999;96:899–902. doi: 10.1073/pnas.96.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hwang HJ, Berry SM, Nilges MJ, Lu Y. J Am Chem Soc. 2005;127:7274–7275. doi: 10.1021/ja0501114. [DOI] [PubMed] [Google Scholar]
- 101.Qi D, Tann CM, Haring D, Distefano MD. Chem Rev. 2001;101:3081–3111. doi: 10.1021/cr000059o. [DOI] [PubMed] [Google Scholar]
- 102.Thomas CM, Ward TR. Chem Soc Rev. 2005;34:337–346. doi: 10.1039/b314695m. [DOI] [PubMed] [Google Scholar]
- 103.Berry SM, Gieselman MD, Nilges MJ, Van der Donk WA, Lu Y. J Am Chem Soc. 2002;124:2084–2085. doi: 10.1021/ja0169163. [DOI] [PubMed] [Google Scholar]
- 104.Berry SM, Ralle M, Low DW, Blackburn NJ, Lu Y. J Am Chem Soc. 2003;125:8760–8768. doi: 10.1021/ja029699u. [DOI] [PubMed] [Google Scholar]
- 105.Ralle M, Berry SM, Nilges MJ, Gieselman MD, Van der Donk WA, Lu Y, Blackburn NJ. J Am Chem Soc. 2004;126:7244–7256. doi: 10.1021/ja031821h. [DOI] [PubMed] [Google Scholar]
- 106.Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Angew Chem, Int Ed. 2003;42:1005–1008. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- 107.Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. J Am Chem Soc. 2004;126:10812–10813. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 108.Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. J Am Chem Soc. 2005;127:6556–6562. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]
- 109.Hwang HJ, Carey JR, Brower ET, Gengenbach AJ, Abramite JA, Lu Y. J Am Chem Soc. 2005;127:15356–15357. doi: 10.1021/ja054983h. [DOI] [PubMed] [Google Scholar]
- 110.Hwang HJ, Nagraj N, Lu Y. Inorg Chem. 2006;45:102–107. doi: 10.1021/ic051375u. [DOI] [PubMed] [Google Scholar]
- 111.Choudhury K, Sundaramoorthy M, Hickman A, Yonetani T, Woehl E, Dunn MF, Poulos TL. J Biol Chem. 1994;269:20239–20249. [PubMed] [Google Scholar]
- 112.Sigman JA, Pond AE, Dawson JH, Lu Y. Biochemistry. 1999;38:11122–11129. doi: 10.1021/bi990815o. [DOI] [PubMed] [Google Scholar]
- 113.Perera R, Sono M, Sigman JA, Pfister TD, Lu Y, Dawson JH. Proc Natl Acad Sci U S A. 2003;100:3641–3646. doi: 10.1073/pnas.0737142100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fujii H, Zhang X, Tomita T, Ikeda-Saito M, Yoshida T. J Am Chem Soc. 2001;123:6475–6484. doi: 10.1021/ja010490a. [DOI] [PubMed] [Google Scholar]
- 115.Blomberg MRA, Siegbahn PEM, Wikstroem M. Inorg Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 116.Cukier RI. Biochim Biophys Acta. 2005;1706:134–146. doi: 10.1016/j.bbabio.2004.10.004. [DOI] [PubMed] [Google Scholar]