

Abstract
Two efficient routes for rapid assembly of the tumor-associated carbohydrate antigen Globo-H hexasaccharide 2 by the pre-activation based iterative one pot strategy are reported. The first method involves the sequential coupling of four glycosyl building blocks, leading to the desired hexasaccharide in 47% overall yield in one-pot. Although model study on constructing the challenging Gal-α-1,4-Gal linkage in Gb3 trisaccharide yielded the desired α linkage almost exclusively, similar approach to assemble the hexasaccharide led to formation of significant amount of β anomer. As an alternative, the second synthesis utilizes three components in one pot with the Gal-α-1,4-Gal linkage pre-formed, producing the desired hexasaccharide in a similar overall yield as the four component approach. Both methods demonstrate that oligosaccharides containing α and β linkages within the same molecule can be constructed in one pot via the pre-activation based approach with higher glyco-assembly efficiencies than the automated solid phase synthesis strategy. Furthermore, because glycosylations can be carried out independent of anomeric reactivities of donors, it is not necessary to differentiate anomeric reactivities of building blocks through extensive protective group adjustment for chemoselective glycosylation. This confers great flexibilities in building block design allowing matching of the donor with the acceptor leading to improved overall yield.
Introduction
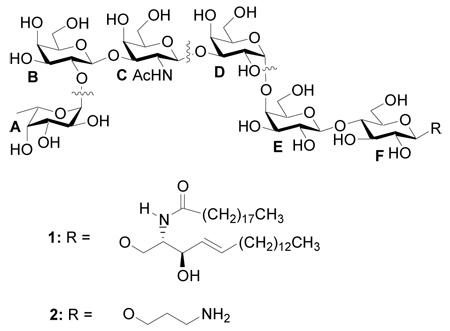
Globo-H 1, a member of the globo series glycolipids, is found over-expressed on many types of human cancer cells including breast cancer, prostate cancer, ovarian cancer and lung carcinomas.1–5 As a distinct tumor-associated antigenic marker, Globo-H has been conjugated to an immunogenic protein carrier, which is effective in generating protective antibodies against cancer cells. Such a construct has shown promising results in clinical trials as an anti-breast cancer vaccine.6–9
Due to its biological significance, Globo-H has attracted considerable attention from the synthetic community. It was first assembled by Danishefsky and coworkers using the glycal strategy,10,11 which was subsequently refined.12 Other elegant syntheses include Schmidt’s trichloroacetimidate method,13 Boon’s two-directional glycosylation,14 reactivity based one-pot method by Wong and coworkers,15–17 linear synthesis18 and automated solid phase synthesis19 by the Seeberger group, as well as syntheses of the non-reducing end fragments.20,21 However, many of the reported methods required various synthetic transformations of the oligosaccharide intermediates. Furthermore, with both α and β linkages in Globo-H, stereochemical control often became a formidable challenge.10,14,18 Therefore, a highly efficient assembly of the Globo-H hexasaccharide is still in great demand.
We have previously developed a pre-activation based iterative one-pot strategy22–24 for construction of complex oligosaccharides. One-pot synthesis refers to the glycosylation processes where multiple step glycosylation reactions can be performed in a single reaction flask without synthetic manipulation and purification of intermediate oligosaccharides, thus overcoming a major hurdle in the conventional stepwise chemical synthesis.25 For a high-yielding one-pot reaction, glycosyl donors and acceptors must be well differentiated, allowing selective donor activation and subsequent glycosylation of the acceptor. This is traditionally accomplished by using building blocks containing different types of activatable aglycons (selective activation),26,27 or carrying out glycosylations in the order of decreasing anomeric reactivities of glycosyl donors (reactivity-based armed-disarmed approach),28,29 or a combination of both strategies.30,31 Besides the integration of several glycosylation processes into a single synthetic operation to furnish the target oligosaccharide in a few hours, the advantages of our pre-activation based one-pot approach are 1) only one type of glycosyl donors, i.e., p-tolyl thioglycosides are used, thus simplifying the synthetic design; and 2) the pre-activation approach allows us to perform glycosylations without the need to follow decreasing anomeric reactivities of donors, thus granting us much freedom in choosing protective groups to match donors and acceptors.23,32,33 As part of the program towards establishing the scope of our method, we explored the synthesis of Globo-H 2 containing both α and β linkages by the pre-activation based multi-component one-pot strategy.
Results and Discussion
Retrosynthetically, we divided Globo-H into four fragments, A, BC, D and EF. Although the exclusive α linkage between A and B units can be produced according to our previous studies,24 an α/β mixture will be formed between the newly formed AB disaccharide and C due to the lack of neighboring group participation.14 In order to circumvent this difficulty, BC disaccharide will be prepared in advance with the desired β linkage. An aminopropyl spacer will be introduced to the reducing end of Globo-H 2, which can be used for future conjugation to an immunogenic carrier protein.8,34
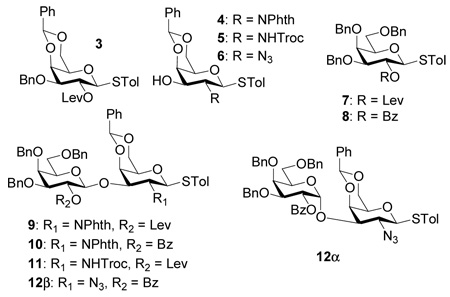
The assembly of BC disaccharide was examined first using several easily prepared glycosyl donor-acceptor pairs and the results are summarized in Table 1. Each glycosylation was carried out by pre-activating the glycosyl donor35 in mixed solvents of dichloromethane and diethyl ether using the promoter p-TolSOTf, formed in situ by reaction of p-TolSCl with AgOTf at −78 °C,36 followed by addition of the acceptor.24 Reaction of 337 and 437 was attempted without success with recovery of acceptor 4 (Table 1, entry 1). Removal of benzylidene in donor 3 and replacing the levulinoyl with benzoyl group led to small improvements with recovery of most acceptors (entries 2,3). We investigated next the effect of substituting the bulky phthalimide (Phth) on the acceptor with more sterically accessible trichloroethoxy carbonyl (Troc) and azido moieties. While the Troc group did not have a significant effect (entry 4), glycosylation of azido containing acceptor 6 by donor 8 produced disaccharide 12β (1H-NMR: δH1’ = 4.79 ppm, 3JH1’,H2’= 7.8 Hz) in 50–70% yield along with 10 – 20% 12α (1H-NMR: δH1’ = 5.57 ppm, 3JH1’,H2’= 4.2 Hz) (entry 5). The structure of 12α was confirmed by the presence of Bz carbonyl in the 13C-NMR spectrum with a chemical shift of δBz = 166.5 ppm, 1JC1’-H1’=174 Hz indicating α linakge,38 and a HMBC correlation between C1’ and H3. The erosion of stereochemical control despite the presence of participating benzoyl group on C2 of the donor is presumably due to the competing α-directing effect of ethereal solvents.22,39–44 We found that by substituting diethyl ether with small amount of acetonitrile to dissolve AgOTf, the formation of disaccharide 12α can be suppressed to negligible amount with 72% yield of the desired β disaccharide 12β (entry 6). The enhancement of stereoselectivity is most likely due to the exclusion of diethyl ether from the reaction and/or the β directing effect of acetonitrile.45 The disaccharide 12β was then converted to 13 through standard transformations with an 85% overall yield (Scheme 1a). The Troc group was introduced to direct the 1,2-trans linkage in future glycosylation.
Table 1.
Evaluation of buiding blocks for BC disaccharide synthesis.
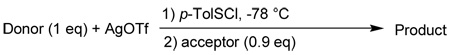 | |||
|---|---|---|---|
| Entry # | Donor | Acceptor | Product (yield) |
| 1 | 3 | 4 | - |
| 2 | 7 | 4 | 9 (10 %)a |
| 3 | 8 | 4 | 10 (30 %)a |
| 4 | 8 | 6 | 11 (30 %)a |
| 5 | 7 | 5 |
12β (50 – 70%)a 12α (10 – 20%)a |
| 6 | 8 | 6 | 12β (72 %)b |
AgOTf was added as a solution in diethyl ether to donor dissolved in dichloromethane. Ratio of diethyl ether to dichloromethane is 1:2
AgOTf was added as a solution in acetonitrile to donor dissolved in dichloromethane. Ratio of acetonitrile to dichloromethane is 1:20.
Scheme 1.

The introduction of the α-(1–4)-linkage at the DE junction of Globo-H is challenging due to the low reactivity of the axial 4-hydroxyl group of the E ring and the difficulty in stereochemical control. As a model, we explored first the formation of DEF trisaccharide, known as Gb3 or Pk trisaccharide, which is also highly enriched on the surface a variety of cancer cells and involved in many carbohydrate-receptor recognition events.46 Many types of glycosyl donors, including fluoride,11,12 chloride,46 trichloroacetimidate, 18,19,47 phosphite,48 phosphate,18,19 thioglycoside,49–51 sulfoxide52 and thioimidates53 have been examined in this reaction. The yield for formation of the Gal-α-(1–4)-Gal linkage is often not high with anomeric mixture of products.11,18,19,48,50 To test whether our glycosylation condition is suitable to construct this key linkage, we examined the glycosylation of lactoside 15 by thiogalactosyl donor 14 (Scheme 1b). Without any optimization, the desired trisaccharide 16 was obtained in 82% yield with only trace amount of the β anomer isolated. The α stereochemistry of the newly formed glycosidic linkage in 16 was confirmed by NMR (1JC1”-H1”=174 Hz).38 Encouraged by this result, we decided to test the possibility of assembling Globo-H using a four component one pot reactions with building blocks 18, 13, 19 and 15.
Pre-activation of the fucosyl donor 18 at − 78 °C with p-TolSCl/AgOTf was followed by addition of the first acceptor 13 (Scheme 2a). A sterically hindered base, 2,4,6-tri-tert-butyl-pyrimidine (TTBP)54 was added with the acceptor to neutralize trifluomethane sulfonic acid generated from glycosylation. The reaction temperature was raised to −20 °C, and the acceptor 13 was completely consumed as judged by TLC analysis. The reaction temperature was cooled back down to −78 °C, followed by sequential addition of AgOTf, p-TolSCl, the second acceptor galactoside 19, TTBP and warming up to − 20°C. After 19 completely disappeared, the reaction temperature was lowered to − 78 °C again, and the last acceptor lactoside 15, TTBP, AgOTf, p-TolSCl were added to the reaction medium. The fully protected Globo-H hexasaccharide 20α were obtained in 47% yield from the four component one-pot reactions within seven hours, which was fully characterized by 1H-NMR, 13C-NMR, gCOSY, gHMQC, gHMBC and HRMS. In addition, the anomer 20β was also produced in 23% yield.55 Both Globo-H anomers will be useful for immunological studies as demonstrated by Danishefsky and coworkers.10 It is noteworthy that thiogalactoside 19 has higher anomeric reactivity than disaccharide 13. This reversal of anomeric reactivity, i.e., a more reactive thioglycosyl acceptor is glycosylated by a less reactive thioglycosyl donor, is not possible with the reactivity based chemoselective glycosylation method.28,29,37 The pre-activation method allowed us to use the readily available building block 19 instead of going through elaborate protective group manipulations to achieve the precise anomeric reactivity.15–17
Scheme 2.

Recently, Seeberger and coworkers have reported an elegant synthesis of Globo-H hexasaccharide19 with an overall yield of 30% for glyco-assembly using the automated solid phase synthesis pioneered by their group.56 Compared to the automated method, our synthesis of the desired Globo-H hexasaccharide achieved higher glyco-assembly yield in shorted time without consuming large excess of precious glycosyl building blocks. This underscores the advantage of the pre-activation based iterative one pot oligosaccharide synthesis method.
Deprotection of the hexasaccharide 20α was performed by first removing the Troc protecting group with 1 M NaOH in THF followed by acetylation. Staudinger reduction of the azide group and subsequent catalytic hydrogenation over Pearlman’s catalyst57 gave the fully deprotected Globo-H 2 in 50% overall yield for all deprotection steps (Scheme 2b). Attempts to reduce the azide and remove benzyl groups simultaneously by hydrogenation failed even in the presence of additives such as di-tbutyl carbonate58 and hydrochloric acid.
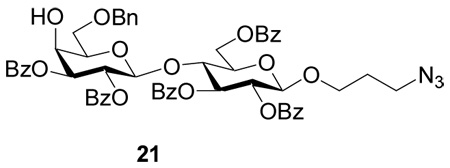
The formation of 20β in the 4+2 glycosylation is surprising in view of our model studies on Gb3. Moreover, previous syntheses of Globo-H hexasaccharides through the 4+2 coupling of tetrasaccharide donors with lactoside acceptors by the Wong group17 and the Schmidt group13 did not report any stereoisomers being formed. This unpredicted discrepancy highlights the challenge of complex oligosaccharide assembly. The generation of 20β may be due to the increased size of the tetrasaccharide donor in our 4+2 coupling as compared to donor 14. We tested next the lactoside acceptor 21 containing multiple electron withdrawing benzoyl groups in the 4+2 glycosylation reaction, as the less reactive acceptor is expected to be more selective. However, the glycosylation yield was low with large amount of acceptor recovered.
Since trisaccharide 16 was synthesized highly stereoselectively, as an alternative to the four component one-pot strategy, we examined a three component approach with the challenging Gal-α-1,4-Gal linkage preformed. The p-methoxybenzyl moiety in trisaccharide 16 was selectively removed to generate acceptor 17 in 80% yield (Scheme 1b). One-pot sequential glycosylation of fucoside 18 with disaccharide 13 and trisaccharide 17 produced hexasaccharide 20α with a 74% overall yield (Scheme 3), which was identical in all aspects to 20α prepared via the four component approach thus further confirming our stereochemical assignment. Starting from monosaccharide and disaccharide building blocks, the overall yield for 20α through the three component approach is similar to that of the four component route.
Scheme 3.

Conclusions
We have developed two routes for rapid assembly of the tumor-associated carbohydrate antigen Globo-H hexasaccharide 2 by the pre-activation based one pot strategy, demonstrating that oligosaccharides containing α and β linkages within the same molecule can be constructed in one pot. Higher glyco-assembly efficiencies have been achieved with only near stoichiometric amount of building blocks via the pre-activation based one-pot method as compared to the automated solid phase synthesis method. However, reliable stereochemical control in glycosylation still remains a challenge, which will require further developments and studies.
Experimental Section
Characterization of anomeric stereochemistry
The stereochemistries of the newly formed glycosidic linkages in Globo-H hexasaccharides and intermediates are determined by 3JH1.H2 through 1H-NMR and/or 1JC1,H1 through gHMQC 2-D NMR (without 1H decoupling). Smaller coupling constants of 3JH1.H2 (around 3 Hz) indicate α linkages and larger coupling constants 3JH1.H2 (7.2 Hz or larger) indicate β linkages. This can be further confirmed by 1JC1,H1 (~170 Hz) for α linkages and 1JC1,H1 (~160 Hz) for β linkages.38
p-Tolyl 2-azido-4,6-O-benzylidene-2-deoxy-1-thio-β-d-galactopyranoside (6)
Trichloroethoxycarbonyl chloride (7.7 mL, 57.2 mmol) was added dropwise over a period of 30 minutes at room temperature to a vigorously stirred solution of d-galactosamine hydrochloride (10 g, 46.3 mmol) and NaHCO3 (11.8 g, 139.9 mmol) in water (90 mL). The mixture was stirred for another 2 hours and then filtered to give a yellowish solid, which was dried under vacuum. The obtained crude solid (15 g) was dissolved in pyridine (50 mL) and then acetic anhydride (30 mL) was added at 0 °C over a period of 30 minutes. The mixture was stirred at room temperature under N2 overnight and then quenched with ethanol (20 mL) at 0 °C. The mixture was concentrated and the resulting residue was diluted with ethyl acetate and washed with saturated aqueous solution of NaHCO3, 10 % HCl, water, and brine. The organic phase was dried over Na2SO4, filtered and concentrated. Without separation, the obtained crude solid (21 g) and p-toluenethiol (5.76 g, 46.3 mmol) were dissolved in CH2Cl2 (60 mL) and the solution was cooled to 0 °C. Boron trifluoride etherate (17.8 mL, 160 mmol) was added dropwise at 0 °C and the mixture was stirred under N2 at room temperature for 6 h. The mixture was diluted with CH2Cl2 (400 mL) and washed with saturated aqueous solution of NaHCO3 until the pH is 7 and then dried over Na2SO4, filtered and concentrated. The obtained crude product was recrystallized from EtOAc/hexanes to afford compound p-tolyl 3,4,6-tri-O-acetyl-2-deoxy-2-N-trichloroethoxycarbonylamino-1-thio-β-d-galactopyranoside59 (S1) as white solid (20.67 g, 76% for three steps). 1H-NMR (600 MHz, CDCl3): δ 1.98, 2.04, 2.13 (3s, 9H, 3 × COCH3), 2.34 (s, 3H, SPhCH3), 3.91–3.95 (m, 2H, H-2, H-5), 4.10–4.20 (m, 2H, H-6a, H-6b), 4.73–4.80 (m, 2H, CH2CCl3), 4.84 (d, 1H, J1,2 = 10.2 Hz, H-1), 5.19 (dd, 1H, J2,3 = 10.8 Hz, J3,4 = 2.4 Hz, H-3), 5.30 (d, 1H, JNH,2 = 9.6 Hz, NH), 5.39 (d, 1H, J3,4 = 2.4 Hz, H-4), 7.10–7.16 (m, 2H), 7.40–7.46 (m, 2H); 13C-NMR (100 MHz, CDCl3): δ 20.9, 20.9, 21.4, 51.4, 62.0, 67.1, 71.2, 74.6, 74.7, 87.8, 95.7, 128.9, 129.9, 133.3, 138.7, 154.3, 170.5, 170.7, 170.8. ESI-MS [M+Na]+ C22H26NaCl3NO9S calcd 608.0, obsd 608.3. p-Tolyl 3,4,6-tri-O-acetyl-2-deoxy-2-N-trichloroethoxycarbonylamino-1-thio-β-d-galactopyranoside S1 (3 g, 5.1 mmol) was dissolved in MeOH (12 mL), AcOH (6 mL) and CH2Cl2 (6 mL). Zn powder (6 g, 92 mmol) was added slowly at 0 °C and the mixture was stirred under N2 at room temperature for 1 hour. The mixture was filtered and concentrated to dryness. The resulting residue was diluted with CH2Cl2 (200 mL) and washed with saturated aqueous solution of NaHCO3 until the pH is 7 and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (EtOAc) afforded p-tolyl 3,4,6-tri-O-acetyl-2-amino-2-deoxy-1-thio-β-d-galactopyranoside as white solid (S2) (1.8 g, 85.7%). 1H-NMR (400 MHz, CDCl3): δ 2.02, 2.05, 2.09 (3s, 9H, 3 × COCH3), 2.35 (s, 3H, SPhCH3), 3.18 (t, 1H, J = 10.0 Hz, H-2), 3.91 (t, 1H, J = 6.2 Hz, H-5), 4.11 (dd, 1H, J = 6.2, 10.8 Hz, H-6a), 4.18 (dd, 1H, J = 6.2, 10.8 Hz, H-6b), 4.49 (d, 1H, J1,2 = 10.0 Hz, H-1), 4.78 (dd, 1H, J2,3 = 10.0 Hz, J3,4 = 3.2 Hz, H-3), 5.36 (d, 1H, J3,4 = 3.2 Hz, H-4), 7.10–7.18 (m, 2H), 7.44–7.50 (m, 2H); 13C-NMR (100 MHz, CDCl3): δ 20.8, 20.9, 20.9, 21.4, 49.9, 62.0, 66.8, 74.4, 75.2, 90.4, 128.4, 129.9, 133.3, 138.6, 170.4, 170.5, 170.7. ESI-MS [M+H]+ C19H26NO7S calcd 412.1, obsd 412.1. p-Tolyl 3,4,6-tri-O-acetyl-2-amino-2-deoxy-1-thio-β-d-galactopyranoside S2 (1.8 g, 4.37 mmol, 1 equiv) was dissolved in MeOH (7.5 mL) and CH2Cl2 (7.5 mL). 1 M NaOMe (2.2 mL, 2.2 mmol) was added and the mixture was stirred at room temperature for 2 h. The mixture was neutralized by conc. HCl until the pH is around 7 and then concentrated and dried under vacuum. The resulting residue, K2CO3 (1.5g, 10.87 mmol) and catalytic amount of ZnCl2 (40 mg, 0.3 mmol) were dissolved in MeOH (12 mL) and H2O (3 mL). Freshly prepared TfN360 (13 mL in CH2Cl2, 13.1 mmol) was added and the mixture was stirred at room temperature overnight. The solvent was evaporated and the resulting residue was diluted with EtOAc (100 mL). The mixture was neutralized by conc. HCl until the pH value is 6–7 and then concentrated to dryness. Silica gel column chromatography (9:1 CH2Cl2–MeOH) afforded p-tolyl 2-azido-2-deoxy-1-thio-β-d-galactopyranoside61 (S3) as a white solid (1.2 g, 88%). 1H-NMR (600 MHz, CD3OD): δ 2.22 (s, 3H, SPhCH3), 3.37 (t, 1H, J = 9.6 Hz, H-2), 3.39–3.40 (m, 2H, H-3, H-5), 3.58–3.68 (m, 2H, H-6a, H-6b), 3.74 (d, 1H, J3,4 = 2.4 Hz, H-4), 4.34 (d, 1H, J1,2 = 9.6 Hz, H-1), 7.01–7.06 (m, 2H), 7.33–7.40 (m, 2H); 13C-NMR (100 MHz, CD3OD): δ 21.3, 62.7, 64.6, 69.8, 75.4, 80.8, 88.5, 130.7, 130.8, 130.9, 133.9, 134.0, 139.3. ESI-MS [M+Na]+ C13H17NaN3O4S calcd 334.1, obsd 334.3. The mixture of compound p-tolyl 2-azido-2-deoxy-1-thio-β-d-galactopyranoside S3 (1.2 g, 3.85 mmol), camphorsulfonic acid (0.27 g, 1.16 mmol) and benzaldehyde dimethylacetal (0.7 mL, 4.62 mmol) in toluene (20 mL) was stirred at room temperature for 1 h and then diluted with EtOAc (200 mL). The mixture was washed with saturated aqueous solution of NaHCO3, water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound 6 as white solid (1.15 g, 75%). [α]d −31.9 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.36 (s, 3H, SPhCH3), 2.62 (d, 1H, J3,3-OH = 10.2 Hz, OH), 3.45 (s, 1H, H-5), 3.47 (t, 1H, J = 10.2 Hz, H-2), 3.59 (dt, 1H, J3,4 = 3.0 Hz. J2,3 = 10.2, J3,3-OH = 10.2 Hz, H-3), 3.99 (dd, 1H, J = 1.8, 12.6 Hz, H-6a), 4.12 (d, 1H, J3,4 = 3.0 Hz, H-4), 4.35 (d, 1H, J1,2 = 9.6 Hz, H-1), 4.37 (dd, 1H, J = 1.8, 12.6 Hz, H-6b), 5.49 (s, 1H, CHPh), 7.11–7.12 (d, 2H, J = 7.8 Hz, aromatic), 7.35–7.41 (m, 5H, aromatic), 7.59–7.66 (m, 2H), 13C-NMR (150 MHz, CDCl3): δ 21.6, 62.2, 69.5, 69.96, 70.00, 73.4, 74.6, 74.7, 85.2, 101.6, 101.7, 126.5, 126.8, 128.5, 129.7, 130.1, 135.0, 137.6, 139.0. ESI-MS [M+Na]+ C20H21NaO4S calcd 422.1, obsd 422.2; Comparison of the NMR data with those reported in the literature37 confirmed the identity of 6.
p-Tolyl 2-O-benzoyl-3,4,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (8)
β-d-Galactopyranosyl pentaacetate (10 g, 25.6 mmol) was dissolved in HBr in acetic acid (30 mL, 33% w/w, 173.6 mmol). After 6 hours, the mixture was diluted with CH2Cl2 (240 mL) and poured into crushed ice in saturated NaHCO3 (600 mL). The organic phase was separated and washed again with saturated NaHCO3 until the pH was about 7 and then dried over Na2SO4, filtered and concentrated. The resulting residue, 2,6-lutidine (11.93 mL, 102.4 mmol) and Bu4NBr (3.3 g, 10.24 mmol) were dissolved in CH2Cl2 (45 mL) and dry EtOH (8.5 mL, 6 equiv.). The mixture was stirred at room temperature under N2 overnight and then concentrated and vacuum dried to afford colorless oil (10g). The obtained oil was dissolved in a mixture of CH2Cl2/MeOH (50 mL each) and 1 M solution of NaOMe (25.6 mL, 25.6 mmol) was added at room temperature. The mixture was stirred for 2 h at room temperature under N2 and then concentrated and vacuum dried. The obtained residue was dissolved in DMF (100 mL) and the solution was cooled to 0 °C. NaH (4 g, 60% NaH in mineral oil, 100 mmol) was added in portions, followed by addition of BnBr (15 mL, 125 mmol). The mixture was stirred at room temperature under N2 for 4 hours and then diluted with EtOAc (300 mL). The mixture was washed with saturated NaHCO3, water and then dried over Na2SO4, filtered and concentrated. The resulting residue (8.5 g), p-toluenethiol (6.7 g, 53.9 mmol) and HgBr2 (0.46 g, 1.28 mmol) were put into CH3CN (20 mL) and the mixture was heated at 60 °C under N2 overnight. The solvent was evaporated and then the residue was diluted with CH2Cl2 (300 mL). The mixture was washed with saturated NaHCO3, water and 10 % HCl and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound p-tolyl 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-β-d-galactopyranoside62 (S4) as white solid (6.4 g, 42% for 5 steps). 1H-NMR (600 MHz, CDCl3): δ 2.04 (s, 3H, COCH3), 2.29 (s, 3H, SPhCH3), 3.54 (dd, 1H, J2,3 = 9.6 Hz, J3,4 = 3.0 Hz, H-3), 3.59–3.67 (m, 3H, H-5, H-6a, H-6b), 3.97 (d, 1H, J3,4 = 3.0 Hz, H-4), 4.41, 4.45, 4.52, 4.56 (4d, 4H, J = 12.0 Hz, CH2Ph), 4.55 (d, 1H, J1,2 = 9.6 Hz, H-1), 4.66 (d, 1H, J = 12.0 Hz, CH2Ph), 4.93 (d, 1H, J = 12.0 Hz, CH2Ph), 5.39 (t, 1H, J = 9.6 Hz, H-2), 6.98–7.06 (m, 2H), 7.25–7.34 (m, 15H, aromatic), 7.37–7.38 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.3, 21.4, 69.0, 70.0, 72.2, 73.0, 73.8, 74.6, 77.8, 81.7, 87.3, 127.67, 127.70, 127.99, 128.03, 128.1, 128.2, 128.4, 128.6, 129.7, 130.0, 132.7, 137.8, 138.09, 138.12, 138.7, 169.7. ESI-MS [M+Na]+ C36H38NaO6S calcd 621.2, obsd 621.5. p-Tolyl 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-β-d-galactopyranoside S4 (3 g, 5 mmol) was dissolved in a mixture of CH2Cl2/MeOH (50 mL each) and 1 M NaOMe (10 mL, 10 mmol) was added at room temperature. The mixture was stirred for 2 h at room temperature under N2 and then was neutralized with Amberlite IR-120 until the pH is around 6–7. The mixture was concentrated and diluted with CH2Cl2 (300 mL) and washed with saturated NaHCO3, 10% HCl and water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound p-tolyl 3,4,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (S5) as white solid (2.56 g, 92%). 1HNMR (600 MHz, CDCl3): δ 2.29 (s, 3H, SPhCH3), 2.42 (d, 1H, J2,2-OH = 2.4 Hz, OH), 3.46 (dd, 1H, J2,3 = 9.6 Hz, J3,4 = 2.4 Hz, H-3), 3.63–3.66 (m, 3H, H-5, H-6a, H-6b), 3.95–3.98 (m, 2H, H-2, H-4), 4.44 (d, 1H, J = 12.0 Hz, CH2Ph), 4.47 (d, 1H, J1,2 = 9.6 Hz, H-1), 4.49, 4.56, 4.67, 4.73, 4.89 (5d, 5H, J = 12.0 Hz, CH2Ph), 6.98–7.05 (m, 2H), 7.25–7.35 (m, 15H, aromatic), 7.44–7.45 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.4, 68.9, 69.3, 72.6, 73.5, 73.8, 74.6, 77.8, 83.4, 89.0, 127.6, 127.9, 127.95, 128.04, 128.05, 128.2, 128.4, 128.7, 128.8, 128.9, 129.8, 133.1, 138.0, 138.1, 138.3, 138.9. ESI-MS [M+Na]+ C34H36NaO5S calcd 579.2, obsd 579.6. p-Tolyl 3,4,6-tri-O-benzyl-1-thio-β-d-galactopyranoside S5 (3.8 g, 6.8 mmol) and N,N-dimethylamino pyridine (DMAP) (0.08 g, 0.68 mmol) were dissolved in pyridine (20 mL) and then benzoyl chloride (2.38 mL, 20.5 mmol) was added. The mixture was stirred at room temperature under N2 for 4 h and then diluted with of CH2Cl2 (200 mL). The mixture was washed with saturated NaHCO3, water and 10 % HCl and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound 8 as white solid (4.2 g, 93%). [α]d +38.2 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.25 (s, 3H, SPhCH3), 3.65–3.69 (m, 4H, H-3, H-5, H-6a, H-6b), 4.03 (d, 1H, J3,4 = 2.4 Hz, H-4), 4.41, 4.45, 4.47, 4.59, 4.61 (5d, 5H, J = 12.0 Hz, CH2Ph), 4.71 (d, 1H, J1,2 = 10.2 Hz, H-1), 4.97 (d, 1H, J = 12.0 Hz, CH2Ph), 5.68 (t, 1H, J = 10.2 Hz, H-2), 6.96–6.97 (d, 2H, J = 7.8 Hz, aromatic), 7.10–7.15 (m, 5H, aromatic), 7.25–7.36 (m, 12H, aromatic), 7.41–7.42 (m, 2H, aromatic), 7.53–7.55 (m, 1H, aromatic), 8.00–8.07 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.5, 69.2, 70.8, 72.1, 73.1, 73.9, 74.7, 78.0, 81.5, 87.5, 127.8, 127.99, 128.00, 128.1, 128.2, 128.4, 128.5, 128.6, 128.7, 128.8, 129.8, 130.0, 130.2, 130.5, 133.0, 133.4, 137.9, 138.0, 138.2, 138.8, 165.6. HRMS: [M+Na]+ C41H40NaO6S calcd 683.2443, obsd 683.2421.
General procedure for single step pre-activation based glycosylation
A solution of donor (0.060 mmol) and freshly activated molecular sieve MS 4 Å (200 mg) in CH2Cl2 (2 mL) was stirred at room temperature for 30 minutes, and cooled to −78 °C, which was followed by addition of AgOTf (47 mg, 0.18 mmol) dissolved in Et2O (1 mL) without touching the wall of the flask. After 5 minutes, orange colored p-TolSCl (9.5 µL, 0.060 mmol) was added through a microsyringe. Since the reaction temperature was lower than the freezing point of p-TolSCl, p-TolSCl was added directly into the reaction mixture to prevent it from freezing on the flask wall. The characteristic yellow color of p-TolSCl in the reaction solution dissipated rapidly within a few seconds indicating depletion of p-TolSCl. After the donor was completely consumed according to TLC analysis (about 5 minutes at −78 °C), a solution of acceptor (0.060 mmol) in CH2Cl2 (0.2 mL) was slowly added dropwise via a syringe. The reaction mixture was warmed to −10 °C under stirring in 2 hours. Then the mixture was diluted with CH2Cl2 (20 mL) and filtered over Celite. The Celite was further washed with CH2Cl2 until no organic compounds were observed in the filtrate by TLC. All CH2Cl2 solutions were combined and washed twice with saturated aqueous solution of NaHCO3 (20 mL) and twice with water (10 mL). The organic layer was collected and dried over Na2SO4. After removal of the solvent, the desired disaccharide was purified from the reaction mixture via silica gel flash chromatography.
p-Tolyl 2-O-benzoyl-3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-1-thio-β-d-galactopyranoside (12β) and p-tolyl 2-O-benzoyl-3,4,6-tri-O-benzyl-α-d-galactopyranosyl-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-1-thio-β-d-galactopyranoside (12α)
Compound 12β and 12α was synthesized from donor 8 and acceptor 6 in 50–70% and 10–20% yield respectively following the general procedure of single step glycosylation. When CH3CN (100 µl) was used instead of diethyl ether to dissolve the AgOTf, the formation of disaccharide 12α can be suppressed to negligible amount with 72% yield of the desired β disaccharide 12β. For 12β: [α]d −28.5 (c = 4.3, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.24 (s, 3H, SPhCH3), 3.31 (s, 1H, H-5), 3.55–3.65 (m, 6H, H-2, H-3, H-3’,H-5’,H-6a’, H-6b’), 3.84 (d, 1H, J = 10.8 Hz, H-6a), 3.96 (d, 1H, J3’,4’ = 2.4 Hz, H-4’), 4.24–4.27 (m, 2H, H-4, H-6b), 4.30 (d, 1H, J1,2 = 9.6 Hz, H-1), 4.44–4.47 (m, 3H, CH2Ph), 4.59–4.62 (m, 2H, J = 12.0 Hz, CH2Ph), 4.79 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 4.98 (d, 1H, J = 12.0 Hz, CH2Ph), 5.37 (s, 1H, CHPh), 5.62 (t, 1H, J1’,2’ = 7.8 Hz, H-2’), 6.83–6.92 (m, 2H), 7.10–7.18 (m, 5H, aromatic), 7.23–7.41 (m, 17H, aromatic), 7.43–7.52 (m, 2H), 7.49–7.59 (m, 1H), 7.94–8.04 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.4, 59.7, 69.2, 69.3, 70.1, 71.7, 72.0, 72.6, 73.7, 74.2, 74.6, 74.9, 79.3, 80.3, 85.8, 100.8, 102.3, 126.2, 126.7, 127.8, 127.9, 128.0, 128.1, 128.2, 128.4, 128.5, 128.6, 128.7, 128.9, 129.9, 130.0, 130.2, 133.1, 134.5, 137.6, 137.9, 138.0, 138.5, 138.6, 165.5; HRMS: [M+Na]+ C54H53N3NaO10S calcd 958.3349, obsd 958.3365. gHMQC (without 1H decoupling): 1JC1’,H1’ = 160.9 Hz, 1JC1,H1 = 159.9 Hz; For 12α: [α]d +70.8 (c = 4.5, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.27 (s, 3H, SPhCH3), 3.26 (s, 1H, H-5), 3.57–3.65 (m, 3H, H-3, H-6a’, H-6b’), 3.79 (t, 1H, J = 9.6 Hz, H-2), 3.86 (d, 1H, J = 12.0 Hz, H-6a), 4.06 (s, 1H, H-4), 4.09 (d, 1H, J3’,4’ = 2.4 Hz, H-4’), 4.17 (dd, 1H, J2’,3’ = 10.8 Hz, J3’,4’ = 2.4 Hz, H-3’), 4.22–4.24 (m, 2H, H-5’, H-6b), 4.32 (d, 1H, J1,2 = 9.6 Hz, H-1), 4.47, 4.51, 4.59, 4.66, 4.68, 4.93 (6d, 6H, J = 12.0 Hz, CH2Ph), 5.16 (s, 1H, CHPh), 5.42 (dd, 1H, J1’,2’ = 3.6 Hz, J2’,3’ = 10.8 Hz, H-2’), 5.57 (d, 1H, J1’,2’ = 3.6 Hz, H-1’), 6.90–6.99 (m, 2H), 6.98–7.02 (m, 4H, aromatic), 7.12–7.44 (m, 19H, aromatic), 7.50–7.59 (m, 2H), 7.72–7.82 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.4, 59.6, 68.9, 69.2, 69.7, 70.0, 70.7, 71.9, 73.2, 73.4, 74.7, 75.0, 76.0, 76.9, 85.7, 93.1, 100.2, 126.1, 126.9, 127.78, 127.80, 127.82, 127.86, 127.87, 128.3, 128.45, 128.49, 128.51, 128.55, 129.68, 129.70, 129.9, 133.0, 134.3, 137.5, 138.2, 138.3, 138.4, 138.5, 166.5. ESI-MS [M+Na]+ C54H53N3NaO10S calcd 958.3, obsd 958.4. gHMQC (without 1H decoupling): 1JC1’,H1’ = 172.4 Hz, 1JC1,H1 = 160.1 Hz.
p-Tolyl 3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-trichloroethoxycarbonylamino)-2-deoxy-1-thio-β-d-galactopyranoside (13)
Compound 12β (1.7 g, 1.81 mmol) was dissolved in a mixture of CH2Cl2/MeOH (25 mL each) and 1 M NaOMe (5.4 mL, 5.4 mmol) was added at room temperature. The mixture was heated at reflux for 4 h under N2 and then was neutralized with conc. HCl until the pH is around 7. The mixture was concentrated and then diluted with of CH2Cl2 (200 mL). The organic phase was washed with saturated aqueous solution of NaHCO3, 10% HCl and water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3:1:1 Hexanes–EtOAc–CH2Cl2) afforded p-tolyl 3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-azido-2-deoxy-1-thio-β-d-galactopyranoside (S6) as white solid (1.5 g, quantitative). 1H-NMR (600 MHz, CDCl3): δ 2.32 (s, 3H, SPhCH3), 2.52 (s, 1H, OH), 3.35 (s, br, 1H, H-5), 3.46 (dd, 1H, J2’,3’ = 9.6 Hz, J3’,4’ = 3.0 Hz, H-3’), 3.51–3.61 (m, 4H, H-3, H-5’, H-6a’, H-6b’), 3.78 (dd, 1H, J1,2 = 10.2 Hz, H-2), 3.85 (d, 1H, J3.4 = 3.0 Hz, H-4’), 3.86–3.89 (dd, 1H, J = 1.8, 12.0 Hz, H-6a), 3.96 (t, 1H, J = 9.6 Hz, H-2’), 4.22 (d, 1H, J3’,4’ = 3.0 Hz, H-4), 4.31 (dd, 1H, J = 1.8, 12.0 Hz, H-6b), 4.35 (d, 1H, J1,2 = 10.2 Hz, H-1), 4.42, 4.46 (2d, 2H, J = 12.0 Hz, CH2Ph), 4.48 (d, 1H, J1’,2’ = 9.6 Hz, H-1’), 4.59, 4.72, 4.77, 4.90 (4d, 4H, CH2Ph), 5.45 (s, 1H, CHPh), 6.98–7.08 (m, 2H), 7.25–7.43 (m, 20H, aromatic), 7.58–7.68 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.5, 60.2, 69.3, 69.4, 70.3, 71.4, 73.3, 73.6, 73.7, 74.3, 74.9, 75.4, 80.4, 82.1, 85.7, 101.3, 105.4, 126.2, 127.0, 127.9, 128.98, 127.99, 128.2, 128.3, 128.5, 128.6, 128.7, 128.8, 129.3, 130.1, 135.0, 138.0, 138.1, 138.5, 138.6, 138.9; HRMS: [M+Na]+ C47H49N3NaO9S calcd 854.3087, obsd 854.3085. p-Tolyl 3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-azido-2-deoxy-1-thio-β-d-galactopyranoside S6 (1.3 g, 1.56 mmol), 1,3-propanedithiol (1.57 mL, 15.6 mmol) and Et3N (1.10 mL, 15.6 mmol) were dissolved in a mixture of CH2Cl2/MeOH (10 mL each). The mixture was heated at reflux overnight under N2 and then concentrated. The resulting residue was diluted with CH2Cl2 (200 mL) and then washed with saturated aqueous solution of NaHCO3 and water, dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (20:1 CH2Cl2–MeOH) afforded p-tolyl 3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-amino-2-deoxy-1-thio-β-d-galactopyranoside (S7) as white solid (1.07 g, 85%). 1H-NMR (600 MHz, CDCl3): δ 2.32 (s, 4H, SPhCH3, OH), 3.25 (t, 1H, J = 9.6 Hz, H-2), 3.37 (s, 1H, H-5), 3.37–3.39 (dd, 1H, J2’,3’ = 9.6 Hz, J3’,4’ = 3.0 Hz, H-3’), 3.53 (dd, 1H, J2,3 = 10.2 Hz, J3,4 = 3.0 Hz, H-3), 3.55–3.59 (m, 3H, H-5’, H-6a’, H-6b’), 3.86 (d, 1H, J3’,4’ = 3.0 Hz, H-4’), 3.86 (d, 1H, J = 12.0 Hz, H-6a), 3.95 (t, 1H, J = 9.6 Hz, H-2’), 4.16 (d, 1H, J3.4 = 3.0 Hz, H-4), 4.29 (d, 1H, J = 12.0 Hz, H-6b), 4.35 (d, 1H, J1,2 = 9.6 Hz, H-1), 4.43–4.44 (m, 3H, J1’,2’ = 10.2 Hz, H-1’, CH2Ph), 4.57, 4.65, 4.68, 4.86 (4d, 4H, J = 12.0 Hz, CH2Ph), 5.43 (s, 1H, CHPh), 6.95–7.08 (m, 2H), 7.23–7.42 (m, 20H, aromatic), 7.50–7.58 (m, 2H); 13C-NMR (100 MHz, CDCl3): δ 21.4, 49.7, 69.0, 69.6, 70.3, 71.5, 72.7, 73.0, 73.7, 74.2, 74.7, 75.4, 82.4, 83.8, 88.4, 101.1, 106.1, 126.6, 126.9, 127.79, 127.83, 127.9, 128.0, 128.2, 128.4, 128.5, 128.66, 128.68, 129.1, 129.9, 134.3, 138.08, 138.14, 138.3, 138.5, 138.6. HRMS: [M+Na]+ C47H51NNaO9S calcd 828.3182, obsd 828.3182. p-Tolyl 3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-amino-2-deoxy-1-thio-β-d-galactopyranoside S7 (1.06 g, 1.31 mmol) and solid NaHCO3 (0.22 g, 2.62 mmol) were put into THF (16 mL) and then TrocCl (0.214 mL, 1.57 mmol) was added. The mixture was stirred at room temperature under N2 for 4 hours and filtrated. The filtrate was concentrated and then diluted with CH2Cl2 (100 mL). The mixture was washed with water and brine, dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound 13 as white solid (1.2 g, 93%); [α]d −13.6 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.32 (s, 3H, SPhCH3), 3.34 (d, 1H, J2’,3’ = 7.8 Hz, J3’,4’ = 3.0 Hz, H-3’), 3.40 (s, 1H, H-5), 3.48–3.61 (m, 3H, H-5’, H-6a’, H-6b’), 3.65 (dd, 1H, J1,2 = 10.2 Hz, JNH,2 = 7.2 Hz, H-2), 3.82 (d, 1H, J3’,4’ = 3.0 Hz, H-4’), 3.87 (d, 1H, J = 12.0 Hz, H-6a), 3.92 (t, 1H, J = 7.8 Hz, H-2’), 4.24–4.29 (m, 3H, H-3. H-4, H-6a), 4.35 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 4.46 (s, 2H, CH2CCl3), 4.58 (d, 1H, J = 12.0 Hz, CH2Ph), 4.66–4.71 (m, 3H, J = 12.0 Hz, CH2Ph), 4.78, 4.88 (2d, 2H, J = 12.0 Hz, CH2Ph), 5.07 (d, 1H, J1,2 = 10.2 Hz, H-1), 5.46 (s, 1H, CHPh), 5.52 (d, 1H, JNH,2 = 7.2 Hz, NH), 7.00–7.09 (m, 2H), 7.21–7.44 (m, 20H, aromatic), 7.50–7.59 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.5, 51.8, 69.2, 69.5, 70.3, 71.3, 73.0, 73.4, 73.8, 74.2, 74.7, 74.8, 76.1, 77.3, 81.9, 84.3, 95.9, 101.1, 105.0, 127.0, 127.4, 127.9, 128.0, 128.0, 128.1, 128.2, 128.3, 128.5, 128.7, 128.76, 128.77, 129.2, 130.0, 134.5, 138.10, 138.12, 138.4, 138.6, 154.3; HRMS: [M+Na]+ C50H52Cl3NNaO11S calcd 1002.2224, obsd 1002.2208.
p-Tolyl 2,4,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (19)
β-d-Galactose pentaacetate (20g, 51.2 mmol) and p-toluenethiol (7.3 g, 58.8 mmol) were dissolved in CH2Cl2. (400 mL). Boron trifluoride etherate (20.15 mL, 153.6 mmol) was added dropwise at 0 °C and the mixture was stirred under N2 at room temperature for 20 h. The mixture was diluted with CH2Cl2 (450 mL) and washed with saturated NaHCO3, dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded p-tolyl 2,3,4,6-tetra-O-acetyl-1-thio-β-d-galactopyranoside (S8) as white solid (21.2 g, 91%). 1H-NMR (600 MHz, CDCl3): δ 1.98, 2.05, 2.10, 2.12 (s, 12H, 4 × COCH3), 2.35 (s, 3H, SPhCH3), 3.92 (t, 1H, J = 6.6 Hz, H-5), 4.10–4.20 (m, 2H, H-6a, H-6b), 4.66 (d, 1H, J1,2 = 10.2 Hz, H-1), 5.04 (dd, 1H, J2,3 = 9.6 Hz, J3.4 = 3.0 Hz, H-3), 5.22 (t, 1H, J = 10.2 Hz, H-2), 5.41 (d, 1H, J3.4 = 3.0 Hz, H-4), 7.08–7.16 (m, 2H), 7.40–7.46 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 20.82, 20.86, 20.90, 21.1, 21.4, 61.8, 67.4, 67.4, 67.5, 72.2, 74.5, 87.1, 128.8, 129.9, 133.3, 138.7, 160.74 169.7, 170.3, 170.4, 170.6. ESI-MS [M+Na]+ C21H26NaO9S calcd 477.1, obsd 477.3; Comparison of the NMR data with those reported in the literature63 confirmed its identity. p-Tolyl 2,3,4,6-tetra-O-acetyl-1-thio-β-d-galactopyranoside S8 (11.5 g, 25.3 mmol) was dissolved in a mixture of CH2Cl2/MeOH (100 mL/70 mL) and 5.14 M NaOMe (2.4 mL, 12.7 mmol) was added. The mixture was stirred at room temperature for 6 h under N2 and then was neutralized with Amberlite IR-120 and concentrated to dryness. Silica gel column chromatography (10:1 CH2Cl2–MeOH) afforded p-tolyl 1-thio-β-d-galactopyranoside (S9) as white solid (7.2 g, quantitative). 1H-NMR (600 MHz, CD3OD): δ 2.28 (s, 3H, SPhCH3), 3.47 (dd, 1H, J2,3 = 9.6 Hz, J3.4 = 3.0 Hz, H-3), 3.51 (t, 1H, J = 6.6 Hz, H-5), 3.55 (t, 1H, J = 9.6 Hz, H-2), 3.67–3.75 (m, 2H, H-6a, H-6b), 3.87 (d, 1H, J3.4 = 3.0 Hz, H-4), 4.49 (d, 1H, J1,2 = 9.6 Hz, H-1), 7.05–7.12 (m, 2H), 7.40–7.47 (m, 2H); 13C-NMR (150 MHz, CD3OD): δ 19.9, 61.4, 69.2, 69.8, 75.1, 79.4, 89.5, 129.4, 130.9, 131.7, 137.2. ESI-MS [M+Na]+ C13H18NaO5S calcd 309.1, obsd 309.1. p-Tolyl 1-thio-β-d-galactopyranoside S9 (3 g, 10.4 mmol) and dibutyltin oxide (2.6 g, 10.4 mmol) were put into MeOH (45 mL). The mixture was heated at reflux for 2 hours and then concentrated to dryness. DMF (30 mL) was added to the resulting residue, p-methoxy benzyl chloride (PMBCl, 1.5 mL, 10.4 mmol) and CsF (1.67 g, 10.4 mmol), which was then stirred under N2 at 50 °C for 2 days and concentrated to dryness. Silica gel column chromatography (1:2 Hexanes–EtOAc) afforded p-tolyl 3-O-p-methoxylbenzyl-1-thio-β-d-galactopyranoside (S10) as white solid (2.34 g, 55% for two steps). 1H-NMR (600 MHz, CD3OD): δ 2.20 (s, 3H, SPhCH3), 3.27 (dd, 1H, J2,3 = 9.6 Hz, J3.4 = 3.0 Hz, H-3), 3.35–3.38 (m, 1H, H-5), 3.58–3.65 (m, 3H, J1,2 = 10.2 Hz, H-2, H-6a, H-6b), 3.67 (s, 3H, OCH3), 3.94 (d, 1H, J3.4 = 3.0 Hz, H-4), 4.41 (d, 1H, J1,2 = 10.2 Hz, H-1), 4.51, 4.58 (2d, 2H, J = 11.4 Hz, CH2PhOCH3), 6.72–6.82 (m, 2H), 6.96–7.05 (m, 2H), 7.20–7.28 (m, 2H), 7.30–7.38 (m, 2H); 13C-NMR (100 MHz, CD3OD): δ 21.3, 55.76, 55.81, 62.7, 67.6, 70.3, 72.5, 80.5, 83.5, 90.8, 114.8, 130.64, 130.68, 130.8, 130.9, 131.9, 132.1, 133.1, 133.2, 138.6, 160.9. ESI-MS [M+Na]+ C21H26NaO6S calcd 429.2, obsd 429.3. p-Tolyl 3-O-p-methoxylbenzyl-1-thio-β-d-galactopyranoside S10 (3.4 g, 8.36 mmol) was dissolved in DMF (50 mL) and the solution was cooled to 0 °C. NaH (1.34 g, 60% NaH in mineral oil, 33.44 mmol) was added in portions, followed by addition of BnBr (4 mL, 33.44 mmol). The mixture was stirred at room temperature under N2 overnight and then diluted with EtOAc (250 mL). The mixture was washed with saturated NaHCO3, water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (8:1 Hexanes–EtOAc) afforded compound 14 p-tolyl 2,4,6-tri-O-benzyl-3-O-p-methoxylbenzyl-1-thio-β-d-galactopyranoside as white solid (4.5 g, 80%); [α]d +3.2 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.27 (s, 3H, SPhCH3), 3.56–3.65 (m, 4H, H-3, H-5, H-6a, H-6b), 3.77 (s, 3H, OCH3), 3.87 (t, 1H, J = 9.6 Hz, H-2), 3.93 (d, 1H, J3.4 = 2.4 Hz, H-4), 4.40, 4.45, 4.58, 4.62, 4.65, 4.72, 4.78, 4.95 (8d, 8H, J = 12.0 Hz, CH2PhOCH3, CH2Ph), 4.58 (d, 1H, J1,2 = 10.2 Hz, H-1), 6.78–6.88 (m, 2H), 6.94–7.02 (m, 2H), 7.24–7.34 (m, 15H, aromatic), 7.35–7.45 (m, 2H), 7.40–7.49 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.4, 55.5, 69.1, 72.7, 73.8, 73.9, 74.7, 75.9, 77.5, 84.2, 88.3, 114.1, 127.7, 127.96, 128.04, 128.1, 128.2, 128.4, 128.57, 128.59, 128.68, 129.5, 129.8, 130.5, 130.7, 132.5, 137.4, 138.2, 138.7, 139.1, 159.5. ESI-MS [M+Na]+ C42H44NaO6S calcd 699.3, obsd 699.5.
Compound 14 (0.3g, 0.44 mmol) was dissolved in a mixture of CH2Cl2/H2O (2.9 mL/0.15 mL) and the solution was cooled to 0 °C. 2,3-Dichloro 5,6-dicyano-1,4-benzoquinone (DDQ, 0.12 g, 0.53 mmol) was added and the mixture was stirred at room temperature for 3 h. The mixture was filtered, diluted with CH2Cl2 (30 mL) and the organic phase was washed with H2O until the solution became colorless. Silica gel column chromatography (4:1 Hexanes–EtOAc) afforded compound 19 as white solid (0.21 g, 85%); [α ]d −3.9 (c = 3.3, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 2.21 (d, 1H, J = 5.4 Hz, OH), 2.30 (s, 3H, SPhCH3), 3.63–3.68 (m, 5H, H-2, H-3, H-5, H-6a, H-6b), 3.89 (d, 1H, J3.4 = 2.4 Hz, H-4), 4.44, 4.50 (2d, 2H, J = 12.0 Hz, CH2Ph), 4.56 (d, 1H, J1,2 = 9.0 Hz, H-1), 4.64 (d, 2H, J = 12.0 Hz, CH2Ph), 4.73, 4.90 (2d, 2H, J = 12.0 Hz, CH2Ph), 6.98–7.08 (m, 2H), 7.28–7.37 (m, 15H), 7.42–7.52 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 21.3, 68.8, 73.7, 75.1, 75.4, 75.9, 76.2, 77.5, 78.5, 87.9, 127.9, 127.98, 128.03, 128.05, 128.2, 128.53, 128.56, 128.60, 128.7, 129.8, 130.4, 132.2, 137.5, 138.0, 138.3, 138.7; HRMS: [M+Na]+ C34H36NaO5S calcd 579.2181, obsd 579.2167.
3-Azidopropyl 2,3,6-tri-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (15)
d-Lactose (5 g, 13.87 mmol) was dissolved in pyridine (30 mL) and then BzCl (20 mL, 172 mmol) was added at 0 °C. The mixture was stirred at room temperature under N2 overnight and then diluted with CH2Cl2 (400 mL). The mixture was washed with saturated NaHCO3, water and then dried over Na2SO4, filtered and concentrated. The resulting residue was dissolved in CH2Cl2 (30 mL) and the solution was cooled to −10 °C. HBr in acetic acid (60 mL, 33% w/w, 104 mmol) was added and the mixture was stirred at room temperature under N2 for 5 hours. The mixture was diluted with CH2Cl2 (200 mL) and poured into crushed ice in saturated NaHCO3 (400 mL). The organic phase was separated and washed again with saturated NaHCO3 until the pH is about 7 and then dried over Na2SO4, filtered and concentrated. The resulting residue (2 g, 1.76 mmol), 1-bromo propanol (2.3 mL, 26.4 mmol) and freshly activated molecular sieve MS 4 Å (300 mg) was put into a 100 mL round-bottomed flask containing CH2Cl2 (20 mL) and the mixture was stirred at room temperature for 30 minutes. AgOTf (0.54 g, 2.11 mmol) was added and the mixture was stirred at room temperature for 1 hour. The mixture was diluted with CH2Cl2 (100 mL) and filtered. The filtrate was washed with saturated aqueous NaHCO3 and H2O. The organic layer was dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3:1 Hexanes–EtOAc) afforded compound 3-bromopropyl 2,3,4,6-tetra-O-benzoyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-β-d-glucopyranoside (S11) as white solid (1.6 g, 65% for 3 steps). 1H-NMR (600 MHz, CDCl3): δ 1.96 (m, 2H, OCH2CH2CH2N3), 3.27 (m, 2H, OCH2CH2CH2N3), 3.64 (m, 1H, CH2N3), 3.73–3.81 (m, 2H, H-5’, H-6a’), 3.88–3.98 (m, 3H, H-5, H-6b’, CH2N3), 4.32 (t, 1H, J = 9.6 Hz, H-4), 4.56 (dd, 1H, J = 4.2, 12.0 Hz, H-6a), 4.67 (d, 1H, J = 12.0 Hz, H-6b), 4.75 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.96 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 5.45 (dd, 1H, J2’,3’ = 10.2 Hz, J3’,4’ = 3.0 Hz, H-3’), 5.51 (t, 1H, J = 9.6 Hz, H-2), 5.78–5.80 (m, 2H, H-2’, H-4’), 5.87 (t, 1H, J = 9.6 Hz, H-3), 7.14–8.04 (m, 35H, aromatic); 13C-NMR (100 MHz, CDCl3): δ 30.1, 32.3, 61.1, 62.4, 67.5, 69.9, 71.4, 71.78, 71.82, 72.83, 73.0, 76.0, 101.1, 101.4, 128.3, 128.4, 128.56, 128.63, 128.8, 129.2, 129.4, 129.5, 129.57, 129.62, 129.68, 129.74, 130.0, 133.2, 133.3, 133.4, 133.6, 164.8, 165.2, 165.3, 165.4, 165.5, 165.8. ESI-MS [M+Na]+ C64H55NaBrO18 calcd 1213.3, obsd 1213.6. 3-Bromopropyl 2,3,4,6-tetra-O-benzoyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-β-d-glucopyranoside S11 (1.2 g, 1 mmol) and NaN3 (0.66 g, 10 mmol) were dissolved in DMF (10 mL). The mixture was stirred at 60 °C for 30 h and then concentrated. The resulting residue was diluted with EtOAc (200 mL), washed with H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3:1 Hexanes–EtOAc) afforded compound 3-azidopropyl 2,3,4,6-tetra-O-benzoyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-β-d-glucopyranoside (S12) as white solid (1.04 g, 90%). 1H-NMR (600 MHz, CDCl3): δ 1.72 (m, 2H, CH2CH2CH2N3), 3.18 (m, 2H, OCH2CH2CH2N3), 3.54 (m, 1H, CH2N3), 3.71–3.78 (m, 2H, H-5’, H-6a’), 3.86–3.95 (m, 3H, H-5, H-6b’, CH2N3), 4.29 (t, 1H, J = 9.0 Hz, H-4), 4.53 (dd, 1H, J = 4.2, 12.0 Hz, H-6a), 4.64 (d, 1H, J = 12.0 Hz, H-6b), 4.71 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.93 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 5.43 (dd, 1H, J2’,3’ = 10.2 Hz, J3’,4’ = 3.0 Hz, H-3’), 5.49 (t, 1H, J = 7.8 Hz, H-2), 5.75–5.78 (m, 2H, H-2’, H-4’), 5.85 (t, 1H, J = 9.6 Hz, H-3), 7.14–8.12 (m, 35H, aromatic); 13C-NMR (100 MHz, CDCl3): δ 29.1, 48.0, 61.2, 62.5, 66.8, 67.7, 70.0, 71.5, 71.9, 72.0, 73.0, 73.2, 76.2, 101.2, 101.4, 128.4, 128.67, 128.71, 128.76, 128.82, 128.99, 129.4, 129.6, 129.66, 129.74, 129.77, 129.84, 129.86, 129.93, 130.18, 130.37, 133.40, 133.5, 133.6, 133.8, 133.9, 165.0, 165.4, 165.58, 165.60, 165.8, 166.0. ESI-MS [M+Na]+ C64H55NaN3O18 calcd 1176.4, obsd 1176.8. 3-Azidopropyl 2,3,4,6-tetra-O-benzoyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-β-d-glucopyranoside S12 (1 g, 0.86 mmol) was dissolved in MeOH (16 mL) and 5.14 M NaOMe (1.67 mL, 8.6 mmol) was added. The mixture was heated at reflux for 6 hours under N2 and then was neutralized with Amberlite IR-120 until pH is around 7. It was filtered and concentrated to dryness. Silica gel column chromatography (4:1 CH2Cl2–MeOH) afforded 3-azidopropyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (S13) as white solid (0.36 g, 97%). 1H-NMR (600 MHz, CD3OD): δ 1.85 (m, 2H, CH2CH2CH2N3), 3.23 (t, 1H, J = 7.8 Hz, H-2’), 3.38–3.40 (m, 1H, H-5), 3.40–3.58 (m, 7H, H-2, H-3, H-6a, H-3’, H-5’, OCH2CH2CH2N3), 3.60–3.64 (m, 1H, CH2N3), 3.68 (dd, 1H, J = 4.8, 12.0 Hz, H-6a’), 3.76 (dd, 1H, J =7.8, 11.4 Hz, H-4), 3.80 (d, 1H, J3’,4’ = 3.0 Hz, H-4’), 3.83 (dd, 1H, J = 3.6, 12.0 Hz, H-6b’), 3.88 (dd, 1H, J = 4.8, 12.0 Hz, H-6b), 3.92–3.96 (m, 1H, CH2N3), 4.27 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 4.34 (d, 1H, J1,2 = 7.8 Hz, H-1); 13C-NMR (150 MHz, CD3OD): δ 29.0, 48.2, 60.7, 61.3, 66.4, 69.1, 71.4, 73.5, 73.6, 75.2, 75.3, 75.9, 79.4, 103.1, 103.9. ESI-MS [M+Na]+ C15H27NaN3O11 calcd 448.2, obsd 448.2. The mixture of 3-azidopropyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside S13 (0.5 g, 1.17 mmol), camphorsulfonic acid (0.14 g, 0.59 mmol), benzaldehyde dimethylacetal (0.2 mL, 1.35 mmol) and DMF (3 mL) was stirred at room temperature under N2 overnight. The mixture was neutralized with solid NaHCO3 (0.98 g, 1.17 mmol) and then concentrated to dryness. Silica gel column chromatography (8:1 CH2Cl2–MeOH) afforded 3-azidopropyl 4,6-O-benzylidene-β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside64 (S14) as white solid (0.49 g, 81%). 1H-NMR (600 MHz, CD3OD): δ 1.80 (m, 2H, CH2CH2CH2N3), 3.21 (t, 1H, J = 7.8 Hz, H-2’), 3.34–3.39 (m, 3H, H-5, OCH2CH2CH2N3), 3.49–3.62 (m, 6H, H-2, H-3, H-6a, H-3’, H-5’, CH2N3), 3.84–3.89 (m, 3H, H-4, H-6b, CH2N3), 4.05 (d, 1H, J = 11.4 Hz, H-6a’), 4.11–4.13 (m, 2H, H-4’, H-6b’), 4.21 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 4.40 (d, 1H, J1,2 = 7.8 Hz, H-1), 5.52 (s, 1H, CHPh), 7.27–7.48 (m, 5H, aromatic); 13C-NMR (100 MHz, CD3OD): δ 30.3, 49.4, 61.8, 67.7, 68.29, 68.33, 70.3, 71.8, 73.5, 74.8, 76.3, 76.5, 77.3, 80.1, 102.2, 104.4, 104.9, 127.6, 129.2, 130.0, 139.6. ESI-MS [M+Na]+ C22H31NaN3O11 calcd 536.2, obsd 536.3. 3-Azidopropyl 4,6-O-benzylidene-β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside S14 (2 g, 3.89 mmol) was dissolved in DMF (25 mL) and the solution was cooled to 0 °C. NaH (0.93 g, 60% NaH in mineral oil, 23.34 mmol) was added in portions, followed by addition of BnBr (2.8 mL, 33.44 mmol). The mixture was stirred at room temperature under N2 for 6 h and then diluted with EtOAc (250 mL). The mixture was washed with saturated aqueous NaHCO3, water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (6:1 Hexanes–EtOAc) afforded 3-azidopropyl 2,3-di-O-benzyl-4,6-O-benzylidene-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside53 (S15) as white solid (3 g, 80%); [α]d +176.4 (c, 0.56, CH2Cl2); [α]d +16.1 (c = 1, CHCl3); 1H-NMR (600 MHz, CDCl3): δ 1.88 (m, 2H, CH2CH2CH2N3), 2.93 (s, 1H, H-5’), 3.33–3.39 (m, 4H, H-3, H-5, OCH2CH2CH2N3), 3.42 (t, 1H, J = 7.8 Hz, H-2’), 3.60–3.64 (m, 2H, H-3’, CH2N3), 3.68–3.70 (d, 1H, J = 10.8 Hz, H-6a), 3.74–3.77 (t, 1H, J = 7.8 Hz, H-2), 3.84 (d, 1H, J3,4 = 12.0 Hz, H-4), 3.86–3.89 (dd, 1H, J5’,6a’ = 4.2 Hz, J6a’,6b’ = 10.8 Hz, H-6a’), 3.96–3.98 (m, 2H, H-6b, CH2N3), 4.02 (d, 1H, J3’,4’ = 3.0 Hz, H-4’), 4.20 (d, 1H, J = 12.0 Hz, H-6b’), 4.32, (d, 1H, J = 12.0 Hz, CH2Ph), 4.36 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 4.44 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.54 (d, 1H, J = 12.0 Hz, CH2Ph), 4.72–4.85 (m, 7H, CH2Ph), 5.19, 5.46 (2d, 2H, J = 12.0 Hz, CH2Ph), 7.17–7.52 (m, 30H, aromatic); 13C-NMR (150 MHz, CDCl3): δ 29.47, 48.56, 66.57, 66.59, 66.78, 68.44, 69.19, 71.85, 73.22, 73.88, 75.33, 75.35, 75.34, 76.07, 77.80, 79.05, 79.86, 82.06, 83.30, 101.61, 103.09, 103.80, 126.81, 127.55, 127.66, 127.70, 127.87, 127.95, 127.99, 128.15, 128.33, 128.38, 128.45, 128.48, 128.59, 128.61, 128.87, 129.10, 138.32, 138.65, 138.72, 138.84, 139.09, 139.13. ESI-MS [M+Na]+ C57H61NaN3O11 calcd 986.4, obsd 986.7. 3-Azidopropyl 2,3-di-O-benzyl-4,6-O-benzylidene-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside S15 (0.6 g, 0.62 mmol) and NaBH3CN (0.35 g, 5.58 mmol) were dissolved in THF (15 mL) and cooled to 0 °C. A solution of HCl in ether (2 M, 3 mL) was added and the mixture was stirred at room temperature for 3 hours and then concentrated to dryness. The obtained residue was diluted with CH2Cl2 and washed with 10% HCl, water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3:1 Hexanes–EtOAc) afforded compound 15 as white solid (0.53 g, 89%); [α]d +16.1 (c = 1, CHCl3); 1H-NMR (600 MHz, CDCl3): δ 1.86 (m, 2H, CH2CH2CH2N3), 2.48 (s, 1H, OH), 3.30–3.41 (m, 6H, H-2’, H-3, H-5, H-5’, OCH2CH2CH2N3), 3.46–3.49 (dd, 1H, J5,6a = 5.4 Hz, J6a,6b = 9.6 Hz, H-6a), 3.55–3.70 (m, 5H, H-2, H-3’, H-6a’, H-6b, CH2N3), 3.79–3.81 (dd, 1H, J5’,6a’ = 4.2 Hz, J6a’,6b’ = 10.8 Hz, H-6b’), 3.94–4.01 (m, 3H, H-4, H-4’, CH2N3), 4.34–4.45 (m, 5H, J1,2 = 7.8 Hz, J1’,2’ = 8.4 Hz, H-1, H-1’, CH2Ph), 4.54, 4.64, 4.69 (3d, 3H, J = 12.0 Hz, CH2Ph), 4.73–4.79 (m, 4H, CH2Ph), 4.83, 4.99 (2d, 2H, J = 12.0 Hz, CH2Ph), 7.19–7.39 (m, 30H, aromatic); 13CNMR (150 MHz, CDCl3): δ 29.6, 48.6, 66.4, 66.8, 68.5, 68.8, 72.3, 73.1, 73.4, 73.8, 75.36, 75.39, 75.6, 75.7, 76.8, 79.7, 81.4, 82.1, 83.2, 89.5, 102.9, 103.9, 127.6, 127.85, 127.88, 127.95, 127.99, 128.09, 128.12, 128.19, 128.26, 128.40, 128.45, 128.61, 128.67, 128.70, 128.80, 138.2, 138.5, 138.6, 138.88, 138.94, 139.4. ESI-MS [M+Na]+ C57H63NaN3O11 calcd 988.5, obsd 988.8;
3-Azidopropyl 2,3-di-O-benzoyl-6-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzoyl-β-d-glucopyranoside(21)
3-Azidopropyl 4,6-O-benzylidene-β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (0.5 g, 0.97 mmol) was dissolved in pyridine (20 mL) and then BzCl (1.1 mL, 9.7 mmol) was added at 0 °C. The mixture was stirred at room temperature under N2 overnight and then diluted with CH2Cl2 (100 mL). The mixture was washed with saturated aqueous solution of NaHCO3, water and then dried over Na2SO4, filtered and concentrated. The mixture of the resulting residue and NaBH3CN (0.6 g, 9.7 mmol) in THF (20 mL) was cooled to 0 °C and then a solution of HCl in ether (2 M) was added until the solution was acidic. The mixture was stirred at room temperature for 3 hours and then concentrated to dryness. The resulting residue was diluted with CH2Cl2 and washed with 10% aqueous HCl, water and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded compound 21 as gel-like solid (0.7 g, 70% for two steps); [α]d +47.9 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 1.70 (m, 2H, CH2CH2CH2N3), 3.01–3.08 (m, 2H, H-6a’, H-6b’), 3.12–3.19 (m, 2H, OCH2CH2CH2N3), 3.45 (t, 1H, J = 6.0 Hz, H-5’), 3.48–3.52 (m, 1H, CH2N3), 3.81–3.87 (m, 2H, H-5, CH2N3), 4.17–4.26 (m, 4H, H-4, H-4’, CH2Ph), 4.42 (dd, 1H, J = 4.8, 12.0 Hz, H-6a), 4.60 (dd, 1H, J = 1.8, 12.0 Hz, H-6b), 4.65 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.76 (d, 1H, J1’,2’ = 7.8 Hz, H-1’), 5.12 (dd, 1H, J2’,3’ = 10.5 Hz, J3’,4’ = 3.0 Hz, H-3’), 5.37 (t, 1H, J = 7.8 Hz, H-2), 5.69–5.75 (m, 2H, H-3, H-2’), 7.20–8.02 (m, 30H, aromatic); 13C-NMR (100 MHz, CDCl3): δ 29.0, 48.0, 62.5, 66.6, 67.2, 67.5, 70.1, 72.0, 73.1, 73.1, 73.4, 73.5, 74.4, 76.4, 101.0, 101.4, 127.7, 128.0, 128.6, 129.0, 129.2, 129.3, 129.7, 129.81, 129.84, 129.91, 129.93, 129.98, 130.01, 133.29, 133.37, 133.47, 137.8, 165.2, 165.43, 165.45, 165.88, 165.93; HRMS: [M+Na]+ C57H53N3NaO16 calcd 1058.3324, obsd 1058.3315.
3-Azidopropyl 2,4,6-tri-O-benzyl-3-O-p-methoxybenzyl-α-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (16)
After the donor 14 (400 mg, 0.59 mmol), acceptor 15 (540 mg, 0.56 mmol) and activated molecular sieve MS-4 Å (500 mg) were stirred for 30 min at room temperature in a mixture solvent of Et2O (8 mL) and CH2Cl2 (16 mL), the mixture was cooled to −78 °C, followed by addition of AgOTf (456 mg, 1.77mmol) in Et2O (12 mL). The mixture was vigorously stirred for 10 min and then p-TolSCl (93.7 µL, 0.59 mmol) was added and the reaction mixture was stirred for 2 h from −78 to −40 °C. (See the general procedure for single step pre-activation based glycosylation for precautions) The mixture was concentrated under vacuum to dryness. The resulting residue was diluted with CH2Cl2 (100 mL), followed by filtration. The organic phase was washed with saturated aqueous NaHCO3 and H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded 16 as gel-like solid (702 mg, 82%); [α]d +37.3 (c = 2.6, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 1.86 (m, 2H, OCH2CH2CH2N3), 3.15 (dd, 1H, J = 4.8, 8.4 Hz, H-6a), 3.28–3.38 (m, 6H, H-2’, H-3, H-5, H-5’, OCH2CH2CH2N3), 3.47–3.50 (m, 2H, H-5”, H-6b), 3.55–3.70 (m, 4H, H-2, H-3’, H-6a’, CH2N3), 3.76 (s, 3H, OCH3), 3.82 (m, 1H, H-6a”), 3.93–3.98 (m, 3H, H-4, H-4’, CH2N3), 4.02–4.07 (m, 5H, H-2”, H-3”, H-6b’, CH2Ph), 4.17 (dd, 1H, H-4”), 4.21–4.28 (2d, 2H, J = 12.0 Hz, CH2Ph), 4.33–4.36 (m, 4H, J1’,2’ = 7.8 Hz, H-1’, H-6b”, CH2Ph), 4.44–4.52 (m, 5H, J1,2 = 7.8 Hz, H-1, CH2Ph), 4.68–4.80 (m, 7H, CH2Ph), 4.85–4.87 (m, 2H, CH2Ph), 5.04 (d, 1H, J1”,2” = 3.0 Hz, H-1”), 5.07 (d, 1H, CH2Ph), 6.77–7.46 (m, 49H, aromatic); 13C-NMR (150 MHz, CDCl3): δ 29.5, 48.6, 55.5, 66.7, 67.9, 68.1, 68.4, 69.7, 72.3, 72.4, 73.26, 73.30, 73.4, 73.50, 74.0, 75.0, 75.13, 75.19, 75.3, 75.50, 75.52, 76.7, 77.4, 79.5, 79.7, 81.9, 82.9, 101.1, 103.1, 103.7, 113.8, 127.62, 127.66, 127.68, 127.72, 127.74, 127.76, 127.83, 127.84, 127.86, 127.89, 127.91, 128.11, 128.21, 128.37, 128.40, 128.42, 128.48, 128.50, 128.51, 128.52, 128.54, 128.56, 128.63, 128.80, 129.08, 138.26, 138.59, 138.66, 138.83, 138.86, 138.99, 139.01, 139.20, 139.33, 159.1; HRMS: [M+Na]+ C92H99N3NaO17 calcd 1540.6872, obsd 1540.6831. gHMQC (without 1H decoupling): 1JC1”,H1” = 169.0 Hz, 1JC1’,H1’ = 160.1 Hz, 1JC1,H1 = 160.0 Hz.
3-Azidopropyl 2,4,6-tri-O-benzyl-α-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside(17)
Compound 16 (0.51g, 0.34 mmol) was dissolved in a mixture of CH2Cl2/H2O (4.9 mL/0.5 mL) and the solution was cooled to 0 °C. DDQ (0.84 g, 0.37 mmol) was added and the mixture was stirred at room temperature for 4 hours. The mixture was filtered, diluted with CH2Cl2 (100 mL) and the organic phase was washed with H2O until the solution became colorless. Silica gel column chromatography (4:1 Hexanes–EtOAc) afforded compound 17 as white solid (0.37 g, 80%). [α]d +34.6 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 1.87 (m, 2H, CH2CH2CH2), 3.17 (dd, 1H, J = 4.8, 8.4 Hz, H-6a), 3.27–3.39 (m, 6H, H-2’, H-3, H-5, H-5’, OCH2), 3.44–3.62 (m, 6H, H-2, H-3’, H-5”, H-6b, H-6a’, CH2N3), 3.69–3.82 (m, 3H, H-2”, H6a”, H-6b’), 3.91–4.14 (m, 9H, H-4, H-4’, H-4”, H-3”, CH2N3, CH2Ph), 4.34–4.39 (m, 3H, J1’,2’ = 7.8 Hz, H-1’, H-6a”, CH2Ph), 4.45 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.47–4.54 (m, 4H, CH2Ph), 4.67–4.84 (m, 8H, CH2Ph), 5.07 (d, 1H, J = 12.0 Hz, CH2Ph), 5.10 (d, 1H, J1”,2” = 3.0 Hz, H-1”), 7.13–7.39 (m, 45H, aromatic); 13CNMR (150 MHz, CDCl3): δ 29.5, 48.6, 66.8, 67.9, 68.0, 68.5, 69.5, 70.3, 72.4, 73.27, 73.33, 73.38, 73.43, 75.1, 75.31, 75.36, 75.47, 75.51, 75.53, 77.2, 77.9, 79.6, 81.8, 81.9, 83.2, 89.5, 99.9, 103.0, 103.8, 127.5, 127.74, 127.76, 127.78, 127.79, 127.85, 127.86, 127.88, 127.92, 127.95, 128.00, 128.02, 128.10, 128.27, 128.34, 128.35, 128.42, 128.44, 128.52, 128.55, 128.56, 128.58, 128.60, 128.71, 128.79, 138.27, 138.48, 138.57, 138.62, 138.82, 138.83, 138.93, 139.02, 139.6; HRMS: [M+Na]+ C84H91N3NaO16 calcd 1420.6297, obsd 1420.6276.
p-Tolyl 2,3,4-tri-O-benzyl-1-thio-β-l-fucopyranoside (18)
l-Fucose (10 g, 60.9 mmol) and DMAP (0.72 g, 6.01 mmol) were dissolved in anhydrous pyridine (100 mL) and acetic anhydride (40 mL) was added at room temperature in a period of 30 minutes. The mixture was stirred at room temperature under N2 overnight and then quenched with ethanol (20 mL) at 0 °C. The mixture was concentrated and the resulting residue was diluted with ethyl acetate (300 mL), washed with water, saturated NaHCO3, 10 % aqueous hydrochloric acid and brine. The organic phase was dried over Na2SO4 and then filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded the 1,2,3,4-tetra-O-acetyl-l-fucopyranoside as white solid (21 g, quantitative, α/β mixtures) with the α isomer (S16) as the major product. 1H-NMR (600 MHz, CDCl3): δ 1.01 (d, 3H, J5,6 = 6.6 Hz, H-6), 1.86, 1.87, 2.01, 2.04 (4s, 12H, 4 × COCH3), 4.15 (m, 1H, H-5), 5.15 (d, 1H, J1,2 = 3.6 Hz, H-2), 5.16–5.20 (m, 2H, H-3, H-4), 6.18 (d, 1H, J1,2 = 3.6 Hz, H-1); 13C-NMR (150 MHz, CDCl3): δ 16.0, 20.6, 20.7, 20.8, 21.0, 66.6, 67.3, 67.9, 70.6, 89.9, 169.2, 170.0, 170.2, 170.6. ESI-MS [M+Na]+ C14H20NaO9 calcd 355.1, obsd 355.3. The obtained α/β mixture of 1,2,3,4-tetra-O-acetyl-l-fucopyranoside (21 g), p-toluenethiol (8.32 g, 67 mmol) were dissolved in CH2Cl2 (180 mL) and cooled to 0 °C. Boron trifluoride etherate (10.5 mL, 83 mmol) was added dropwise at 0 °C and the mixture was stirred under N2 at room temperature overnight. The mixture was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3 until the pH around 7 and then dried over Na2SO4, filtered and concentrated. The obtained crude product was recrystallized by EtOAc/hexanes to give p-tolyl 2,3,4-tri-O-acetyl-1-thio-β-l-fucopyranoside63 (S17) as white solid (11.6g, 48%). 1H-NMR (600 MHz, CDCl3): δ 1.15 (d, 3H, J5,6 = 6.6 Hz, H-6), 1.90, 2.01, 2.07 (3s, 9H, 3 × COCH3), 2.26 (s, 3H, SPhCH3), 3.74 (m, 1H, H-5), 4.58 (d, 1H, J1,2 = 10.2 Hz, H-1), 4.98 (dd, 1H, J2,3 = 9.6 Hz, J3,4 = 3.0 Hz, H-3), 5.13 (t, 1H, J = 9.6 Hz, H-2), 5.18 (d, 1H, J3,4 = 3.0 Hz, H-4), 7.00–7.09 (m, 2H), 7.30–7.39 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 16.7, 20.8, 20.9, 21.1, 21.3, 67.5, 70.5, 72.6, 73.2, 86.9, 129.3, 129.8, 133.0, 138.3, 169.6, 170.3, 170.8. ESI-MS [M+Na]+ C19H24NaO7S calcd 419.1, obsd 419.2. p-tolyl 2,3,4-tri-O-acetyl-1-thio-β-l-fucopyranoside S17 (9.2 g, 23.2 mmol) was dissolved in a mixture of CH2Cl2/MeOH (70 mL /50 mL) and 1 M NaOMe (12 mL, 11.6 mmol) was added at room temperature. The mixture was stirred for 2 h at room temperature under N2, neutralized with Amberlite IR-120, concentrated and vacuum dried. The obtained residue (6 g) was dissolved in DMF (100 mL) and cooled to 0 °C. NaH (3.6 g, 60% NaH in mineral oil, 88 mmol) was added in portions, followed by addition of BnBr (10.5 mL, 88 mmol) 30 minutes later. The mixture was stirred at room temperature under N2 for 2 hours and then diluted with EtOAc (300 mL). The mixture was washed with saturated aqueous solution of NaHCO3, water and then dried over Na2SO4, filtered and concentrated. The obtained crude product was recrystallized by EtOAc/Hexanes to give compound 1837 as white solid (9.4 g, 75% for two steps); [α]d −9.4 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 1.25 (d, 3H, J5,6 = 6.6 Hz, H-6), 2.29 (s, 3H, SPhCH3), 3.50 (m, 1H, H-5), 3.58 (dd, 1H, J2,3 = 9.6 Hz, J3,4 = 2.4 Hz, H-3), 3.62 (d, 1H, J3,4 = 2.4 Hz, H-4), 3.89 (t, 1H, J = 9.6 Hz, H-2), 4.55 (d, 1H, J1,2 = 10.2 Hz, H-1), 4.66 (d, 1H, J = 12.0 Hz, CH2Ph), 4.72–4.81 (m, 4H, J = 12.0 Hz, CH2Ph), 5.00 (d, 1H, J = 12.0 Hz, CH2Ph), 6.94–7.08 (m, 2H), 7.27–7.40 (m, 15H, aromatic), 7.44–7.56 (m, 2H); 13C-NMR (150 MHz, CDCl3): δ 17.6, 21.4, 73.1, 74.8, 75.8, 76.8, 77.4, 84.8, 88.1, 127.7, 127.8, 127.95, 127.97, 128.2, 128.4, 128.59, 128.62, 128.70, 129.78, 130.7, 132.4, 137.4, 138.6, 138.7, 139.0. ESI-MS [M+Na]+ C34H36NaO4S calcd 563.2, obsd 563.5.
3-Azidopropyl 2,3,4-tri-O-benzyl-α-l-fucopyranosyl-(1→2)-3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-d-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-α-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (20α) and 3-Azidopropyl 2,3,4-tri-O-benzyl-α-l-fucopyranosyl-(1→2)-3,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→3)-4,6-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamino)-2-deoxy-β-d-galactopyranosyl-(1→3)-2,4,6-tri-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (20β)
After the donor 18 (50 mg, 92.47 µmol) and freshly activated molecular sieve MS-4 Å (500 mg) were stirred for 30 minutes at room temperature in Et2O (4 mL), the solution was cooled to −78 °C, followed by addition of AgOTf (72 mg, 277.4 µmol) in Et2O (1.5 mL). The mixture was stirred for 5 minutes at −78 °C and then p-TolSCl (14.7 µL, 92.47 µmol) was added into the solution. (See the general procedure for single step pre-activation based glycosylation for precautions) The mixture was vigorously stirred for 5 minutes, followed by addition of a solution of acceptor 13 (77.1 mg, 78.60 µmol) and TTBP (23 mg, 92.47 µmol) in CH2Cl2 (1 mL). The reaction mixture was stirred for 2 hour from −78 to −20 °C and then the mixture was cooled down to −78 °C, followed by addition of AgOTf (24 mg, 92.47 µmol) in Et2O (1 mL). The mixture was stirred for 10 minutes at −78 °C and then p-TolSCl (12.5 µL, 78.60 µmol) was added into the solution. After stirred for 5 minutes, a solution of acceptor 19 (30.9 mg, 55.48 µmol) and TTBP (23 mg, 92.47 µmol) in CH2Cl2 (1 mL) was added slowly along the flask wall into the mixture and the reaction mixture was stirred for 2 h from −78 to −20 °C. The mixture was cooled down again to −78 °C, followed by sequential additions of AgOTf (24 mg, 92.47 µmol) in Et2O (1 mL), the last acceptor 15 (35.7 mg, 36.99 µmol) and TTBP (23 mg, 92.47 µmol) in CH2Cl2 (1 mL). The mixture was stirred for 10 minutes at −78 °C and then p-TolSCl (12.5 µL, 78.60 µmol) was added into the solution. The reaction mixture was stirred for 2 hours from −78 to 10 °C and then was quenched with Et3N (40 µL), concentrated under vacuum to dryness. The resulting residue was diluted with CH2Cl2 (30 mL), followed by filtration. The organic phase was washed with saturated aqueous solution of NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded 46.6 mg of 20α (47%) and 22.4 mg of 20β (23%) respectively as colorless gel. For 20α: [α]d −18.0 (c = 2.4, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 0.42 (d, 3H, J = 6.6 Hz, H-6””’), 1.84 (m, 2H, OCH2CH2CH2N3), 2.80 (s, 1H), 3.19 (m, 1H), 3.27–3.39 (m, 7H), 3.46–3.68 (m, 11H), 3.76–3.86 (m, 5H), 3.92–4.25 (m, 17H), 4.29–4.59 (m, 18H), 4.64–4.87 (m, 13H), 4.94 (d, 1H, J1”,2” = 3.0 Hz, H-1”), 5.06 (d, 1H, J = 11.4 Hz), 5.24 (d, 1H, J =11.4 Hz), 5.42 (s, 1H), 5.55 (d, 1H, J1””’,2””’ = 3.6 Hz, H-1””’), 7.00–7.45 (m, 80H, aromatic); 13C-NMR (150 MHz, CDCl3): δ 16.1, 29.4, 29.9, 48.5, 54.4, 66.2, 66.6, 66.7, 67.7, 68.1, 68.4, 69.0, 69.2, 69.4, 72.0, 72.1, 72.5, 72.7, 72.9, 73.1, 73.22, 73.24, 73.28, 73.57, 73.63, 73.77, 73.9, 74.57, 74.67, 74.76, 74.78, 74.94, 75.13, 75.18, 75.27, 75.4, 76.2, 76.3, 76.8, 77.6, 78.6, 79.2, 79.7, 80.4, 81.5, 81.7, 82.0, 84.1, 95.8, 97.5, 100.8, 101.8, 102.6, 102.7, 102.8, 103.6, 126.6, 127.03, 127.06, 127.24, 127.28, 127.31, 127.41, 127.44, 127.55, 127.61, 127.67, 127.68, 127.70, 127.76, 127.79, 127.81, 127.93, 128.01, 128.02, 128.11, 128.16, 128.18, 128.20, 128.34, 128.38, 128.42, 128.45, 128.50, 128.56, 128.65, 128.68, 128.72, 128.76, 128.77, 129.1, 129.9, 137.9, 138.2, 138.3, 138.44, 138.45, 138.47, 138.67, 138.81, 138.94, 138.99, 139.1, 139.4, 139.7, 139.8, 154.0; HRMS: [M+Na]+ C154H163Cl3N4O31 calcd 2692.0265, obsd 2692.0332. gHMQC (without 1H decoupling): 1JC1””’,H1””’ = 171.9 Hz, 1JC1”,H1” = 169.8 Hz, other four 1JC1,H1 = 160.1 Hz, 160.1 Hz, 162.6 Hz, 162.6 Hz.
For 20β: [α]d −4.2 (c = 1, CH2Cl2); 1H-NMR (600 MHz, CDCl3): δ 0.60 (d, 3H, J = 6.6 Hz, H-6””’), 1.85 (m, 2H, OCH2CH2CH2N3), 3.22–3.63 (m, 21H), 3.66–3.79 (m, 6H), 3.86–3.99 (m, 6H), 4.05–4.24 (m, 7H), 4.28–4.51 (m, 18H), 4.55–4.60 (m, 4H), 4.63–4.79 (m, 8H), 4.85–4.94 (m, 3H), 5.09–5.13 (m, 2H), 5.22 (d, 1H, J = 12.0 Hz), 5.49 (s, 1H), 5.55 (d, 1H, J1””’,2””’ = 3.6 Hz, H-1””’), 6.92–7.43 (m, 80H, aromatic); 13C-NMR (150 MHz, CDCl3): δ 16.4, 29.5, 29.9, 48.5, 54.7, 66.6, 67.1, 68.2, 68.8, 69.0, 69.2, 69.3, 70.0, 72.3, 72.68, 72.74, 73.0, 73.3, 73.67, 73.69, 73.75, 73.80, 73.96, 74.75, 74.99, 75.12, 75.2, 75.4, 75.8, 76.2, 76.3, 76.6, 78.4, 79.3, 79.9, 80.6, 81.4, 81.8, 82.6, 83.2, 83.7, 96.0, 97.4, 101.6, 101.9, 102.8, 103.1, 103.7, 126.8, 126.92, 126.96, 127.1, 127.2, 127.36, 127.40, 127.45, 127.48, 127.51, 127.53, 127.67, 127.69, 127.73, 127.76, 127.83, 127.87, 127.90, 127.93, 128.01, 128.07, 128.10, 128.16, 128.27, 128.28, 128.34, 128.35, 128.38, 128.44, 128.50, 128.52, 128.53, 128.60, 128.64, 128.67, 128.71, 129.2, 138.1, 138.2, 138.3, 138.43, 138.47, 138.51, 138.57, 138.86, 138.88, 138.97, 139.06, 139.24, 139.51, 139.8, 140.0, 154.1; HRMS: [M+Na]+ C154H163Cl3N4NaO31 calcd 2692.0265, obsd 2692.0254. 1JC1””’,H1””’ = 170.9 Hz, other five 1JC1,H1 = 162.0 Hz, 162.0 Hz, 162.0 Hz, 162.0 Hz. 159.4 Hz.
Three component one-pot synthesis procedure:
After the donor 18 (50 mg, 92.47 µmol) and activated molecular sieve MS-4 Å (500 mg) were stirred for 30 minutes at room temperature in Et2O (6 mL), the solution was cooled to −78 °C, followed by addition of AgOTf (72 mg, 277.4 µmol) in Et2O (1.5 mL). The mixture was stirred for 5 minutes at −78 °C and then p-TolSCl (14.7 µL, 92.47 µmol) was added into the solution. (See the general procedure for single step pre-activation based glycosylation for precautions) The mixture was vigorously stirred for 10 minutes, followed by addition of a solution of acceptor 13 (81.7 mg, 83.22 µmol) and TTBP (23 mg, 92.47 µmol) in CH2Cl2 (1 mL). The reaction mixture was stirred for 2 hours from −78 to −20 °C and then the mixture was cooled down to −78 °C, followed by sequential additions of AgOTf (24 mg, 92.47 µmol) in Et2O (1 mL), acceptor 17 (90.5 mg, 64.73 µmol) and TTBP (23 mg, 92.47 µmol) in CH2Cl2 (1 mL). The mixture was stirred for 5 minutes at −78 °C and then p-TolSCl (13.2 µL, 83.22 µmol) was added into the solution. The reaction mixture was stirred for 3 hours from −78 to 10 °C and then was quenched with Et3N (40 µL), concentrated under vacuum to dryness. The resulting residue was diluted with CH2Cl2 (30 mL), followed by filtration. The organic phase was washed with saturated aqueous NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded 20α as colorless gel (128mg, 74%).
3-Azidopropyl α-l-fucopyranosyl-(1→2)-β-d-galactopyranosyl-(1→3)-2-acetamido-2-deoxy-β-d-galactopyranosyl-(1→3)-α-d-galactopyranosyl-(1→4)-β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (2)
The mixture of compound 20α (0.075g, 0.028 mmol), 1 M NaOH (0.56 mL, 0.56 mmol) and THF (4 mL) was stirred at 50 °C overnight and then concentrated to dryness. The resulting residue was diluted with CH2Cl2 (50 mL) and the organic phase was washed by H2O and then dried over Na2SO4, filtered and concentrated to dryness. The resulting residue was dissolved in pyridine (2 mL) and catalytic amount of DMAP was added. Acetic anhydride (0.5 mL, 5.3 mmol) was added dropwise and the mixture was stirred at room temperature under N2 for 4 hours. The reaction was quenched by adding a few drops of H2O and then diluted with EtOAc (20 mL). The organic phase was washed with saturated aqueous solution of NaHCO3, H2O and then dried over Na2SO4, filtered and concentrated to dryness. Silica gel column chromatography (2:1 Hexanes–EtOAc) afforded the N-acetylation product as white solid. The mixture of the N-acetylation product, 1 M of PMe3 in THF (56 µL, 0.056 mmol), 0.1 M NaOH (0.5 mL, 0.05 mmol) and THF (3 mL) was stirred at 60 °C under N2 overnight. The mixture was concentrated and the resulting residue was diluted with CH2Cl2 (50 mL). The organic phase was washed with H2O and then dried over Na2SO4, filtered and concentrated to dryness. The resulting residue was purified by quickly passing through a short silica gel column (10:1, CH2Cl2–MeOH). The mixture of the obtained solid and Pd(OH)2 in MeOH/H2O/HOAc (3 mL/1 mL/1 mL) was stirred under H2 at room temperature overnight and then filtered. The filtrate was concentrated to dryness under vacuum and then was co-evaporated with H2O (10 mL) three times to remove the AcOH. The aqueous phase was further washed with CH2Cl2 (5 mL × 3) and EtOAc (5 mL × 3) and then the aqueous phase was dried under vacuum to afford compound 2 (acetate salt) as white solid (16.2 mg, 50% for three steps). [α]d +27.8 (c = 1.4, H2O); 1H-NMR (600 MHz, D2O): δ 1.00 (d, 3H, J = 6.6 Hz, H-6””’), 1.74 (s, 3H), 1.79 (m, 2H), 1.83 (s, 3H), 2.95 (t, 2H, J = 6.6 Hz), 3.12 (t, 1H, J = 8.4 Hz), 3.36–3.89 (m, 32H), 4.02–4.03 (m, 2H), 4.18 (t, 1H, J = 6.0 Hz), 4.29 (d, 2H, J = 7.8 Hz), 4.32 (d, 1H, J = 7.8 Hz), 4.40 (d, 1H, J = 7.8 Hz), 4.69 (d, 1H, J = 3.6 Hz), 5.02 (d, 1H, J = 3.6 Hz); 13C-NMR (150 MHz, D2O): δ 15.4, 22.3, 23.4, 26.8, 37.7, 51.8, 60.1, 60.5, 61.1, 66.9, 67.9, 68.1, 68.6, 69.2, 69.3, 69.6, 70.2, 71.0, 72.0, 72.2, 73.0, 73.7, 74.5, 74.7, 75.0, 75.2, 75.6, 76.2, 76.4, 77.3, 78.3, 78.8, 99.4, 100.6, 102.2 (×2), 103.5, 104.1, 174.4; HRMS: [M+Na]+ C41H72N2NaO30 calcd 1095.4068, obsd 1095.4048. 1JC1””’,H1””’ = 170.0 Hz, 1JC1”,H1” = 171.2 Hz, Other four 1JC1,H1 = 162.6 Hz, 163.9 Hz, 162.4 Hz, 162.4 Hz.
Supplementary Material
Selected 1H-, 13C- and 2D NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We are grateful for financial supports from the University of Toledo, the National Institutes of Health (R01-GM-72667) and the Pardee foundation. We dedicate this work to Prof. Nina Berova, Columbia University, for her guidance and continual encouragements over the years.
References
- 1.Zhang S, Zhang HS, Reuter VE, Slovin SF, Scher HI, Livingston PO. Clin. Cancer Res. 1998;4:295–302. [PubMed] [Google Scholar]
- 2.Hakomori S, Zhang Y. Chem. Biol. 1997;4:97–104. doi: 10.1016/s1074-5521(97)90253-2. [DOI] [PubMed] [Google Scholar]
- 3.Zhang S, Cordon-Cardo C, Zhang HS, Reuter VE, Adluri S, Hamilton WB, Lloyd KO, Livingston PO. Int. J. Cancer. 1997;73:42–49. doi: 10.1002/(sici)1097-0215(19970926)73:1<42::aid-ijc8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Kannagi R, Levery SB, Ishijamik F, Hakomori S, Schevinsky LH, Knowles BB, Solter D. J. Biol. Chem. 1983;258:8934–8942. [PubMed] [Google Scholar]
- 5.Bremer EG, Levery SB, Sonnino S, Ghidoni R, Canevari S, Kannagi R, Hakomori S. J. Biol. Chem. 1984;259:14773–14777. [PubMed] [Google Scholar]
- 6.Gilewske T, Ragupathi G, Bhuta S, Williams LJ, Musselli C, Zhang X-F, Bencsath KP, Panageas KS, Chin J, Hudis CA, Norton L, Houghton AN, Livingston PO, Danishefsky SJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3270–3275. doi: 10.1073/pnas.051626298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang ZG, Williams LJ, Zhang XF, Zatorski A, Kudrya-shov V, Ragupathi G, Spassova M, Bornmann W, Slovin SF, Scher HI, Livingston PO, Lloyd KO, Danishefsky SJ. Proc. Natl. Acad. Sci. U. S. A. 2000;97:2719. doi: 10.1073/pnas.97.6.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danishefsky SJ, Allen JR. Angew. Chem. Int. Ed. 2000;39:836–863. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.Slovin SF, Ragupathi G, Adluri S, Ungers G, Terry K, Kim S, Spassova M, Bornmann WG, Fazzari M, Dantis L, Olkiewicz K, Lloyd KO, Livingston PO, Danishefsky SJ, Scher HI. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5710. doi: 10.1073/pnas.96.10.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park TY, Kim IJ, Hu S, Bilodeau MT, Randolph JT, Kwon O, Danishefsky SJ. J. Am. Chem. Soc. 1996;118:11488–11500. [Google Scholar]
- 11.Bilodeau MT, Park TK, Hu S, Randolph JT, Danishefsky SJ, Livingston PO, Zhang S. J. Am. Chem. Soc. 1995;117:7840–7841. [Google Scholar]
- 12.Allen JR, Allen JG, Zhang X-F, Williams LJ, Zatorski A, Ragupathi G, Livingston PO, Danishefsky SJ. Chem. Eur. J. 2000;6:1366–1375. doi: 10.1002/(sici)1521-3765(20000417)6:8<1366::aid-chem1366>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 13.Lassaletta JM, Schmidt RR. Liebigs Ann.Chem. 1996:1417–1423. [Google Scholar]
- 14.Zhu T, Boons GJ. Angew. Chem. Int. Ed. 1999;38:3495–3497. doi: 10.1002/(sici)1521-3773(19991203)38:23<3495::aid-anie3495>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 15.Huang C-Y, Thayer DA, Chang AY, Best M, Hoffmann L, Head S, Wong C-H. Proc. Natl. Acad. Sci. U. S. A. 2006;100:15–20. doi: 10.1073/pnas.0509693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wacowich-Sgarbi S, Rabuka D, Sgarbi PWM, Ichikawa Y. In: ACS Symposium Series 873; Synthesis of Carbohydrates through Biotechnology. Wang PG, Ichikawa Y, editors. Washington, DC: American Chemical Society; 2004. pp. 23–38. [Google Scholar]
- 17.Burkhart F, Zhang Z, Wacowich-Sgarbi S, Wong C-H. Angew. Chem. Int. Ed. 2001;40:1274–1277. doi: 10.1002/1521-3773(20010401)40:7<1274::aid-anie1274>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 18.Bosse F, Marcauelle LA, Seeberger PH. J. Org. Chem. 2002;67:6659–6670. doi: 10.1021/jo025834+. [DOI] [PubMed] [Google Scholar]
- 19.Werz DD, Castagner B, Seeberger PH. J. Am. Chem. Soc. 2007;129:2770–2771. doi: 10.1021/ja069218x. [DOI] [PubMed] [Google Scholar]
- 20.For an example, see: Adinolfi M, Iadonisi A, Ravidá A, Schiattarella M. J. Org. Chem. 2005;70:5316–5319. doi: 10.1021/jo050301x.
- 21.Tanaka H, Matoba N, Takanishi T. Chem. Lett. 2005;34:400–401. [Google Scholar]
- 22.Huang L, Huang X. Chem. Eur. J. 2007;13:529–540. doi: 10.1002/chem.200601090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang L, Wang Z, Li X, Ye X-S, Huang X. Carbohydr. Res. 2006;341:1669–1679. doi: 10.1016/j.carres.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Huang L, Wang H, Ye X-S. Angew. Chem. Int. Ed. 2004;42:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 25.Yu B, Yang Z, Cao H. Curr. Org. Chem. 2005;9:179–194. and references cited therein. [Google Scholar]
- 26.Pornsuriyasak P, Demchenko AV. Tetrahedron: Asymmetry. 2005;6:433–439. and references cited therein. [Google Scholar]
- 27.Kanie O. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Weinheim: Wiley-VCH; 2000. pp. 407–426. [Google Scholar]
- 28.Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J. Org. Chem. 1990;55:6068–6070. [Google Scholar]
- 29.Koeller KM, Wong C-H. Chem. Rev. 2000;100:4465–4493. doi: 10.1021/cr990297n. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka H, Adachi M, Takahashi T. Chem. Eur. J. 2005;11:849–862. doi: 10.1002/chem.200400840. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka H, Adachi M, Tsukamoto H, Ikeda T, Yamada H, Takahashi T. Org. Lett. 2002;4:4213–4216. doi: 10.1021/ol020150+. [DOI] [PubMed] [Google Scholar]
- 32.Fraser-Reid B, López JC, Gómez AM, Uriel C. Eur. J. Org. Chem. 2004;2004:1387–1395. [Google Scholar]
- 33.Fraser-Reid B, López JC, Radhakrishnan KV, Mach M, Schlueter U, Gomez AM, Uriel C. J. Am. Chem. Soc. 2002;124:3198–3199. doi: 10.1021/ja012383m. [DOI] [PubMed] [Google Scholar]
- 34.Jennings HJ, Snood RK. In: Neoglycoconjugates, Preparation and Application. Lee YC, Lee RT, editors. San Diego: Academic Press; 1994. p. 325. [Google Scholar]
- 35.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 36.Martichonok V, Whitesides GM. J. Org. Chem. 1996;61:1702–1706. doi: 10.1021/jo951711w. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Ollman IR, Ye X-S, Wischnat R, Baasov T, Wong C-H. J. Am. Chem. Soc. 1999;121:734–753. [Google Scholar]
- 38.Bock K, Pedersen C. J. Chem. Soc. Perkin Trans. 1974;2:293–297. [Google Scholar]
- 39.Demchenko AV. Synlett. 2003:1225–1240. and references cited therein. [Google Scholar]
- 40.Tokimoto H, Fujimoto Y, Fukase K, Kusumoto S. Tetrahedron Asym. 2005;16:441–447. [Google Scholar]
- 41.Chiba H, Funasaka S, Mukaiyama T. Bull. Chem. Soc. Jpn. 2003;76:1629–1644. [Google Scholar]
- 42.Adinolfi M, Barone G, Iadonisi A, Schiattarella M. Tetrahedron Lett. 2002;43:5573–5577. [Google Scholar]
- 43.Jona H, Mandai H, Chavasiri W, Takeuchi K, Mukaiyama T. Bull. Chem. Soc. Jpn. 2002;75:291–309. [Google Scholar]
- 44.Demchenko AV, Stauch T, Boons G-J. Synlett. 1997:818–819. [Google Scholar]
- 45.Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]
- 46.Kamath VP, Yeske RE, Gregson JM, Ratcliffe RM, Fang YR, Palcic MM. Carbohydr. Res. 2004;339:1141–1146. doi: 10.1016/j.carres.2003.12.027. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, Zhao X-E, Lai D, Song Z, Kong F. Carbohydr. Res. 2006;341:1174–1180. doi: 10.1016/j.carres.2006.03.029. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh S-Y, Jan M-D, Patkar LN, Chen C-T, Lin C-C. Carbohydr. Res. 2005;340:49–57. doi: 10.1016/j.carres.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Li Q, Wang H, Zhang L, Ye X-S. Tetrahedron. 2006;62:11657–11662. [Google Scholar]
- 50.Dohi H, Nishida Y, Takeda T, Kobayashi K. Carbohydr. Res. 2002;337:983–989. doi: 10.1016/s0008-6215(02)00093-9. [DOI] [PubMed] [Google Scholar]
- 51.Bhattacharyya S, Magnusson BG, Wellmar U, Nilsson UJ. J. Chem. Soc. Perkin Trans. 2001;1:886–890. [Google Scholar]
- 52.Sarkar AK, Matta KL. Carbohydr. Res. 1992;233:245–250. doi: 10.1016/s0008-6215(00)90937-6. [DOI] [PubMed] [Google Scholar]
- 53.Pornsuriyasak P, Demchenko AV. Carbohydr. Res. 2006;341:1458–1466. doi: 10.1016/j.carres.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 54.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 55.To determine the configuration of newly formed glycosyl bonds, we once isolated the intermediate oligosaccharides during one-pot synthesis and confirmed the linkages via NMR analysis.
- 56.Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Wang J, Czyryca PG, Chang H, Orsak TW, Evanson R, Chang C-WT. Org. Lett. 2004;6:1381–1384. doi: 10.1021/ol0497685. [DOI] [PubMed] [Google Scholar]
- 58.Cheshev PE, Khatuntseva EA, Gerbst AG, Tsvetkov YE, Shashkov AS, Nifantiev NE. Russ. J. Bioorg. Chem. 2003;29:372–381. [Google Scholar]
- 59.Mong TK-K, Lee H-K, Duron SG, Wong C-H. Proc. Natl. Acad. Sci. U. S. A. 2003;100:797–802. doi: 10.1073/pnas.0337590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nyffeler PT, Liang C-H, Koeller KM, Wong C-H. J. Am. Chem. Soc. 2002;124:10773–10778. doi: 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- 61.Luening B, Norberg T, Tejbrant J. Glycoconj. J. 1989;6:5–19. doi: 10.1007/BF01047886. [DOI] [PubMed] [Google Scholar]
- 62.Lee J-C, Wu C-Y, Apon JV, Siuzdak G, Wong CH. Angew. Chem. Int. Ed. 2006;45:2753–2757. doi: 10.1002/anie.200504067. [DOI] [PubMed] [Google Scholar]
- 63.Tai C-A, Kulkarni SS, Hung S-C. J. Org. Chem. 2003;68:8719–8722. doi: 10.1021/jo030073b. [DOI] [PubMed] [Google Scholar]
- 64.Yang Z-Q, Puffer EB, Pontrello JK, Kiessling LL. Carbohydr. Res. 2002;337:1605–1613. doi: 10.1016/s0008-6215(02)00270-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Selected 1H-, 13C- and 2D NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.