

Abstract
The ability of a drug or probe to cross a biological barrier has historically been viewed to be a function of its intrinsic physical properties. This view has largely restricted drug design and selection to agents within a narrow log P range. Molecular transporters offer a strategy to circumvent these restrictions. In the case of guanidinium-rich transporters (GRTs), a typically highly water-soluble conjugate is found to readily pass through the non-polar membrane of a cell and for some across tissue barriers. This activity opens a field of opportunities for the use of GRTs to enable delivery of polar and non-polar drugs or probes as well as to enhance uptake of those of intermediate polarity. The field of transporter enabled or enhanced uptake has grown dramatically in the last decade. Some GRT drug conjugates have been advanced into clinical trials. This review will provide an overview of recent work pertinent to the design and mechanism of uptake of GRTs.
Keywords: Molecular transporters, Cell-penetrating peptides, Oligoarginine, Bidentate hydrogen bond, Endocytosis, Adaptive translocation, HIV Tat, Dendrimers, Drug delivery
1. Introduction
The intent of this collection of reviews edited by Professor Futaki is to update research advances on molecular transporters of which cell-penetrating peptides (CPPs) are an important subclass. Molecular transporters [1] are agents which, when attached to an otherwise poorly bioavailable drug, drug candidate or probe, enhance or enable its passage through biological barriers. The intent of this specific chapter is twofold: to discuss the design criteria that have been successfully used to advance a rather robust class of guanidinium-rich molecular transporter-drug conjugates, including a conjugate that has entered Phase II clinical trials, and to address the mechanisms of the entry of such conjugates into cells and tissue.
At the outset, it is important to note, especially for those new to this field, that understanding biological barriers is of great fundamental, applied and philosophical significance. Life itself depends on such barriers whether they be the plasma membrane of a cell or the skin that envelops our bodies. Understanding the origins and functions of these barriers and their evolution is thus a prerequisite to understanding the origins of life itself and its progression to more complex living systems. At a fundamental level there is hardly an area of chemistry, physics, biology, or medicine that is not connected to this multifaceted and indeed grand challenge. From a purely molecular perspective, membranes represent dynamic, heterogeneous, self-assembled systems, held together principally by weak forces. In general, they keep the outside world out and the inside world in. They provide compartmentalization and are critical to the selective import, concentration, and export of compounds needed for sustenance, protection, movement, adherence, and replication. These very functions are of course what often result in the failure or reduced efficacy of many therapeutic candidates. Indeed, drug design and selection have historically been dominated by the conventional but reasonable view that only compounds with a certain log P would be properly solubilized in the mostly polar, water-based biological fluids of the body and at the same time would also be able to pass through the relatively non-polar membrane of a cell. The use of many polar (e.g., nucleic acids, siRNA, etc.) and non-polar (e.g., taxol, camptothecin, etc.) molecules would thus be precluded by adherence to this view or require that special effort be taken to address formulation, distribution or bioavailability problems arising from their inherent physical properties. However, even compounds with the preferred log P characteristics often are poor drug candidates because of other factors (e.g., metabolism, side effects, distribution) also intrinsic to their structures that attenuate their efficacy.
A solution to the problems noted above is to modify the drug or drug candidate to change its inherent physical properties. This approach has been heavily used in industry, allowing one to tune the properties of a candidate to meet performance criteria. Often this takes the form of changing the lipid characteristics (e.g., methyl to ethyl esters) of a drug lead to achieve the right balance of water and membrane solubility and passage without compromising its potency or selectivity. It is not uncommon to make hundreds if not thousands of such modifications to a lead to find the optimally tuned clinical candidate.
Another approach to improving the performance characteristics of an agent that is difficult to formulate, exhibits poor bioavailability, or suffers from metabolism, distribution or excretion problems is to attach the agent to a molecular transporter. The properties of the drug-transporter conjugate could then be influenced, if not dominated, by the properties of the transporter. For example, conjugation of poorly water-soluble taxol to an octaarginine transporter produces a conjugate that is freely water soluble and for reasons to be discussed later readily enters cells [2]. For many applications, the transporter-drug conjugate can be designed as a prodrug, i.e., with a linker that is chemically or biochemically cleaved after passage through a barrier, allowing for release of the free drug in targeted cells or tissue.
There are many types of “molecular transporters”. The term itself is connected to function and thus applies to all structural varieties of agents that enable or enhance passage across biological barriers. This classification by “function” (i.e., transporter) that we introduced is important because the frequently used classification by “structure” (e.g., peptide) is limiting and often misleading, especially with respect to design, one focus of this chapter. For example, the term “cell-penetrating peptides” (CPPs), while accurate in part with respect to function, would be technically and obviously limited to “peptides”. However, as discussed herein, systems have been designed based on peptide leads which, while emulating the cell-penetrating function of the leads, are not themselves peptides. For example, our realization that the key to the uptake exhibited by a peptide derived from the protein Tat (a.k.a. Tat 9-mer: RKKRRQRRR) is simply a function of its array of guanidinium ions led to our preparation of guanidinium-rich peptides (octaarginine or R8), peptoids [1], oligocarbamates [3], and dendrimers [4]. All exhibit similar function – high water solubility and rapid entry into cells – but they differ structurally in many significant respects except for the key conserved oligoguanidinium array. In addition to peptides, peptoids, and oligocarbamates, many other types of molecular transporters have been identified that cover a range of structural classes, including polyamines [5,6], polysaccharides [7], steroids [8], cationic lipids [9,10], guanidinoglycosides [11], and even nanotubes [12], the last shown recently to transport proteins into cells [13]. The term molecular transporters thus fully covers these structural variants and their similar function. In addition, many of these transporters can be hybridized (e.g., steroid-modified oligoguanidines [14]) to create new transporter types. Our focus here will be on guanidinium-rich transporters (GRTs).
In considering the mechanism of uptake of GRTs it is important to take stock of the variables that influence passage through a barrier including, in addition to the transporter structure itself, temperature, incubation time, assay solution or conditions, cell type, tissue type, counterions, cargo size and type, and linker amongst others. This complexity has contributed to the view by some that the mechanism of uptake is “controversial”. Still others have argued for a single mechanistic interpretation. These positions are not readily sustained for any one transporter class given the range of variations in transporters, cargoes, cell types, assay conditions and the general lack of systematic analysis in this emerging field. For example, it should neither be surprising nor controversial that a Tat 9-mer with a protein cargo and a Tat 9-mer with a peptide cargo, being very different structures, exhibit different uptake characteristics [15]. Similarly, cellular uptake of an octaarginine attached to fluorescein is very rapid whereas changing the cargo from fluorescein to a polycarboxylate completely shuts down uptake [16,17]. The same transporter is used but its uptake is clearly influenced by the cargo in ways that are quite reasonable mechanistically and certainly not controversial. Similarly, some attempts to make comparisons between uptake in adherent and non-adherent cells or in different cells types are fraught with problems that arise from non-systematic “comparative” analyses. The attempt to fit such complex behavior into one mechanistic box is neither sustained by the current state of analysis nor by historical mechanistic analyses. For example, on the more fundamental issue of what constitutes a mechanism, it is now widely understood that the mechanism of even simple substitutions reactions – often the first reaction studied in organic chemistry courses - can be unimolecular or bimolecular depending on conditions, substrate structure, leaving group, nucleophile, solvent, temperature, and many other variables. Indeed, it is observed in some cases that both mechanisms can operate concurrently. Moreover, new substitution mechanisms such as single electron transfer have been uncovered. While seemingly far afield from the subject of transporter uptake mechanisms, this caveat on mechanistic analyses merits attention in considering how GRTs enter cells. In short, while a given system (transporter, linker, cargo, cell type, assay conditions, etc) might exhibit behavior consistent with a single mechanism, it is unlikely that a single mechanism will apply to all systems and it is possible that more than one mechanism might operate concurrently.
During the past decade, many significant and exciting advances have been reported from our and other laboratories related to the mechanisms of uptake of GRTs and especially to their use in mediating cell and tissue uptake. An octaarginine transporter linked to Cyclosporin A has also been advanced to phase II clinical trials for dermatological applications [18] and a Tat 9-mer linked to a biologically active peptide cargo has been advanced clinically for the treatment of ischemia [19]. In this chapter we provide an overview of mechanistic studies pertinent to the uptake of GRTs and of how these studies have influenced the design of new GRTs. An emphasis will be placed on the most recent literature.
1. Design and synthesis of guanidinium-rich transporters (GRTs)
2.1. Structure-function relationships of fluorescently labeled peptides inspired by Tat49–57
In 1996, we started a program directed at designing molecular transporters that would be superior to the Tat 9-mer (RKKRRQRRR or Tat49–57) in performance and in cost, thereby allowing for broader exploitation of transporter-based drug delivery for therapeutic applications. Our selection of the Tat 9-mer as a starting point relative to other transporter leads was influenced by the rather paradoxical but potentially therapeutically valuable behavior of this lead: it is highly polar and thus readily soluble in water – a factor that could be exploited in therapeutic administration – but unlike most polar systems it readily passes through the non-polar membrane of cells. At the time, little was known about the mechanism of uptake of the Tat 9-mer. Our initial efforts were thus focused on elucidation of the structural features of the Tat 9-mer that influence its ability to enter cells. A rigorously systematic and quantifiable assay, involving changes in only one variable at a time, was developed in which uptake into Jurkat cells of transporters conjugated to fluorescein was measured by confocal microscopy and FACS analysis (fluorescence-activated cell-sorting).
A review covering some of this work has recently appeared [20] and thus only a summary of findings will be presented here to provide a context for understanding more recent studies. Comparison of the uptake of the Tat 9-mer conjugated by an aminohexanoic acid linker (ahx) to fluorescein (Fl), i.e., Tat49–57-ahx-Fl (1, Fig. 1), to the uptake of N- and C-truncated Tat variants (Tat50–57-ahx-Fl, Tat51–57-ahx-Fl, Tat49–56-ahx-Fl, and Tat49–55-ahx-Fl) revealed that truncations lead to less effective uptake and that deletions of arginines cause greater reduction in uptake than deletions of lysines, an important early clue on mechanism. In a second comparative study, the uptake of Tat49–57-ahx-Fl 1 was compared to the uptake of nine Tat variants differing systematically only by the replacement of each residue in the Tat 9-mer by an alanine (e.g., AKKRRQRRR-ahx-Fl, RAKRRQRRR-ahx-Fl, etc.). All 9 variants performed less well than the Tat 9-mer lead but interestingly the one in which a non-charged glutamine residue is replaced by an alanine (i.e., RKKRRARRR-ahx-Fl) was the least affected [1].
Fig. 1.

Structure-function studies with fluorescently labeled peptides inspired by Tat49–57 (counterions not specified).
The above results suggested that charge is important for uptake, leading to the preparation of a lysine 9-mer (K9-ahx-Fl, 2). This construct performed less well than the Tat 9-mer lead. Significantly, examination of the corresponding arginine 9-mer (R9-ahx-Fl, 3) provided a breakthrough in our work as it proved to be superior in uptake to the Tat 9-mer reference. Both enter cells in minutes, but the arginine 9-mer enters more quickly, thus allowing for a greater amount of internalization. A hypothesis was entertained that the arginine-rich transporter might be a substrate for a cell surface convertase, enzymes which recognize, cleave, and internalize arginine-rich precursor proteins [21]. However, it was subsequently found that an oligomer of the non-natural arginine stereoisomer (D-arginine), namely r9-ahx-Fl (4), works as well if not slightly better than the natural isomer 3, suggesting that enzyme recognition, a process usually very sensitive to chirality, is not at play. The slightly better performance of the D oligomer is an expected consequence of its greater stability towards proteolysis. As will be discussed later, the finding that both D and L oligomers work similarly was of enormous importance in the design of new transporters as it suggested that the backbone of the transporter is not critical to uptake.
In the course of the above studies it was determined that uptake varies with the number of arginines in an oligomer. Uptake is observed for oligomers of 6–20 arginine residues with a maximum around 15, yet another finding which at the time required mechanistic reconciliation. The observation that shorter oligomers of arginine do not readily enter cells provided the basis for selecting the tetraarginine system as a negative control. Our eventual selection of an octaarginine transporter for many fundamental and clinical studies is a reflection of the interplay between design, cost considerations, and mechanism. For reasons that go beyond the scope of this review, construction of an 8-mer (r8) through a segment doubling strategy can be conducted more cost effectively, on scale, and with greater step economy than resin-based syntheses [22]. As our goals from the outset were to improve the performance and cost of transporters, the arginine 8-mer became the preferred candidate to move forward, representing a compromise between best performance and lowest cost.
2.2. Linear GRTs
The key finding that the transporter backbone stereochemistry is not critical for uptake opened a wide range of design opportunities and indeed allowed for a transition from peptide-based transporters to non-peptidic systems. It also called increasing attention to the view that uptake might be predominantly influenced by the guanidinium groups. To further test this hypothesis, the first oligoarginine peptoids were prepared (5, Fig. 2) [1]. These preserve the 1,4-spacing of the backbone but move the side chain attachment from carbon to nitrogen. The resultant peptoids significantly outperform the oligoarginine transporters in uptake experiments, pointing again to the importance of the guanidinium group and now to the apparent lack of a secondary structure requirement based on hydrogen bonding of the amide functionality. Of further mechanistic and design importance was the finding that peptoids with longer side chains (5, m=5, n=9), and thus greater flexibility, perform better in uptake. This again teaches away from a receptor-mediated uptake process.
Fig. 2.

Representative linear GRTs (counterions not specified).
The above studies revealed that backbone stereochemistry is not critical for uptake and that the side chain could be attached on carbon as in the arginine oligomers or on nitrogen as in the peptoid oligomers (5). In order to explore whether the backbone itself could be changed and whether the side chain spacing is important, a series of guanidinium-rich oligocarbamates were prepared (6, Fig. 2) [3]. These incorporate carbamate groups connected by a 2-carbon spacer to which is attached a side chain terminated with a guanidinium group. In contrast to the peptides and peptoids in which side chains are 1,4-spaced along the backbone, these oligomers have side chains 1,6-spaced. These proved to be very effective transporters. For example, the carbamate 9-mer 6 (n=9) translocates into cells 2.3 times faster than the D-arg 9-mer (4, n=9) which itself is taken up into cells approximately three times faster than Tat49–57. While many studies address only cellular uptake, this study also showed that the oligocarbamate transporters penetrate mouse skin, a finding of much significance as the skin represents a much more formidable barrier than the plasma membrane and is the key barrier for many dermatological applications.
A more ambitious design modification of the backbone led to the synthesis and study of a library of “spaced” arginine-rich transporters (7, Fig. 2) [23]. There are 64 permutations involving insertion of a single aminohexanoic acid spacer (ahx) between consecutive arginines (R7, R-ahx-R6, R2- ahx-R5, up to R-[ahx-R]6). Interestingly, the maximally spaced construct (R-[ahx-R]6), containing seven arginines, outperforms unspaced heptaarginine in uptake by six-fold. Conversely, when octaarginine is replaced with two tetraarginines separated by a rigid, substituted benzene spacer (8), a 3.8-fold decrease in cellular translocation is observed at 25 μM (T. Pillow, unpublished results). Consistent with results from peptoids, these studies again suggest that greater flexibility leads to faster uptake. This is inconsistent with a receptor-mediated pathway but consistent with a dynamic pathway that requires weak associations and fast turnover.
A further creative variation of the transporter backbone was reported by Seebach and coworkers in impressively comprehensive studies using β-peptides [24]. They showed that a series of fluorescein-labeled β-peptides readily enter 3T3 mouse fibroblast cells. Localization in the nucleus is found. Uptake is observed only for polycations, the β-oligolysine 9 and β-oligoarginine 10, with the latter being more effective. It is noteworthy that, like the arginine-based systems, acute toxicity is not observed and uptake is rapid, with fluorescence as intense after 5 minutes as after 40 minutes for the arginine analog 10.
Gellman and coworkers have recently reported a study in which they investigated the effect of the spatial arrangement of guanidinium groups and helix stability on cellular uptake [25]. They have prepared a series of fluoresceinated arginine rich β-peptide heptamers and studied their uptake in HeLa cells by FACS and confocal microscopy (cells were not fixed) by incubation with the peptide for 15, 30, 60 and 120 min at 8 μM. Their results indicate that the uptake of the β-peptide that forms a stable 14-helix in aqueous solution with all six arginines clustered along one side is about 6-fold higher than the uptake of other analogs after 30 min incubation. Interestingly, the peptides are similar in cell surface binding with the best performing peptide being only 2-fold better than the rest. All of the peptides produced an endosomal-like punctate pattern of distribution when imaged by confocal microscopy after a 2-hr incubation.
Collectively, these studies show that uptake of the Tat 9-mer is principally determined by its arginine content and that homo-oligomers of arginine outperform the Tat 9-mer. An especially important finding is that the backbone chirality, backbone types, spacing, and hydrogen bonding can be varied indicating that the key to uptake is the singularly conserved element, specifically the guanidinium group.
2.3. Dendrimeric and branched GRTs
The preceding studies illustrate the range of design options available using a linear backbone scaffold to which guanidinium-terminated side chains are attached. Another design motif would involve branched or dendrimeric scaffolds. Goodman and coworkers reported a series of polyguanidinylated dendritic structures that readily enter cells (11–15, Fig. 3) [26]. The dendritic oligoguanidines were conjugated to fluorescein and to a mutant green fluorescent protein (GFP). Both the small molecule and the protein conjugates translocate efficiently into HeLa S3 cells as quantified by FACS analysis. The conjugates 14–15, possessing 9 (G9) and 12 (G12) guanidinium groups, respectively, exhibit better uptake than 12 and 13 with 3 and 6 guanidinium groups. The authors indicate that 9 guanidinium groups appear to be optimal. It is noteworthy mechanistically that the enhanced uptake observed when the number of guanidinium groups was increased to 9 is similar to that found with peptoids. To investigate the localization of the conjugates, the authors also treated HeLa S3 cells individually with G9-Fl (14), G9-GFP, and Tat(49–57)-GFP. The cells were fixed and visualized by deconvolution microscopy. The resultant images of G9-Fl (14) and G9-GFP indicate that they enter cells and localize in the nucleus, cytoskeleton and cytoplasm. While not necessarily relevant to this study, caution is often recommended in analyzing results from cell fixation because of its ability to produce artifacts in the cellular distribution of GRTs [27].
Fig. 3.

Representative dendrimeric and branched GRTs (counterions not specified).
We have also reported the synthesis and study of guanidinium-rich dendrimers (16, Fig. 3) [4]. While we have noted previously that uptake is principally determined by the number of guanidinium groups, the scaffold does play a role. For example, even though all of the dendrimeric transporters 16 have the same number of guanidinium groups, they exhibit different rates of uptake into Jurkat cells as measured by FACS analysis after a 3-minute incubation time. The hexyl-hexyl (16, n=k=5) and hexyl-propyl (16, n=5, k=2) dendrimers are the most effective while the ethyl-ethyl (16, n=k=1) dendrimer is not detected intracellularly during the time course of the experiment. The most effective dendrimers outperform the nonaarginine system.
Recently, Harth and coworkers have reported the design, synthesis and assay of two “Newkome-type” dendrimers with guanidinium head groups (17 and 18, Fig. 3) that exhibit subcellular targeting [28]. Internalization into mammalian cells was visualized with a fluorescein label. The subcellular distribution varies according to structure with 17 concentrating in the nucleus and 18 in the cytosol. Similar results are obtained with NIH-3T3 fibroblasts and with human microvascular endothelial cells. The authors report an influence of the alkyl spacer on localization, though they do not provide an explanation. It is noteworthy but not surprising that the transcription factor Tat, the Tat 9-mer, and some other GRTs localize in the nucleus. Collectively, this earlier work and the more recent dendrimer studies suggest that molecular transporters can be used to not only enhance cellular uptake but also to target subcellular destinations.
The Futaki group has reported another type of cleverly designed branched transporter using lysine residues to create nodes of divergency, glycines to incorporate flexibility and extension, and arginines to supply key guanidinium residues (19–21, Fig. 3)[29]. Uptake into HeLa cells was studied using fluorescence microscopy. The (R2)4 peptide 19 that has arginine residues on four branched chains exhibits the most efficient translocation. Not unlike linear transporters, uptake is found to be a function of the number of arginine residues.
2.4. Carbohydrate-based GRTs
Goodman and coworkers reported the preparation of guanidinoglycosides, in which the amine groups of natural aminoglycosides (kanamycin A, kanamycin B, tobramycin, neomycin B, and paromomycin) are converted into guanidinium groups [30]. A solid-phase assay method used to evaluate the RNA specificity of the guanidinylated compounds revealed between 5- and 10-fold increases in inhibitory activity for the guanidinoglycosides. Also, they reported that guanidinotobramycin and guanidinoneomycin B inhibit the replication of the HIV virus with activities approximately 100 times greater than the parent aminoglycosides [31]. However, no comparative cell uptake study has been reported.
Tor and coworkers reported a noteworthy study on guanidinylated aminoglycosides [32]. They prepared a series of BODIPY-conjugated aminoglycosides and guanidinoglycosides and evaluated their cellular uptake properties by FACS. This study revealed that when the amine groups in tobramycin and neomycin B (22a and 23a, Fig. 4, TFA = trifluoroacetic acid) are converted into guanidine groups (22b and 23b), cellular uptake of the resultant vectors are enhanced by 10- and 20-fold, respectively. A comparison of their uptake relative to a fluorescently labeled R9 (positive control) demonstrated that guanidinoneomycin B (23b) exhibits a highly efficient uptake, superior to that of the positive control [11]. The authors also noted that the cellular uptake of fluorescent R9 is inhibited by guanidinoneomycin B, suggesting some similarity in the uptake mechanism for both the arginine-rich peptides and the guanidinoglycosides.
Fig. 4.

Representative GRTs based on carbohydrate scaffolds.
In a subsequent study, the authors explored the cellular requirements for uptake of guanidinoneomycin B as well as its delivery potential [11]. Biotinylated neomycin B (24a), biotinylated guanidinoneomycin B (24b), and biotinylated R9 were conjugated to streptavidin-PE-Cy5 (>300 kDa) and their uptake was studied in wild type HeLa and CHO cells and four CHO mutant cell lines (two glycosaminoglycan-deficient, heparan sulfate-deficient, and sialic acid-deficient lines). To test the dependence of guanidinoglycoside uptake on heparan sulfate, the experiments in wild type cells were also done in the presence and absence of heparan sulfate lyases. A significant observation in this study is that at nanomolar concentrations, the uptake of the guanidinoneomycin B depends entirely on cell surface heparan sulfate proteoglycans while the uptake of an R9 system follows both heparan sulfate-dependent and -independent pathways. As noted before in connection with the role of scaffold, this indicates that, for a given number of guanidinium groups, their spatial array can influence uptake and that multiple mechanisms of uptake can operate. The high selectivity of guanidinoneomycin B for heparan sulfate suggests that these systems can be used to selectively target cells based on proteoglycan composition differences.
It is also noteworthy that these systems can be used to carry large (>300 kDa) bioactive molecules into cells. Conjugation of guanidinoneomycin B 24b to the streptavidin-labeled plant toxin saporin, a ribosome-inactivating agent, results in proteoglycan-dependent cell toxicity. Notably, saporin itself does not kill cells due to the lack of cell surface receptors [33]. However, conjugation of saporin to a ligand for which receptors exist leads to cell death. The complex formed from guanidinoneomycin and streptavidin-labeled saporin kills the wild type CHO cells with an IC50 of ~2 nM. No toxicity is observed for unconjugated guanidinoneomycin B 24b or for free saporin. Also, mutant cells are not affected by saporin up to a concentration of 100 nM. Significantly, this proteoglycan-dependent behavior of the guanidinoneomycin transporter in comparison to R9 could provide the basis for the development of more selective cellular delivery vectors.
Recently, Chung and coworkers reported a novel class of octaguanidinium transporters (25–31, Fig. 5) based on an inositol dimer scaffold [34,35]. The dimers are prepared by linking two units of myo- or scyllo-inositol, and multiple guanidine functionalities are introduced through a peracylation of the inositol scaffold with ω-aminocarboxylate derivatives of varying length. The cellular uptake of these transporters was studied by confocal microscopy in three cell lines (simian kidney COS-7, mouse microphage RAW264.7, and HeLa cells) using a 3–5 minute incubation time. The results are compared with dansyl- and Fl-R9 peptides. The authors observed that conjugates 25–27 have similar uptake to dansyl-R9 in COS-7 cells with conjugates 26 and 27 exhibiting slightly better uptake than 25. Comparison of myo- and scyllo-inositol scaffolds (26 and 28) shows similar uptake performance in RAW264.7 cells. For HeLa cells, the transporters based on scyllo-inositol linked via an amide exhibit greater uptake with longer chain length (31>30>Fl-R9>29). In HeLa cells, 31 and 30 show 2.5 and 1.8 times greater uptake than Fl-R8, respectively, whereas 29 is half of that for Fl-R8. While the uptake mechanism for these transporters is unclear, their localization is different from that observed for other GRTs, suggesting again that the spatial orientation of guanidinium groups could be used to confer selectivity in performance or localization.
Fig. 5.

Representative GRTs based on inositol dimer scaffolds.
In vivo biodistribution studies of conjugate 31 based on ip (intraperitoneal) injection into mice reveal differences relative to Tat-related peptides, the latter distributing relatively uniformly in the liver, kidney, lung, heart muscle and spleen [36]. Conjugate 31 demonstrates much higher distribution in heart, lung, and brain tissues with only a weak signal observed in liver, kidney and spleen. In a second tissue distribution experiment the authors observed a strong signal from the brain cortex 20 min after ip injection, consistent with the passage of conjugate 31 across the blood-brain barrier. Lastly, a derivative of 31 in which Fl is replaced with doxorubicin (an anticancer antibiotic) was prepared and shown to effectively translocate into the cytoplasm of HeLa cells producing a stronger cytotoxic effect than doxorubicin itself. Ip injection of the conjugate also produces a strong signal in the brain cortex relative to free doxorubicin. These studies suggest that variations in the transporter conjugate could result in selective tissue and cell targeting [35].
Chung et al. also studied two types of branched transporters based on sorbitol and a bis-guanidine unit (32a and 32b, Fig. 6) [37]. Sorbitol (D-glucitol) was selected in part because it is safe, being found in fruit and also readily available from D-glucose or D-glucono-1,4-lactone. Conjugate 32a (n=5 and 7) and fluorescently labeled R8 (at 10 μM) entered cells with comparable efficiencies as evaluated by confocal microscopy and FACS analysis. The internalization of 32b was less efficient. Uptake of 32a increases as its concentration is increased up to 20 μM, reaching a plateau after approximately 3 hr.
Fig. 6.

Representative GRTs based on sorbitol scaffolds.
Conjugates 32a (n=5 and 7) exhibited similar cytosolic distributions while 32b differed in its intensity. Internalization of 32a is inhibited at 4 °C, suggesting entry by an energy-dependent path. However, 32a (n=5) did not colocalize with tetramethylrhodamine-labeled Tat peptide or transferrin, suggesting that different mechanisms of uptake might be involved. It is interesting that 32a colocalizes with a mitochondria-specific dye (MitoTracker Red) in HeLa and in KG1a leukemia cells, suggesting mitochondrial accumulation and a possible use of this or related systems for detection or treatment of mitochondrial-based diseases. The tissue distribution of 32a (n=5) was also studied. While Tat-based systems are found to distribute widely in the liver, kidneys, lungs, heart muscle and spleen, 32a shows higher selectivity for lung, heart muscle and brain tissues. These findings are of potential significance for targeted therapy based on GRTs.
2.5. GRTs based on a polyproline helix and other scaffolds
Another fascinating class of cell-penetrating vectors, reported by Chmielewski and coworkers, uses trans-proline residues known to adopt a well-defined left-handed type II helix in polar solvents (33–34, Fig. 7) [38]. The designed transporters have repeating hydrophobic and hydrophilic faces. The hydrophobic face consists of a proline-leucine mimetic and the hydrophilic face consists of either a proline-arginine or a proline-lysine mimetic. Several fluoresceinated control compounds were prepared, including Tat peptide (Fl-G4YGRKKRRQRRR-NH2), tetra- and hexalysine (Fl-K4 or 6-NH2) and tetra- and hexaarginine (Fl-R4 or 6-NH2) peptides and scrambled versions of their proline analogs with arginine and lysine mimetics mixed together. All compounds (at 50 μM) exhibited uptake in MCF-7 breast cancer cell lines as determined by FACS after an incubation time of 6 hr. A much higher cellular uptake is achieved with the guanidinium-containing compounds (33) in comparison to the amine analogs (34). Secondly, the cellular uptake increased with increasing number of cationic groups (33(n=3)>33(n=2) and 34(n=3)>34(n=2)). Both observations are consistent with the previously reported behavior of peptoids and oligoarginines [1]. In addition, the proline tetramer and heptamer of arginine 33 (n=2 and 3, respectively) are 1.5 and 7.7 times more effective in entering MCF-7 cells than their peptidic analogs (Fl-R4-NH2 and Fl-R6-NH2). The most potent conjugate, 33 (n=3), is also almost an order of magnitude more effective than Tat peptide. However, when the amine analogs (34 (n=2–3) and Fl-K4 or 6-NH2) are compared in the same fashion, no such enhancement is observed in comparison to the peptidic analogs, with 34 (n=3) being only 1.7 times better than Fl-K6-NH2. Interestingly, the enhancement effect is lost when the uptake of R6 is compared to the “scrambled” version of 33 (n=3) that had the same number of arginines, but no amphiphilic character. Based on these results, the authors conclude that the amphiphilic character of the transporter plays an important role in the enhancement of cellular uptake. The authors also demonstrate that in both fixed and living MCF-7 cells, proline-based analogs localize in the cytoplasm and nucleus. Finally, based on an MTT assay, no toxicity is observed for the new conjugates at the concentration used for the study (50 μM).
Fig. 7.

GRTs based on a polyproline helix and other scaffolds (counterions not specified).
Mendoza and coworkers recently reported a new family of tetraguanidinium compounds 35, in which bicyclic guanidinium subunits are linked by thioether spacers [39]. The cell-penetrating ability of the conjugates was studied in HeLa cells by confocal microscopy (both in living and fixed cells) and FACS analysis after a 1-hour incubation. The uptake was compared with fluorescently labeled Tat49–57 9-mer and Antennapedia (RQIKIWFQNRRMKWKK-NH2). Interestingly, conjugate 35 (R=OH) outperforms its silylated analog 35 (R=SiPh2tBu) and peptidic controls at low concentrations (0.1–1 μM). The compound was found to be toxic in an MTT assay with 72% reduction in cell viability at 10 μM concentration.
Lipidated oligoarginine systems have been prepared by Tung and coworkers and their uptake has been studied in HeLa cells by incubating with the conjugate for 1.5 hr [40]. In this study, the most effective conjugate (11-mer of arginine conjugated to myristic acid) is an order of magnitude better than Tat 9-mer or R7. Acylation of R7 with the same lipid also improves its uptake by about 6-fold.
2.6. GRTs based on peptide nucleic acids
The seminal recognition of the key importance of guanidinium groups in mediating cellular uptake and the lower sensitivity of the system to chirality, scaffold, and spacing, allowed for the dramatic expansion of the field of GRTs to many other biooligomers and scaffolds. Oligonucleic acids have emerged as another fascinating class of GRTs. In general, there are two main reasons why connecting guanidinium groups to oligonucleotides (ONs) could be advantageous. First, it could enhance solubility because, in comparison to their amino analogs, guanidine ONs would remain protonated under a wider range of physiological pHs due to the highly basic character of the guanidinium group (pKa=12.5). Second, and of great significance, it could enable the uptake of ONs or ON functional equivalents into cells.
In a key study, Ly and coworkers reported guanidine nucleic acids, based on the insertion of arginines into the nucleic acid backbone [41,42]. Cellular uptake was studied by confocal microscopy in HCT116 (colon) and Sao-2 (osteosarcoma) cell lines by incubation of cells with the conjugate (1 μM) for 10 min. The cellular uptake of guanidine peptide nucleic acid 36 is found to be similar to that of the Tat 9-mer peptide. The amino analog of 36 does not enter cells, suggesting again the importance of the guanidinium group. Interestingly, the authors also show that the guanidino nucleic acids maintain Watson-Crick recognition with complementary DNA strands and the unnatural analog (D-Arg) 36 is more biologically active than its natural analog (L-Arg). The spaced arginine peptide nucleic acids (NH2-Arg-T-Arg-G-Arg-T-Arg-A-Arg-C-Arg-G-Arg-T-Arg-C-Arg-A) show similar uptake properties to non-spaced systems.
Ohmichi et al. have also demonstrated the importance of the guanidinium groups for cellular uptake of DNA using deoxyuridine analog 37 that contains guanidinium groups at the C5 position as an example [43]. A 20-mer DNA oligomer was labeled with fluorescein at the 5′ end (5-Fl-dTGTGAAGTGTCCCAGCCTGT) and repeating sequences were added to the 3’ end by primer extension. The uptake of the resulting 20-mer was studied in HeLa and RAW264.7 cells by confocal microscopy and FACS after a 48-hr incubation period. The results reveal that introduction of the guanidinium groups does not inhibit duplex stability but does enhance uptake. Interestingly, the 20-mer containing four guanidinium groups is found to be the most effective construct, followed by systems with three and then six guanidinium groups.
Lebleu and coworkers recently reported the cellular uptake of new fluorescently labeled guanidinylated DNA analogs (38) [44]. The results of their study are consistent with previous work, showing that the guanidinylated 12-mer of 38 is taken up into HeLa cells six times more efficiently than its unmodified analog. The authors observed mainly cytosolic distribution of the construct.
Multiple phosphate-modified deoxynucleic guanidines (39) in which negative phosphodiester linkages of DNA are replaced with guanidinium groups have been reported by Bruice and coworkers. In addition to being highly nuclease resistant, these compounds also have high binding affinities and specificities for various duplex formations [45,46,47]. While they enter cells, their cellular uptake has not yet been quantified.
While many of the above studies are based on covalent attachment of a GRT to its cargo, Khavari, Wender, and coworkers reported that an arginine-rich cysteine-flanked peptide (CG(RHGH)5RGC), when templated with DNA can be used to form a disulfide-linked polymer that non-covalently associates with DNA plasmids and enhances their uptake into cells and tissue [48]. The plasmid encoded for luciferase was used, allowing for an optical real-time readout when the cells or animals are treated with luciferin. Uptake of the DNA plasmid is observed and is found to be more significant than naked DNA alone. The transfection efficiency in cells is found to be greater than that observed with commercial transfection agents (e.g., lipofectin). It is noteworthy that in this case the more arginine-rich system worked less well. It was reasoned that the better performance of the preferred histidine and arginine-rich peptide is due to the role of the histidines in enabling escape from the endosome for an endosomal uptake path [49]. This represents one of the largest molecular cargoes carried into cells and animal tissue. It is also an encouraging starting point for the development of non-viral gene vectors for gene therapy.
3. Internalization mechanisms of GRTs
The preceding section provides examples of the wide range of GRTs that have been shown to enter cells and in some cases animal tissue. As noted at the outset, given the structural variety of GRTs and cargoes, the latter running from small molecule probes, metals, and drugs to peptides, oligosaccharides, proteins and plasmid DNA, a universal GRT uptake mechanism is unlikely. However, there are common aspects to many mechanistic possibilities and these derive from the special properties of the guanidinium group. In this section some general observations pertinent to the mechanism of cellular entry of GRTs and thus the design of new GRTs are presented.
3.1. Association
The generally agreed upon first step of the translocation mechanism involving GRTs is the association of the oligoguanidinium transporter with the membrane. Each positively-charged guanidinium head group features a specialized, rigid planar array of hydrogen bond donors that allows for highly effective bidentate hydrogen bond formation with negatively-charged carboxylates, phosphates, and sulfates (Fig. 9). These negatively-charged functional groups are subunits of many cell membrane constituents such as phospholipids, fatty acids, proteins, and heparan sulfate proteoglycans (HSPGs). As charged (polar) groups they often extend beyond the cell surface or membrane inner leaf into the surrounding polar milieu. Relative to the single hydrogen bond that would be possible with the ammonium group in lysine, the bidentate hydrogen bond network formed from guanidinium groups is stronger and thus provides a structural rationalization for the differing abilities of oligolysines and oligoarginines to associate with and cross cellular membranes [50]. The decreased translocation ability of lysine and ornithine relative to arginine also supports the observation that, while charge is necessary, it is not sufficient, as both the guanidinium and the ammonium group have a single positive charge. Obviously, the degree of protonation of the systems under assay conditions is another factor that could be of consequence as oligomers of the more basic guanidinium group would be more protonated (have more cationic sites) relative to lysine analogs.
Fig. 9.
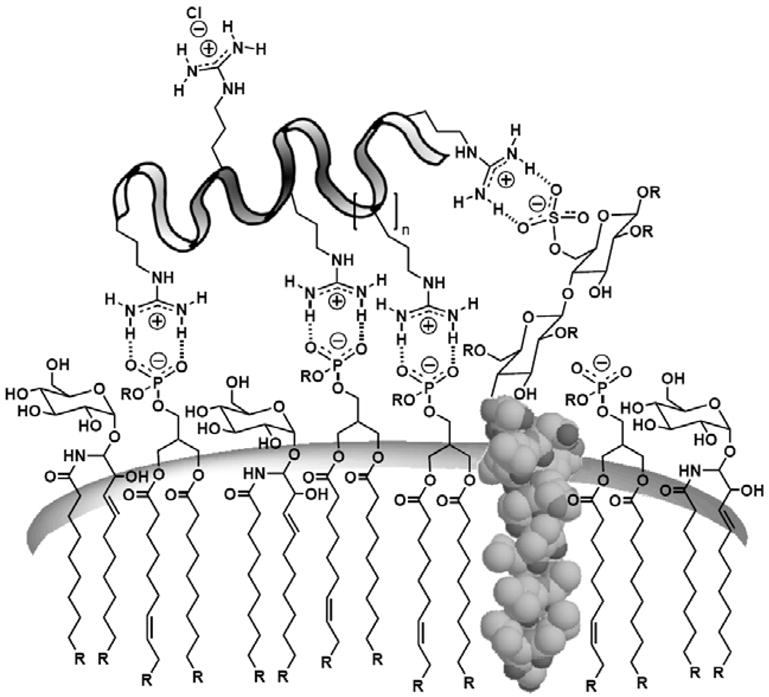
Representative associations of a polycationic guanidinium transporter with anionic cell membrane constituents.
Combining the positive charge with a bidentate hydrogen bond donor results in a functional moiety ideally poised to interact with the negatively charged cell surface constituents. The driving force for this pairing is the associated decrease in free energy in going from solution to the surface of a membrane. While a guanidinium group has no recognizable hydration shell [51] and a very weak association with a counterion such as a phosphate group in an aqueous environment (~ 0.2 kcal/mol) [52], as the guanidinium group approaches the surface of a lipid bilayer and local polarity decreases, its association with a phosphate counterion increases dramatically (~2.6 – 6.0 kcal/mol) [53]. This association is further strengthened as the complex moves into the less polar membrane. The magnitude of this association is expected to increase with guanidinium content. Indeed, arginine oligomers containing fewer than six amino acids are relatively ineffective in entering cells, while oligomers of six or more arginine residues enter readily. Maximal uptake is seen for an oligomer of 15 arginines [1]. The inefficient entry of longer oligomers (>20 arginines) is also explained by this association as the increased interaction resulting from additional guanidinium groups suppresses release of the oligomer from the membrane.
To further explore the importance of this bidentate hydrogen bond, oligomers of arginine containing monomethyl- and dimethylguanidinium head groups were synthesized and attached to fluorescein [54]. A single methyl substitution on each guanidinium group of an oligomer resulted in a compound with conserved charge but with a reduced ability to form a bidentate hydrogen bond. This compound, when assayed for cellular uptake, is 80% less effective than the unalkylated arginine oligomer. Furthermore, dimethylation of each guanidinium group provides a compound unable to form a bidentate hydrogen bond. Significantly, the cellular translocation of this compound is reduced by greater than 95% (Fig. 10). In short, the decreased ability of mono- and dimethylated oligomers of arginines to form hydrogen bonds correlates with their decreased ability to enter cells, suggesting that hydrogen bond formation plays an important role in the entry process.
Fig. 10.

The effect of mono and dimethylation of the guanidinium group on cellular uptake.
Another parameter to consider in the association of a GRT with the membrane is the spatial orientation of the guanidinium head groups. Due to electrostatic repulsion, there is a low probability of having two adjacent anionic cell membrane constituents. Consequentially, an improved association would be found in transporters with a similar spacing in their cationic functionalities. Additionally, an increase in the flexibility of the scaffold presenting the guanidiniums would increase the percentage of guanidiniums capable of interacting with species of complementary charge on the surface of the cell. The hypothesis that increases in guanidinium group spacing and flexibility would improve cell membrane association and cellular uptake is supported by experiments using oligomers of arginine interdigitated with a series of non α-amino acids of increasing length (glycine, β-alanine, 4-aminobutyric acid, 6-aminohexanoic acid). For these systems, an increase in the spacing between arginine residues results in a concomitant increase in cellular uptake, with a maximal level observed with 6-aminohexanoic acid [23]. This increase in cellular uptake is also observed for oligocarbamate transporters with increased spacing of the guanidinium-containing sidechains along the linear backbone [3], as well as in peptoid transporters, where a lengthened spacing between the guanidinium head group and the backbone results in an increase in uptake [1]. Dendrimer transporters also exhibit this property: branches of increasing length and thus flexibility show greater uptake [4].
The favorably aligned bidentate hydrogen bond between a guanidinium-containing transporter and a carboxylate, sulfate, or phosphate on the surface of a cell and the electrostatic attraction of such charged species is expected to produce an association between the transporter and the lipid bilayer. This association strengthens with increasing numbers of and spacing (up to a point) between guanidiniums. With a stronger association between a membrane and a transporter, there is a greater chance of the transporter being internalized, whether through an active or passive transport mechanism. At the same time, this association must be reversible to allow release after uptake. In short, guanidinium groups are well-suited for adherence to cell surfaces bearing negatively-charged groups. The association would allow for increased residence time and therefore favor internalization. The association, while relatively strong, must be reversible to allow for release after entry as discussed below.
3.2. Mechanisms of Uptake
CPPs, and more specifically GRTs, can enter cells through multiple mechanisms. Confocal microscopy is often used to visualize their uptake. It has been proposed that the observed punctate staining is due to endocytosis and that the diffuse cytosolic staining arises from nonendosomal uptake. Other studies suggest that the diffuse cytosolic staining cannot be due solely to escape from the endosomes as it occurs more quickly than the punctate endosomal fluorescence [55]. Maiolo et al. reported that at 4 °C, while there is a significant decrease in the amount of punctate staining of U2OS cells with fluoresceinated R7W, there is an increase of diffuse cytosolic staining. They suggest that the lack of punctate staining indicates an inhibition of endocytosis and that the diffuse cytosolic staining, not inhibited by the cold, is due to a second, non-endocytotic mechanism at work [55]. They have also observed a different degree of endosomal uptake in different cell types with some cell types displaying more diffuse staining than others.
Though there are many possible mechanisms for GRT uptake, there is little doubt that endocytosis is involved with several systems, as many have observed a decrease in cellular uptake at 4 °C, a condition known to inhibit endocytosis. This has been observed with cargoes such as fluorescent tags [27,56,57,67], peptide cargoes [58], fusion proteins [59,60], DNA-Tat complexes [61], gold particles, [62] liposomes [63] and FITC-avidin complexed CPPs [64], though in the last case the authors state that Tat conjugates were less impacted by the low temperature than the other CPPs studied. It has also been shown that chemical means to induce energy depletion result in decreased uptake of CPPs [27,56,61,63,64,65,67], adding further evidence that a significant portion of uptake could involve an energy-dependent mechanism. This energy dependence appears to be cargo sensitive. Exemplifying this, in a comparison between a DNA-Tat complex and a much smaller fluorescently labeled Tat conjugate, the Tat complex exhibited an almost complete loss of uptake into cells pretreated with sodium azide [61], while the smaller conjugate showed no such sensitivity. This will be more fully discussed later in the review.
Within the large category of endocytosis there are several possible mechanisms of uptake (Fig. 11). The most studied form of endocytosis is clathrin-mediated endocytosis. Several authors have proposed that this process is the primary mechanism of uptake of arginine-rich transporters. It has been reported that in HeLa cells, labeled CPPs colocalize with transferrin [27,65] a glycoprotein marker for endocytosis, while others have reported that fluorescently labeled r8 conjugates [57,66] and fusion proteins [59,60] do not colocalize with transferrin. Inhibition of endocytosis was also attempted with varying results. A Tat-avidin conjugate shows only a modest decrease in uptake upon treatment with hyperosmolar medium, a condition that was shown to decrease clathrin-dependent endocytosis. This led the authors to suggest that other mechanisms might be at play [64]. Other authors report that treatment with the endocytosis inhibitor chlorpromazine results in a 50% decrease of uptake of fluorescently-tagged Tat, while a potassium-free buffer results in a 40% decrease in HeLa cells [67].
Fig. 11.
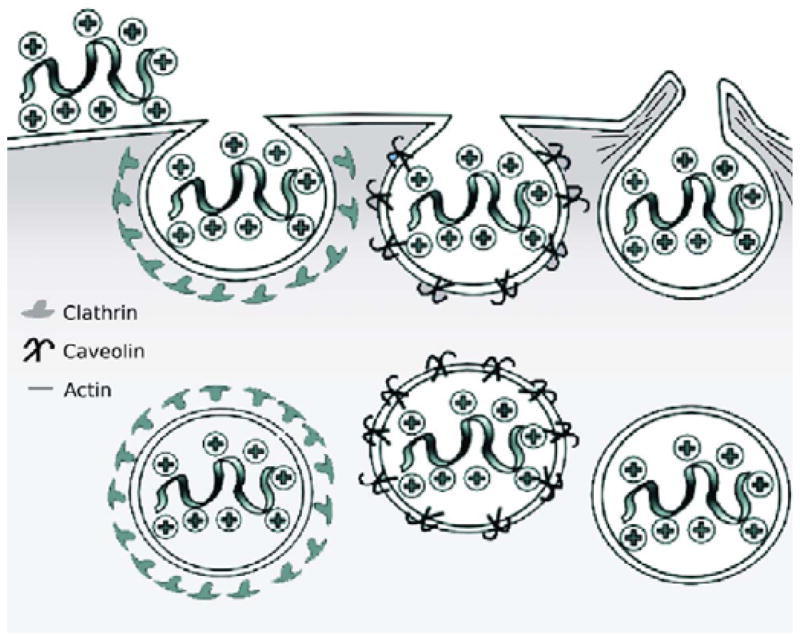
Proposed endocytotic mechanisms of uptake, from left to right: clathrin-mediated endocytosis, caveolin-mediated endocytosis, macropinocytosis.
Other forms of endocytosis have also been implicated with a great deal of current interest being directed toward macropinocytosis. Macropinocytosis is a type of endocytosis that is mediated by lipid rafts and is clathrin-, caveolae-, and receptor-independent. The specific process involves the enclosing of actin-containing membrane protrusions to form vesicles called macropinosomes [68]. These vesicles can often be greater than 1 μm in size and it has been suggested that they are leaky [69]. Specific monitoring of this endocytotic pathway can be achieved by pretreatment with amiloride, an inhibitor of the Na+/H+ exchange that is required in macropinocytosis [70]. Additionally, an F-actin elongation inhibitor, cytochalasin D, can also be used [71]. A number of research groups have proposed macropinocytosis as the mechanism of uptake for CPPs. Dowdy and coworkers have recently reported that both a Tat-Cre fusion protein [58] and FITC-labeled Tat [72] enter cells through lipid raft-mediated macropinocytosis, which is supported by the dose-dependent inhibition of uptake observed when cells are pretreated with amiloride or cholesterol is removed with β-cyclodextrin. However, cargo and cell type must be considered when examining mechanisms of uptake. For example, Ferrari and coworkers observed uptake into 3T3 cells (in contrast to the HeLa and CHO cells in Dowdy’s work) of a different Tat construct, a GST-Tat-GFP fusion protein, through a caveolae-mediated pathway [60], observing colocalization with the caveolar endocytosis marker, caveolin-1. In an excellent mechanistic study of fluorescently labeled octaarginine, Futaki et al. observed a considerable role of macropinocytosis in peptide uptake into HeLa cells, finding that both cytochalasin-D and the macropinocytosis inhibitor ethylisopropylamiloride significantly suppress uptake of the peptide into HeLa cells [57]. Interestingly, in a more recent study, Futaki and Harashima discovered that when octaarginine-modified liposomes containing a rhodamine dye are incubated with NIH3T3 cells, the mechanism of uptake depends upon the density of the peptide on the surface of the liposome [73]. The authors observed a shift from clathrin-mediated endocytosis to a macropinocytosis uptake mechanism as the density of the octaarginine increased. In contrast to the aforementioned groups, Shen et al. reported that the translocation of I125-labeled YGR9 in HeLa cells is not inhibited by amiloride pre-treatment or incubation at 16 °C, which argues for an uptake mechanism distinct from macropinocytosis [74]. They support their claim with a subcellular fractionation method they developed to separate the vesicular from the cytosolic compartments [75]. Additionally, the authors found that coincubation with EGF, a known stimulator of macropinocytosis, does not significantly increase the amount of oligoarginine found in the cytosol.
In addition to macropinocytosis, other lipid raft-mediated forms of endocytosis have been implicated, in particular caveolin-dependent endocytosis. Tat-GFP in HeLa [59] cells and in CHO-K1 and HL3T1 cells [60] have been shown to colocalize with caveolin-1. Both Tat-rhodamine [66] and Tat-GFP complexes [60] have also been shown to colocalize with cholera toxin. Cholera toxin is known to proceed through a caveolin-dependent pathway [76]. β-cyclodextrin, which is known to disrupt lipid rafts, has been reported to have differing effects on uptake. FITC-avidin complexed with biotin-CPP exhibits only a 20% drop in uptake [64] upon treatment with β-cyclodextrin, while the uptake of a Tat-GFP fusion peptide is shown to be significantly inhibited by methyl β-cyclodextrin [59] as was a Tat-rhodamine compound whose uptake is substantially reduced in HeLa cells [66]. Also, nystatin, a compound known to inhibit caveolae formation, only decreased the amount of uptake in some cell lines (CHO1) and not others (BGM) and only for some cargoes such as a large DNA-Tat complex and not for fluorescently labeled Tat alone, suggesting that not only is caveolae-mediated uptake cell dependent, but also cargo dependent [61]. Other authors have found that nystatin and filipin III have little impact on the uptake of fluorescently labeled Tat into HeLa cells or CHO cells [67]. Some have proposed that whether a specific conjugate is taken up by clathrin or caveolae endocytosis is highly dependent on cargo and cell type. Indeed, a clathrin-mediated endocytosis mechanism predominates with fluorescent beads less than 200 nm in diameter, but as the size of the beads increases the mode of uptake becomes increasingly caveolae-mediated. For beads of 500 nm in diameter, the caveolae pathway dominates uptake [77].
Another type of mechanism that has been proposed for cellular entry is non-endocytotic, direct diffusion through the membrane. The uptake of a charged (polar) species into a highly lipophilic (non-polar) environment such as a membrane would seem to oppose dogma. However, natural examples of such a process do exist as demonstrated in the groundbreaking research by MacKinnon on potassium ion channels, in which four highly conserved arginine residues in the channel’s voltage sensor are shown to move into the lipid environment of the membrane [84,85]. Numerous models for understanding how direct diffusion of GRTs could occur have been suggested. Derossi et al. proposed an inverted micelle model where positively-charged peptides interact with negatively-charged membrane constituents to form an inverted micelle structure which can open on the inside or outside of the cell [86]. A carpet model, in which aggregates of peptides coat the cell surface and disrupt its structure, has also been proposed [87]. While pore formation in the membrane is also possible, this mechanism can be ruled out by testing for membrane leakage of lactate dehydrogenase or propidium iodide staining [88]. Our group has proposed a mechanism of uptake called adaptive translocation, which involves the recruitment of negatively-charged cell surface constituents by the positively-charged GRT peptide as the latter contacts a cell surface, transiently forming an ion pair complex with attenuated polarity that is able to adaptively diffuse into the membrane and subsequently into the cell (Fig. 12) [54]. This mechanism is supported by a simple water-octanol partitioning experiment in which it is shown quantifiably that highly water-soluble arginine oligomers partition into the non-polar membrane surrogate phase (octanol) upon addition of sodium laurate, a surrogate of a membrane constituent. Solubilization is readily quantified by phase separation and weighing of the dissolved residue. Remarkably, the once water soluble (>95%) GRT becomes membrane soluble (>95%) by forming a complex with membrane constituents. This does not require micelles or vesicles, as only 1.2 laurate counterions are needed on average per guanidinium group.
Fig. 12.

Adaptive translocation mechanism of uptake and octanol-water partitioning.
The driving force for the passage of this ion pair complex across the cell membrane is proposed to be the membrane potential. Since this potential favors the movement of positively-charged species into a cell, if the ion pair complex formed from the polyguanidinium transporter retains any time-averaged cationic character its passage across the membrane would be favorable. Since the ability of a cell to maintain a membrane potential is dependent on ATP, this mechanism is consistent with studies that show an energy dependence on cellular uptake. To probe this mechanism, it was shown that the cellular uptake of Fl-r8 was reduced by more than 90% by eliminating the membrane potential with an extracellular buffer containing a concentration of potassium ion equivalent to that found intracellularly. Additionally, varying the membrane potential by the use of potassium ion buffers with different concentrations results in differing amounts of cellular uptake. Notably, cellular uptake possessed a linear relationship with the potassium Nernst potential calculated based on the extracellular potassium ion concentration. Finally, treatment of cells with gramicidin A, known to reduce the membrane potential through its pore-forming activity, reduces cellular uptake of guanidinium-based transporters by greater than 90%. Conversely, hyperpolarizing the cells with valinomycin, a peptide that selectively shuttles potassium into the cell, significantly increases cellular uptake.
These mechanistic explanations for direct penetration arose from the observation by many groups that uptake of the CPPs was not significantly reduced either by incubation at 4 °C or in the presence of metabolic inhibitors, both of which suggest a non-endocytotic mechanism [1,50,55,86,89,90,91,92,93]. This low temperature, non-endocytotic pathway has been demonstrated with multiple peptides, including truncated Tat peptide sequences [89], HIV-1 Tat [90,91,92], octaarginine [1,50,90], R7W [55], and Tat-decorated liposomes [93]. It has been reported, however, that cell fixation leads to artifacts in both uptake and intracellular distribution of peptides [27,94], leading some researchers to propose that the observed results need to be reevaluated and endocytosis is the actual uptake pathway. It should be noted, however, that a number of groups have reported non-endocytotic uptake with live (unfixed) cells [1,50,55,91,95]. Thoren et al. studied the uptake of fluorescent analogs of Tat(48–60) and heptaarginine in live PC-12 cells, reporting that R7W is efficiently internalized at both 37 °C and 4°C [91]. Additionally, the authors note that depletion of ATP with rotenone and 2-deoxy-D-glucose inhibited entry of FM 4–64 (a commercial marker of endocytosis), but not the uptake of R7W. Rothbard et al. reported that Fl-R7 uptake into Jurkat cells is inhibited by pre-incubation with sodium azide, but rapid and efficient uptake at 3 °C along with the absence of fluorescence in endocytotic vesicles provided support for a mechanism distinct from endocytosis [50]. Representative of how the various pathways are not mutually exclusive, a study by Jones et al. with fluorescently tagged R8 in KG1a cells reported staining in both endocytotic vesicles and the cytoplasm at intermediate temperatures (12–30 °C), supporting the idea that multiple pathways of uptake for a given GRT can occur simultaneously [95].
As has already been alluded to above, uptake of a given GRT is dependent on a variety of factors, all of which could influence and direct the uptake towards a certain pathway. This interplay of conditions should be considered when attributing a mechanism to a given CPP. The size of the cargo attached to the transporter is one factor that affects the mechanism. It has been shown that complexes or fusions of Tat with large 20 nm quantum dots or proteins (>50 amino acids) enter cells primarily through an endocytotic, vesicle-associated pathway whereas a small peptide (<50 amino acids) cargo can transduce cells through an additional, non-endocytotic pathway [15]. Another study has shown that fluorescently labeled, unconjugated R7 and R7W are taken up efficiently by A431 and U2OS cells, with both diffuse and vesicular staining observed; however, conjugating negatively-charged amino acid sequences to the GRTs dramatically reduces uptake and the little staining observed is solely vesicular [55]. A temperature dependence on the mechanism of uptake has also been demonstrated. While some studies have shown inhibition of uptake at 4 °C, others have proposed a temperature-independent mechanism for GRT translocation. A combination of the two has also been reported in a study of the uptake of Alexa488-R8 (both L and D stereoisomers) into KG1a cells. At temperatures between 4 and 12 °C, fluorescence is observed in both the cytoplasm and the nucleus. At 12–30 °C, however, additional labeling is observed in endocytotic vesicles. Finally, when the cells are warmed to 37 °C fluorescence is observed solely in vesicles [95]. GRTs have also been shown to enter one cell type selectively over others. In a study involving both ex vivo and in vivo applications, a modified Tat peptide conjugated to Alexa594 is taken up by renal ganglion cells and a subset of inner nuclear layer cells of the retina, but no fluorescence is observed in any outer retina cells [96]. As further evidence that not all cell types are equivalent, one research group observed different amounts of uptake for biotinylated R9 in glycosaminoglycan-deficient CHO cells and in wild-type cells [11]. Furthermore, in contrast to the R9 case, internalization of a guanidinylated aminoglycoside is inhibited in the former cell type as compared to the normal uptake in cells containing glycosaminoglycans. Yet another factor to consider when examining the uptake of charged GRTs involves the associated counterions. In the absence of the aromatic counteranion pyrenebutyrate, Alexa488-R8 is internalized into HeLa cells primarily by endocytosis; however, preincubation with pyrenebutyrate provides rapid, diffuse cytosolic labeling, arguing for direct membrane translocation of the peptide [97]. These examples call attention to only a few of the many factors that can affect the contribution of a given internalization pathway to the overall uptake of a GRT.
Elucidating the mechanism of uptake is of more than just scholarly importance. The mechanism of uptake impacts intracellular trafficking, which in turn determines how quickly a cargo is either degraded or delivered to various organelles. These various fates will have a large impact on the therapeutic efficiency of the cargo. Fischer et al. have demonstrated colocalization of a fluoresceinated Tat with BODIPY, a Golgi tracer, and failed to see any colocalization with Lysotracker [98], a lysosome tracer. Fretz et al. have observed colocalization with Lysotracker in the case of a Tat-modified liposome [63]. Fuchs et al. have also observed colocalization of fluoresceinated R9 with FM 1–43, a marker for endocytotic vesicles, in CHO cells [100]. Geisler and Chmielewski have also reported colocalization of an arginine-rich CPP with Lysotracker in MCF-7 cells, though they do report extensive cytosolic staining that does not colocalize with the lysosomes, suggesting either endosomal escape or a different mode of uptake [99]. Al-Taei et al. found that disruption of the Golgi in K562 with brefeldin A resulted in no difference in staining with fluorescently labeled Tat [90]. However, the authors performed a dextran pulse-chase experiment and observed colocalization of Tat and R8 with dextran, suggesting that both peptides are trafficked to lysosomes. Still others have observed no colocalization of a Tat-GFP fusion protein with Lysotracker or with early endosome antigen-1 in HeLa cells but it does colocalize with caveolae-associated markers [59]. These studies suggest that not only mechanism, but also trafficking, is cargo- and cell type-dependent.
In the cases where endosomal uptake predominates, in order for the cargo to be therapeutically active, it is necessary for the GRT conjugate to escape the endosomes. Attempts have been made to study endosomal release by inhibiting it. Potocky et al. demonstrated that if HeLa cells are incubated with NH4Cl little to no fluorescence is observed in the cytoplasm compared to untreated cells, suggesting that acidification of the endosomes is necessary for escape [65]. They also found that at low concentrations (1.5 μM) Fl-Tat has mainly vesicular staining while at higher concentration (7 μM) cytosolic staining is observed, which they propose is due to a concentration dependence of endosomal escape. Fuchs et al. have also suggested that there is a concentration dependence on endosomal escape, observing that fluorescently labeled R9-induced leakage from vesicles occurs only at high concentration [100]. Some have built endosomal escape into their conjugates. Dowdy et al. reported that treatment with a Tat fusogenic peptide to facilitate escape from the endosomes increases the amount of recombination with a Tat-Cre fusion protein in NIH3T3 cells [58]. Still others have found if they treat cells with a soluble photosensitizer that creates reactive oxygen species a significant increase in endosomal release of various GRTs is observed. They found in the case of Tat a greater than 75-fold increase in escape when the cells are treated with the photosensitizer and light [101]. Once endosomal escape has been achieved in many cases it is still necessary to release the cargo from the transporter. This has been achieved, through a variety of mechanisms including disulfide reduction that selectively releases the cargo inside of the cell [102].
As described in the example above, for many conjugates it is necessary to release the drug/probe cargo from the transporter after uptake to achieve the activity of the free cargo. Recently, we have also reported an imaging method that provides, for the first time, quantification of transporter-conjugate uptake and cargo release in real-time in both cells and animal models [103]. This method uses luciferase-transfected cells and transgenic (luciferase) reporter mice and whole-body imaging, allowing non-invasive quantification of transporter-conjugate uptake and probe (luciferin) release in real-time. This process also effectively emulates drug-conjugate delivery, drug release, and drug turnover by an intracellular target, providing a facile method to evaluate comparative uptake of new transporters and efficacy and selectivity of linker release as required for fundamental studies and therapeutic applications. In our study we selected a disulfide linker because its cleavage would occur only after cell entry upon encountering a high glutathione concentration (15 mM inside vs. 15 μM outside [104]) (Fig. 13). The resultant thiol would then undergo cyclization, releasing free luciferin which would be converted by luciferase to oxyluciferin and a photon of light [105]. Because luciferin derivatives alkylated on the phenolic oxygen do not generate light [106], only free luciferin is measured. Importantly, the signal-to-noise ratios are excellent relative to fluorescence because there is essentially no background tissue luminescence. The approach is readily applied to cells and to live animals [107]. This new procedure allows one to quantitatively compare the performance of different transporters for a constant linker design or different linkers and their release kinetics for a given transporter system. Because signal arises only when free drug surrogate is turned over by its intracellular target (luciferase), it is a true measure of effective cargo delivered.
Fig. 13.

Scheme for the measurement of the uptake of GRTs and the intracellular release of their cargo.
3.3. Tissue Uptake
As seen in the preceding paragraphs, cells and even artificial membranes provide a variety of intriguing, though sometimes conflicting, mechanistic insights regarding the uptake of cell-penetrating peptides and their conjugates. In addition to all of the variables previously discussed affecting uptake, the mechanism of uptake and its relevancy certainly vary with the complexity of the overall system, ranging from artificial membranes to cells to tissue. While most uptake studies have focused on cell entry, the ability of GRTs to enter tissue is of critical importance, and is required for the advancement of this class of molecular transporters toward therapeutic applications. Research into the tissue distribution of cell-penetrating peptides has yielded promising results. Intravenous injection of the Tat peptide labeled with 99mTc-tricarbonyl and fluorescein showed high levels of uptake in the kidney and liver, followed closely by bone and muscle tissue [108]. These data were supported by studies of Tat conjugated to the proteinaceous Fab fragment. The 125I-labeled Fab fragment showed high concentrations in the liver as well as the spleen [109]. Further studies with oligoarginine transporters demonstrated penetration into endothelial and smooth muscle cells when injected intravenously [110].
To investigate the potential of utilizing polyguanidinium transporters to enhance oral bioavailability, the small and large intestines of mice were injected with solutions of biotinylated oligoarginine. While the small intestine showed low levels of uptake, the large intestine showed high concentrations, and in some cases, penetration across the entire thickness of the sectioned tissue. Similar studies showed that uptake was significant in both the tongue and cheek [110].
GRTs have been shown to penetrate many types of tissue, calling attention then to whether tissue entry could be selective. One strategy to achieve selectivity is the use of local administration. This has potentially wide application as lung, buccal, ocular, skin and many other sites are readily reached by direct administration of a drug. GRTs show uptake in lung tissue when inhaled, as well as penetration of both the epidermal and dermal layers when applied topically. In addition to local delivery, a targeted delivery system can also be utilized to achieve specificity. This approach has been demonstrated by using a prodrug strategy in which a transporter is transiently disabled by intramolecular interaction with a polycarboxylate connected to the transporter through a peptide link. The peptide is selected as a substrate for an extracellular protease that is localized in target tissue. Upon encounter with the protease, the peptide linker is cleaved, thereby activating the transporter and allowing its entry into surrounding tissue. This tissue-specific cleavage then promotes dissociation of the transporter from its suppressing domain and subsequently the active transporter translocates the nearby tissue [16,17]. This concept has potentially wide application for targeted therapy.
4. Conclusion
The ability of a drug or probe to cross a biological barrier has historically been viewed to be a function of its intrinsic physical properties. This view has largely restricted drug design and selection to agents within a narrow log P range. Molecular transporters offer a strategy to circumvent these restrictions. In the case of guanidinium-rich transporters, a typically highly water-soluble conjugate is found to readily pass through the non-polar membrane of a cell and for some across the stratum corneum barrier of the skin and barriers of other tissue. This activity opens a field of opportunities for the use of GRTs to enable delivery of polar and non-polar agents as well as to enhance uptake of those of intermediate polarity. The penetration of GRTs has been found to be quite general, working effectively for a variety of cells. Impressive work has been reported on mechanisms of GRT uptake, collectively suggesting, not surprisingly for systems whose function is a consequence of numerous variables, that multiple mechanisms might be involved. For a given system, a single mechanism might operate but it is unlikely that a single mechanism will be universal. Evidence has also been presented for the concurrent operation of more than one mechanism of entry. Much noteworthy work has been done with cells while less attention has been directed to tissue uptake, a subject of critical importance for therapeutic applications. This direction is increasingly important as molecular transporters not only allow one to utilize new cargoes, including biologicals (e.g., proteins, nucleic acids, etc.), they also provide a means to control formulation, change distribution, alter metabolism, circumvent side effects, suppress pathways that might compromise the efficacy of a drug, and achieve targeted therapy. While some GRTs have been advanced into clinical trials, further research is critically important to the realization of the enormous potential of molecular transporters in both fundamental research and human therapy.
Fig. 8.

Representative guanidine-based peptide nucleic acids (B = nucleobase, counterions not specified).
Table 1.
Studies on the mechanism of GRT uptake.
| Cell Line | GRT | Cargo | Proposed Mechanism | Factors studied | Ref |
|---|---|---|---|---|---|
| HeLa | Tat peptide | Fl | Endocytosis | NH4Cl, colocalize with transferrin, sodium azide, concentration dependence | [65] |
| K562 | RGGRLAYLRRR WAVLGR | TAMRA | Endocytosis | 4 °C, sodium azide, NH4Cl, cytochalasin D | [56] |
| HeLa, CHO | Tat peptide | Alexa 488 | Endocytosis | 4 °C, sodium azide, colocalize with transferrin | [27] |
| Jurkat | Tat protein | 125I | Clathrin endocytosis | Chlorpromazine, wortmannin, nocodazole, colocalize with transferrin | [78] |
| HeLa | Tat peptide | Fl-avidin | Lipid raft endocytosis | 4 °C, serum, hyperosmolar medium, sodium azide, β-CD | [64] |
| MC57, HeLa | Tat, R9 | Fl | Endocytosis | Colocalize with BODIPY, wortmannin, balfilomycin A1, chloroquine, lysotracker | [98] |
| CHO-K1 | R9 | TAMRA | Endocytosis | Heparin, concentration, colocalize with FM 1–43 | [100] |
| HeLa, NIH3T3, HepG2 | Tat peptide | Gold nanoparticle | Endocytosis | 4 °C | [62] |
| HUVEC, CHO-K1, pgsA-745 | Tat peptide | Fl, Alexa Fl | Clathrin endocytosis | 4 °C, no heparan, nystatin, chloropromazine, K+ free, filipin III, sodium azide | [67] |
| U2OS, A431 | R7, R7W | Fl | Endocytosis and other | 4 °C | [55] |
| HepG2, CHO1 | Tat | Fl, DNA | Endocytosis and other | Sodium azide, 4 °C, Ca2+, BSA, nystatin | [61] |
| OVCAR-3 | Tat | liposomes | Endocytosis | Colocalize with dextran, Lysotracker, 4 °C, cytochalasin D iodoacetamide, heparin | [63] |
| KG1a, K562 | Tat, R8 | Alexa | Endocytosis | Colocalize with transferrin and dextran, wortmannin, 4 °C, nocodazole, Brefeldin A, amiloride, cytochalasin D | [90] |
| CHO | Tat, R8 | GFP | Endocytosis | Heparin, 4 °C | [79] |
| HeLa, B16F1, CHO K1, proteoglycan -deficient CHO | Tat | Biotin, liposomes | Endocytosis | 0 °C, TRITC-dextran, colocalize with transferrin, heparin, dextran sulfate, other glycosaminoglycans | [80] |
| HeLa | Tat(48–60), oligoarginine | Fl-Peptide nucleic acids | Endocytosis | Ca2+, colocalize with transferrin, chloroquine, dextran | [81] |
| Skin fibroblasts, RBL-2H3 | Tat | Lipophilic cation, Oregon green | Endocytosis | MitoTracker, colocalize with transferrin, concentration | [82] |
| KG1a | R8, r8 | Alexa | Endocytosis and direct translocation | 4 °C, concentration, β-CD | [95] |
| HL3T1, CHO-K1 | Tat | GFP | Caveolae endocytosis | 4 °C, colocalize with transferrin, caveolin-1, cholera toxin, cytochalasin D, nocodazole | [60] |
| HeLa, Jurkat, HL3T1 | Tat | GFP | Caveolae endocytosis | Colocalize with transferrin, Lysotracker, cholera toxin, β-CD, 4 °C, brefeldin, cytochalasin D, nocodazole, taxol | [59] |
| HeLa, CHO, A549 | Tat, polyarg. | Rhodamine, 11 aa’s | Caveolae endocytosis | β-CD, colocalize with cholera toxin, transferrin | [66] |
| COS-7, Namalwa | Tat, YGRKARRQRRR, YGRKARRQARR, YGKKKKKQKKK | Fl | Lipid-raft macropinocytosis | 4 °C, heparin, β-CD, cytochalasin D, EIPA, colocalize with dextran | [72] |
| NIH3T3 | R8-modified liposomes | Rhodamine, Fl-DNA | Clathrin endocytosis and macropinocytosis | Heparin, sodium azide, sodium fluoride, antimycin A, hypertonic medium, amiloride, filipin, cytochalasin D, nystatin, colocalize with transferrin, neutral dextran, and BODIPY-LacCer, LysoSensor | [73] |
| HeLa, COS- 7, RAW264.7 | Tat(48–60), R8, R16 | Fl, Rhodamine | Multiple energy- independent pathways | Concentration, colchicine, taxol, nocodazole, cytochalasin D, brefeldin A, wortmannin, chloroquine, sodium azide, rotenone, nystatin, LDH assay, heparin, chondroitin sulfate, heparinase III | [88] |
| HeLa | R8, r8, R4, R16 | Texas red, Fl | Macropinocytosis and others | 4 °C, colocalize with transferrin, MitoTracker, EIPA, phalloidin-TRITC, cytochalasin D, nocodazole | [57] |
| V79 and PC- 12 | R7W, TatP59W, TatLysP59W | Fl | Energy-indep, non- endocytotic | FM 4–64, concentration, 4 °C, rotenone | [91] |
| BT20, LLC, H9C2 | Tat | liposomes | Reverse micelles, energy-indep | 4 °C, sodium azide, iodoacetamide, FITC-dextran | [93] |
| HeLa | R9 | 125I-Tyr | Not macropinocytosis | EGF, 16 °C, amiloride, FITC-dextran | [74] |
| EGFP reporter T cells | Tat | Cre- Alexa488 | Lipid raft macropinocytosis | β-CD, nystatin, cavoelin-1, dynamin, amiloride, cytochalasin D, chloroquine, HA2 peptide | [58] |
| HeLa, CHO, proteoglycan -deficient CHO | Tat | Fl, Rhodamine, GFP | Peptide: non- endocytosis, HS- independent
Protein: energy- and HS-dependent |
Proteoglycan-deficient cells, concentration, 4 °C, heparinase III | [92] |
| C2C12, NIH3T3 reporter | Tat | Fl- or TAMRA- labeled peptides and proteins | Peptides: direct translocation
Proteins: Endocytosis |
Concentration, HA2 peptide, cytochalasin D, sodium azide, high K+ | [15] |
| HeLa | R8, Tat | Alexa488 | Direct translocation | Pyrenebutyrate counteranion, concentration, LDH assay, 4 °C | [97] |
| Jurkat | D- and L-Arg homopolymers | Fl | Adaptive translocation | Sodium azide, no serum, concentration, 3 °C | [50] |
| Jurkat | R8, Tat(49–57) | Fl | Adaptive translocation | High K+, gramicidin A, valinomycin | [54] |
| CHO | R9 | 125I-Tyr | Direct translocation | 4 °C, 16 °C, FITC-dextran, NH4Cl, hypertonic medium, filipin, colocalize with transferrin or BODIPY- lactosylceramide, amiloride | [83] |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirschberg TA, VanDeusen CL, Rothbard JB, Yang M, Wender PA. Arginine-based molecular transporters: The synthesis and chemical evaluation of releasable taxol-transporter conjugates. Org Lett. 2003;5:3459–3462. doi: 10.1021/ol035234c. [DOI] [PubMed] [Google Scholar]
- 3.Wender PA, Rothbard JB, Jessop TC, Kreider EL, Wylie BL. Oligocarbamate molecular transporters: Design, synthesis, and biological evaluation of a new class of transporters for drug delivery. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]
- 4.Wender PA, Kreider E, Pelkey ET, Rothbard J, VanDeusen CL. Dendrimeric molecular transporters: Synthesis and evaluation of tunable polyguanidino dendrimers that facilitate cellular uptake. Org Lett. 2005;7:4815–4818. doi: 10.1021/ol051496y. [DOI] [PubMed] [Google Scholar]
- 5.Wang CJ, Delcros JG, Cannon L, Konate F, Carias H, Biggerstaff J, Gardner RA, Phanstiel O. Defining the molecular requirements for the selective delivery of polyamine conjugates into cells containing active polyamine transporters. J Med Chem. 2003;46:5129–5138. doi: 10.1021/jm030223a. [DOI] [PubMed] [Google Scholar]
- 6.Reguera RM, Tekwani BL, Balana-Fouce R. Polyamine transport in parasites: A potential target for new antiparasitic drug development. Comp Biochem Physiol Part C Toxicol Pharmcol. 2005;140:151–164. doi: 10.1016/j.cca.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Alaimo C, Catrein I, Morf L, Marolda CL, Callewaert N, Valvano MA, Feldman MF, Aebi M. Two distinct but interchangeable mechanisms for flipping of lipid-linked oligosaccharides. EMBO (Eur Mol Biol Organ) J. 2006;25:967–976. doi: 10.1038/sj.emboj.7601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuji A, Tamai I. Carrier-mediated intestinal transport of drugs. Pharm Res. 1996;13:963–977. doi: 10.1023/a:1016086003070. [DOI] [PubMed] [Google Scholar]
- 9.Rao NM, Gopal V. Cationic lipids for gene delivery in vitro and in vivo. Expert Opin Ther Pat. 2006;16:825–844. [Google Scholar]
- 10.Eichhorn ME, Becker S, Strieth S, Werner A, Sauer B, Teifel M, Ruhstorfer H, Michaelis U, Griebel J, Brix G, Jauch KW, Dellian M. Paclitaxel encapsulated in cationic lipid complexes (MBT-0206) impairs functional tumor vascular properties as detected by dynamic contrast enhanced magnetic resonance imaging. Cancer Biology & Therapy. 2006;5:89–96. doi: 10.4161/cbt.5.1.2346. [DOI] [PubMed] [Google Scholar]
- 11.Elson-Schwab L, Garner OB, Schuksz M, Crawford BE, Esko JD, Tor Y. Guanidinylated neomycin delivers large, bioactive cargo into cells through a heparan sulfate-dependent pathway. J Biol Chem. 2007;282:13585–13591. doi: 10.1074/jbc.M700463200. [DOI] [PubMed] [Google Scholar]
- 12.Kam NWS, Jessop TC, Wender PA, Dai HJ. Nanotube molecular transporters: Internalization of carbon nanotube-protein conjugates into mammalian cells. J Am Chem Soc. 2004;126:6850–6851. doi: 10.1021/ja0486059. [DOI] [PubMed] [Google Scholar]
- 13.Kam NWS, Liu ZA, Dai HJ. Carbon nanotubes as intracellular transporters for proteins and DNA: An investigation of the uptake mechanism and pathway. Angew Chem, Int Ed. 2006;45:577–581. doi: 10.1002/anie.200503389. [DOI] [PubMed] [Google Scholar]
- 14.Vigneron JP, Oudrhiri N, Fauquet M, Vergely L, Bradley JC, Basseville M, Lehn P, Lehn JM. Guanidinium-cholesterol cationic lipids: Efficient vectors for the transfection of eukaryotic cells. Proc Natl Acad Sci U S A. 1996;93:9682–9686. doi: 10.1073/pnas.93.18.9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F, Cardoso MC. Cargo-dependent mode of uptake and bioavailability of Tat-containing proteins and peptides in living cells. FASEB J. 2006;20:1775–1784. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- 16.Goun EA, Shinde R, Dehnert KW, Adams-Bond A, Wender PA, Contag CH, Franc BL. Intracellular cargo delivery by an octaarginine transporter adapted to target prostate cancer cells through cell surface protease activation. Bioconjug Chem. 2006;17:787–796. doi: 10.1021/bc0503216. [DOI] [PubMed] [Google Scholar]
- 17.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat Med. 2000;6:1253–1257. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly-Rosen D. Molecular transporters for peptides: Delivery of a cardioprotective epsilon PKC agonist peptide into cells and intact ischemic heart using a transport system, r-7. Chem Biol. 2001;8:1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 20.Goun EA, Pillow TH, Jones LR, Rothbard JB, Wender PA. Molecular transporters: Synthesis of oligoguanidinium transporters and their application to drug delivery and real-time imaging. Chembiochem. 2006;7:1497–1515. doi: 10.1002/cbic.200600171. [DOI] [PubMed] [Google Scholar]
- 21.Seidah NG, Prat A. Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Proteases in Biology and Medicine. 2002;38:79–94. doi: 10.1042/bse0380079. [DOI] [PubMed] [Google Scholar]
- 22.Wender PA, Jessop TC, Pattabiraman K, Pelkey ET, VanDeusen CL. An efficient, scalable synthesis of the molecular transporter octaarginine via a segment doubling strategy. Org Lett. 2001;3:3229–3232. doi: 10.1021/ol0161108. [DOI] [PubMed] [Google Scholar]
- 23.Rothbard JB, Kreider E, Vandeusen CL, Wright L, Wylie BL, Wender PA. Arginine-rich molecular transporters for drug delivery: Role of backbone spacing in cellular uptake. J Med Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 24.Rueping M, Mahajan Y, Sauer M, Seebach D. Cellular uptake studies with beta-peptides. Chembiochem. 2002;3:257–259. doi: 10.1002/1439-7633(20020301)3:2/3<257::AID-CBIC257>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 25.Potocky TB, Menon AK, Gellman SH. Effects of conformational stability and geometry of guanidinium display on cell entry by ss-peptides. J Am Chem Soc. 2005;127:3686–3687. doi: 10.1021/ja042566j. [DOI] [PubMed] [Google Scholar]
- 26.Chung HH, Harms G, Seong CM, Choi BH, Min CH, Taulane JP, Goodman M. Dendritic oligoguanidines as intracellular translocators. Biopolymers - Peptide Science Section. 2004;76:83–96. doi: 10.1002/bip.10597. [DOI] [PubMed] [Google Scholar]
- 27.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides: a reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 28.Huang K, Voss B, Kumar D, Hamm HE, Harth E. Dendritic molecular transporters provide control of delivery to intracellular compartments. Bioconjug Chem. 2007;18:403–409. doi: 10.1021/bc060287a. [DOI] [PubMed] [Google Scholar]
- 29.Futaki S, Nakase I, Suzuki T, Zhang YJ, Sugiura Y. Translocation of branched-chain arginine peptides through cell membranes: Flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry. 2002;41:7925–7930. doi: 10.1021/bi0256173. [DOI] [PubMed] [Google Scholar]
- 30.Luedtke NW, Baker TJ, Goodman M, Tor Y. Guanidinoglycosides: A novel family of RNA ligands. J Am Chem Soc. 2000;122:12035–12036. [Google Scholar]
- 31.Baker TJ, Luedtke NW, Tor Y, Goodman M. Synthesis and anti-HIV activity of guanidinoglycosides. J Org Chem. 2000;65:9054–9058. doi: 10.1021/jo001142e. [DOI] [PubMed] [Google Scholar]
- 32.Luedtke NW, Carmichael P, Tor Y. Cellular uptake of aminoglycosides, guanidinoglycosides, and poly-arginine. J Am Chem Soc. 2003;125:12374–12375. doi: 10.1021/ja0360135. [DOI] [PubMed] [Google Scholar]
- 33.Flavell DJ. Saporin immunotoxins. Clinical Applications of Immunotoxins. 1998;234:57–61. doi: 10.1007/978-3-642-72153-3_4. [DOI] [PubMed] [Google Scholar]
- 34.Maiti KK, Jeon OY, Lee WS, Kim DC, Kim KT, Takeuchi T, Futaki S, Chung SK. Design, synthesis, and membrane-translocation studies of inositol-based transporters. Angew Chem, Int Ed. 2006;45:2907–2912. doi: 10.1002/anie.200600312. [DOI] [PubMed] [Google Scholar]
- 35.Maiti KK, Jeon OY, Lee WS, Chung SK. Design, synthesis, and delivery properties of novel guanidine-containing molecular transporters built on dimeric inositol scaffolds. Chemistry - A European Journal. 2007;13:762–775. doi: 10.1002/chem.200600898. [DOI] [PubMed] [Google Scholar]
- 36.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 37.Maiti KK, Lee WS, Takeuchi T, Watkins C, Fretz M, Kim DC, Futaki S, Jones A, Kim KT, Chung SK. Guanidine-containing molecular transporters: Sorbitol-based transporters show high intracellular selectivity toward mitochondria. Angew Chem, Int Ed. 2007;46:5880–5884. doi: 10.1002/anie.200701346. [DOI] [PubMed] [Google Scholar]
- 38.Fillon YA, Anderson JP, Chmielewski J. Cell penetrating agents based on a polyproline helix scaffold. J Am Chem Soc. 2005;127:11798–11803. doi: 10.1021/ja052377g. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Carneado J, Van Gool M, Martos V, Castel S, Prados P, de Mendoza J, Giralt E. Highly efficient, nonpeptidic oligoguanidinium vectors that selectively internalize into mitochondria. J Am Chem Soc. 2005;127:869–874. doi: 10.1021/ja044006q. [DOI] [PubMed] [Google Scholar]
- 40.Pham W, Kircher MF, Weissleder R, Tung CH. Enhancing membrane permeability by fatty acylation of oligoarginine peptides. Chembiochem. 2004;5:1148–1151. doi: 10.1002/cbic.200400063. [DOI] [PubMed] [Google Scholar]
- 41.Dragulescu-Andrasi A, Zhou P, He GF, Ly DH. Cell-permeable gpna with appropriate backbone stereochemistry and spacing binds sequence-specifically to rna. Chem Commun. 2005:244–246. doi: 10.1039/b412522c. [DOI] [PubMed] [Google Scholar]
- 42.Zhou P, Wang MM, Du L, Fisher GW, Waggoner A, Ly DH. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (gpna) J Am Chem Soc. 2003;125:6878–6879. doi: 10.1021/ja029665m. [DOI] [PubMed] [Google Scholar]
- 43.Ohmichi T, Kuwahara M, Sasaki N, Hasegawa M, Nishikata T, Sawai H, Sugimoto N. Nucleic acid with guanidinium modification exhibits efficient cellular uptake. Angew Chem, Int Ed. 2005;44:6682–6685. doi: 10.1002/anie.200500904. [DOI] [PubMed] [Google Scholar]
- 44.Deglane G, Abes S, Michel T, Prevot P, Vives E, Debart F, Barvik I, Lebleu B, Vasseur JJ. Impact of the guanidinium group on hybridization and cellular uptake of cationic oligonucleotides. Chembiochem. 2006;7:684–692. doi: 10.1002/cbic.200500433. [DOI] [PubMed] [Google Scholar]
- 45.Park M, Toporowski JW, Bruice TC. Ribonucleic guanidine demonstrates an unexpected marked preference for complementary DNA rather than rna. Bioorg Med Chem. 2006;14:1743–1749. doi: 10.1016/j.bmc.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 46.Dempcy RO, Almarsson O, Bruice TC. Design and synthesis of deoxynucleic guanidine: A polycation analogue of DNA. Proc Natl Acad Sci U S A. 1994;91:7864–7868. doi: 10.1073/pnas.91.17.7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park M, Bruice TC. Binding studies of cationic uridyl ribonucleic guanidine (rng) to DNA. Bioorg Med Chem Lett. 2005;15:3247–3251. doi: 10.1016/j.bmcl.2005.04.073. [DOI] [PubMed] [Google Scholar]
- 48.Siprashvili Z, Scholl FA, Oliver SF, Adams A, Contag CH, Wender PA, Khavari PA. Gene transfer via reversible plasmid condensation with cysteine-flanked, internally spaced arginine-rich peptides. Hum Gene Ther. 2003;14:1225–1233. doi: 10.1089/104303403767740768. [DOI] [PubMed] [Google Scholar]
- 49.Midoux P, Monsigny M. Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem. 1999;10:406–411. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 51.Mason PE, Neilson GW, Dempsey CE, Barnes AC, Cruickshank JM. The hydration structure of guanidinium and thiocyanate ions: Implications for protein stability in aqueous solution. Proc Natl Acad Sci U S A. 2003;100:4557–4561. doi: 10.1073/pnas.0735920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Springs B, Haake P. Equilibrium-constants for association of guanidinium and ammonium-ions with oxyanions - effect of changing basicity of oxyanion. Bioorg Chem. 1977;6:181–190. [Google Scholar]
- 53.Onda M, Yoshihara K, Koyano H, Ariga K, Kunitake T. Molecular recognition of nucleotides by the guanidinium unit at the surface of aqueous micelles and bilayers. A comparison of microscopic and macroscopic interfaces. J Am Chem Soc. 1996;118:8524–8530. [Google Scholar]
- 54.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 55.Maiolo JR, Ferrer M, Ottinger EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712:161–172. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 56.Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem. 2003;278:31192–31201. doi: 10.1074/jbc.M303938200. [DOI] [PubMed] [Google Scholar]
- 57.Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, Takehashi M, Tanaka S, Ueda K, Simpson JC, Jones AT, Sugiura Y, Futaki S. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011 – 1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proeins after lipid raft macropinocytosis. Nature Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 59.Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, et al. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 60.Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F. Caveolae-mediated internalization of extracellular HIV-1 Tat fusion proteins visualized in real time. Mol Ther. 2003;8:284–294. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 61.Ignatovich IA, Dizhe EB, Pavlotskaya AV, Akifi ev BN, Burov SV, Orlov SV, et al. Complexes of plasmid DNA with basic domain 47–57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J Biol Chem. 2003;278:42625–42636. doi: 10.1074/jbc.M301431200. [DOI] [PubMed] [Google Scholar]
- 62.Tkachenko AG, Xie H, Liu Y, Coleman D, Ryan J, Glomm WR, et al. Cellular trajectories of peptide-modified gold particle complexes: comparison of nuclear localization signals and peptide transduction domains. Bioconjug Chem. 2004;15:482–490. doi: 10.1021/bc034189q. [DOI] [PubMed] [Google Scholar]
- 63.Fretz MM, Koning GA, Mastrobattista E, Jiskoot W, Storm G. OVCAR-3 cells internalize TAT-peptide modified liposomes by endocytosis. Biochim Biophys Acta. 2004;1665:48–56. doi: 10.1016/j.bbamem.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 64.Saalik P, Elmquist A, Hansen M, Padari K, Saar K, Viht K, Langel U, Pooga M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjug Chem. 2004;15:1246–1253. doi: 10.1021/bc049938y. [DOI] [PubMed] [Google Scholar]
- 65.Potocky TB, Menon AK, Gellman SH. Cytoplasmic and nuclear delivery of a TAT-derived peptide and a beta-peptide after endocytic uptake into HeLa cells. J Biol Chem. 2003;278:50188–50194. doi: 10.1074/jbc.M308719200. [DOI] [PubMed] [Google Scholar]
- 66.Jones SW, Christison R, Bundall K, Voyce C, Brockbank S, Newham P, Lindsay M. Characterisation of cell-penetrating peptide-mediated peptide devlivery. British journal of Pharmacology. 2005;145:1093–1102. doi: 10.1038/sj.bjp.0706279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 68.Nichols BJ, Lippincott-Schwartz J. Endocytosis without clathrin coats. Trends Cell Biol. 2001;11:406–412. doi: 10.1016/s0962-8924(01)02107-9. [DOI] [PubMed] [Google Scholar]
- 69.Meier O, Boucke K, Vig Hammer S, Keller S, Stidwill RP, Hemmi S, Greber UF. Adenovirus triggers macropinocytosis and endosomal leakage together with clathrin-mediated uptake. J Cell Biol. 2002;158:1119–1131. doi: 10.1083/jcb.200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.West MA, Bretscher MS, Watts C. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J Cell Biol. 1989;109:2731–2739. doi: 10.1083/jcb.109.6.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sampath P, Pollard TD. Effects of cytochalasin, phalloidin, and pH on the elongation of actin filaments. Biochemistry. 1991;30:1973–1980. doi: 10.1021/bi00221a034. [DOI] [PubMed] [Google Scholar]
- 72.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Controlled Release. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 73.Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 74.Zaro JL, Rajapaksa TE, Okamoto CT, Shen WC. Membrane transduction of oligoarginine in HeLa cells is not mediated by macropinocytosis. Mol Pharm. 2006;3:181–186. doi: 10.1021/mp0500869. [DOI] [PubMed] [Google Scholar]
- 75.Zaro JL, Shen W. Quantitative comparison of membrane transduction and endocytosis of oligopeptides. Biochem Biophys Res Comm. 2003;307:241– 247. doi: 10.1016/s0006-291x(03)01167-7. [DOI] [PubMed] [Google Scholar]
- 76.Orlandi PA, Fishman PH. Filipin-dependent Inhibition of Cholera Toxin: Evidence for Toxin Internalization and Activation through Caveolae-like Domains. J Cell Biol. 1998;141:905–915. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell. 2004;15:2347–2360. doi: 10.1091/mbc.E03-12-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lundberg M, Wikstrom S, Johansson M. Cell surface adherence and endocytosis of protein transduction domains. Mol Ther. 2003;8:143–150. doi: 10.1016/s1525-0016(03)00135-7. [DOI] [PubMed] [Google Scholar]
- 80.Console S, Marty C, Garcia-Echeverria C, Schwendener R, Ballmer-Hofer K. Antennapedia and HIV Transactivator of Transcription (Tat) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J Biol Chem. 2003;278:35109–35114. doi: 10.1074/jbc.M301726200. [DOI] [PubMed] [Google Scholar]
- 81.Shiraishi T, Pankratova S, Nielsen PE. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic Tat and oligoarginine peptides. Chem Biol. 2005;12:923–929. doi: 10.1016/j.chembiol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 82.Ross MF, Filipovska A, Smith RA, Gait MJ, Murphy MP. Cell-penetrating peptides do not cross mitochondrial membranes even when conjugated to a lipophilic cation: evidence against direct passage through phospholipids bilayers. Biochem J. 2004;383:457–468. doi: 10.1042/BJ20041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zaro JL, Shen WC. Evidence that membrane transduction of oligoarginine does not require vesicle formation. Exper Cell Res. 2005;307:164–173. doi: 10.1016/j.yexcr.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 84.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 85.Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 86.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 87.Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipids membranes. Biochemistry. 1992;31:12416–12423. doi: 10.1021/bi00164a017. [DOI] [PubMed] [Google Scholar]
- 88.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. 2002;277:2437– 2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 89.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 90.Al-Taei S, Penning NA, Simpson JC, Futaki S, Takeuchi T, Nakase I, Jones AT. Intracellular traffic and fate of protein transduction domains HIV-1 TAT peptide and octaarginine: implications for their utilization as drug delivery vectors. Bioconj Chem. 2006;17:90–100. doi: 10.1021/bc050274h. [DOI] [PubMed] [Google Scholar]
- 91.Thoren PE, Persson D, Isakson P, Goksor M, Onfelt A, Norden B. Uptake of analogs of penetratin, Tat(48–60) and oligoarginine in live cells. Biochem Biophys Res Commun. 2003;307:100–107. doi: 10.1016/s0006-291x(03)01135-5. [DOI] [PubMed] [Google Scholar]
- 92.Silhol M, Tyagi M, Giacca M, Lebleu B, Vives E. Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. Eur J Biochem. 2002;269:494–501. doi: 10.1046/j.0014-2956.2001.02671.x. [DOI] [PubMed] [Google Scholar]
- 93.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. Tat peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Nat Acad Sci. 2001;98:8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lundberg M, Johansson M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem Biophys Res Commun. 2002;291:367–371. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 95.Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I, Storm G, Jones AT. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem J. 2007;403:335–342. doi: 10.1042/BJ20061808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Barnett EM, Elangovan B, Bullok KE, Piwnica-Worms D. Selective cell uptake of modified Tat peptide-fluorophore conjugates in rat retina in ex vivo and in vivo models. Invest Opthalmol Vis Sci. 2006;47:2589–2595. doi: 10.1167/iovs.05-1470. [DOI] [PubMed] [Google Scholar]
- 97.Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, Sakai N, Matile S, Futaki S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 98.Fischer R, Karsten K, Mariola F, Roland B. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J Biol Chem. 2004;279:12625–12635. doi: 10.1074/jbc.M311461200. [DOI] [PubMed] [Google Scholar]
- 99.Geislera I, Chmielewski J. Probing length effects and mechanism of cell penetrating agents mounted on a polyproline helix scaffold. Bioorg Med Chem Lett. 2007;17:2765–2768. doi: 10.1016/j.bmcl.2007.02.077. [DOI] [PubMed] [Google Scholar]
- 100.Fuchs SM, Raines RT. Pathway for polyarginine entry into mammalian cells. Biochemistry. 2004;43:2438 –2444. doi: 10.1021/bi035933x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shiraishi T, Nielsen P. Photochemically enhanced cellular delivery of cell penetrating peptide-PNA conjugates. FEBS Letters. 2006;580:1451–1456. doi: 10.1016/j.febslet.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 102.Jones LR, Goun EA, Shinde R, Rothbard JB, Contag CH, Wender PA. Releasable luciferin-transporter conjugates: Tools for the real-time analysis of cellular uptake and release. J Am Chem Soc. 2006;128:6526–6527. doi: 10.1021/ja0586283. [DOI] [PubMed] [Google Scholar]
- 103.Wender PA, Goun EA, Jones LR, Pillow TH, Rothbard JB, Shinde R, Contag CH. Real-time analysis of uptake and bioactivatable cleavage of luciferin-transporter conjugates in transgenic reporter mice. Proc Natl Acad Sci U S A. 2007;104:10340–10345. doi: 10.1073/pnas.0703919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv Drug Delivery Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 105.Jenkins DE, Oei Y, Hornig YS, Yu SF, Dusich J, Purchio T, Contag PR. Bioluminescent imaging (bli) to improve and refine traditional murine models of tumor growth and metastasis. Clin Exp Metastasis. 2003;20:733–744. doi: 10.1023/b:clin.0000006815.49932.98. [DOI] [PubMed] [Google Scholar]
- 106.Denburg JL, Lee RT, McElroy WD. Substrate binding properties of firefly luciferase part 1 luciferin binding site. Arch Biochem Biophys. 1969;134:381–394. doi: 10.1016/0003-9861(69)90297-5. [DOI] [PubMed] [Google Scholar]
- 107.Cao YA, Wagers AJ, Beilhack A, Dusich J, Bachmann MH, Negrin RS, Weissman IL, Contag CH. Shifting foci of hematopoiesis during reconstitution from single stem cells. Proc Natl Acad Sci U S A. 2004;101:221–226. doi: 10.1073/pnas.2637010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bullok KE, Dyszlewski M, Prior JL, Pica CM, Sharma V, Piwnica-Worms D. Characterization of novel histidine-tagged Tat-peptide complexes dual-labeled with tc-99m-tricarbonyl and fluorescein for scintigraphy and fluorescence microscopy. Bioconjug Chem. 2002;13:1226–1237. doi: 10.1021/bc025573a. [DOI] [PubMed] [Google Scholar]
- 109.Kameyama S, Horie M, Kikuchi T, Omura T, Takeuchi T, Nakase I, Sugiura Y, Futaki S. Effects of cell-permeating peptide binding on the distribution of I-125-labeled Fab fragment in rats. Bioconjug Chem. 2006;17:597–602. doi: 10.1021/bc050258k. [DOI] [PubMed] [Google Scholar]
- 110.Wright LR, Rothbard JB, Wender PA. Guanidinium rich peptide transporters and drug delivery. Curr Protein Pept Sci. 2003;4:105–124. doi: 10.2174/1389203033487252. [DOI] [PubMed] [Google Scholar]