

Abstract
Chiral α-tertiary hydroxyaldehydes are very versatile building blocks in synthetic chemistry. Herein, we reported the first examples of a catalytic asymmetric protocol for the synthesis of such compounds from readily available α-halo or α-hydroxy ketones or enol silyl ethers with excellent yields and enantioselectivity. Its synthetic utility is demonstrated in the short, efficient formal synthesis of (S)-oxybutynin. In this process, the chiral ligand controls with the regioselectivity as well as enantioselectivity.
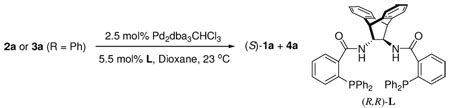
α-Hydroxyaldehydes are very versatile building blocks for the synthesis of natural products as well as clinical drugs.1 Chiral α-hydroxyaldehydes enjoy the added benefit of being a potential source of introducing other stereogenic centers. Up to now, the access of such compounds in enantiomerically enriched form can be classified as a chiral pool approach1, a chiral auxiliary approach2 or a transformation from other enantio-enriched compounds, such as 1,2-diols,3a–c α-hydroxy acids3d and cyanohydrins,3e synthesized by other enantioselective methods. However, to our knowledge catalytic enantioselective synthesis of α-tertiary hydroxyaldehydes directly from prochiral precursors has not been reported.4 In the course of studying palladium-catalyzed asymmetric allylic alkylation (AAA) of simple ketone enolates,5 we postulated that treatment of enol carbonate 2 or 3 bearing a shiftable OR1 group with a proper chiral palladium catalyst presumably could regio- and enantioselectively generate R1 protected α-tertiary hydroxyaldehydes 1 (eq. 1). Substrates 2 and 3 can be made from readily available α-halo or α-hydroxy ketones.6 Herein, we report the first example of a palladium catalyzed highly enantioselective synthesis of α-tertiary hydroxyaldehydes resulting from a novel competition and demonstrated its synthetic utility in a formal synthesis of (S)-oxybutynin.7

We initially subjected 2a-1 and 3a-1 respectively to our previously reported conditions (2.5 mol% Pd2(dba)3CHCl3 and 5.5 mol% (R,R)-L in 1,4-dioxane at 23 °C).5 Although the reaction of 2a-1 was significantly faster, the only product from either was aldehyde (S)-1a with excellent yields and ee’s (Table 1, entry 1 and 2).8 As summarized in Table 1, the scope of the R1 group was explored. With few exceptions, the reaction favored the formation of aldehyde 1a independent of the O- substituent while, in several cases, reactions with achiral ligand 1,2-bis(diphenylphosphino)ethane (dppe) as ligand the major product was ketone 4a (Table 1, entry 6, 8 and 10). The reaction of 3a-3 with a TIPS or 3a-8 with a mesitoyl group was slower than that with a less bulky group in the same class; in both cases a significant amount of ketone 4a was isolated (Table 1, entry 4 and 12). Presumably if the equilibrium between enolate I and II is faster than the allylation, the reaction should proceed through the former since it is more stable than II both electronically and sterically. On the other hand, if the rate of allylation became faster than the rate of enolate equilibration as may be occurring in the case of acetyl (Table 1, entry 7) or with dppe as ligand, then an increasing amount of ketone is observed (Table 1, entry 6, 8 and 10). Substrate 3a-9 with an un-shiftable methoxymethyl (MOM) group had very poor conversion under the same conditions, perhaps due to the chelation of the intermediate enolate with the Pd catalyst (Table 1, entry 13). Support for this contention derives from the observation that the reaction of 3a-1 was severely inhibited by the addition of an equal amount of 3a-9 (6% conversion in contrast to a full conversion in 1 h in the absence of 3a-9. In addition, The E-enolate generated from 5 can not chelate to the catalyst and reacted readily (eq. 2).
Table 1.
Various Hydroxy Protecting Groups are Suitable.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | SM | R1 | time | yield% of 1a | ee% of 1a | yield% of 4a |
| 1 | 2a-1 | TBDMS | 1/4 h | 93 | 92 | 0 |
| 2 | 3a-1 | TBDMS | 1 h | 86 | 91 | 0 |
| 3 | 2a-2 | TMS | 1/4 h | 99 | 92 | 0 |
| 4 | 3a-3 | TIPS | 5 h | 67 | 91 | 9 |
| 5 | 2a-4 | Benzoyl | 2 h | 93 | 81 | 0 |
| 6b | 2a-4 | Benzoyl | 2 h | 11 | 75 | |
| 7 | 3a-5 | Acetyl | 7 h | 40 | 91 | 60c |
| 8b | 3a-5 | Acetyl | 1/2 h | 24 | 67 | |
| 9 | 2a-6 | Piv | 4 h | 90 | 91 | 0 |
| 10b | 2a-6 | Piv | 4 h | 36 | 55 | |
| 11 | 2a-7 | CO2Me | 4 h | 71d | 86 | 0 |
| 12 | 3a-8 | Mesitoyl | 36 h | 10 | 91 | 36e |
| 13 | 3a-9 | MOM | 16 h | <5 | - | - |
The structure of the substrates were confirmed with HMBC and NOE NMR data; unless otherwise indicated, all reactions were performed at 23 °C on a 0.2 mmol scale at 0.1 M using 2.5 mol% 2 and 5.5 mol% ligand; yields were isolated yields; ee’s of 1a and 4a were determined by chiral HPLC.
5.5 mol% dppe was used as the ligand
With 21% ee
Product partially hydrolyzed on silica gel column
With 13% ee.

Although various R1 groups are suitable, we selected the most commonly used TBDMS as the hydroxy protecting group and investigated the scope of the nucleophilic moiety (Table 2). In general, excellent yields and ee’s were obtained with R as different as aryl, alkenyl or alkynyl groups (Table 2). For substrates where R are alkenyl groups, only the α-alkylated aldehydes are generated, no γ-alkylated enal is observed (entry 9 and 10).9 Since the silicon migration of trans enediolate is not likely as in substrate 11, it reacted to afford ketone 4i in 95% yields and 99% ee while its cis isomer 2i converted to aldehyde 1i in 76% yield and 89% ee under the same conditions (entry 12 and 15). In the case of tetra-substituted enol carbonate 7 (entry 13), α-tertiary siloxy ketone 8 generated from the corresponding E-enolate favored formation of the R-enantiomer.10 The reaction also works for the synthesis of a cyclic α-tertiary siloxy ketone 10 with a moderate ee in favor of the R- enantiomer (entry 14).11
Table 2.
Reactions with Different Nucleophilic Moieties.a
| substrate | product | time | yield | ee | |
|---|---|---|---|---|---|
| 1 | 2a (R=Ph) | (S)-1a | 1/4 h | 93% | 92% |
| 2 | 3a (R=Ph) | (S)-1a | 1 h | 89% | 91% |
| 3 | 2b (R=p-MeOPh) | 1b | 1/4 h | 94% | 92% |
| 4 | 3b (R=p-MeOPh) | 1b | 2 h | 86% | 92% |
| 5 | 2c (R=2-Naphthyl) | 1c | 1/4 h | 92% | 85% |
| 6 | 3c (R=2-Naphthyl) | 1c | 1/2 h | 94% | 85% |
| 7 | 2d (R=o-NO2Ph) | 1d | 12 h | 69% | 79% |
| 8 | 3d (R=o-NO2Ph) | 1d | 12 h | 69% | 72% |
| 9 | 2e (R=2-furyl) | 1e | 1/2 h | 81% | 93% |
| 10 | 2f (R=1-cyclohexenyl) | 1f | 7 h | 93% | 98% |
| 11 | 2g (R=2-methyl-1-propenyl) | 1g | 5 h | 89% | 98% |
| 12 | 2i (R=PhC C) | 1i | 1/4 h | 76% | 89% |
| 13 | 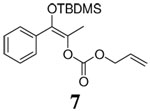 |
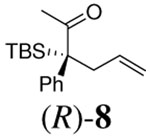 |
12 h | 94% | 80% |
| 14 | 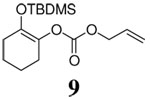 |
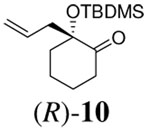 |
10 h | 96% | 64% |
| 15 | 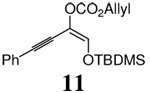 |
4i | 2 h | 95% | 99% |
All reactions were performed on a 0.2 mmol scale at 0.1 M in dioxane at 23 °C using 2.5 mol% Pd2(dba)3CHCl3 and 5.5 mol% ligand L; the yields were isolated yields and ee values were determined by chiral HPLC.
Variation of the allyl moiety to cycloalkenyl as in 12 led to 14 quantitatively (eq. 3). The regioisomer 13b reacted slower than 12b but still in quantitative yield; whereas, the reaction of 13c proceeded only in 30% conversion. The dr of 14 increased from 2.5:1 to over 50:1 with increasing cycloalkenyl ring size. Removal of one of the stereogenic centers by hydrogenation of the C=C double bond gave compounds 15 a-c (n =1,2, or 3). The ee of 15 reflected the dr of 14. 15b was converted into the key intermediate 17 for the synthesis of (S)-oxybutynin in 95% yield, wherein one recrystallization increased the ee to over 99% (Scheme 1).
Scheme 1.

Formal Synthesis of (S)-Oxybutynin.a
a Reagents and conditions: (a) NaH, CO2, THF, then PhCOCH2Br, DMF, 23 °C, 42%; (b) NaHMDS, TBSCl, THF, −78 °C to 23 °C, 83%; (c) 2.5 mol% Pd2(dba)3CHCl3, 5.5 mol% L, 1,4-dioxane, 23 °C, 99% (dr 11:1); (d) H2, cat. Pd/C, EtOH, 23 °C, 96%, 84% ee; (e) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH, H2O, 23 °C, 95% (recrystallization from hexane/DCM, >99% ee).

In summary, we report the first catalytic asymmetric synthesis of α-tertiary hydroxyaldehydes by palladium catalyzed allylic alkylation of siloxy enol carbonates. The excellent selectivity towards aldehyde was achieved by using chiral ligand L, which is in stark contrast to dppe which favors ketone formation. The reaction proceeds under very mild conditions and generates an α-tetra-substituted stereogenic center with excellent yield and enantiomeric excess. Further investigation of the mechanism, reaction scope and its application in organic synthesis are ongoing.
Supplementary Material
Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Table 3.
Reactions with different electrophilic moieties.
| substrate | time | yield | dr | ee of major | ee of minor | ee of 15 | |
|---|---|---|---|---|---|---|---|
| 1 | 13a (n=1) | 1 h | quant. | 2.5:1 | 92% | 87% | 43% |
| 2 | 12b (n=2) | 2 h | quant. | 11:1 | >99% | - | 84% |
| 3 | 13b (n=2) | 16 h | quant. | 11:1 | >99% | - | |
| 4 | 12c (n=3) | 16 h | quant. | 50:1 | >99% | - | 99% |
| 5 | 13c (n=3) | 16 h | 30% | 50:1 | >99% | - |
Acknowledgement
We thank the National Science Foundation and the National Institutes of Health, General Medical Sciences Grant GM13598, for their generous support of our programs. J. Xu has been supported by Abbott Laboratories Fellowships. Mass spectra was provided by the Mass Spectrometry Regional Center of the University of California–San Francisco supported by the NIH Division of Research Resources. We thank Chirotech (now Dow) for their generous gifts of ligands and Johnson Matthey for gifts of palladium salts.
References
- 1.Coppola GM, Schuster HF. α-Hydroxy Acids in Enantioselective Syntheses. Weinheim: Wiley-VCH; 1997. [Google Scholar]
- 2.a Seyden-Penne J. Chiral Auxiliaries and Ligands in Asymmetric Synthesis. New York, NY: John Wiley & Sons; 1995. [Google Scholar]; b Ruano JLG, Barros D, Maestro MC, Alcudia A, Fernández I. Tetrahedron: Asymmetry. 1998;9:3445. [Google Scholar]; c Tamura Y, Kondo H, Annoura H, Takeuchi R, Fujioka H. Tetrahedron Lett. 1986;27:81. [Google Scholar]; d Guanti G, Narisano E, Pero F, Banfi L, Scolastico C. J. Chem. Soc. Perkin Trans. 1. 1984:189. [Google Scholar]; e Guanti G, Narisano E. Tetrahedron Lett. 1983;24:817. [Google Scholar]; f Mukaiyama T. Tetrahedron. 1981;37:4111. [Google Scholar]
- 3.a Hughes DL. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Berlin: Springer; 1999. p. 1273. [Google Scholar]; b Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483. [Google Scholar]; c Hof RR, Kellogg RM. J. Chem. Soc. Perkin Trans.1. 1996:2051. [Google Scholar]; d Ooi T, Fukumoto K, Maruoka K. Angew. Chem. Int. Ed. 2006;45:3839. doi: 10.1002/anie.200600470. [DOI] [PubMed] [Google Scholar]; e Brunel JM, Holmes IP. Angew. Chem. Int. Ed. 2004;43:2752. doi: 10.1002/anie.200300604. [DOI] [PubMed] [Google Scholar]
- 4.Enantioseletive aldol reactions of α-oxyaldehydes catalyzed by organocatalysts have been reported. SeeNorthrup AB, Mangion IK, Hettche F, MacMillian DWC. Angew. Chem. Int. Ed. 2004;43:2152. doi: 10.1002/anie.200453716.
- 5.Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:17180. doi: 10.1021/ja055968f.Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:2846. doi: 10.1021/ja043472c.c. Similar results were reported independently by Stoltz et. al. using t-Bu-PHOX ligands. SeeBehenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044. doi: 10.1021/ja044812x.
- 6.See Supporting Information for the examples of the synthesis of siloxy enol carbonates.
- 7.Thompson IM, Lauvetz R. Urology. 1976;8:452–454. doi: 10.1016/0090-4295(76)90273-9.For the synthesis of (S)-Oxybutynin see:Tokuda O, Kano T, Gao WG, Ikemoto T, Maruoka K. Org. Lett. 2005;7:5103. doi: 10.1021/ol052164w.Gupta P, Fernandes RA, Kumar P. Tetrahedron Lett. 2003;44:4231.Masumoto S, Suzuki M, Kanai M, Shibasaki M. Tetrahedron Lett. 2002;43:8647.Grover PT, Bhongle NN, Wald SA, Senanayake CH. J. Org. Chem. 2000;65:6283. doi: 10.1021/jo005562f.Senanayake CH, Fang K, Crover P, Bakale RP, Vandenbossche CR, Wald SA. Tetrahedron Lett. 1999;40:819.
- 8.The absolute configuration of 1a was determined by reduction of CHO to CH2OH followed by removal of TBDMS and comparison of the optical rotation of the product 1,2-diol ([α]D 24 = − 45.2 (c 1.2, CHCl3)) with the reported data for (2R)-1,2-dihydroxy-2-phenylpent-4-ene ([α]D 20 = +43.4 (c 1.2, CHCl3)).Agami C, Couty F, Lequesne C. Tetrahedron. 1995;51:4043.
- 9.Waetzig SR, Rayabarapu DK, Weaver JD, Tunge JA. Angew. Chem. Int. Ed. 2006;45:4977. doi: 10.1002/anie.200600721. [DOI] [PubMed] [Google Scholar]
- 10.The absolute configuration of 8 was determined by removal of TBDMS and comparison of the optical rotation of the product ([α]D 24 = −91.3 (c 1.05, benzene)) with the reported data for (S)-3-hydroxy-3-phenyl-hex-5-en-2-one ([α]D 26 = +124.7 (c 0.635, benzene)).Soai K, Ishizaki M. J. Org. Chem. 1986;51:3290.
- 11.The absolute configuration of 10 was determined by removal of TBDMS and comparison of the optical rotation of the product ([α]D 23 = −73.4 (c, 2.6, CHCl3)) with the reported data for (S)-2-allyl-2-hydroxy-cyclohexanone ([α]D = +136.6 (c 2, CHCl3)).Compain P, Goré J, Vatèle JM. Tetrahedron. 1996;52:6647.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.