

Abstract
The Waardenburg syndromes (WS) account for approximately 2% of congenital sensorineural deafness. This heterogeneous group of diseases currently can be categorized into four major subtypes (WS types 1-4) on the basis of characteristic clinical features. Multiple genes have been implicated in WS, and mutations in some genes can cause more than one WS subtype. In addition to eye, hair and skin pigmentary abnormalities, dystopia canthorum and broad nasal bridge are seen in WS type 1. Mutations in the PAX3 gene are responsible for the condition in the majority of these patients. In addition, mutations in PAX3 have been found in WS type 3 that is distinguished by musculoskeletal abnormalities, and in a family with a rare subtype of WS, craniofacial-deafness-hand syndrome (CDHS), characterized by dysmorphic facial features, hand abnormalities, and absent or hypoplastic nasal and wrist bones. Here we describe a woman who shares some, but not all features of WS type 3 and CDHS, and who also has abnormal cranial bones. All sinuses were hypoplastic, and the cochlea were small. No sequence alteration in PAX3 was found. These observations broaden the clinical range of WS and suggest there may be genetic heterogeneity even within the CDHS subtype.
Keywords: Waardenburg syndrome, craniofacial, deafness, dysmorphology
INTRODUCTION
Hearing impairment is a common condition with a prevalence of 1/1000 [Smith and VanCamp, 2007]. There are hundreds of different syndromes that include an auditory phenotype as one symptom [Reardon et al., 2004; Gürtler and Lalwani, 2002]. It has many recognized genetic and environmental causes. Approximately 50% of profound hearing impairment may have a genetic etiology [Nance, 2003; Smith and VanCamp, 2007]. Most inherited hearing loss is transmitted as a monogenic trait [Friedman and Griffith, 2003] that may be autosomal recessive (77-88%), autosomal dominant (10-20%) or X-linked (1-2%) [Nance, 2003]. Most cases of hearing loss are nonsyndromic, but in approximately 30% there are additional physical findings [Reardon et al., 2004; Friedman and Griffith 2003; Shear et al., 2004]. The accompanying abnormalities range from subtle to obvious and may be present at birth or delayed in appearance.
Waardenburg syndrome (WS) is one of the more common genetic causes of deafness and accounts for about 2% of the congenitally deaf [Milunsky, 2007]. This autosomal disorder is characterized by varying degrees of hearing loss associated with pigmentary disturbances of the eye, skin and skin appendages [Milunsky et al., 2007; Waardenburg, 1951]. One of the more distinctive features is a prominent white forelock. The presentation of WS is quite variable and attempts have been made to categorize subtypes on the basis of clinical features. It was first divided into two major subgroups based on the presence (WS type I) or absence (WS type II) of dystopia canthorum (lateral displacement of the inner canthi) [Arias, 1971]. As additional clinical features that distinguished different families were described, the classification scheme was further broadened to include four subtypes. WS type 2 remains the “pure” subtype. The presence of musculoskeletal abnormalities separates WS type 3 (Klein-Waardenburg syndrome) from WS type 1 [Klein, 1983]. Skeletal malformations in WS type 3 range from minimal contractures of the fingers to hypoplasia of the upper limbs and/or pectoral girdle [Hoth et al., 1993; Klein 1983; Senrui 1984; Tekin et al., 2001], and in some cases there is mental retardation. WS type 4 (Shah-Waardenburg syndrome) combines the features of WS type 2 with the presence of Hirschsprung disease [Shah et al., 1981]. A rare subtype, craniofacial-deafness-hand syndrome (CDHS, OMIM 122880) [Sommer et al., 1983; Asher et al., 1996; Sommer and Bartholomew, 2003] was described in a three-generation family that was evaluated several times over a period of 24 years. Affected individuals have severe sensorineural hearing impairment and show clinical features of flat facial profile, hypertelorism and dystopia canthorum, downslanting palpebral fissures, depressed nasal bridge, small “pursed” mouth, ulnar deviation and contractures of the hand, and unique imaging findings of absence or hypoplasia of the nasal bones and a hypoplastic ulnar styloid. The imaging findings and distinct facial features was felt to distinguish CDHS from WS type 3.
Six genes responsible for WS have been identified, that allow objective identification of affected individuals. Heterozygous mutations in the paired box gene 3 (PAX3) were first reported in both familial and sporadic cases of WS type 1 [Baldwin et al., 1992; Hoth et al., 1993; Wollnik et al., 2003] and are responsible for the majority of cases. It was later shown that WS type 3 is an allelic disorder caused by heterozygous, homozygous or compound heterozygous mutations in PAX3 (e.g., [Hoth et al., 1993; Read and Newton, 1997; Zlotogora et al., 1995; Tassabehji et al., 1995; Tekin et al., 2001; Wollnik et al., 2003]. CDHS is also an allelic variant, and may be caused by PAX3 mutations affecting the paired box domain [Asher et al., 1996]. More than 50 point mutations and 20 unique deletions and insertions spread throughout the PAX3 gene have been reported (http://www.hgmd.cf.ac.uk/docs/login.html accessed February 4, 2008). The majority of these mutations are unique to a specific family or person. Even within a single family there can be marked clinical variability.
WS type 2 has been associated with abnormalities in the microphthalmia-associated transcription factor (MITF) gene [Hughes et al., 1994; Liu et al., 1995] or the encoding snail homolog 2 (SNAI2) gene [Sanchez-Martin et al., 2002]. Missense, splicing and small deletion mutations but no rearrangements have been detected in MITF (http://www.hgmd.cf.ac.uk/docs/login.html accessed February 4, 2008) and homozygous deletions of SNAI2 have been found in two families [Sanchez-Martin et al., 2002], although the molecular etiology of most cases of WS type 2 is unknown. WS type 4 is heterogeneous and is associated with mutations in EDNRB (endothelin receptor type B) [Puffenberger et al., 1994; Syrris et al., 1999], EDN3 (endothelin-3) [Baynash et al., 1994; Edery et al., 1996], and SOX10 (sex determining region y-box-10) [Pingault et al., 1998]. Deletions in SOX10 were recently identified in patients with WS types 2 and 4 [Bondurand et al., 2007].
In this paper, we report on a patient with congenital sensorineural hearing loss, facial features that include dystopia canthorum, ptosis, downsloping palpebral fissures, retrognathia and microgenia, and unique facial bone and sinus abnormalities. As far as we can ascertain, the clustering of these findings has not been previously reported.
CLINICAL REPORT
The patient is a 37-year-old woman who presented to the University of Washington Medical Genetics Clinic for evaluation of bilateral sensorineural hearing loss and facial abnormalities. Informed consent for participation in genetic studies and permission to publish identifiable photographs was obtained under a protocol approved by the University of Washington Institutional Review Board. She was the younger of two children of non-consanguineous parents, born by normal vaginal delivery at full term after a normal pregnancy during which the mother consumed moderate amounts of alcohol (2-3 beers several times a week). Motor milestones were apparently normal. Bilateral hearing loss was diagnosed in infancy, worse on the right, and she was fitted with a unilateral hearing aid at the age of 6. She noticed acute worsening of hearing in her left ear when she was 12 years old, and at that time was provided hearing aids for both ears. Serial audiograms revealed moderate-to-severe bilateral sensorineural hearing loss. At age 29, there was an overlying 55 to 5 dB conductive hearing loss from 250 through 1000 Hz in the right ear. On the left, there was an overlying 70 to 10 dB conductive hearing loss from 250 to 4000 Hz. Intellectual development was normal; she graduated from high school and is an accomplished artist.
She underwent several surgeries including tonsillectomy and adenoidectomy at the age of 5, plastic surgery on her eyelids to correct ptosis at the age of 6 and repair of a septal nasal deformity and airway obstruction at the age of 26. Presurgical imaging studies and medical records were not available for review. She denied constipation or other gastrointestinal problems suggestive of Hirschsprung disease. Family history revealed no mental retardation, skeletal abnormalities or white forelock and no other cases of hearing loss in three generations.
On physical examination her height was 174 cm and her weight was 65.7kg, both above the 75th centile. Her physical examination was remarkable for a flat facial profile, a short broad nose, depressed nasal bridge, microgenia and retrognathia, and hypertelorism; the innercanthal distance was 4 cm, i.e. 2 standard deviations above the mean (Figs 1A,B), and she had dystopia canthorum with a W index of 2.27 cm. Her philtrum was normal and not smooth. There was no white forelock or evidence of any skin depigmentation. Her hair showed some graying at the age of 34 (within the normal range for Caucasians). Her eyes were normal, without heterochromia of the irides. Her ears were of normal shape and position without pits or tags. She had a high arched palate with normal uvula. The upper front teeth were replaced by a bridge to correct severe malocclusion (not shown). No goiter or other palpable thyroid abnormality was detected. There was limited movement of the wrist with ulnar deviation of the hands and mild flexion contractures of digits 2 through 5 (Fig 1C). In addition, there was clindactaly of the 5th digits. At the time of consultation, she had profound hearing loss and was adept at lip-reading. Her speech cadence and tone was monotonic and metallic.
Figure 1.

Clinical features of the patient at age 35 years. A. Frontal view demonstrating hypertelorism, down slanting palpebral fissures, depressed nasal bridge, frontal bossing and post surgical residual ptosis. B. lateral view shows flat profile. C. Lateral view of the right hand demonstrates mild flexion contractures of digits 2-5; she could not fully extend her fingers.
Pediatric clinical records were not available. However, family photographs during childhood showed striking frontal bossing, depressed nasal bridge, ptosis and apparent hypertelorism, and small downturned corners of the mouth (Figs 2A-C).
Figure 2.

Clinical features of the patient during infancy and early childhood. A. and B. Frontal and lateral views at approximately 8 months. C. Frontal view at approximately 20 months demonstrating moderate ptosis prior to surgery.
Molecular Analysis
Genomic DNA was prepared from peripheral leukocytes of the proband by standard procedure. All 9 exons and 20 bp of the flanking introns of the PAX3 gene were amplified by polymerase chain reaction (PCR) using primers designed by PRIMER 3 software [Rozen and Skaletsky, 2000] and standard conditions. PCR products were directly sequenced on an ABI 3130×l Genetic Analyzer by a modification of previously described methods [Chen et al., 2003].
DNA studies
Because of some similarities in facial and hand features between our patient and affected members of a family with craniofacial deafness hand syndrome who carry a mutation in exon 2 of the PAX3 gene [Asher et al., 1996] we first sequenced this exon. However, no mutation was found in it or the remainder of the coding sequence and intron-exon boundaries. Heterozygosity for known single nucleotide polymorphisms was detected in exon 8, ruling out a whole gene deletion.
Radiographic Imaging
AP and lateral hand and wrist radiographs were normal. The patient underwent a craniofacial computed tomography (CT) scan with axial thin section images and coronal reformatted images from below the hard palate to the supraorbital region. This provided only a limited view of the anterior two thirds of the brain to a level almost to the top of the lateral ventricles. There were no congenital brain anomalies or other brain abnormalities within the visualized portion of the cerebrum.
The craniofacial structures showed hypertelorism along with markedly underdeveloped paranasal sinuses (Fig 3). Only the maxillary sinuses showed near normal development although mildly small in size. The frontal and sphenoid sinuses showed no development. The ethmoid complex was markedly hypoplastic with only a very small amount of pneumatization.
Figure 3.

CT scan of the face at age 35 years. A. Axial image shows markedly hypoplastic ethmoid sinuses bilaterally (arrows). The sphenoid sinus is aplastic. Note the hypertelorism. B. Aplasia of frontal sinuses. The hypertelorism is again seen. C. Sagittal midline image again shows aplasia of the frontal and sphenoid sinuses. The laterally placed hypoplastic ethmoid air cells are not visualized on this midline image.
The temporal bones were also abnormal in appearance. The mastoid sinuses were underdeveloped with only the mastoid antra showing pneumatization. There was no pneumatization of mastoid cells within the mastoid processes on either side. The cochlea were small on both sides and contained only 1.5 turns although the vestibule and semicircular canals were normal in appearance (Fig 4). The internal auditory canals were normally formed and the facial nerve canals were normal in course and appearance. The middle ear and ossicles were normally formed.
Figure 4.
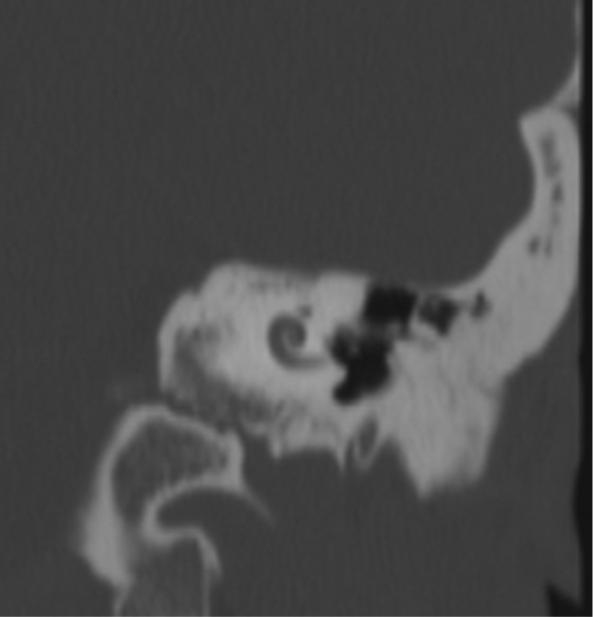
CT scan of the left cochlea demonstrating Mondini deformity with only 1.5 turns instead of the normal 2.5 turns. The same cochlear abnormality was present on the right.
DISCUSSION
Waardenburg syndrome type I is a dominantly inherited and clinically variable syndrome of deafness, pigmentary abnormalities and characteristic facial features [Farrer et al., 1994; Waardenburg, 1951]. The number of disorders belonging the Waardenberg syndromes continues to grow and there is heterogeneity on both clinical and molecular grounds. Currently six autosomal genes have been implicated in WS, with dominant mutations in PAX3 responsible for the majority of the cases for which the gene is known. There are also several reports of families displaying an unusual cluster of clinical features that resemble to WS. A family with two children was reported to have conductive deafness, unusual faces, severe ptosis, mild ectodermal dysplasia, recurrent ear infections and skeletal abnormalities, including internal rotation of the hips, with awkward toe-in gait, and restriction of pronation-supination, with subluxation of the radial head. The hands showed clindactaly of the 5th digits. Both children had very small and narrow external auditory canals with a narrow space between the dural plate, temporomandibular joint and sigmoid sinus [Jackson and Barr, 1978]. DNA studies were not performed on this family. Our patient had ptosis but lacks the specific skeletal and hair abnormalities seen in this family. Another paper described three families with a combination of early graying, sensorineural hearing loss and essential tremor [Karmody et al., 2005]. No sequence change was identified in PAX3 or MITF. The authors raised the question of whether this represents a new syndrome or is a subtype of WS. A third paper presented a girl with WS type I and bilateral renal anomaly [Ekinci et al., 2005]. Molecular studies were not performed.
Does our patient have a variant of WS? She shares some but not all the characteristics of CDHS. Her hand and wrist radiographs are normal and CT scan of the face did not show absence or hypoplasia of the nasal bones, but revealed underdevelopment of the sinuses ranging from mild hypoplasia to complete aplasia, features not previously associated with WS. To our knowledge only one family with CDHS has been published previously [Asher et al., 1996] and CT imaging was not performed in any of the affected individuals. Therefore, it is not possible to know whether the bone abnormalities observed in our patient are also present in them.
A search of the English literature did not yield previous reports of a syndrome with the combination of abnormalities seen in our patient. The cochlear malformation observed in our patient is a form of Mondini deformity that is associated with sensorineural hearing loss in Pendred syndrome, but is more often seen as an isolated abnormality. Although similar abnormalities have been described in WS (e.g., [Oysu et al., 2001], most of these patients do not have macroscopic inner ear malformations that are visualized by CT or MR imaging. There is one report of a man with similar sinus abnormalities discovered during evaluation of sinusitis symptoms [Haktanir et al., 2005]. No other description of the patient was provided beyond the statement that “no skeletal, systemic or hematological abnormality was detected on clinical and laboratory examinations.”
As is increasingly found for heterogeneous disorders in general, categorization on the basis of molecular etiology will likely be more useful than eponymous terminology or grouping by clinical features. For example, different PAX3 mutations in the paired box domain have been identified in the previously reported CDHS family [Asher et al., 1996] and in families with WS type 3 [Tekin et al., 2001]. Our patient does not carry these particular mutations or another coding or splice site sequence alteration in PAX3. Point mutations have been identified in more than 90% of people with WS type 1 or WS type 3 [DeStefano et al., 1998; Hoth et al., 1993], but a recent study found whole or partial PAX3 gene deletions in approximately 10% of affected individuals [Milunsky et al., 2007]. Partial or full SOX10 deletions have also been identified in WS types 2 and 4 [Bondurand et al., 2007]. The presence of heterozygosity for a PAX3 polymorphism rules out a full gene deletion, but a partial deletion remains a possible disease mechanism in our patient. PAX3 cDNA could be examined to look for deletion of several exons or abnormal splice forms and a copy number variation might be detected by array technology.
The pathway of melanocyte formation and differentiation is slowly being unveiled through mouse models and genetic syndromes. The majority of known dominant mutations that cause WS occur in one of four transcription factors: SOX10, PAX3, MITF and SNAI2 [Cook et al., 2005; Potterf et al., 2000]. In addition, mutations in either EDN3 or EDNRB may have dominant or recessive inheritance. SOX10 and PAX3 may activate MITF separately or in synergy [Bondurand et al., 2000; Cook et al., 2005; Potterf et al., 2000]. Downstream targets of MITF include SNAII2 among other genes. It is possible that a different gene in these pathways is responsible for the syndrome in our patient. Identification and further studies of the causative gene will explain the constellation of abnormalities affecting the function of the ear, facial abnormalities, and abnormal sinus development.
ACKNOWLEDGMENTS
We are grateful to the patient whose intellectual curiosity about her disorder was the impetus for this study. This research was supported in part by a training grant to AG (NIH 5T32GM007454).
REFERENCES
- Arias S. Genetic heterogeneity in the Waardenburg syndrome. Birth Defects Orig Art Ser. 1971;VII:87–101. [PubMed] [Google Scholar]
- Asher JJ, Sommer A, Morell R, Friedman T. Missense mutation in the paired domain of PAX3 causes craniofacial-deafness-hand syndrome. Hum Mutat. 1996;7:30–35. doi: 10.1002/(SICI)1098-1004(1996)7:1<30::AID-HUMU4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Baldwin C, Hoth C, Amos J, da-Silva E, Milunsky A. An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature. 1992;355:637–638. doi: 10.1038/355637a0. [DOI] [PubMed] [Google Scholar]
- Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A, Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M, Pingault V. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet. 2007;81:1169–1185. doi: 10.1086/522090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondurand N, Pingault V, Goerich DE, Lemort N, Sock E, Le Caignec C, Wegner M, Goossens M. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet. 2000;9:1907–1917. doi: 10.1093/hmg/9.13.1907. [DOI] [PubMed] [Google Scholar]
- Chen D-H, Brkanac Z, Verlinde C, Tan X-J, Bylenok L, Nochlin D, Matsushita M, Lipe H, Wolff J, Fernandez M, Cimino PJ, Bird TD, Raskind WH. Missense mutations in the regulatory domain of PKCg: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet. 2003;72:839–849. doi: 10.1086/373883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AL, Smith AG, Smit DJ, Leonard JH, Sturm RA. Co-expression of SOX9 and SOX10 during melanocytic differentiation in vitro. Exp Cell Res. 2005;308:222–235. doi: 10.1016/j.yexcr.2005.04.019. [DOI] [PubMed] [Google Scholar]
- DeStefano AL, Cupples LA, Arnos KS, Asher JH, Jr., Baldwin CT, Blanton S, Carey ML, da Silva EO, Friedman TB, Greenberg J, Lalwani AK, Milunsky A, Nance WE, Pandya A, Ramesar RS, Read AP, Tassabejhi M, Wilcox ER, Farrer LA. Correlation between Waardenburg syndrome phenotype and genotype in a population of individuals with identified PAX3 mutations. Hum Genet. 1998;102:499–506. doi: 10.1007/s004390050732. [DOI] [PubMed] [Google Scholar]
- Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, Martelli H, Bidaud C, Munnich A, Lyonnet S. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome) Nat Genet. 1996;12:442–444. doi: 10.1038/ng0496-442. [DOI] [PubMed] [Google Scholar]
- Ekinci S, Ciftci AO, Senocak ME, Buyukpamukcu N. Waardenburg syndrome associated with bilateral renal anomaly. J Pediatr Surg. 2005;40:879–881. doi: 10.1016/j.jpedsurg.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Arnos KS, Asher JH, Jr., Baldwin CT, Diehl SR, Friedman TB, Greenberg J, Grundfast KM, Hoth C, Lalwani AK, Landa B, Leverton K, Milunsky A, Morell R, Nance WE, Newton V, Ramesar R, Rao VS, Reynolds JE, San Agustin TB, Wilcox ER, Winship I, Read AP. Locus heterogeneity for Waardenburg syndrome is predictive of clinical subtypes. Am J Hum Genet. 1994;55:728–737. [PMC free article] [PubMed] [Google Scholar]
- Friedman R, Griffith A. Human nonsyndromic sensorineural deafness. Annu Rev Genomics Hum Genet. 2003;4:341–402. doi: 10.1146/annurev.genom.4.070802.110347. [DOI] [PubMed] [Google Scholar]
- Gürtler N, Lalwani A. Etiology of syndromic and nonsyndromic sensorineural hearing loss. Otolaryngol Clin North Am. 2002;35:891–908. doi: 10.1016/s0030-6665(02)00053-1. [DOI] [PubMed] [Google Scholar]
- Haktanir A, Acar M, Yucel A, Aycicek A, Degirmenci B, Albayrak R. Combined sphenoid and frontal sinus aplasia accompanied by bilateral maxillary and ethmoid sinus hypoplasia. Br J Radiol. 2005;78:1053–1056. doi: 10.1259/bjr/38163950. [DOI] [PubMed] [Google Scholar]
- Hoth C, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin C. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I) Am J Hum Genetics. 1993;52:455–462. [PMC free article] [PubMed] [Google Scholar]
- Hughes AE, Newton VE, Liu XZ, Read AP. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat Genet. 1994;7:509–512. doi: 10.1038/ng0894-509. [DOI] [PubMed] [Google Scholar]
- Jackson L, Barr M. Conductive deafness with ptosis and skeletal malformations in sibs: a probably autosomal recessive disorder. Birth Defects Orig Art Ser. 1978;14:199–204. [PubMed] [Google Scholar]
- Karmody CS, Blevins NH, Lalwani AK. Sensorineural hearing loss, early greying, and essential tremor: a new hereditary syndrome? Otolaryngol Head Neck Surg. 2005;133:94–99. doi: 10.1016/j.otohns.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Klein D. Historical background and evidence for dominant inheritance of the Klein-Waardenburg syndrome (type III) Am J Med Genet (Neuropsych Genet) 1983;14:231–239. doi: 10.1002/ajmg.1320140205. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55:95–100. doi: 10.1002/ajmg.1320550123. [DOI] [PubMed] [Google Scholar]
- Milunsky J. GeneReviews at GeneTests: Medical Genetics Information Resource [database online] Copyright, University of Washington; Seattle: 2007. Waardenburg Syndrome Type I. 1997-2008. Last revised 19 April 2007. [Google Scholar]
- Milunsky JM, Maher TA, Ito M, Milunsky A. The value of MPLA in Waardenburg Syndrome. Genet Testing. 2007;11:179–182. doi: 10.1089/gte.2006.0531. [DOI] [PubMed] [Google Scholar]
- Nance W. The genetics of deafness. Ment Retard Dev Disabil Res Rev. 2003;9:109–119. doi: 10.1002/mrdd.10067. [DOI] [PubMed] [Google Scholar]
- Oysu C, Oysu A, Aslan I, Tinaz M. Temporal bone imaging findings in Waardenburg’s syndrome. Int J Pediatr Otorhinolaryngol. 2001;58:215–221. doi: 10.1016/s0165-5876(01)00443-8. [DOI] [PubMed] [Google Scholar]
- Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu MO, Puliti A, Herbarth B, Hermans-Borgmeyer I, Legius E, Matthijs G, Amiel J, Lyonnet S, Ceccherini I, Romeo G, Smith JC, Read AP, Wegner M, Goossens M. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18:171–173. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- Potterf SB, Furumura M, Dunn KJ, Arnheiter H, Pavan WJ. Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum Genet. 2000;107:1–6. doi: 10.1007/s004390000328. [DOI] [PubMed] [Google Scholar]
- Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, Chakravart A. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell. 1994;79:1257–1266. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34:656–665. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon W, Toriello HV, Downs CA. Epidemiology, Etiology, Genetic Patterns and Genetic Counseling. In: Toriello HV, Reardon, Gorlin RJ, editors. Hereditary Hearing Loss and its Syndromes. Oxford University Press; New York: 2004. [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet. 2002;11:3231–3236. doi: 10.1093/hmg/11.25.3231. [DOI] [PubMed] [Google Scholar]
- Senrui H. Congenital clasped thumb combined with Waardenburg syndrome in three generations of one family: an undescribed congenital anomalies complex. J Pediatr Orthop. 1984;4:472–476. doi: 10.1097/01241398-198408000-00017. [DOI] [PubMed] [Google Scholar]
- Shah K, Dalal S, Desai M, Sheth P, Joshi N, Ambani L. White forelock, pigmentary disorder of irides, and long segment Hirschsprung disease: possible variant of Waardenburg syndrome. J Pediatr. 1981;99:432–435. doi: 10.1016/s0022-3476(81)80339-3. [DOI] [PubMed] [Google Scholar]
- Shears D, Conlon H, Murakami T, Fukai K, Alles R, Trembath R, Bitner-Glindzicz M. Molecular heterogeneity in two families with auditory pigmentary syndromes: the role of neuroimaging and genetic analysis in deafness. Clin Genet. 2004;65:384–389. doi: 10.1111/j.0009-9163.2004.00235.x. [DOI] [PubMed] [Google Scholar]
- Smith R, VanCamp G. GeneReviews at GeneTests: Medical Genetics Information Resource. Copyright, University of Washington; 2007. Deafness and Hereditary Hearing Loss Overview. 1997-2008. Last revised 30 January 2007. [Google Scholar]
- Sommer A, Bartholomew DW. Craniofacial-deafness-hand syndrome revisited. Am J Med Genet Part A. 2003;123A:91–94. doi: 10.1002/ajmg.a.20501. [DOI] [PubMed] [Google Scholar]
- Sommer A, Young-Wee T, Frye T. Previously undescribed syndrome of craniofacial, hand anomalies, and sensorineural deafness. Am J Med Genet. 1983;15:71–77. doi: 10.1002/ajmg.1320150109. [DOI] [PubMed] [Google Scholar]
- Syrris P, Carter ND, Patton MA. Novel nonsense mutation of the endothelin-B receptor gene in a family with Waardenburg-Hirschsprung disease. Am J Med Genet. 1999;87:69–71. [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Liu XZ, Brady A, Donnai D, Krajewska-Walasek M, Murday V, Norman A, Obersztyn E, Reardon W, Rice JC, Trembath R, Wieacker P, Whiteford M, Winter R, Read AP. The mutational spectrum in Waardenburg syndrome. Hum Mol Genet. 1995;4:2131–2137. doi: 10.1093/hmg/4.11.2131. [DOI] [PubMed] [Google Scholar]
- Tekin M, Bodurtha JN, Nance WE, Pandya A. Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3: a simple variant or a true syndrome? Clinical Genetics. 2001;60:301–304. doi: 10.1034/j.1399-0004.2001.600408.x. [DOI] [PubMed] [Google Scholar]
- Waardenburg P. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genetics. 1951;3:195–253. [PMC free article] [PubMed] [Google Scholar]
- Wollnik B, Tukel T, Uyguner O, Ghanbari A, Kayserili H, Emiroglu M, Yuksel-Apak M. Homozygous and heterozygous inheritance of PAX3 mutations causes different types of Waardenburg syndrome. Am J Med Genet Part A. 2003;122:42–45. doi: 10.1002/ajmg.a.20260. [DOI] [PubMed] [Google Scholar]
- Zlotogora J, Lerer I, Bar-David S, Ergaz Z, Abeliovich D. Homozygosity for Waardenburg syndrome. Am J Hum Genet. 1995;56:1173–1178. [PMC free article] [PubMed] [Google Scholar]