

Abstract

Dehydrochlorination of methyl 2-chloroaziridine-2-carboxylates generates the first examples of enantiopure 2-substituted 2H-azirine-3-carboxylates which undergo the aza Diels-Alder with dienes to give bicyclic and tricyclic aziridines in good yields.
The chemistry of 2H-azirines, the smallest unsaturated nitrogen heterocycle, has been extensively explored.1 The C=N π-bond and nitrogen lone pair, activated by the high ring strain, partake in a variety of reactions with nucleophiles. 2H-Azirines also undergo Diels-Alder reactions, but are generally poor dienophiles reacting only with highly reactive dienes. However, Gilchrist and co-workers have shown that 2H-azirine-3-carboxylates such as (±)-1, available only in racemic form, are effective aza dienophiles and participate in a variety of cycloaddition reactions (Scheme 1).2 These additions are highly regio- and stereoselective and consistent with endo addition of the azirine to the diene. Unfortunately, attempts to control the absolute stereochemistry by installing chiral auxiliaries (chiral ester and amide) in 1 were unsucessful.3 However, using the same approach, Somfai and co-workers later found that Diels-Alder reactions with unsubstituted chiral aziridine 2 and Lewis acids resulted in bi- and tricyclic aziridines in good yields and in high selectivity.4 In 2002 we disclosed the first asymmetric synthesis of 2H-azirine-3-phosphonates (+)-3 by Swern oxidation of NH-aziridine- 2-phosphonates and demonstrated their utility as new chiral aza dienophiles.5 Here we describe a new method for the asymmetric synthesis of 2-substituted 2H-azirines-3- carboxylates 1, where the chirality resides within the azirine ring, and their Diels-Alder reactions.
Scheme 1.

Methods currently available for the preparation of azirines, including the thermal decomposition of vinyl azides and elimination reactions of functionalized aziridine, are not suitable for the asymmetric synthesis of 2H-azirine -3-carboxylates.1 Vinyl azides lack chirality1 and Swern oxidation of NH aziridine esters6 and elimination of sulfenic acid from N-sulfinyl aziridine esters always produce 2H-azirine-2-carboxylates.7 Conceptually, dehydrochlorination of a chiral 2-chloroaziridine-2-carboxylate 5 would have to give the desired enantiopure 2H-azirine-3-carboxylate 4. We describe here the realization of this concept resulting in the first asymmetric synthesis of 4 where the chirality resides only at the 2-position (Scheme 1).
Although 2-halo 2H-azirines are known1d methods for the synthesis of 2-haloaziridines usually result in low yields and mixtures of cis and trans isomers.8 Recently, De Kimpe and co-workers described the selective synthesis of N-tosyl 2-chloro-2-imidoylaziridines via the addition of dichloro-aza-allylic anions to N-sulfonyl imines.8 However, there are no reports of the asymmetric syntheses of 2-haloaziridines.
Earlier studies from our laboratory demonstrated that the one-pot, highly diastereoselective aza-Darzens reaction of lithium α-bromo enolates with enantiopure sulfinimines (N-sulfinyl imines) is an important method for the asymmetric construction of aziridine-2-carboxylates.9.10 For the asymmetric synthesis methyl 2-chloroaziridine-2-carboxylates 9, we examined the addition of the lithium enolate of methyl dichloroacetate 7 to sulfinimines (S)-(+)-6 (Scheme 2). The dichloro enolate 7 was generated at −78 °C by treatment of methyl dichloroacetate with LiHMDS.
Scheme 2.

The enolate was added to the appropriate sulfinimines (S)-(+)-6, affording the corresponding 2,2-dichloro-β-amino esters (SS,S)-(+)-8 in good to excellent yield as single enantiomers. The (S)-stereochemistry was assigned to the stereocenter at C-3 in 8 based on our earlier proposed six memberd chair like transition state model for the addition of enolates to sulfinimines.11
Cyclization of the β-amino esters 8 to the 2-chloroaziridines 9 was next explored. Treatment of 8 with NaH in THF gave low yields, ca 20% of the corresponding aziridines 9b and 9c along with recovered starting material and decomposition products. Attempts to improve the yields by increasing the amount of hydride and increasing the reaction temperature were unsuccessful. With 2 equiv of KH at 0 °C in THF, (+)-8b and (+)-8c gave (SS,2R,3S)-(+)-9b and (SS,2R,3S)-(+)-9c in 88 and 92% yields, respectively, (Scheme 2). Under similar conditions, (+)-8a (R = Ph) gave the retro-Mannich product sulfinimine (S)-(+)-6a in 72% isolated yield. Retro-Mannich fragmentations of sulfinimine-derived sulfinamide products are usually rare because the N-sulfinyl group stabilizes anions at nitrogen.12 However, in 8a the combination of steric inhibition to chloride displacement and the stability of the dichloro enolate 7 apparently favors the retro-Mannich fragmentation.13
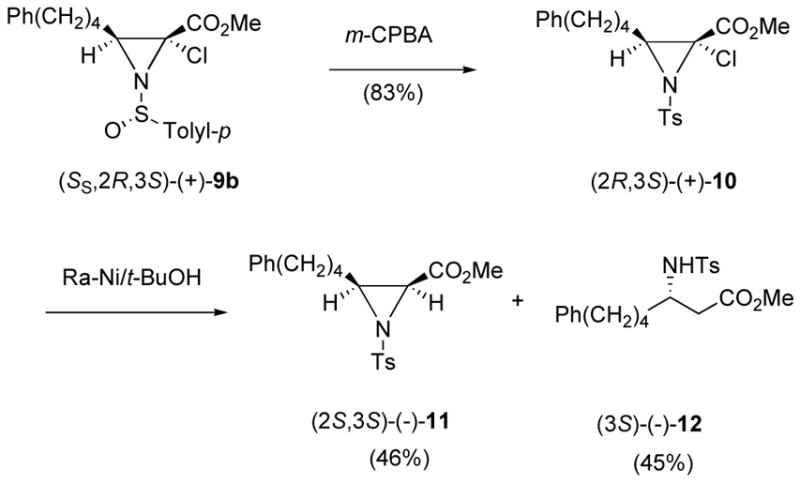
The absolute stereochemistry of methyl 2-chloroaziridine-2-carboxylate (+)-9b was assigned by transforming it into products of known absolute configuration as shown in Scheme 3. Oxidation of (+)-9b with m-CPBA gave the N-tosyl 2-chloroaziridine (+)-10 in 83% isolated yield. When (+)-10 was subjected to catalytic ring opening with Raney-Nickel in t-BuOH two products, aziridine (2S,3S)-(−)-11 and β-amino ester (S)-(−)-12, were isolated in 45 and 46 % yields, respectively. Under these conditions the C-2 chloro group is stereoselectivity replaced with a hydrogen atom, and aziridine ring opening gives the β-amino ester. Both (−)-11 and (−)-12 have previously been prepared9c which confirms the stereochemistry at C-3 in (+)-8 as (S) and two stereocenters at C-2 and C-3 in the 2-chloroaziridines (+)-9 as (2R) and (3S).
Scheme 3.
To induce dehydrochlorination in (+)-9 it is necessary to first remove the N-sulfinyl group. Not unexpectedly, attempts to eliminate this group in the usual manner with TFA-MeOH or HCl-Et2O resulted in decomposition. 2H-Azirines-3-carboxylates are very reactive toward nucleophilic addition2,3 and would not be expected to survive these conditions. When we employed our MeMgBr tactic for selective removal of aziridine N-sulfinyl groups with (+)-9b, 2-methylaziridine-2-carboxylate (2S,3S)-14a was isolated as a single isomer in 58% yield (Scheme 4).7c This result provides strong evidence for the formation of 2H-azirine-3-carboxylate (S)-13 as an intermediate where the nitrogen anion induces the loss of the chloride anion (Scheme 4). The stereochemistry of the product results from addition of the Grignard reagent from the least hindered direction. The stereochemistry of 14 was determined by removal of the N-sulfinyl group in aziridine (+)-15,9c a known compound, as shown in Scheme 4. Similar results were observed for the addition of phenylmagnesium bromide and vinylmagnesium chloride to 9b and 9c. This procedure represents a new way of preparing 2-substituted aziridine-2-carboxylates which are valuable precursors of enantiopure quaternary α-amino acids.7b
Scheme 4.

It is not possible to carry out aza Diels-Alder reactions with 2H-azirine (S)-13 because this highly reactive azirine does not survive the conditions currently employed for removal of the N-sulfinyl group. A new method, that does not require acids, bases, or protic solvents, is needed for elimination of the N-sulfinyl group in 9. Photodesulfinylation is a solution to this problem. Although the selective photocleavage of the N-tosyl protecting group is well known,14 related photo cleavage of the N-sulfinyl groups has not been described.
Photolysis of (+)-9b in ether degassed with argon in a Pyrex vial at 3000 Å in a Rayonet photochemical reactor for 10 h gave 2-chloroaziridine (−)-16 in 70% yield following chromatography (Scheme 5). Significantly, (−)-16 is the first stable example of an N-unprotected α-halo-α-amino ester.15 The reluctance of (−)-16 to eliminate HCl is likely due to the added strain energy that results in formation of the C-N double bond in the three-membered aziridine ring. Attempts at chromatographic isolation of azirine (S)-13 by reaction of 16 with Et3N or Hunig’s base were unsuccessful, resulting in decomposition.
Scheme 5.
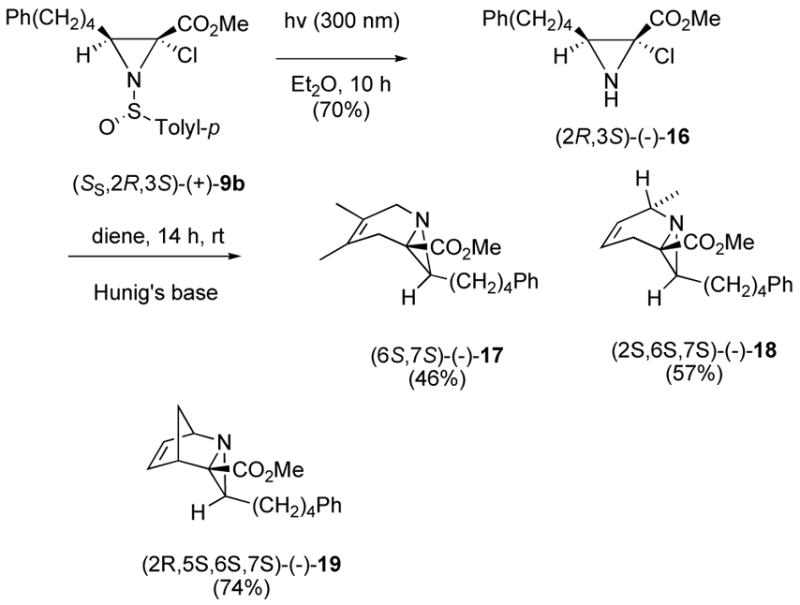
It was found to be more efficient to conduct the aza Diels-Alder reaction with crude (−)-16. Following the photodesulfinylation of (+)-9b, the crude reaction mixture was treated with 100 equivalents of the appropriate diene, 2,3-dimethyl-1,3-butadiene, trans-piperylene, or cyclopentadiene, and a few drops of Hunig’s base. After stirring for 8 h at rt, the enantiopure bicyclic and tricyclic aziridines carboxylates (−)-17, (−)-18, and (−)-19 were isolated as single isomers in 46 to 74% yield for the two steps by chromatography (Scheme 5). The structures assigned to these aziridines are based on endo addition of the 2H-azirine-3-carboxylated aza dienophile (S)-13 as previously reported.2,3,5
In summary, the first asymmetric synthesis of 2-substituted 2H-azirine-3-carboxylates has been accomplished by dehydrochlorination of 2-chloroaziridine 2-carboxylates. These azirines readily undergo the aza Diels-Alder reaction with dienes to give enantiomerically pure bi- and tricyclic aziridine carboxylates in good yields. Highlights of this methodology include: 1) a new method for the asymmetric synthesis of 2-substituted aziridine-2-carboxylates; and 2) the first synthesis of a stable N-unprotected α-halo amino acid. Photodesulfinylation of sulfinamides represents an important new procedure, not requiring acids or bases, for removal of the valuable amine N-sulfinyl protecting group.
Supplementary Material
Experimental details and 1H and 13C NMR spectra for all new compounds. This materiual is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Fang Yu (Temple University) for aid with the photochemical apparatus. This work was supported by grants from the NIH-NIGMS (GM578970 and GM51982).
References
- 1.For recent reviews on 2H-azirines see Pearson WH, Lian BW, Bergmeier SC. In: Comprehensive Heterocyclic Chemistry II. Padwa A, editor. Chapter 1 Pergamon Press; Oxford: 1966. Palacios F, Ochoa de Retana AM, de Marigorta ZM, Manuel de los Santos J. Eur J Org Chem. 2001:2401.Pinho e Melo TMVD, Rocha Gonsalves ADD’a. Curr Org Syn. 2004;1:275.
- 2.Gilchrist TL. Aldrichimica Acta. 2001;34:51. [Google Scholar]
- 3.(a) Alves MJ, Gilchrist TL. J Chem Soc, Perkin Trans 1. 1998:229. [Google Scholar]; (b) Alvares YSP, Alves MJ, Azoia NG, Bickley JF, Gilchrist TL. J Chem Soc, Perkin Trans 1. 2002:1911. [Google Scholar]
- 4.Timen AS, Somfai P. J Org Chem. 2003;68:9958. doi: 10.1021/jo0352326. [DOI] [PubMed] [Google Scholar]
- 5.Davis FA, Wu Y, Yan H, Prasad KR, McCoull W. Org Lett. 2002;4:655. doi: 10.1021/ol017289p. [DOI] [PubMed] [Google Scholar]
- 6.Gentilucci L, Grijzen Y, Thijs L, Zwanenburg B. Tetrahedron Lett. 1995;36:4665. [Google Scholar]
- 7.(a) Davis FA, Reddy GV, Liu H. J Am Chem Soc. 1995;117:3651. [Google Scholar]; (b) Davis FA, Liang CH, Liu H. J Org Chem. 1997;62:3796. [Google Scholar]; (c) Davis FA, Liu H, Liang CH, Reddy GV, Zhang Y, Fang T, Titus DD. J Org Chem. 1999;64:8929. doi: 10.1021/jo991389f. [DOI] [PubMed] [Google Scholar]
- 8.For leading references to 2-haloaziridines see: Giubellina N, Mangelinckx S, Tornroos KW, De Kimpe N. J Org Chem. 2006;71:5881. doi: 10.1021/jo060241a.
- 9.(a) Davis FA, Liu H, Zhou P, Fang T, Reddy GV, Zhang Y. J Org Chem. 1999;64:7559. [Google Scholar]; (b) Davis FA, Zhang Y, Rao A, Zhang Z. Tetrahedron. 2001;57:6345. [Google Scholar]; (c) Davis FA, Deng J, Zhang Y, Haltiwanger RC. Tetrahedron. 2002;58:7135. [Google Scholar]
- 10.For a review on the synthesis of chiral aziridines see: McCoull W, Davis FA. Synthesis. 2000:1347.
- 11.Davis FA, Reddy RT, Reddy RE. J Org Chem. 1992;57:6387. [Google Scholar]
- 12.For leading references see Zhou P, Chen BC, Davis FA. Tetrahedron. 2004;60:8003.Davis FA. J Org Chem. 2006;71:8993. doi: 10.1021/jo061027p.
- 13.For a recent example of the sulfinimine retro-Mannich fragmentation see: Davis FA, Zhang Y, Qiu H. Org Lett. 2007;9 doi: 10.1021/ol063058c. ASAP.
- 14.(a) Hamada T, Nishida A, Yonemitsu O. J Am Chem Soc. 1986;108:140. [Google Scholar]; (b) Art JF, Kestemont JP, Soumillion JPh. Tetrahedron Lett. 1991;32:1425. [Google Scholar]; (c) Abad A, Mellier D, Pete JP, Portella C. Tetrahedron Lett. 1971;47:4555. [Google Scholar]
- 15.For leading references to attempts to prepare α-fluoro-α-amino acids see: Huber DP, Stanek K, Togni A. Tetrahedron: Asymmetry. 2006;17:658.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and 1H and 13C NMR spectra for all new compounds. This materiual is available free of charge via the Internet at http://pubs.acs.org.