

Abstract
We previously reported that only a subpopulation of PAR-1-stimulated platelets binds coagulation factor IXa, since confirmed by other laboratories. Since calcium changes have been implicated in exposure of procoagulant aminophospholipids, we have now examined calcium fluxes in this subpopulation by measuring fluorescence changes in Fura Red/AM-loaded platelets following PAR-1 stimulation. While fluorescence changes in all platelets indicated calcium release from internal stores and influx of external calcium, a subpopulation of platelets displayed a pronounced increase in calcium transients by 15 seconds and positive factor IXa binding by 2 minutes, with calcium transients sustained for 45 minutes. Pretreatment of platelets with Xestospongin C to inhibit IP3-mediated dense tubule calcium release, and the presence of impermeable calcium channel blockers nifedipine, SKF96365 or LaCl3, inhibited PAR-1-induced development of a subpopulation with pronounced calcium transients, factor IXa binding, and platelet support of FXa generation, suggesting the importance of both release of calcium from internal stores and influx of extracellular calcium. When platelets were stimulated in EDTA for 5 to 20 minutes before addition of calcium, factor IXa binding sites developed on a smaller subpopulation but with unchanged rate indicating sustained opening of calcium channels and continued availability of signaling elements required for binding site exposure. While pretreatment of platelets with 100 μM BAPTA/AM (Kd 160 nM) had minimal effects, 100 μM 5, 5′-dimethylBAPTA/AM (Kd 40 nM) completely inhibited the appearance and function of the platelet subpopulation, indicating the importance of minor increases of cytoplasmic calcium. We conclude that PAR-1-stimulated development of factor IXa binding sites in a subpopulation of platelets is dependent upon release of calcium from internal stores leading to sustained and pronounced calcium transients.
An essential event in the hemostatic response to vascular injury is the assembly of the factor X (FX)1 activating complex on the surface of activated platelets (1, 2). The assembly of this important enzymatic complex requires the exposure of coagulation protein binding sites on the surface of platelets activated with thrombin or collagen but not with adenosine diphosphate (3–6). All proteins required for physiologically relevant platelet-supported FX-activation, including the enzyme factor IXa (FIXa), the cofactor factor VIIIa (FVIIIa) and the substrate factor X (FX), must be bound to their respective receptors on activated platelets (7). Thus, the zymogen FIX binds with high affinity (Kd ~2.5 nM) to a discreet number of platelet receptors that can also be occupied by the enzyme FIXa (8), which binds to an additional class of sites specific for the enzyme whose binding is enhanced (Kd ~2.5 nM becomes 0.5 nM) in the presence of both FVIIIa and FX. FVIII (Kd ~3.0 nM) (4, 9, 10) and FVIIIa (Kd ~0.8 nM) (4) also bind to specific, high-affinity sites on activated platelets. Although FX and prothrombin occupy a high-capacity, low-affinity (Kd ~300 nM) shared site (5), FX also occupies a specific binding site (Kd ~5 nM), consisting of bound FVIIIa (4, 6, 11). The physiological relevance of these interactions is emphasized by the fact that occupancy of these binding sites on activated platelets is closely correlated with enhanced rates of FX activation leading to an increase in catalytic efficiency (kcat/Km) of >2 × 107-fold in the presence of the assembled complex (7), and the fact that severe, spontaneous and post-traumatic bleeding complications occur in patients with deficiencies of FIX (12), FVIII (13), FX (14) and platelet receptors for FVIIIa (15, 16).
Although platelets respond to many agonists with functional endpoints required for primary hemostasis including adhesion to subendothelial matrix, secretion of granule contents, aggregation and platelet plug formation to stop the flow of blood through breaches of the vasculature (17, 18), exposure to strong agonists, such as collagen and thrombin, results as well in membrane surface changes allowing binding and complexation of the coagulation proteins responsible for physiologically relevant intrinsic FXa and thrombin generation (19–21). Little is known of the platelet signal transduction mechanisms resulting in these membrane surface changes.
All platelet agonists induce changes in cytoplasmic calcium that are usually followed using UV-excitable calcium indicators like Fura 2/AM (22). Increased intracellular calcium has been shown to be required for platelet adhesion (23), secretion of dense- and α-granule contents (24, 25), and for platelet shape change and aggregation (26–28). A requirement for increased cytoplasmic calcium has been demonstrated for platelet procoagulant surface changes. Thapsigargin, an inhibitor of the intracellular sarco(endo)plasmic reticulum Ca/Mg/ATPase (SERCA), has been shown to increase cytoplasmic calcium by depleting internal stores of calcium and initiating capacitative entry of extracellular calcium (29, 30), and capacitative calcium entry has been shown to lead to exposure of phosphatidylserine, a requirement for binding of coagulation factors to activated cell surfaces (31–33). However, chelation of cytoplasmic calcium with internalized BAPTA/AM has been reported not to inhibit phospholipid scrambling (34). Requirements for capacitative calcium entry, termed variously Store Operated Calcium Entry (SOCE) or Store Mediated Calcium Entry (SMCE) have been intensely investigated (35–43).
We recently showed that only a subpopulation of PAR-1 stimulated platelets binds FIXa, although the remainder of stimulated platelets shows other endpoints of PAR-1 activation including α-granule secretion and aggregation (44). To further explore the requirements for surface changes in this specific subpopulation leading to exposure of FIXa binding sites, we investigated the calcium changes post PAR-1 stimulation in both the bulk of platelets shown previously to be negative for FIXa binding sites, and in the subpopulation of platelets that binds FIXa.
EXPERIMENTAL PROCEDURES
Reagents
Bovine serum albumin, buffer reagents, disodium EDTA, dimethylsulfoxide (DMSO), calcium chloride, Sepharose 2B-CL, and lanthanum chloride were obtained from Sigma Chemical Company (St. Louis, MO). Electrophoresis reagents were from Bio-Rad Laboratories, Inc. (Melville, NY). Chromogenic substrates N-α-Benzyloxycarbonyl-D-arginyl-L-glycyl-L-arginine-p-nitroanaline-dihydrochloride (S-2765) and H-D-Phenylalanyl-L-pipecolyl-L-arginine-p-nitroanaline dihydrochloride (S-2238) were purchased from DiaPharma (West Chester, OH). The thrombin receptor agonist peptide, SFLLRN-amide, was synthesized using 9–fluorenylmethyloxycarbonyl (FMOC) chemistry on an Applied Biosystems 430A synthesizer and reverse-phase HPLC purified to greater than 99% homogeneity. FITC-labeled CD42b against glycoprotein Ibα (GPIbα) was purchased from BD-PharMingen (Los Angeles, CA). [1,2–bis(o–Aminophenoxy)ethane-N,N,N′,N′-tetraacetic Acid Tetra(acetoxymethyl) Ester] (BAPTA/AM), thapsigargin, nifedipine, SKF96365, Xestospongin C and Fura Red/AM were purchased from Calbiochem (La Jolla, CA). 5,5′-dimethylBAPTA/AM was purchased from Molecular Probes (Eugene, OR).
Proteins
FIX, FX, and FXIa were obtained from Enzyme Research Laboratories, Inc. (South Bend, IN) or Haematologic Technologies, Inc. (Burlington, VT). Coagulation proteins were obtained in or dissolved in 20 mM Tris-hydroxymethane, 150 mM NaCl and dialyzed against the same buffer to remove inhibitors. Concentrations were determined by bicinchoninic acid assay (Pierce Chemical Co. Rockford, IL) and purity was assessed by SDS-PAGE visualized with Coomassie Brilliant Blue staining or by Western blot using specific antibodies. Baxter Healthcare Corporation (Duarte, CA) generously supplied recombinant FVIII. The monoclonal antibody C10D raised against the catalytic domain of FIX (45) was fluoresceinated as previously reported (44). Protein modifications did not interfere with the ability of the antibody to inhibit clotting of normal human plasma. FIX was activated to FIXa by FXIa as previously described (46).
Platelet Preparation
Washed, aspirin-treated, gel-filtered platelets were prepared from human whole blood as described previously (46). Platelet rich plasma was incubated with 1 mM acetylsalicylic acid for 30 minutes to inhibit feedback activation through prostaglandin synthesis, and with prostaglandin E1 (PGE1) (20 μM) for 15 minutes, before layering over the bovine serum albumin gradient for the wash step and/or gel filtration.
Treatment of Platelets with Inhibitors
SKF96365 was dissolved in water, BAPTA/AM, 5, 5′-dimethylBAPTA/AM, nifedipine, and Xestospongin C were dissolved in DMSO to 10 mM, thapsigargin to 2 mM, and all were aliquoted into dark tubes at 10–30 μl/tube, and kept at −20° or −80°C until use. Thawed aliquots of inhibitors were maintained covered on ice until warmed just before addition to platelet suspensions. Stock solutions were diluted in DMSO (SKF96365 in buffer) so that addition to platelets constituted 0.5–1% DMSO (v/v). This DMSO concentration was determined not to interfere with FXa generation. Platelets were agitated gently upon addition of inhibitors and incubated covered at 37°C for 10 minutes prior to functional testing. Thapsigargin was added with or without agonist just before testing. To test the ability of cell permeant calcium chelators to remove free calcium, fluorescence emission (λex = 380 nm; λem = 510 nm) of Fura 2/AM-loaded control platelets and Fura 2/AM-loaded platelets treated for 10 minutes with 100 μM BAPTA/AM were followed before and after stimulation with 1U/ml thrombin. Treatment with 100 μM BAPTA/AM was found to completely suppress fluorescence changes seen in control platelets.
FXa Generation
FXa was generated as described previously (47) with the following modifications. Washed, aspirin-treated, gel-filtered platelets (5 × 107/ml f.c.) were treated with DMSO or inhibitors for 10 minutes at 37°C before dilution into wells of a microtiter plate. FIXa and varying concentrations of agonists (thrombin or SFLLRN-amide) and/or thapsigargin were added and activation proceeded for 10 minutes before addition of cofactor and substrate. FVIII (200 U/ml) was incubated 1 minute at 37°C with thrombin (0.5 U/ml) and an additional minute with hirudin (5 U/ml) immediately before addition to reaction mixtures. Reactions were stopped after 2–3 minutes and were analyzed for FXa generated by chromogenic substrate S-2765 hydrolysis as described (47). Negative controls included reactions missing the enzyme, reactions missing platelets, and reactions using unstimulated platelets.
Data Analysis
Results from enzyme assays were converted to FXa formed per minute by comparison to a standard curve of FXa cleavage of S-2765 and plotted using KaleidaGraph software (Synergy, Reading, PA) to derive kinetic parameters. Results from multiple experiments were pooled and analyzed for means and standard errors.
Flow Cytometry
PRP was incubated with 10 μM Fura Red/AM for 45 minutes at room temperature in the dark with aspirin and PGE1 added 15 minutes before gel filtration. Gel-filtered Fura Red-loaded control platelets, inhibitor-treated or DMSO-treated platelets, were incubated (2 × 107/ml) with 10 nM FIXa and FITC-labeled monoclonal antibody C10D in reaction buffer containing 2.5 mM calcium or 2.5 mM calcium with 5 mM EDTA. Reactions were maintained at 37°C and sampled at time intervals before and after addition of 125 μM SFLLRN-amide or 25 nM thapsigargin or both. All timed samples were diluted 10-fold immediately and analyzed on a FACSCalibur flow cytometer (BD-PharMingen, Los Angeles, CA) equipped with a 488 nm-emitting laser. Data were collected using E00 voltage settings for forward scatter sensitivity, with a threshold of 2 to eliminate analysis of debris. Compensations were set to minimize fluorescence channel 1 (FL-1) spillover into fluorescence channel 2 (FL-2) and spillover of fluorescence channel 3 (FL-3) into FL-2. Using CellQuest Pro software (BD-PharMingen), platelets were defined by their characteristic forward vs. side scatter pattern as well as by positive staining with fluorescein-CD42b (anti-GPIbα). Negative controls included unlabeled platelets, excess unlabeled CD42b for FITC-CD42b, and incubation with FITC-C10D in the absence of FIXa. Gated platelets were analyzed for fluorescein emission in FL-1, and for Fura Red emission in FL-3 that were displayed together on log scales as density plots or individually as histograms. All reactions contained the FITC-labeled anti-FIX antibody.
Data Analysis
Quadrants (density plots) or markers (histograms) were established with negative control samples to determine the statistics of positive and negative populations. Events measured with buffer alone were subtracted from events measured with platelet samples. Fluorescence data from both density plots and histograms were averaged wherever possible. Data from different experiments collected under the same conditions were averaged and statistically analyzed.
RESULTS
Correlation of Procoagulant Subpopulation Size with Platelet Support of FXa Generation
Platelets prepared from individual donor whole blood were stimulated with 125 μM SFLLRN-amide and analyzed for both FIXa binding by flow cytometry and for support of FXa generation in assays containing thrombin-activated FVIII (Figure 1). PAR-1-stimulated platelets showed a wide range of FIXa-binding subpopulation size (2.4–28%) and a wide range of maximal FXa generation (14–64 nM FXa/min) calculated from FIXa titration studies. When the subpopulation size of individual blood donors (Figure 1, abscissa) was plotted against the maximal platelet-supported FXa generation (Figure 1, ordinate) the correlation coefficient was 0.83.
Figure 1. Correlation of FIXa-binding subpopulation with platelet support of FXa generation.

Whole blood drawn from 15 individuals was processed to gel-filtered platelets as described in EXPERIMENTAL PROCEDURES. Platelets adjusted to 2 × 107/ml were incubated with 2.5 mM CaCl2, 10 nM FIXa and stimulated with 125 μM SFLLRN-amide for 10 minutes at 37°C before addition of FITC-labeled anti-FIX. After 15 minutes at room temperature, reactions were diluted for flow cytometry analysis. The percent of stimulated platelets binding FIXa is shown on the abscissa. Gel-filtered platelets were adjusted to 5 × 107/ml for FXa generation reactions that included 5 mM CaCl2, various concentrations of FIXa, thrombin-activated FVIII, and 250 nM FX. Reactions were analyzed for FXa as described in EXPERIMENTAL PROCEDURES. Data were plotted in KaleidaGraph to determine maximum rates of FXa generation shown on the ordinate. KaleidaGraph was used to generate the correlation coefficient of 0.83.
Thapsigargin Effect on FXa Generation
Since previous reports demonstrated a calcium requirement for exposure of phosphatidylserine (31) using thapsigargin, a SERCA pump inhibitor, we investigated a requirement for calcium for development of a FIXa binding subpopulation. Thapsigargin was used alone or in combination with 50 μM SFLLRN-amide to stimulate platelet support of FXa generation (Figure 2). As little as 8 nM thapsigargin stimulated the platelets to the same degree as 50 μM SFLLRN-amide. Increasing thapsigargin produced increasing FXa generation. Stimulating platelets with both thapsigargin and SFLLRN-amide increased the platelet response to SFLLRN-amide. These data suggested that increased cytoplasmic calcium via both emptying of internal calcium stores and entry of external calcium via Store Operated Calcium Entry (SOCE) stimulated platelet membrane changes that supported FXa generation.
Figure 2. Thapsigargin as stimulant or costimulant of platelet procoagulant changes.
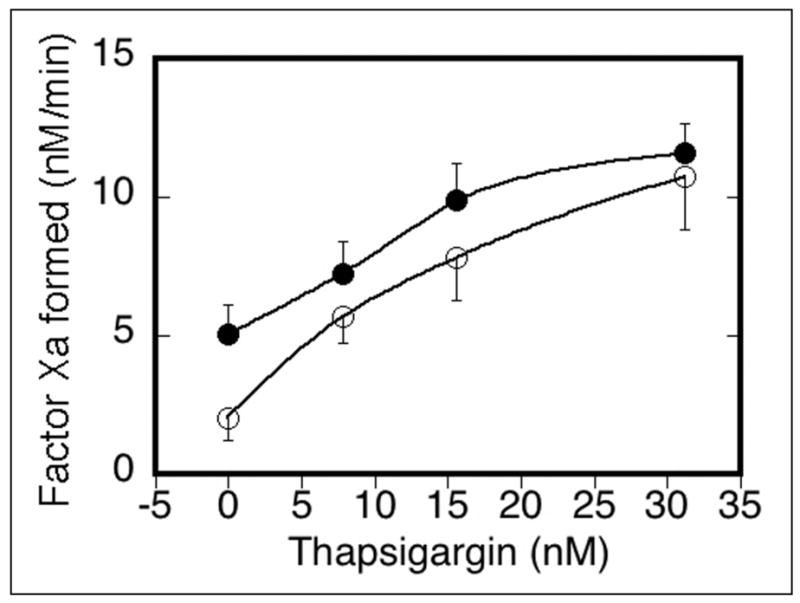
Platelets were stimulated with either thapsigargin alone (❍), or with 50 μM SFLLRN-amide and thapsigargin (●) for 10 minutes at 37°C before addition to FX activation reactions containing 5 mM calcium chloride, 1 nM FIXa, FVIIIa (6 U/ml) and 250 nM FX. Results of chromogenic substrate S-2765 cleavage assays converted to nM FXa formed per minute were collected from six experiments and analyzed for means and S.E.M. before graphing.
Visualizing Calcium Fluxes Preceding FIXa Binding
To ascertain the relationship between calcium fluxes and exposure of FIXa binding sites on a subpopulation of platelets, platelets were loaded with Fura Red/AM, a cell-permeant calcium indicator (48, 49). Excited at 488 nm, Fura Red fluoresces at 580 nm in the absence of calcium, but is quenched by its presence. Fura Red-loaded platelets were stimulated with saturating amounts of SFLLRN-amide (125 μM) in the presence of 10 nM FIXa and FITC-labeled monoclonal antibody C10D raised to the FIX catalytic domain, in the presence or absence of sufficient external free calcium (2.5 mM) for FIXa binding. Aliquots were removed at time intervals up to 45 minutes post stimulation for dilution and analysis by flow cytometry. FVIIIa was not added since, at saturating levels of FIXa (10 nM), the presence of FVIIIa was not associated with increased FIXa binding (44).
Within 15 seconds of agonist stimulation (Figure 3b), the earliest time point sampled, a subpopulation of platelets emerged from the bulk of platelets showing decreased Fura Red fluorescence relative to the bulk of stimulated platelets, indicating calcium fluxes in this subpopulation (lower left quadrant) that are greater than seen in the remainder of the platelets (lower right quadrant). Two minutes post stimulation (Figure 3e) this subpopulation was becoming FIXa positive with low fluorescence intensity (rising into the upper left quadrant). During the course of activation, increasing fluorescein fluorescence indicated increasing numbers of binding sites for FIXa per platelet (Figure 3f–j, upper left quadrants). By 10 minutes post stimulation (Figure 3j), this agonist-dependent subpopulation that is positive for FIXa had joined the <1% of platelets showing FIXa binding without agonist stimulation (Figure 3a). These platelets maintained their calcium signal as well as their equilibrium binding of FIXa for at least 45 minutes (Figure 3l).
Figure 3. PAR-1-stimulated fluorescence changes in Fura Red/AM-loaded platelets in the presence of FIXa and 2.5 mM calcium chloride.

Fura Red-loaded platelets in 2.5 mM calcium chloride combined with 10 nM FIXa and FITC-labeled antibody C10D were sampled for flow cytometry analysis before and after stimulation with 125 μM SFLLRN-amide. Fura Red fluorescence changes are shown as a left shift on the abscissas, and FIXa binding changes are shown as upward shifts on the ordinates. Platelets positive for differential calcium increases and FIXa binding are seen in the upper left quadrants. Duration of stimulation: a. 0, b. 0.25, c. 0.5, d. 1, e. 2, f. 3, g. 4, h. 5, i. 10, j. 20, k. 30, l. 45 minutes post stimulation.
Data from seven experiments were combined to analyze calcium changes in both the FIXa-positive and FIXa-negative populations over time in response to agonist stimulation. Figure 4a depicts the Fura Red quenching changes in both populations, normalized, separately for each experiment, to the pre-activation Fura Red fluorescence to track calcium changes with time. The inset in Figure 4a expands the time scale between 0 and 10 minutes for FIXa-negative platelets in the presence and absence of external calcium. In the presence of EDTA (open circles, Figure 4a and inset), the small decrease of Fura Red fluorescence at 30 seconds post stimulation (10% change) (seen better in the inset) indicated release of calcium from intracellular stores. In the presence of 2.5 mM external calcium (closed circles, Figure 4a and inset), the decrease of Fura Red fluorescence at 15 seconds post stimulation was more pronounced (35% change) and represented both release from intracellular stores and influx of external calcium through plasma membrane divalent cation channels. In both cases, Fura Red fluorescence in the FIXa-negative populations returned to baseline values by 8 minutes as subcellular and plasma membrane calcium pumps restored cytoplasmic calcium to resting levels. The continual action of calcium pumps contributed to an overshoot seen at 20 minutes. Thus, platelets showed calcium fluxes in response to activation by PAR-1 agonist.
Figure 4. PAR-1 stimulated calcium changes and platelet binding of FIXa.
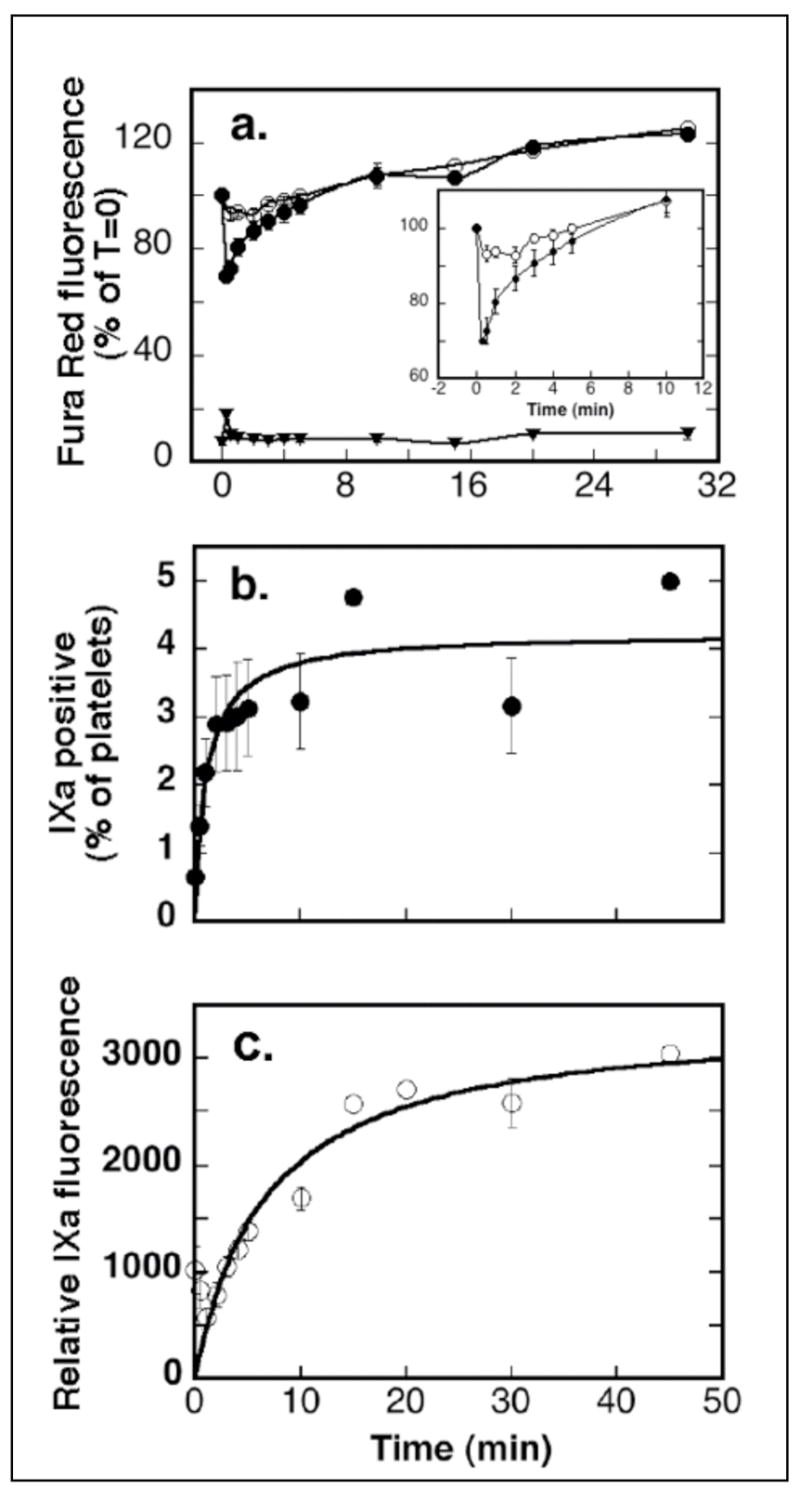
Seven experiments, described in Figure 3, were performed in parallel in either 2.5 mM calcium chloride or in a combination of calcium/EDTA (2.5 mM/5 mM). Positive gates were set for each experiment and fluorescence statistics from CyQuest Pro software were combined from seven experiments and analyzed for means and S.E.M. a: Calcium changes: (❍) negative platelets in EDTA, (●) negative platelets in 2.5 mM calcium, (▼) platelets in 2.5 mM calcium and positive for increased calcium and FIXa. b and c: FIXa binding: (●) the percent of platelets positive for FIXa, (❍) increasing FIXa fluorescence.
A subpopulation of platelets (Figure 4a, closed triangles) responded to PAR-1 stimulation with a far greater degree of Fura Red fluorescence quenching (90%), indicating that they had accumulated more intracellular calcium. This subpopulation retained the increased calcium signal (quenched Fura Red), contrasted with the FIXa negative population, for at least 45 minutes post stimulation since the Fura Red fluorescence did not return to resting levels. The pronounced and sustained calcium transients may be the result of sustained opening of calcium channels, or a decreased activity of calcium pumps or a combination of these.
Following stimulation, the population of platelets positive for increased calcium becomes positive for FIXa (Figure 3). The size of this population increased (Figure 4b) as platelets positive for increased calcium developed surface changes resulting in FIXa binding, with half-maximal population size at 1.1 (+/−0.46) minutes post-stimulation. In seven experiments, 4.2 (+/−0.36)% of platelets became FIXa positive under the conditions of these kinetic experiments that permitted no incubation period for antibody recognition of FIXa and that were performed in 2.5mM calcium chloride. The gradually increased mean fluorescein fluorescence (Figure 4c) of this subpopulation reflected the equilibrium binding of increased numbers of FIXa molecules to an increased density of exposed FIXa binding sites, with half maximal fluorescence occurring at 6.7 (+/−0.45) minutes post stimulation.
Dependence on Calcium Release from Internal Stores
To identify the signaling requirements for the calcium changes noted, platelets were pretreated with Xestospongin C, an inhibitor of inositol-1,4,5 trisphosphate (IP3)-induced release of calcium from dense tubules (Figure 5). When the FIXa binding capacity of platelets treated with 25 μM Xestospongin C (Figure 5aB) was compared with controls (Figure 5aA), it was noted that the FIXa-binding subpopulation (upper left quadrants) was decreased by 75%, and the calcium flux (degree of Fura Red quenching) in FIXa-negative platelets was decreased by 60%. In FX-activation assays (Figure 5b), treatment of platelets with 25 μM and 50 μM Xestospongin C decreased maximal FXa generation by 71% and 90%, respectively. Thus, release of calcium from internal stores was required for PAR-1-stimulated development of a subpopulation of platelets showing FIXa binding sites and support of FXa generation.
Figure 5. Release of calcium from intracellular stores is required for development of FIXa binding sites and for platelet support of FXa generation.

a.) Fura Red-loaded platelets were pretreated with DMSO (A and C) or 25 μM Xestospongin C (B and D) for 20 minutes at 37°C before addition to reaction mixtures containing 10 nM FIXa, FITC-labeled antibody to FIX, and 5 mM calcium chloride. The reactions, maintained at 37°C, were sampled before and after addition of either 125 μM SFLLRN-amide (A and B) or 25 nM thapsigargin (C and D) and diluted for flow cytometry data acquisition. Platelets positive for calcium appear in the left quadrants; platelets positive for FIXa and calcium appear in the upper left quadrant. b.) FXa generation. Platelets were pretreated with DMSO (■) or with 10 (◆), 25 (▲) or 50 μM (×) Xestospongin C for 20 minutes at 37°C before no stimulation (❍) or stimulation with 125 μM SFLLRN in reactions containing various concentrations of FIXa. FXa formed was tested 3 minutes after addition of thrombin-activated FVIII and 250 nM FX as described in EXPERIMENTAL PROCEDURES. Data shown are means of two experiments.
As a control, and since thapsigargin was shown to increase FXa generation, FIXa binding studies by flow cytometry were performed on platelets stimulated with 25nM thapsigargin (Figure 5aC). These thapsigargin-stimulated platelets showed a slowly emerging subpopulation (22%) becoming markedly positive for calcium transients and for FIXa binding. In contrast to PAR-1-stimulated platelets, by 10 minutes post stimulation with thapsigargin, platelets were still emerging from the bulk platelet population. Pretreatment with 25 μM Xestospongin C before thapsigargin stimulation (Figure 5aD) neither inhibited cytoplasmic calcium transients nor decreased the size of the FIXa-binding subpopulation, although emergence of the calcium and FIXa-positive subpopulation was delayed relative to the thapsigargin control. The inability of Xestospongin C to inhibit emergence of a calcium and FIXa-positive subpopulation was expected since PAR-1 signaling (IP3-receptor mediated) and thapsigargin stimulation (unrestored calcium leakage) operate through different mechanisms of internal calcium store emptying.
Effect of Inhibiting an Influx of Extracellular Calcium
To determine whether development of FIXa binding sites depended on an influx of external calcium or only on emptying of internal stores, platelets were stimulated with 125μM SFLLRN-amide in the presence of 5mM calcium chloride and either nifedipine (a reputed voltage-dependent calcium channel blocker), or SKF96365 (an activation-dependent calcium channel blocker) or 100 μM LaCl3 (a non-specific calcium channel blocker) to block plasma membrane calcium channels (Figure 6). Increasing concentrations of nifedipine decreased Fura Red quenching (calcium transients) in negative platelets, decreased the size of the FIXa-binding subpopulation (Figure 6a) and decreased platelet-supported FXa generation (Figure 6c), with 25 μM and 50 μM nifedipine resulting in 50% and 80% inhibition, respectively. Inclusion of SKF96365 was inhibitory (up to 50%) of all these effects of PAR-1 stimulation but only at lower concentrations (10–25 μM), and inhibition decreased at higher concentrations (25–100 μM) with no inhibition seen in either FIXa-binding subpopulation size (Figure 6a) or FXa generation (Figure 6d) when SKF96365 was used at 100 μM. Platelet reactions containing 100 μM LaCl3 developed no increase of platelets above the unstimulated control that were either positive for increased calcium transients or for FIXa (Figure 6a), and while 10 μM LaCl3 inhibited FXa generation by 25%, 100 μM LaCl3 completely eliminated agonist stimulated platelet support of FXa generation (Figure 6b). While the presence of calcium channel blockers had a dampening effect on Fura Red quenching in the FIXa-negative platelets, quenching always exceeded the degree seen with stimulation in the presence of EDTA, indicating both emptying of internal stores and a small influx of external calcium (data not shown). Thus, emptying of internal stores of calcium was required but insufficient for PAR-1-induced procoagulant surface changes, and a vigorous influx of external calcium was necessary to recruit a platelet subpopulation to develop FIXa binding sites and assemble a FXa-generation complex.
Figure 6. Calcium influx is required for PAR-1-stimulated development of FIXa binding sites and platelet support of FXa generation.
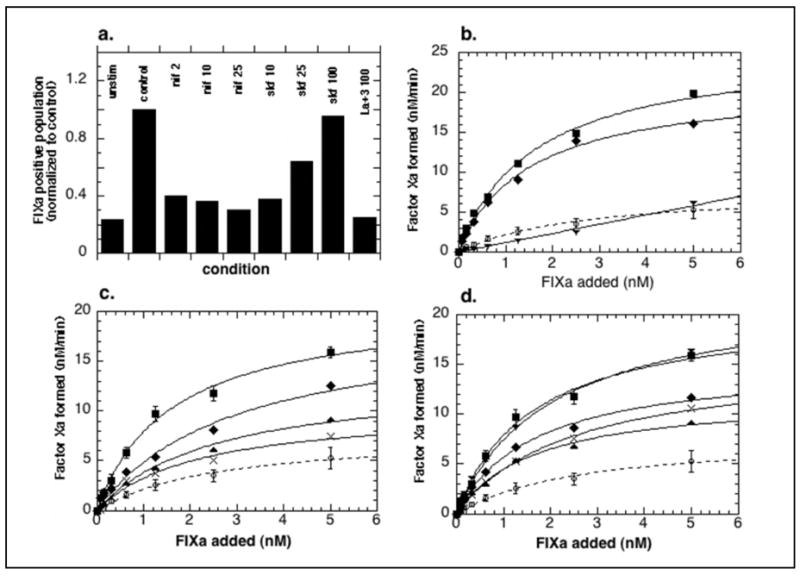
a. Flow cytometry: Fura Red-loaded platelets were added to reaction mixtures containing 10 nM FIXa, FITC-labeled anti-FIXa, and either 5 mM calcium chloride (control) alone or in the presence of 2, 10, or 25 μM nifedipine, or 10, 25, or 100 μM SKF96365, or 100 μM LaCl3. The reactions were sampled at 37°C before and after addition of 125 μM SFLLRN-amide and diluted for flow cytometry analysis. Shown is the percent of total platelets showing FIXa binding at 10 minutes after stimulation. b., c., and d. FXa generation: platelets stimulated in the presence of LaCl3 (b.), nifedipine (c.) or SKF96365 (d.) as described in EXPERIMENTAL PROCEDURES. Concentrations are 10 (◆), 25 (▲), 50 (◆) and 100 μM (▼). Shown are means of two or means (+/−SEM) of three experiments.
Duration of PAR-1-induced calcium channel opening
Since entry of external calcium was required and calcium changes were sustained in the subpopulation of platelets positive for FIXa binding, we explored the duration of activation-dependent opening of calcium channels and the continued availability of signaling machinery necessary for development of FIXa binding sites. Fura Red-loaded platelets were stimulated with 125 μM SFLLRN-amide for 0 to 20 minutes in the presence of 10nM FIXa/FITC-C10D and in the absence of external free calcium (2 mM CaCl2/5 mM EDTA) before addition of calcium to 2.5 mM and timed detection of fluorescence changes. From 5 to 20 minutes post PAR-1 stimulation, platelets (Figure 7a) responded with Fura Red quenching to addition of calcium to the external medium. With calcium addition from 5 to 20 minutes post PAR-1 stimulation, a subpopulation of platelets (Figure 7b) showed differentially pronounced calcium transients and FIXa binding, although with reduced size (50%) relative to the control samples stimulated in calcium. The rate of binding site exposure in these subpopulations (Figure 7c), and the maximal FIXa-fluorescence of the positive platelet subpopulations (Figure 7c) were the same as seen in control platelets stimulated in the presence of calcium. These results suggested that calcium channels in the PAR-1-stimulated procoagulant platelet subpopulation remain sufficiently open for at least 20 minutes post stimulation to allow calcium entry to trigger exposure of FIXa binding sites in a subpopulation of platelets.
Figure 7. Sustained opening of calcium channels in a subpopulation of PAR-1- stimulated platelets.
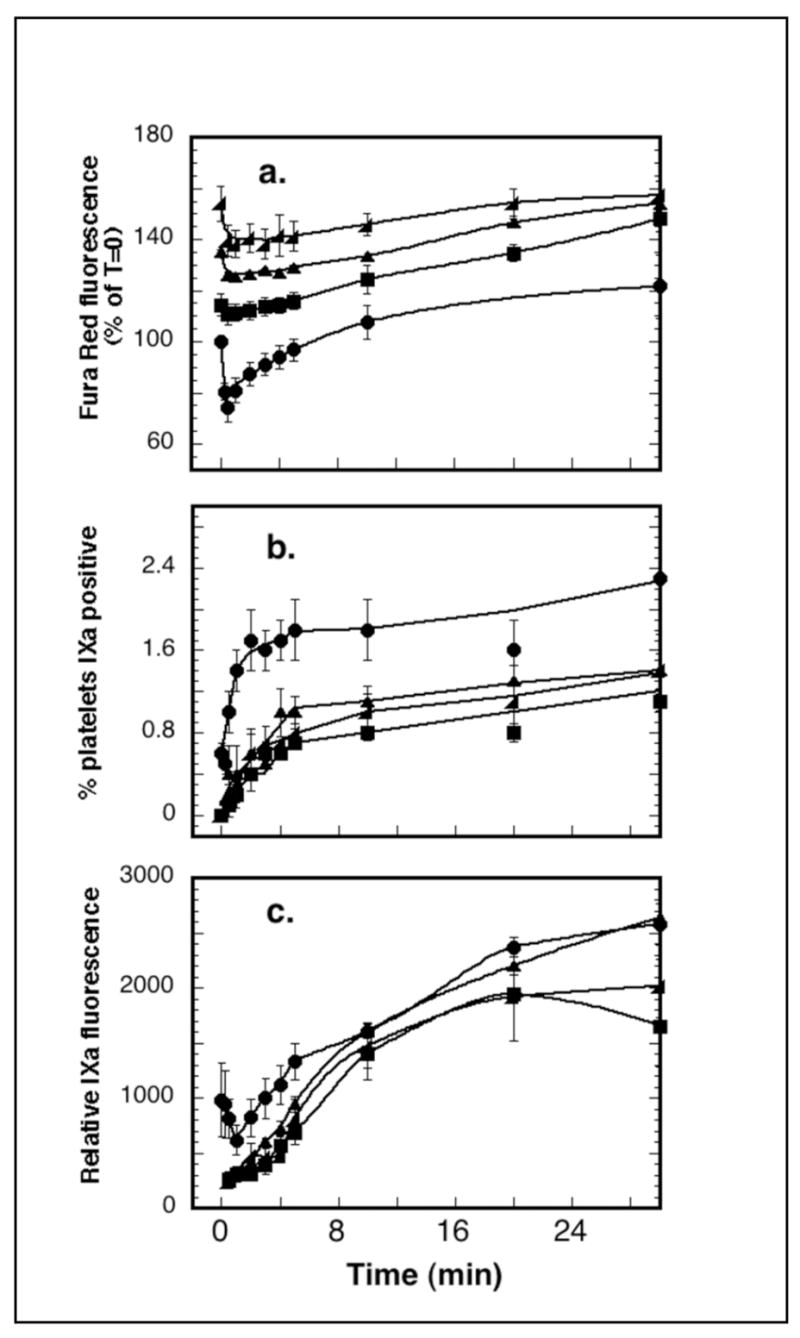
Fura Red-loaded platelets in reaction mixtures containing 10 nM FIXa, FITC-labeled anti-FIXa, and calcium/EDTA (2.0 mM/5 mM) were stimulated with 125 μM SFLLRN-amide. At 0, 5, 10 or 20 minutes post stimulation, CaCl2 was added to 2.5 mM free ionized calcium. Reactions were sampled before and after calcium addition and diluted for flow cytometry acquisition of calcium and FIXa-binding changes. Data from three experiments were combined and analyzed for a. Fura Red/calcium changes, b. percent FIXa-positive, and c. mean population fluorescein intensity or FIXa binding site density. Symbols: (●) calcium added during stimulation, or 5 minute (■), 10 minutes (▲), or 20 minutes (◆) post stimulation.
Cytoplasmic Calcium Transients
To distinguish between a requirement for internal store emptying and a requirement for increased cytoplasmic calcium in triggering the emergence of the FIXa-binding subpopulation, platelets were pretreated with one of two membrane-permeable calcium chelators, either BAPTA/AM or 5,5′-dimethylBAPTA/AM. While BAPTA/AM (Kd 160 nM) at 100 μM, determined to completely inhibit fluorescence changes in thrombin-stimulated Fura 2/AM-loaded platelets (data not shown), showed minor (20%) inhibition of both the proportion of platelets stimulated to bind FIXa (Figure 8a) and platelet-supported FXa generation (Figure 8b), 100 μM 5, 5′-dimethyl BAPTA/AM (Kd 40 nM) inhibited both parameters, causing a 70% decrease in the size of the FIXa-binding subpopulation (Figure 8a) and a 95% decrease in FXa generation (Figure 8b). Thus, more efficient chelation of cytoplasmic calcium was inhibitory, while less efficient chelation of cytoplasmic calcium allowed procoagulant surface changes to occur. Therefore, emptying of internal calcium stores, small cytoplasmic calcium fluxes and vigorous external calcium entry are required for PAR-1-induced platelet procoagulant surface changes.
Figure 8. Effect of chelating cytoplasmic calcium on the platelet subpopulation supporting FX activation.
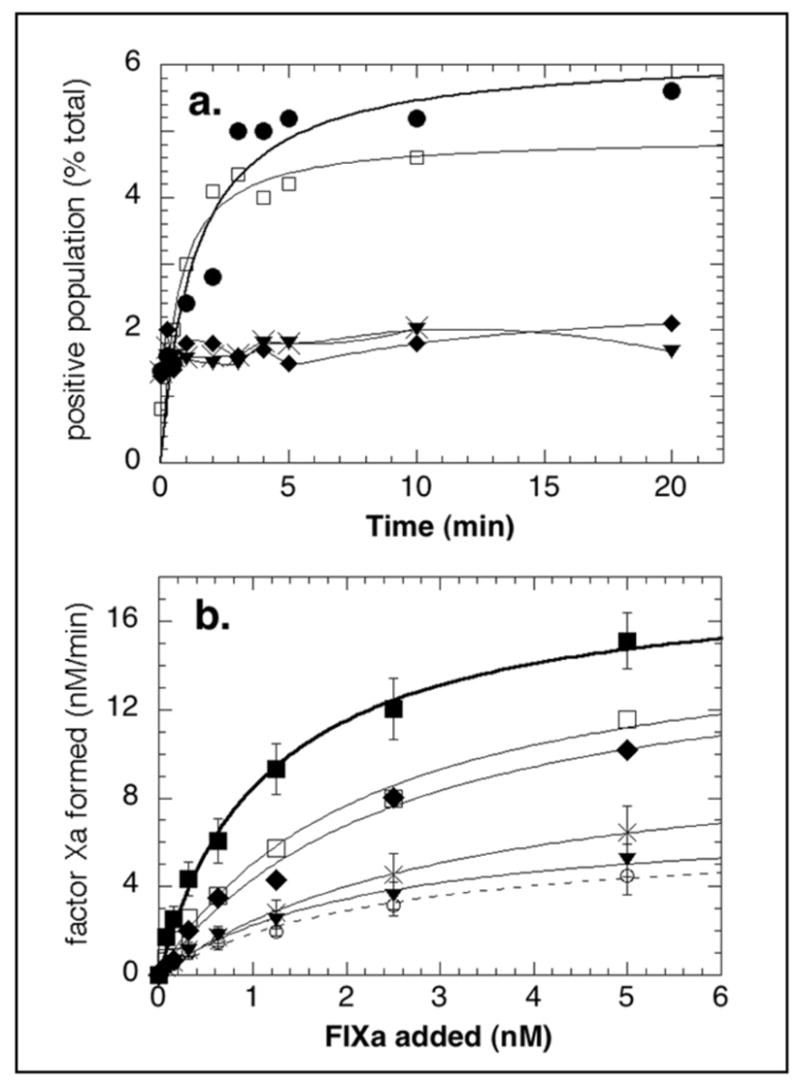
a. Percent of platelets binding FIXa. Fura Red-loaded platelets were treated with DMSO (●) or with 10 (◆), 50 (×), or 100 μM (▼) dimethylBAPTA/AM, or with 100 μM BAPTA/AM (❑) for 10 minutes at 37°C before stimulation with 125 μM SFLLRN-amide in the presence of 5 mM calcium chloride, 10 nM of FIXa, and FITC-labeled anti-FIX, as described in EXPERIMENTAL PROCEDURES. Data shown are the means of two experiments. b. FXa generation. Platelets were treated with DMSO (■) or with 10 (◆), 50 (×), or 100 μM (▼) dimethylBAPTA/AM, or with 100 μM BAPTA/AM (❑) for 10 minutes at 37°C before no stimulation (❍) or stimulation with 125 μM SFLLRN-amide in FX activation reactions containing 5mM calcium chloride, various concentrations of FIXa, FVIIIa 6 U/ml, and 250 nM FX, as described in EXPERIMENTAL PROCEDURES. Data shown are either means of two or means +/−SEM of 3–5 experiments.
DISCUSSION
Previously we reported (44) that only a subpopulation of platelets responded to PAR-1 stimulation by either SFLLRN-amide or by thrombin with exposure of binding sites for FIXa, or for annexin V. The size of the subpopulation was dose-dependent using either SFLLRN-amide or thrombin. The density of bound fluorophore increased with increasing concentrations of FIXa. While the presence of FVIIIa increased FIXa binding at low concentrations, FVIIIa had no effect at saturating FIXa (10 nM). FIX also bound to a subpopulation with approximately half the density of binding sites, consistent with previous equilibrium binding studies (8). Currently we show that the size of the FIXa-positive subpopulation varied widely with individual donors and correlated well with the degree to which single donor platelets supported maximal FXa generation. This PAR-1-stimulated FIXa-binding subpopulation correlated with a PAR-1-stimulated platelet subpopulation that differentially increased calcium flux within 15 seconds of stimulation, prior to development of FIXa binding sites. These calcium changes were sustained during the entire course of FIXa-binding site exposure, and both parameters were retained for at least 45 minutes. On the contrary, the bulk of the stimulated platelets, entirely negative for FIXa binding, showed only modest and transient cytoplasmic calcium increases that were reversed within 5 minutes of stimulation, presumably due to the combined actions of calcium channel closure and increased calcium pump activity. This is the first report of differential calcium responses in platelet subpopulations and the first to identify both sustained calcium changes and exposure of FIXa binding sites in the same subpopulation.
Because pronounced calcium transients accompany development of FIXa binding sites in the same subpopulation of platelets, we investigated a possible mechanistic link between them. The PAR-1 receptor is known to signal through Gq protein to phospholipase Cβ leading to IP3-mediated release of calcium from internal stores. Inhibiting IP3-mediated calcium release with Xestospongin C-pretreatment of platelets inhibited both PAR-1-induced development of pronounced, sustained calcium transients, development of FIXa-binding sites, and PAR-1-stimulated platelet-supported FXa generation.
The emptying of internal calcium stores caused by thapsigargin is known to trigger an influx of calcium through store-operated calcium channels. Use of thapsigargin has established the role of increased calcium in activation of platelets and in expression of a procoagulant surface (31). Thapsigargin in conjunction with thrombin stimulation has been shown to result in platelet exposure of aminophospholipids (50). Those reports and our FXa-generation and FIXa-binding data prove that, in the presence of external calcium, thapsigargin-mediated emptying of dense tubule calcium stores can, in the absence of agonist stimulation, activate platelets to potentiate FIXa-catalyzed FX activation, and can also potentiate PAR-1 stimulated platelet responses. Its mechanism of calcium store emptying leads to emergence of a subpopulation, larger than stimulated by PAR-1 activation, showing differentially increased calcium fluxes and developing subsequently the capacity to bind FIXa. Xestospongin C, however, had no effect on thapsigargin-induced emptying of internal calcium stores, since this occurs through thapsigargin inhibition of SERCA pumps preventing the refilling of leaky calcium stores (51). Therefore, store emptying and increased calcium transients were both implicated in development of a FIXa-binding platelet subpopulation.
Since store emptying has been previously implicated in development of platelet procoagulant activity through store operated calcium entry (SOCE), we investigated the necessity for calcium entry in development of the FIXa-binding subpopulation. Three calcium channel blockers nifedipine, SKF96365 and LaCl3 all inhibited appearance of a subpopulation showing pronounced and sustained calcium transients and FIXa-binding and inhibited platelet-supported FXa generation, proving that emptying of internal calcium stores was required but insufficient and that calcium influx was also necessary for development of the subpopulation. Although calcium channel blockers reduced the Fura Red quenching at 15 seconds (18–25%) in the FIXa-negative platelet population relative to controls (30%), an expected outcome from reduced calcium entry, quenching was not reduced to that in the presence of EDTA (7–15% quenching). So none of the three calcium channel blockers prevented emptying of calcium stores or completely eliminated external calcium entry. Interestingly, the reduced subpopulation that emerged calcium- and FIXa-positive from platelets with partially inhibited Fura Red quenching achieved the same pronounced degree of Fura Red quenching (90%) as in matched controls, demonstrating that platelets are heterogeneous in their sensitivity to agonist stimulation and heterogeneous in their sensitivity to the presence of calcium channel blockers. From these data, both internal calcium store emptying and external calcium entry are essential for development of a FIXa-binding subpopulation.
Two of the calcium channel blockers were originally reported to display some specificity: nifedipine for voltage-dependent (52), SKF96365 for activation-dependent or receptor-operated (or SOCE) calcium channels (53, 54), while LaCl3 is known to be non-specific (55). SKF96365 has been reported to have overlapping specificity (56). This could be one possible explanation for the curious biphasic pattern the SKF96365 data displayed whereby up to 25 μM caused up to 50% inhibition of FIXa binding and FXa generation, while increasing concentrations beyond 25 μM showed less inhibitory activity with 100 μM producing no inhibition at all. Higher concentrations may target another process, such as that involved in negative feedback that regulates the responses to agonist stimulation. Some SKF96365-sensitive channels might provide the calcium-dependent down-regulation of organelle and plasma membrane calcium or potassium channels noted in other cell types (57, 58). As more of these channels are targeted by SKF96365, the reduced calcium-down-regulation of calcium-sensitive calcium channels would lead to their increased calcium influx, noted as a loss of inhibition. One of the channels found to be regulated by calcium negative feedback is the L-type calcium channel (57, 59, 60). While there is no evidence for platelets possessing voltage-dependent L-type calcium channels (61), nifedipine was an effective inhibitor in these studies: nifedipine at 50 μM resulted in 80% inhibition of FXa generation. Since SOCE is thought to operate through a non-voltage-dependent pathway (62), this may be attributable to an inhibition of potassium channels reported for higher concentrations of nifedipine (>30 μM) (63, 64), or to an effect of nifedipine on SOCE itself (65). It is clear that at the concentration of PAR-1 agonist used (3-fold above EC50), there is more than one type of calcium channel involved in agonist-stimulated calcium influx, with SOCE or ROCC providing some of the stimulation, and a nifedipine-sensitive channel providing the rest.
The sustained calcium channel availability seen in the subpopulation of platelets that developed FIXa binding sites suggested that, once opened, these channels remained open for at least 45 minutes. Since, upon calcium addition even at 20 minutes post agonist stimulation, a platelet subpopulation developed that was positive for both increased calcium and FIXa binding changes, signaling elements dependent upon calcium and required for the procoagulant surface changes must remain available for at least 20 minutes post stimulation. These results support a hypothesis that a vigorous influx of calcium is utilized for membrane changes exposing FIXa binding sites.
Finally, we determined that increases in cytoplasmic calcium were indeed required for development of a FIXa-binding subpopulation. Pretreatment of platelets with 100μM BAPTA/AM caused only minor decreases in the size of a PAR-1-stimulated subpopulation with FIXa binding, and minor inhibition of FXa generation, and did not decrease the pronounced cytoplasmic calcium fluxes in this population. This same pretreatment of Fura 2/AM-loaded platelets caused total inhibition of thrombin-induced Fura 2 fluorescence changes seen in control Fura 2/AM-loaded platelets. The literature contains conflicting reports on the necessity for cytoplasmic calcium transients in development of platelet procoagulant surface changes. BAPTA chelation of cytoplasmic calcium sufficient to inhibit tyrosine kinases had no inhibitory effect on SOCE-triggered calcium entry (66) or on phospholipid scrambling (34). However, when 5, 5′-dimethylBAPTA/AM was used at the same concentration (a calcium chelator with 4-fold increased affinity for calcium), it completely inhibited both development of the FIXa-binding subpopulation and its resulting support of FXa-generation. Thus, the higher affinity of 5, 5′-dimethylBAPTA was more efficient at chelating sufficient cytoplasmic calcium to be inhibitory, suggesting that residual or local cytoplasmic calcium fluxes available in the presence of 100 μM BAPTA were sufficient to allow PAR-1-induced signaling to stimulate development of a procoagulant surface.
All PAR-1-stimulated platelets displayed transiently increased cytoplasmic calcium achieved by the combined release of calcium from internal stores and influx of external calcium, balanced by the combined actions of calcium pumps in internal organelles and the plasma membrane. Yet these transient and well-modulated calcium changes were insufficient to generate PAR-1-triggered membrane changes in all platelets. Inhibiting internal calcium release, reducing external calcium entry, or effective reduction of cytoplasmic free calcium decreased calcium changes in all platelets and also reduced the FIXa-binding subpopulation. Therefore, the transient calcium fluxes are necessary but become vigorous and sustained in only the subpopulation of agonist-stimulated platelets that develops FIXa binding sites. The transition to a procoagulant subpopulation must represent a PAR-1-signaling event that is facilitated in only that subpopulation.
In conclusion, a subpopulation of platelets responds to PAR-1 stimulation with a differentially pronounced and sustained influx of external calcium leading to development of FIXa binding sites. Agonist stimulation of this subpopulation sensitizes them for at least 20 minutes post stimulation to respond to an influx of calcium with development of FIXa binding sites. These data are consistent with the hypothesis that the facilitating calcium entering through cation channels is utilized with high affinity by elements close to these membrane channels, such that inefficient cytoplasmic chelating agents have little effect on the signaling mechanism leading to exposure of FIXa binding sites.
Acknowledgments
We are grateful for the secretarial and administrative assistance of Patricia Pileggi.
Abbreviations
- PAR-1
Protease-activated receptor-1
- F
factor
- FITC
fluorescein isothiocyanate
- SERCA
sarco(endo)plasmic reticulum Ca/Mg/ATPase
- DMSO
dimethylsulfoxide
- S-2765
N-α-Benzyloxycarbonyl-D-arginyl-L-glycyl-L-arginine-p-nitroanaline-dihydrochloride
- SFLLRN-amide
thrombin receptor agonist peptide
- GPIbα
glycoprotein Ibα
- BAPTA/AM
[1,2–bis(o–Aminophenoxy)ethane-N,N,N′,N′-tetraacetic Acid Tetra(acetoxymethyl) Ester]
- PGE1
prostaglandin E1
- SOCE
Store Operated Calcium Entry
- IP3
inositol 1, 3, 5-phosphate
Footnotes
This study was supported by research grants from the National Institute of Health: HL70683, HL46213 and HL74124.
References
- 1.Tracy PB. Role of platelets and leukocytes in coagulation. In: Colman RWHJ, Marder VJ, Clowes AW, George JN, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 575–596. [Google Scholar]
- 2.Walsh PN. Platelet Coagulation-Protein Interactions. Semin Thromb Hemost. 2004;30:461–471. doi: 10.1055/s-2004-833481. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad SS, Rawala-Sheikh R, Walsh PN. Platelet receptor occupancy with factor IXa promotes factor X activation. J Biol Chem. 1989;264:20012–20016. [PubMed] [Google Scholar]
- 4.Ahmad SS, Scandura JM, Walsh PN. Structural and functional characterization of platelet receptor-mediated factor VIII binding. J Biol Chem. 2000;275:13071–13081. doi: 10.1074/jbc.275.17.13071. [DOI] [PubMed] [Google Scholar]
- 5.Scandura JM, Ahmad SS, Walsh PN. A binding site expressed on the surface of activated human platelets is shared by factor X and prothrombin. Biochemistry. 1996;35:8890–8902. doi: 10.1021/bi9525029. [DOI] [PubMed] [Google Scholar]
- 6.Scandura JM, Walsh PN. Factor X bound to the surface of activated human platelets is preferentially activated by platelet-bound factor IXa. Biochemistry. 1996;35:8903–8913. doi: 10.1021/bi9525031. [DOI] [PubMed] [Google Scholar]
- 7.Rawala-Sheikh R, Ahmad SS, Ashby B, Walsh PN. Kinetics of coagulation factor X activation by platelet-bound factor IXa. Biochemistry. 1990;29:2606–2611. doi: 10.1021/bi00462a025. [DOI] [PubMed] [Google Scholar]
- 8.Ahmad SS, Rawala-Sheikh R, Walsh PN. Comparative interactions of factor IX and factor IXa with human platelets. J Biol Chem. 1989;264:3244–3251. [PubMed] [Google Scholar]
- 9.Nesheim ME, Pittman DD, Wang JH, Slonosky D, Giles AR, Kaufman RJ. The binding of 35S-labeled recombinant factor VIII to activated and unactivated human platelets. J Biol Chem. 1988;263:16467–16470. [PubMed] [Google Scholar]
- 10.Saenko EL, Scandella D. A mechanism for inhibition of factor VIII binding to phospholipid by von Willebrand factor. J Biol Chem. 1995;270:13826–13833. doi: 10.1074/jbc.270.23.13826. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad SS, Walsh PN. Coordinate binding studies of the substrate (factor X) with the cofactor (factor VIII) in the assembly of the factor X activating complex on the activated platelet surface. Biochemistry. 2002;41:11269–11276. doi: 10.1021/bi025785v. [DOI] [PubMed] [Google Scholar]
- 12.Thompson A. Molecular Biology of Factor IX. In: Colman RW, editor. Hemostasis and Thrombosis; Basic Principles and Clinical Practice. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 123–134. [Google Scholar]
- 13.Kauffman RJ, Antonarakis SE, Fay PJ. Factor VIII and Hemophilia A. In: Colman RW, editor. Hemostasis and Thrombosis; Basic Principles and Clinical Practice. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 135–156. [Google Scholar]
- 14.Greenberg DL, Davie EW. Blood Coagulation Factors: Their Complementary DNAs, Genes, and Expression. In: Colman RW, editor. Thrombosis and Hemostasis: Basic Principles and Clinical Practice. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 21–58. [Google Scholar]
- 15.Ahmad SS, Rawala-Sheikh R, Ashby B, Walsh PN. Platelet receptor-mediated factor X activation by factor IXa. High- affinity factor IXa receptors induced by factor VIII are deficient on platelets in Scott syndrome. J Clin Invest. 1989;84:824–828. doi: 10.1172/JCI114242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dachary-Prigent J, Pasquet JM, Fressinaud E, Toti F, Freyssinet JM, Nurden AT. Aminophospholipid exposure, microvesiculation and abnormal protein tyrosine phosphorylation in the platelets of a patient with Scott syndrome: a study using physiologic agonists and local anaesthetics. Br J Haematol. 1997;99:959–967. doi: 10.1046/j.1365-2141.1997.5003302.x. [DOI] [PubMed] [Google Scholar]
- 17.Colman RW, Clowes AW, George JN, Hirsh J, Marder VJ. Overview of Hemostasis. In: Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN, editors. Hemostasis and Thrombosis, Basic Principles & Clinical Practice. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 3–16. [Google Scholar]
- 18.George JN, Colman RW. Overview of Platelet Structure and Function. In: Colman RW, editor. Thrombosis and Hemostasis: Basic Principles and Clinical Practice. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 381–386. [Google Scholar]
- 19.Ahmad SS, London FS, Walsh PN. The assembly of the factor X-activating complex on activated human platelets. J Thromb Haemost. 2003;1:48–59. doi: 10.1046/j.1538-7836.2003.00020.x. [DOI] [PubMed] [Google Scholar]
- 20.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 21.London FS. The protein kinase C inhibitor RO318220 potentiates thrombin-stimulated platelet-supported prothrombinase activity. Blood. 2003;102:2472–2481. doi: 10.1182/blood-2003-03-0734. [DOI] [PubMed] [Google Scholar]
- 22.Heemskerk JW, Feijge MA, Henneman L, Rosing J, Hemker HC. The Ca2+-mobilizing potency of alpha-thrombin and thrombin-receptor-activating peptide on human platelets -- concentration and time effects of thrombin-induced Ca2+ signaling. Eur J Biochem. 1997;249:547–555. doi: 10.1111/j.1432-1033.1997.00547.x. [DOI] [PubMed] [Google Scholar]
- 23.Mazzucato M, Pradella P, Cozzi MR, De Marco L, Ruggeri ZM. Sequential cytoplasmic calcium signals in a 2-stage platelet activation process induced by the glycoprotein Ibalpha mechanoreceptor. Blood. 2002;100:2793–2800. doi: 10.1182/blood-2002-02-0514. [DOI] [PubMed] [Google Scholar]
- 24.Shaw JO, Lyons RM. Requirements for different Ca2+ pools in the activation of rabbit platelets. I. release reaction and protein phosphorylation. Biochim Biophys Acta. 1982;714:492–499. doi: 10.1016/0304-4165(82)90159-3. [DOI] [PubMed] [Google Scholar]
- 25.Lemons PP, Chen D, Whiteheart SW. Molecular mechanisms of platelet exocytosis: requirements for alpha-granule release. Biochem Biophys Res Commun. 2000;267:875–880. doi: 10.1006/bbrc.1999.2039. [DOI] [PubMed] [Google Scholar]
- 26.Paul BZ, Daniel JL, Kunapuli SP. Platelet shape change is mediated by both calcium-dependent and -independent signaling pathways. Role of p160 Rho-associated coiled-coil-containing protein kinase in platelet shape change. J Biol Chem. 1999;274:28293–28300. doi: 10.1074/jbc.274.40.28293. [DOI] [PubMed] [Google Scholar]
- 27.Jin J, Daniel JL, Kunapuli SP. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J Biol Chem. 1998;273:2030–2034. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- 28.Ohlmann P, Eckly A, Freund M, Cazenave JP, Offermanns S, Gachet C. ADP induces partial platelet aggregation without shape change and potentiates collagen-induced aggregation in the absence of Galphaq. Blood. 2000;96:2134–2139. [PubMed] [Google Scholar]
- 29.Kunzelmann-Marche C, Freyssinet JM, Martinez MC. Regulation of phosphatidylserine transbilayer redistribution by store-operated Ca2+ entry: role of actin cytoskeleton. J Biol Chem. 2001;276:5134–5139. doi: 10.1074/jbc.M007924200. [DOI] [PubMed] [Google Scholar]
- 30.Stuart MC, Bevers EM, Comfurius P, Zwaal RF, Reutelingsperger CP, Frederik PM. Ultrastructural detection of surface exposed phosphatidylserine on activated blood platelets. Thromb Haemost. 1995;74:1145–1151. [PubMed] [Google Scholar]
- 31.Smeets EF, Heemskerk JW, Comfurius P, Bevers EM, Zwaal RF. Thapsigargin amplifies the platelet procoagulant response caused by thrombin. Thromb Haemost. 1993;70:1024–1029. [PubMed] [Google Scholar]
- 32.Rosing J, van Rijn JL, Bevers EM, van Dieijen G, Comfurius P, Zwaal RF. The role of activated human platelets in prothrombin and factor X activation. Blood. 1985;65:319–332. [PubMed] [Google Scholar]
- 33.Bevers EM, Rosing J, Zwaal RF. Membrane phospholipids are the major determinant of the binding site for factor X activating--and prothrombinase complexes at the surface of human platelets. Agents Actions Suppl. 1986;20:69–75. [PubMed] [Google Scholar]
- 34.Wurth GA, Zweifach A. Evidence that cytosolic calcium increases are not sufficient to stimulate phospholipid scrambling in human T-lymphocytes. Biochem J. 2002;362:701–708. doi: 10.1042/0264-6021:3620701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diver JM, Sage SO, Rosado JA. The inositol trisphosphate receptor antagonist 2-aminoethoxydiphenylborate (2-APB) blocks Ca2+ entry channels in human platelets: cautions for its use in studying Ca2+ influx. Cell Calcium. 2001;30:323–329. doi: 10.1054/ceca.2001.0239. [DOI] [PubMed] [Google Scholar]
- 36.Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS. Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J Biol Chem. 2003;278:27208–27215. doi: 10.1074/jbc.M301118200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hassock SR, Zhu MX, Trost C, Flockerzi V, Authi KS. Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood. 2002;100:2801–2811. doi: 10.1182/blood-2002-03-0723. [DOI] [PubMed] [Google Scholar]
- 38.Rosado JA, Sage SO. Activation of store-mediated calcium entry by secretion-like coupling between the inositol 1,4,5-trisphosphate receptor type II and human transient receptor potential (hTrp1) channels in human platelets. Biochem J. 2001;356:191–198. doi: 10.1042/0264-6021:3560191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosado JA, Sage SO. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Trends Cardiovasc Med. 2000;10:327–332. doi: 10.1016/s1050-1738(01)00073-1. [DOI] [PubMed] [Google Scholar]
- 40.Rosado JA, Sage SO. The ERK cascade, a new pathway involved in the activation of store-mediated calcium entry in human platelets. Trends Cardiovasc Med. 2002;12:229–234. doi: 10.1016/s1050-1738(02)00161-5. [DOI] [PubMed] [Google Scholar]
- 41.Redondo PC, Harper AG, Salido GM, Pariente JA, Sage SO, Rosado JA. A role for SNAP-25 but not VAMPs in store-mediated Ca2+ entry in human platelets. J Physiol. 2004;558:99–109. doi: 10.1113/jphysiol.2004.064899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosado JA, Redondo PC, Sage SO, Pariente JA, Salido GM. Store-operated Ca(2+) entry: Vesicle fusion or reversible trafficking and de novo conformational coupling? J Cell Physiol. 2005 doi: 10.1002/jcp.20399. [DOI] [PubMed] [Google Scholar]
- 43.Redondo PC, Lajas AI, Salido GM, Gonzalez A, Rosado JA, Pariente JA. Evidence for secretion-like coupling involving pp60src in the activation and maintenance of store-mediated Ca2+ entry in mouse pancreatic acinar cells. Biochem J. 2003;370:255–263. doi: 10.1042/BJ20021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.London FS, Marcinkiewicz M, Walsh PN. A subpopulation of platelets responds to thrombin- or SFLLRN-stimulation with binding sites for factor IXa. J Biol Chem. 2004;279:19854–19859. doi: 10.1074/jbc.M310624200. [DOI] [PubMed] [Google Scholar]
- 45.Frazier D, Smith KJ, Cheung WF, Ware J, Lin SW, Thompson AR, Reisner H, Bajaj SP, Stafford DW. Mapping of monoclonal antibodies to human factor IX. Blood. 1989;74:971–977. [PubMed] [Google Scholar]
- 46.London F, Walsh PN. The role of electrostatic interactions in the assembly of the factor X activating complex on both activated platelets and negatively-charged phospholipid vesicles. Biochemistry. 1996;35:12146–12154. doi: 10.1021/bi960097v. [DOI] [PubMed] [Google Scholar]
- 47.London F, Ahmad SS, Walsh PN. Annexin V inhibition of factor IXa-catalyzed factor X activation on human platelets and on negatively-charged phospholipid vesicles. Biochemistry. 1996;35:16886–16897. doi: 10.1021/bi960712v. [DOI] [PubMed] [Google Scholar]
- 48.Yip KP, Marsh DJ. An Arg-Gly-Asp peptide stimulates constriction in rat afferent arteriole. Am J Physiol. 1997;273:F768–776. doi: 10.1152/ajprenal.1997.273.5.F768. [DOI] [PubMed] [Google Scholar]
- 49.Kuwahara M, Sugimoto M, Tsuji S, Miyata S, Yoshioka A. Cytosolic calcium changes in a process of platelet adhesion and cohesion on a von Willebrand factor-coated surface under flow conditions. Blood. 1999;94:1149–1155. [PubMed] [Google Scholar]
- 50.Williamson P, Bevers EM, Smeets EF, Comfurius P, Schlegel RA, Zwaal RF. Continuous analysis of the mechanism of activated transbilayer lipid movement in platelets. Biochemistry. 1995;34:10448–10455. doi: 10.1021/bi00033a017. [DOI] [PubMed] [Google Scholar]
- 51.Malcolm KC, Fitzpatrick FA. Epoxyeicosatrienoic acids inhibit Ca2+ entry into platelets stimulated by thapsigargin and thrombin. J Biol Chem. 1992;267:19854–19858. [PubMed] [Google Scholar]
- 52.Toll L. Calcium antagonists High-affinity binding and inhibition of calcium transport in a clonal cell line. J Biol Chem. 1982;257:13189–13192. [PubMed] [Google Scholar]
- 53.Weber C, Kruse HJ, Sellmayer A, Erl W, Weber PC. Platelet activating factor enhances receptor-operated Ca(++)-influx and subsequent prostacyclin synthesis in human endothelial cells. Biochem Biophys Res Commun. 1993;195:874–880. doi: 10.1006/bbrc.1993.2126. [DOI] [PubMed] [Google Scholar]
- 54.Ufret-Vincenty CA, Short AD, Alfonso A, Gill DL. A novel Ca2+ entry mechanism is turned on during growth arrest induced by Ca2+ pool depletion. J Biol Chem. 1995;270:26790–26793. doi: 10.1074/jbc.270.45.26790. [DOI] [PubMed] [Google Scholar]
- 55.Li HF, Shen AY, Jan CR, Wu SN. Co-activation of nonselective cation channels by store depletion and oxidative stress in monocytic U937 cells. Chin J Physiol. 1998;41:113–119. [PubMed] [Google Scholar]
- 56.Rosado JA, Meijer EM, Hamulyak K, Novakova I, Heemskerk JW, Sage SO. Fibrinogen binding to the integrin alpha(IIb)beta(3) modulates store-mediated calcium entry in human platelets. Blood. 2001;97:2648–2656. doi: 10.1182/blood.v97.9.2648. [DOI] [PubMed] [Google Scholar]
- 57.Rabl K, Thoreson WB. Calcium-dependent inactivation and depletion of synaptic cleft calcium ions combine to regulate rod calcium currents under physiological conditions. Eur J Neurosci. 2002;16:2070–2077. doi: 10.1046/j.1460-9568.2002.02277.x. [DOI] [PubMed] [Google Scholar]
- 58.Smith MR, Nelson AB, Du Lac S. Regulation of firing response gain by calcium-dependent mechanisms in vestibular nucleus neurons. J Neurophysiol. 2002;87:2031–2042. doi: 10.1152/jn.00821.2001. [DOI] [PubMed] [Google Scholar]
- 59.Guo J, Duff HJ. Inactivation of ICa-L is the major determinant of use-dependent facilitation in rat cardiomyocytes. J Physiol. 2003;547:797–805. doi: 10.1113/jphysiol.2002.033340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poteser M, Wakabayashi I, Rosker C, Teubl M, Schindl R, Soldatov NM, Romanin C, Groschner K. Crosstalk between voltage-independent Ca2+ channels and L-type Ca2+ channels in A7r5 vascular smooth muscle cells at elevated intracellular pH: evidence for functional coupling between L-type Ca2+ channels and a 2-APB-sensitive cation channel. Circ Res. 2003;92:888–896. doi: 10.1161/01.RES.0000069216.80612.66. [DOI] [PubMed] [Google Scholar]
- 61.Doyle VM, Ruegg UT. Lack of evidence for voltage dependent calcium channels on platelets. Biochem Biophys Res Commun. 1985;127:161–167. doi: 10.1016/s0006-291x(85)80139-x. [DOI] [PubMed] [Google Scholar]
- 62.Koch BD, Faurot GF, Kopanitsa MV, Swinney DC. Pharmacology of a Ca(2+)-influx pathway activated by emptying the intracellular Ca2+ stores in HL-60 cells: evidence that a cytochrome P-450 is not involved. Biochem J. 1994;302(Pt 1):187–190. doi: 10.1042/bj3020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhabyeyev P, Missan S, Jones SE, McDonald TF. Low-affinity block of cardiac K(+) currents by nifedipine. Eur J Pharmacol. 2000;401:137–143. doi: 10.1016/s0014-2999(00)00413-1. [DOI] [PubMed] [Google Scholar]
- 64.Teramoto N, Brading AF. The effects of nifedipine and other calcium antagonists on the glibenclamide-sensitive K+ currents in smooth muscle cells from pig urethra. Br J Pharmacol. 1998;123:1601–1608. doi: 10.1038/sj.bjp.0701777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu SN, Jan CR, Li HF. Characteristics of store-operated Ca(2+)-permeable current in monocytic U937 cells. Chin J Physiol. 1997;40:115–120. [PubMed] [Google Scholar]
- 66.Vostal JG, Shafer B. Thapsigargin-induced calcium influx in the absence of detectable tyrosine phosphorylation in human platelets. J Biol Chem. 1996;271:19524–19529. doi: 10.1074/jbc.271.32.19524. [DOI] [PubMed] [Google Scholar]