

Abstract
Picromycin/methymycin synthase (PICS) is a modular polyketide synthase (PKS) that is responsible for the biosynthesis of both 10-deoxymethynolide (1) and narbonolide (2), the parent 12- and 14-membered aglycone precursors of the macrolide antibiotics methymycin and picromycin, respectively. PICS module 2 is a dehydratase (DH)-containing module that catalyzes the formation of the unsaturated triketide intermediate using malonyl-CoA as the chain extension substrate. Recombinant PICS module 2+TE, with the PICS thioesterase domain appended to the C-terminus to allow release of polyketide products, was expressed in Escherichia coli. Purified PICS module 2+TE converted malonyl-CoA and 4, the N-acetylcysteamine thioester of (2S,3R)-2-methyl-3-hydroxy-pentanoic acid, to a 1:2 mixture of the triketide acid (4S,5R)-4-methyl-5-hydroxy-2-heptenoic acid (5) and (3S,4S,5R)-3,5-dihydroxy-4-methyl-n-heptanoic acid-δ-lactone (10) with a combined kcat of 0.6 min−1. The triketide lactone 10 is formed by thioesterase-catalyzed cyclization of the corresponding D-3-hydroxyacyl-SACP intermediate, a reaction which competes with dehydration catalyzed by the dehydratase domain. PICS module 2+TE showed a strong preference for the syn-diketide-SNAC 4, with a 20-fold greater kcat/Km than the anti-(2S,3S)-diketide-SNAC 14, and a 40-fold advantage over the syn-(2R,3S)-diketide-SNAC 13. PICS module 2(DH0)+TE, with an inactivated DH domain, produced exclusively 10, while three PICS module 2(KR0)+TE mutants, with inactivated KR domains, produced exclusively or predominantly the unreduced triketide ketolactone, (4S,5R)-3-oxo-4-methyl-5-hydroxy-n-heptanoic acid-δ-lactone (7). These studies establish for the first time the structure and stereochemistry of the intermediates of a polyketide chain elongation cycle catalyzed by a DH-containing module, while confirming the importance of key active site residues in both KR and DH domains.
Picromycin/methymycin synthase (PICS) is a typical modular polyketide synthase (PKS) that is responsible for the biosynthesis of both 10-deoxymethynolide (1) and narbonolide (2), the parent 12- and 14-membered macrolactone aglycone precursors of the macrolide antibiotics methymycin and picromycin, respectively (Figure 1A).1 PICS is a large, multifunctional enzyme that catalyzes the formation of its natural polyketide products from the simple building blocks acetyl- and propionyl-CoA by way of their activated derivatives malonyl-CoA and methylmalonyl-CoA.2 PICS is organized into 6 functional protein assemblies known as modules, in which each homodimeric module (subunit MW>150 kDa) is responsible for a single round of polyketide chain elongation and functional group modification. Within each module are several catalytic domains of 100–400 amino acids each that are analogous in structure, function, and organization to the corresponding enzymes of fatty acid biosynthesis. Each PICS module consists of a core set of three domains: a β-ketoacyl-SACP synthase (ketosynthase, KS) domain, an acyltransferase (AT) domain, and an acyl carrier protein (ACP) domain. Additional combinations of ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains are found in individual modules, corresponding to the required degree of functional group modification associated with each polyketide chain elongation cycle. A dedicated thioesterase (TE) domain at the C-terminus of the most downstream biosynthetic module is responsible for cyclization and release of the mature polyketide product. The PICS TE domain is unusual in naturally mediating the formation of both 10-deoxymethynolide (1) and narbonolide (2).2,3 Such thioesterase domains have been found to have broad substrate specificity and can be artificially attached to the C-termini of numerous PKS modules in order to promote the release of the derived polyketide chain elongation products by cyclization or hydrolysis.4,5
Figure 1.

Modular organization of (A) picromycin/methymycin synthase (PICS) and (B) 6-deoxyerythronolide B synthase (DEBS). In addition to the three core catalytic domains – the ketosynthase (KS), acyl transferase (AT), and acyl carrier protein (ACP) domains – individual modules carry specific combinations of auxiliary ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains. Loading tri- or didomains prime module 1 of each synthase with the propionyl starter unit and dedicated thioesterase (TE) domains cyclize the mature polyketide with release of the corresponding macrolide aglycone.
PICS is closely related to the prototypical modular PKS, 6-deoxyerythronolide B synthase (DEBS), which is responsible for the biosynthesis of 3, the parent macrolide aglycone of the broad spectrum antibiotic erythromycin A (Figure 1B).6 Although both polyketide synthases are composed of 6 elongation modules that mediate a closely related series of biochemical reactions, there are several key differences between the two PKS systems: 1) While DEBS consists of three bimodular proteins, PICS is organized into two bimodular proteins (PikAI and PikAII) plus two monomodular subunits (PikAIII and PikAIV).2 2) PICS is unusual in mediating the formation of both 12- and 14-membered ring macrolactones. 3) The loading didomain for DEBS, located at the N-terminus of DEBS1, consists of an AT and ACP domain utilizing propionyl-CoA as the polyketide chain primer. By contrast, the PICS loading domain is composed of three domains, an AT and an ACP domain as well as a modified ketosynthase domain with a glutamine residue at its active site (KSQ) that together use methylmalonyl-CoA to generate the propionyl starter unit. 4) Although both DEBS module 2 and PICS module 2 utilize the same diketide substrate, (2S,3R)-2-methyl-3-hydroxypentanoyl-SACP, generated by DEBS or PICS module 1: a) PICS module 2 uses malonyl-CoA (acetate) rather than methylmalonyl-CoA (propionate) as the chain extension unit1a,7 and b) PICS module 2 contains an additional dehydratase (DH) domain, resulting in the formation of the trans-(E)-unsaturated triketide acylthioester product.
Our current understanding of the sequence of reactions catalyzed by PKS modules is based largely on analogy to the known mechanism of action of the biochemically and genetically closely related enzymes of fatty acid biosynthesis. According to this model, the AT domain of PICS module 2 uses malonyl-CoA to generate malonyl-SACP (Figure 2). The KS-catalyzed reaction involves two consecutive chemical steps –the formation of a covalent acyl-enzyme intermediate between an active site Cys thiol and the (2S,3R)-2-methyl-3-hydroxypentanoyl diketide intermediate donated by PICS module 1, and subsequent decarboxylative condensation of this acylthioester with the malonyl-SACP chain extension unit – resulting in formation of the triketide β-ketoacyl-SACP product.8 This 3-ketopentanoyl acylthioester then undergoes stereospecific reduction by the KR domain, followed by DH domain-catalyzed dehydration. The resultant unsaturated triketide then serves as the substrate for the downstream PICS module 3
Figure 2.

Sequence of reactions catalyzed by PICS module 2.
Although it is well accepted that polyketide double bonds are generated by the action of DH domains, the transient, covalently bound β-hydroxyacyl-SACP intermediates that serve as substrates for these dehydratases have never been observed. As a consequence, nothing is known about the stereochemistry of these cryptic β-hydroxyacyl thioester intermediates, the substrate specificity of the DH domains that process them, nor the stereochemistry of the actual PKS dehydration reaction. To address these issues, we have constructed and expressed in Escherichia coli recombinant PICS module 2+TE, a hybrid protein expected to convert diketide-SNAC 4 to the unsaturated triketide acid 5 corresponding to the normally enzyme-bound acyclic triketide intermediate of picromycin/methymycin biosynthesis. We report below detailed biochemical analysis of the reactions catalyzed by PICS module 2, and describe the generation and analysis of PICS module 2 mutants with specifically inactivated DH and KR domains that provide the first direct evidence for the structure and stereochemistry of the previously cryptic 3-keto- and 3-hydroxyacyl-thioester intermediates of polyketide enoyl thioester biosynthesis.
Results
PICS module 2+TE
We have previously expressed a variety of individual PKS modules in E. coli.9 Experience has shown that a short, 50–100 amino acid linker peptide or docking domain located at the N-terminus of the recombinant module is required for catalytic activity.9a For PKS modules normally found at the N-terminus of a monomodular or multimodular synthase subunit, it is sufficient to utilize the native N-terminal linker peptide. On the other hand, expression of active internal modules requires appending a natural N-terminal linker peptide upstream of the KS domain. For the construction of the expression plasmid for PICS module 2+TE, we chose to use the previously engineered plasmid pKW119 in which DNA encoding the N-terminal linker peptide of DEBS module 3 (eryL3; bp 14–119 of eryAII) had been fused upstream of the open reading frame corresponding to PICS module 2 obtained from pikAI.9d Ordinarily a functional thioesterase domain is also appended downstream of the ACP domain of the recombinant module in order to facilitate release of the ACP-bound polyketide chain-elongation product and to regenerate free enzyme. For this purpose we chose to use the previously described PICS TE domain that we had cloned from the 3′-end of PICS module 6 (pikAIV).10
Plasmid pHL4–65b, which encodes recombinant PICS TE plus a portion of the interdomain linker peptide that begins 28 amino acids downstream of PICS ACP6 (bp 3145–4038 of PikAIV),10 served as the template for PCR amplification of the PICS TE gene and upstream linker with the concomitant introduction of flanking HindIII and XhoI restriction sites. The resulting amplicon was digested with HindIII and XhoI and ligated into the corresponding sites of the T7-based, pET28a(+) expression vector to give plasmid pTJZ41. This plasmid was digested in turn with NdeI and HindIII and ligated to an NdeI-HindIII fragment of pKW119 corresponding to eryL3 fused to the 5′-end of PICS module 2. The resultant plasmid, pTJZ61, served as the source of (eryL3)PICS module 2+TE with an appended N-terminal His6-tag.
Expression of functional PKS modules in E. coli requires posttranslational modification of the apo-ACP domain by 4′-phosphopantetheinylation.11 Plasmid pTJZ61 was therefore used to transform E. coli BAP1, a derivative of the E. coli expression host BL21(DE3) that carries a chromosomally integrated copy of the sfp gene encoding the requisite 4′-phosphopantetheinyl transferase.12 Induction at reduced IPTG concentration (0.1 mM) and lower temperature (18 °C) enhanced the proportion of soluble protein and minimized the generation of insoluble protein inclusion bodies. A single step metal affinity chromatography using Ni2+-NTA resin combined with gradient elution afforded PICS module 2+TE protein of >95% purity, as judged by SDS-PAGE and densitometry, in a yield of 7 mg protein/L culture (Figure 3).
Figure 3.
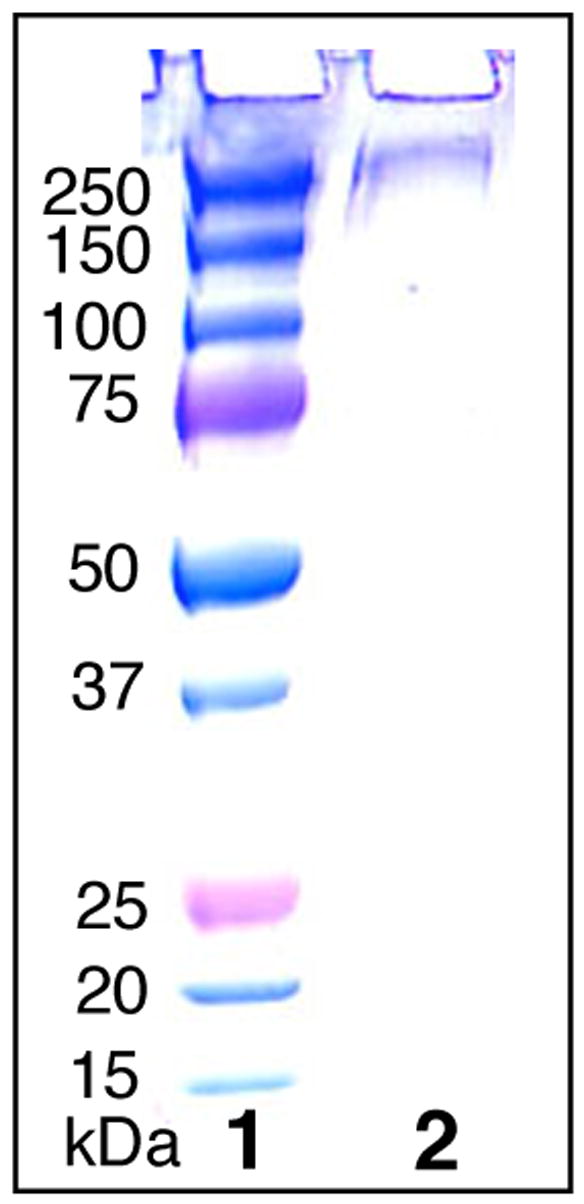
SDS-PAGE of purified PICS module 2+TE. Lane 1, MW markers. Lane 2, PICS module 2+TE.
Synthesis of triketide standards
The Evans oxazolidinone 6 can be used to prepare the diketide-SNAC substrate 413 as well as each of the requisite samples of the expected triketide products (Scheme 1). An authentic sample of (4S,5R)-4-methyl-5-hydroxy-2-heptenoic acid (5), the predicted product of PICS module 2+TE, was prepared as previously described starting with 6.13 The triketide ketolactone, (4S,5R)-3-oxo-4-methyl-5-hydroxy-n-heptanoic acid-δ-lactone (7), was synthesized from the β-acetoxy imide 8 by anion-accelerated Claisen rearrangement, using lithium hexamethyldisilazide to generate the enolate.14 Reduction as described by Hinterding with t-BuNH2-BH3 and citric acid gave a 5:1 mixture of the corresponding (3R,4S,5R)- and (3S,4S,5R)-3,5-dihydroxy-4-methyl-n-heptanoic acid-δ-lactones 9 and 10.14b Although these two diastereomers have been previously separated chromatographically,14b the individual compounds were readily resolved by 1H NMR and could therefore be unambiguously assigned by comparison with literature values for each of the pure components prepared by alternative methods.15
Scheme 1.
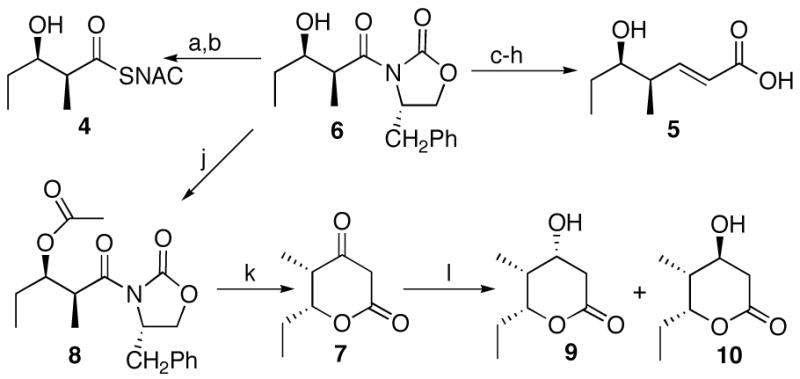
Synthesis of diketide 4 and triketides 5, 10, and 7. a. 30% H2O2, LiOH; b. i. (PhO)2PON3, ET3N, ii. HSNAC; c. Me3Al, MeONHMe; d. TBDMSOTf, Et3N; e. DiBALH; f. Ph3PCHCO2Me; g. K2CO3, MeOH; h. HF, CH3CN/H2O; j. Ac2O, Et3N, DMAP; k. LIHMDS; l. t-BuNH2-BH3, citric acid.
Activity of PICS module 2+TE
We have previously shown that intact cells of S. venezuelae convert both (2S-3R)-4 and (4S,5R)-4-methyl-5-hydroxy-2-heptenoyl-SNAC (11), the N-acetylcysteamine thioester analogues of the natural diketide substrate and product of PICS module 2, intact into methymycin (12) (Scheme 2).7 We have also shown that 4 can serve as a surrogate substrate for a variety of DEBS modules,9b as well as for PICS modules 5 and 6.9c,16 Incubation of PICS module 2+TE with (2S,3R)-4 in the presence of [2-14C]malonyl-CoA and NADPH gave a 1:2 mixture of two products, as detected by TLC-phosphorimaging: the predicted unsaturated triketide acid 5 (Rf 0.30; CH2Cl2:CH3OH:CH3CO2H 95:5:0.1) and, unexpectedly, triketide lactone 10 (Rf 0.30), each of which co-chromatographed with the corresponding synthetic reference compound (Scheme 3). No product was detected when [2-14C]methylmalonyl-CoA was substituted for malonyl-CoA, consistent with the expected strict specificity of the AT domain for the malonyl substrate.17
Scheme 2.
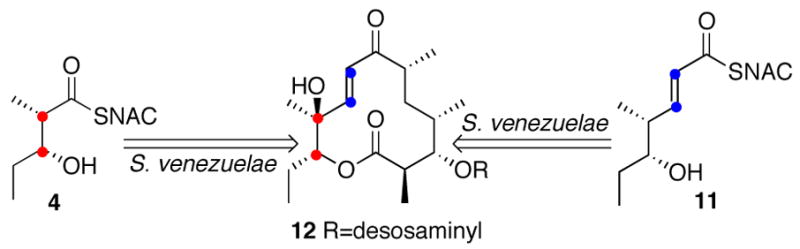
Incorporation of [2,3-13C2]-4 and [2,3-13C2]-11 into methymycin.
Scheme 3.
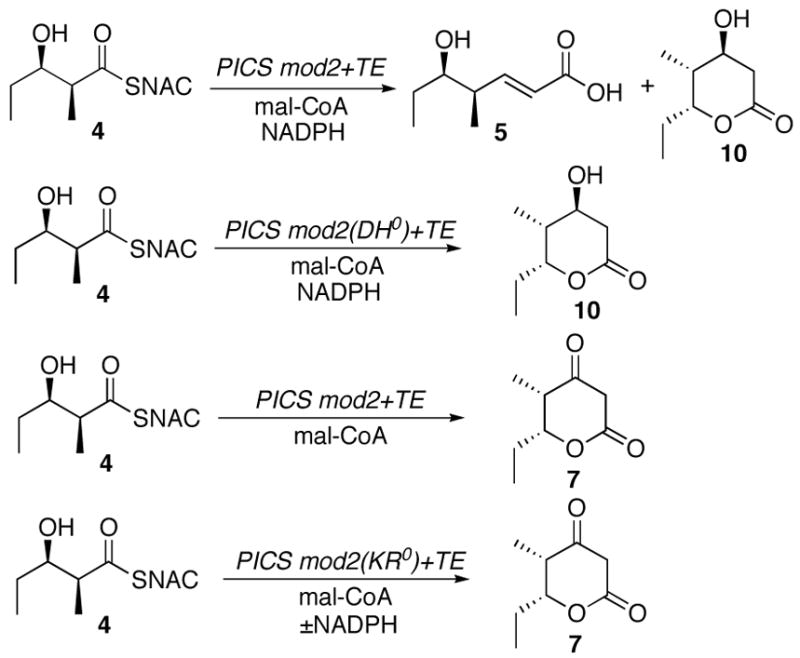
Enzymatic conversion of diketide 4 to triketides 5, 10, and 7 by PICS module 2+TE and mutants.
To confirm the structure and stereochemistry of each of the two observed enzymatic reaction products, a preparative scale incubation of PICS module 2+TE with 4, malonyl-CoA, and NADPH was carried out. Since control experiments established that formation and hydrolysis of the malonyl-SACP intermediate was 1-2 orders of magnitude faster than acylation of the KS domain by the diketide, (see below) malonyl-CoA was added to the incubation mixture in three separate batches to compensate for competing non-productive consumption of this latter substrate. The individual products were purified from the organic extract of the reaction mixture and subjected to rigorous characterization by 1H NMR spectroscopy and GC-mass spectrometry. The component of Rf 0.30 displayed a 1H NMR spectrum identical with that of synthetic acid 5 (Figure 4). Derivatization of 5 with bis(trimethylsilyl)trifluoroacetamide (BSTFA) gave a mixture of mono- and bis(trimethylsilyl)ethers that were identical by GC-MS retention time and fragmentation pattern to the corresponding derivatives of authentic (4S,5R)-4-methyl-5-hydroxy-2-heptenoic acid 5 (See Supporting Information). The purified Rf 0.56 enzymatic reaction product was identical by 1H NMR with the (3S,4S,5R)-triketide lactone 10. In particular, enzymatically-generated 10 displayed characteristic resonances at δ 4.61 (H-6, m, 1 H) and 4.04 (H-4, m, 1 H), readily distinguished from the corresponding signals of the (3R)-diastereomer, which appear at δ 4.24 and 4.13, respectively (Figure 5). The derived 3-trimethylsilyl ether, 10-TMS, was also identical by GC-MS comparison with the minor component of the trimethylsilylated mixture of synthetic diastereomers, in both retention time (6.43 min) and mass spectrum ([M-CH3] m/z 214.8, in contrast with the TMS derivative of the major diastereomer, (3R)-9-TMS, which had a retention time of 6.50 min and a distinct mass fragmentation pattern ([M-CH3] m/z 214.8) (See Supporting Information).
Figure 4.
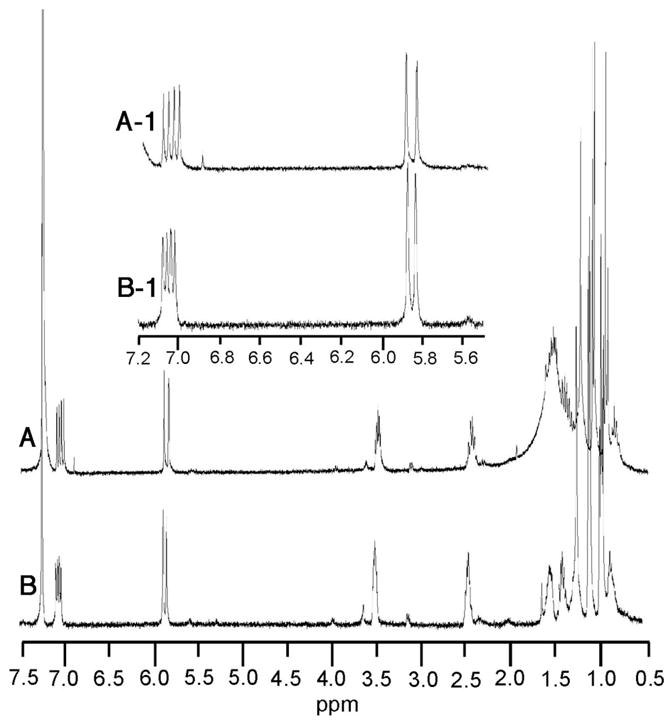
1H NMR comparison of A) enzymatically generated triketide acid 5 and B) synthetic 5.
Figure 5.
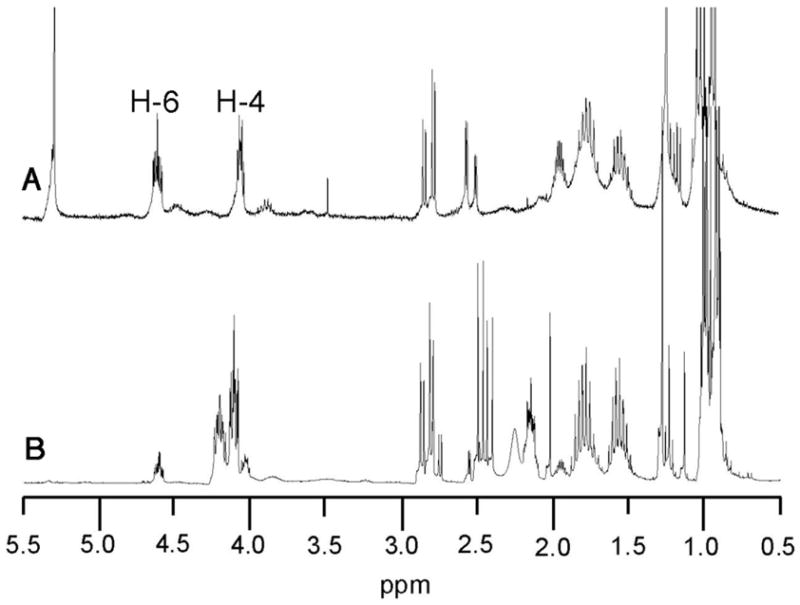
A. 1H NMR comparison of A) enzymatically generated triketide lactone 10 and B) synthetic 5:1 mixture of 9 and 10.
PICS module 2(DH0)+TE
The formation of triketide lactone 10 by PICS module 2+TE results from TE-catalyzed cyclization of the transiently generated (3S,4S,5R)-3,5-dihydroxy-4-methylheptanoyl-SACP intermediate, in competition with normal processing of the same substrate by the DH domain. In order to eliminate the dehydration reaction, we constructed a mutant PICS module 2(DH0)+TE with an inactive DH domain. Sequence alignments of modular PKS and fatty acid synthase DH domains have revealed the presence of a highly conserved His residue, corresponding to His3611 of PikAI, located in the DH domain of PICS module 2 (Figure 6A).18 Consequently, we used site-directed mutagenesis on the pTJZ61 expression plasmid to replace His3611 by Phe. The resultant plasmid pTJZ61-H3611F was used to transform E. coli BAP1 and the mutant PICS module 2(DH0)+TE was expressed and purified to near homogeneity as before (~4 mg protein/liter LB culture).
Figure 6.

Sequence alignments A) PICS module 2 DH domain, conserved His in boldface: PICS_DH2, PikAI, aa 3588–3647, UniProtKB/TrEMBL entry Q9ZGI5_9ACTO; DEBS_DH4, EryAII, aa 2386–2445, Q5UNP5_SACER; RAPS_DH7, RapB, aa 4130–4390, Q54296_STRHY; RIFS_DH4, RifB, aa 932–992, O52545_AMYMD; PICS_DH4, PikAII, aa 2437–2497, Q9ZGI4_9ACTO; TYLS_DH2, TylG, aa 3523–3583, O33954_STRFR. B) PICS module 2 KR domain, conserved Lys, Ser, and Tyr in boldface: PICS_KR2, PikAI, aa 4202–4260, Q9ZGI5_9ACTO; TYLS_KR2, TylG, aa 4119–4175, O33954_STRFR; RIFS_KR3, RifA, aa 1927–1983, O54666_AMYMD; DEBS_KR4, EryAII, aa 3242–3297, EryAII, Q5UNP5_SACER; PICS_KR5, PikAIII, aa 1217–1273, Q9ZGI3_9ACTO; DEBS_KR1, EryAI, aa 1767–1823, Q5UNP6_SACER. Alignments generated using CLUSTALW at PBIL (http://pbil.univ-lyon1.fr/) (Ref. Combet, C.; Blanchet,C.; Geourjon, C.; Deléage, G. TIBS 2000, 25, 147–150.)
Incubation of PICS module 2(DH0)+TE with diketide 4 and [2-14C]malonyl-CoA in the presence of NADPH and assay by TLC-phosphorimaging confirmed that the major product of the DH mutant enzyme corresponded to the expected triketide lactone 10 (Rf 0.56), accompanied by no more than a trace amount (~1%) of dehydration product 5 (Scheme 3). Preparative scale incubation with purified PICS module 2(DH0)+TE, diketide 4, malonyl-CoA, and NADPH gave the D-3-hydroxy-10, whose structure and stereochemistry were confirmed directly by 1H NMR.
PICS module 2(KR0)+TE
The D-3-hydroxyacyl-SACP triketide is generated by KR-catalyzed reduction of the corresponding β-ketoacyl-SACP intermediate (Figure 2). Indeed, incubation of purified recombinant PICS module 2+TE with diketide 4 and [2-14C]malonyl-CoA in the absence of the NADPH cofactor suppressed the KR reaction and gave rise to the formation a single radioactive product, as monitored by TLC-phosphorimaging, with the same Rf (0.65, CH2Cl2:CH3OH:CH3CO2H 95:5:0.1) as synthetic triketide ketolactone 7 (Scheme 3). The identity of the enzymatic reaction product was confirmed by preparative scale incubation with 4 and malonyl-CoA and isolation of the product (Rf 0.65) that displayed both 1H NMR and 1H-1H COSY spectra that were identical to those of synthetic 7. Both the enzymatic reaction product and synthetic 7 exhibited the same parent MS peak at m/z 157.08 [M+ H]+.
To eliminate the KR reaction constitutively, we used site-directed mutagenesis to engineer a set of three PICS module 2(KR0)+TE mutants with inactivated KR domains. Recent independent bioinformatic analysis by both Reid19 and Caffrey20 has identified a triad of highly conserved Lys, Tyr, and Ser residues at or near the active sites of PKS KR domains. Analogous sequence alignments allowed us to locate the corresponding triad of amino acid residues in the KR domain of PICS module 2, consisting of Lys4211, Ser4237, and Tyr4250 (Figure 6B). Using plasmid pTJZ61 as template, three single amino acid substitutions - K4211Q, S4237A, and Y4250F – were generated by PCR mutagenesis. Each of the corresponding mutant proteins was expressed in E. coli BAP1 and purified to near homogeneity by Ni2+-affinity chromatography. Incubation of each of the PICS module 2(KR0)+TE mutants with diketide 4, [2-14C]malonyl-CoA, and NADPH and analysis by TLC-phosphorimaging established that all three mutants catalyzed the formation of ketolactone 7 (Scheme 3, Figure 7). While 7 was the sole product observed for both the Y4250F and S4237A KR0 mutants, both with and without NADPH, the K4211Q mutant produced 7 along with both the reduced and dehydrated triketides, 10 and 5. In the absence of NADPH, the latter mutant produced only 7.
Figure 7.
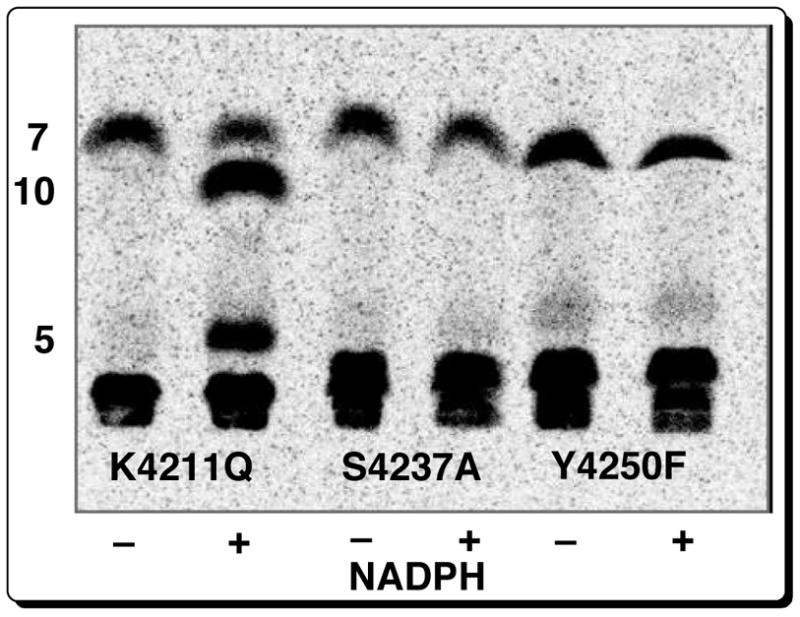
TLC-phosphorimaging of incubation of 4 and [2-14C]malonyl-CoA with PICS module 2(KR0)+TE mutants, with and without NADPH.
Kinetic analysis of PICS module 2+TE and mutants
We determined the steady-state kinetic parameters of “wild-type” PICS module 2+TE and each of the inactivated DH and KR mutants. Incubations were carried out at 30 °C using wild-type or mutant protein in the presence of NADPH and variable concentrations of diketide 4 or [2-14C]malonyl-CoA, while holding the concentration of the second substrate fixed. The reactions were quenched with HCl after 40–60 min and the individual reaction products were quantitated by TLC-phosphorimaging. The kcat and Km were calculated by direct fitting of the plots of ν vs [S] to the Michaelis-Menten equation. The results are summarized in Table 1. For PICS module 2+TE, the kcat values for the production of unsaturated acid 5 and triketide lactone 10 were 0.23 min−1 and 0.49 min−1, respectively. The two substrates, (2S,3R)-4 and malonyl-CoA had average Km values of 1.4 mM and 0.40 mM. When the incubations were carried out in the absence of NADPH, the observed rate of formation of the unreduced triketide ketolactone 7 was reduced by ~4-fold compared to the combined rate of formation of 10 and 5. The S4237A and Y4250F KR0 mutants also showed similar minor decreases in kcat for formation of triketide ketolactone 7 accompanied by no more than 2-fold increases in Km for the substrate 4. The DH0 mutant on the other hand exhibited an insignificant change in kcat, but an apparent 10-fold increase in Km for the diketide substrate, with no significant change in Km for malonyl-CoA.
Table 1.
Steady-state kinetic parameters for PICS module 2+TE and mutants.
| enzyme/mutant | product | substrate | Km (mM) | kcat (min−1) |
|---|---|---|---|---|
| PICS module 2+TE | 5 | 4 | 1.6±0.3 | 0.23±0.02 |
| malonyl-CoA | 0.44±0.06 | |||
| 10 | 4 | 1.2±0.4 | 0.49±0.04 | |
| malonyl-CoA | 0.35±0.07 | |||
| PICS module 2+TE
(-NADPH) |
7 | 4 | 7.7±4.5 | 0.15±0.04 |
| malonyl-CoA | 0.40±0.17 | |||
| DH0 - H3611F | 10 | 4 | 17.0±3.5 | 0.44±0.06 |
| malonyl-CoA | 0.25±0.07 | |||
| KR0 - S4237A | 7 | 4 | 2.9±2.0 | 0.31±0.19 |
| KR0 – Y4250F | 7 | 4 | 3.9±1.3 | 0.32±0.06 |
| KR0 – K4211Q | 7 | 4 | 12.7±4.5 | 0.11±0.04 |
| 10 | 4 | 12.7±4.5 | 0.20±0.09 | |
| 5 | 4 | 12.7±4.5 | 0.12±0.04 |
Using procedures that we had previously developed for the study of the acylation of DEBS module 2,8 we analyzed the rate of acylation of the KS and ACP domains of PICS module 2 using a variant of PICS module 2+TE lacking the TE domain. Purified PICS module 2 (14 μM) was incubated at 22 °C with 1–8 mM [1-14C]-4. The reaction was quenched with ice-cold acetone at times ranging from 1–20 min and the washed, pelleted protein was analyzed by liquid scintillation counting to quantitate the amount of covalently bound diketide. The data were fit to the relevant equations for active-site acylation,8,21 giving a first-order rate constant for acylation, kacyln, of 0.32±0.05 min−1 and a dissociation constant, KS, of 2.9±1.0 mM, corresponding to a kcat/KS for KS acylation of 1.1×10−4 μM−1min−1 (Scheme 4). The acylation endpoint corresponded to a stoichiometry of 1:1 diketide:protein. The much more rapid acylation by malonyl-CoA was analyzed by quenching with trichloroacetic acid in a rapid chemical quench apparatus at reaction times ranging from 50 ms to 5 s. Malonylation of the ACP domain was complete within ~5 s, with a kacyln of 0.38±0.10 s−1 (Scheme 4). The kcat/Km determined for AT-catalyzed transfer of the malonyl chain extension unit from malonyl-CoA to the ACP domain of PICS module 2 was estimated to be 2.3±0.1 μM−1 min−1. In the absence of diketide cosubstrate the malonylated protein species decayed rapidly, presumably by AT-catalyzed hydrolysis, with essentially no enzyme-bound radioactivity remaining after 5 min.
Scheme 4.
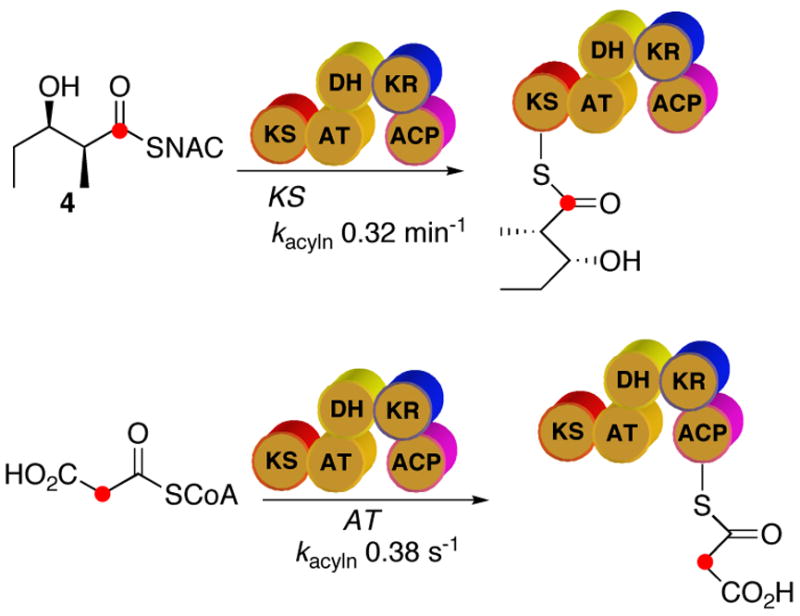
Acylation of PICS module 2 by diketide 4 and malonyl-CoA
Substrate specificity of PICS module 2+TE
Although PKS modules usually show relatively broad substrate tolerance, they also can exhibit considerable stereochemical specificity.5a,9b,13b We have determined the substrate specificity of PICS module 2+TE towards each of the four diastereomeric SNAC esters 4, 13, 14, and 15 (Figure 8). The diketide diastereomers were first screened by incubating 8 mM of each substrate with PICS module 2+TE in the presence of [2-14C]methylmalonyl-CoA and NADPH for 1 h at 30 °C. The resultant triketide lactone and triketide acid were separated by HPLC and the yields were measured by liquid scintillation counting of the combined triketide products. Under these conditions, PICS module 2+TE processed the natural syn-(2S,3R)-diketide-SNAC 4 more than 40 times more rapidly than the enantiomeric syn-(2R,3S)-diketide-SNAC 13 and 20 times faster than the diastereomeric anti-(2S,3S)-diketide-SNAC 14. The anti-(2R,3R)-diketide-SNAC 15 was not converted in detectable yields to either chain elongation product. PICS module 2+TE also showed low level activity toward (2E,4R,5R)-4-methyl-5-hydroxy-2-heptenoyl-SNAC (11), the analogue of its normal triketide chain elongation product, but at no more than 5% the rate for the natural diketide-SNAC substrate 4. We next determined the steady-state kinetic parameters for each of the three most active substrates, 4, 13, and 14, as summarized in Table 2.
Figure 8.
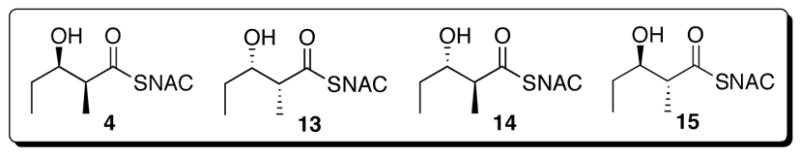
Diketide diastereomers incubated with PICS module 2+TE
Table 2.
Steady-state kinetic parameters for diketide diastereomers and PICS module 2+TEa
| Substrate | kcat (min−1) | Km (mM) | kcat/Km (M−1min−1) |
|---|---|---|---|
| syn-(2S,3R)-4 | 0.61±0.15 | 4.8±0.19 | 127±60 |
| anti-(2S,3S)-14 | 0.027±0.007 | 7.9±3.2 | 3.4±1.7 |
| syn-(2R,3S)-13 | 0.0067±0.0010 | 2.6±1.2 | 2.6±1.3 |
Monitored by liquid scintillation counting of combined HPLC-purified triketide acid and triketide lactone products of each incubation.
Discussion
Unsaturated Polyketides
Among the more than 2000 known macrolides, the vast majority contain one or more double bonds.22 The 12- and 14-membered aglycones, 10-deoxymethynolide (1) and narbonolide (2) as well as the 16-membered macrolide aglycone tylactone (16), are typical examples of unsaturated macrolactones (Figure 9). The broader class of polyketide metabolites, including the immunosupressants rapamycin and FK-506, the antiparasitic polyketide avermectin, and the antifungal polyene antibiotics such as amphotericin B also contain multiple double bonds. The best studied macrolide antibiotic, erythromycin A, and its aglycone, 6-deoxyerythronolide B (6-dEB, 3), are therefore atypical in that they do not carry any double bonds (although a double bond is transiently generated and then reduced by DEBS module 4 during 6-EB biosynthesis.) In spite of the prevalence of unsaturated features in polyketide metabolites, most of what we know about polyketide double bond biosynthesis has been inferred by structural comparisons among known metabolites and analysis of modular domain compositions. For example numerous modular polyketide synthases have been found to harbor one or more DH domains, most often, but not always, corresponding to the presence of double bonds in the corresponding polyketide metabolites.18d Explicit mechanistic information about the action of DH domains has come from studies of classical fatty acid biosynthesis. Thus Sedgwick demonstrated nearly 30 years ago that the dehydration catalyzed by the yeast fatty acid synthase involves a net syn elimination of water from a D-(3R)-hydroxyacylthioester substrate.23
Figure 9.
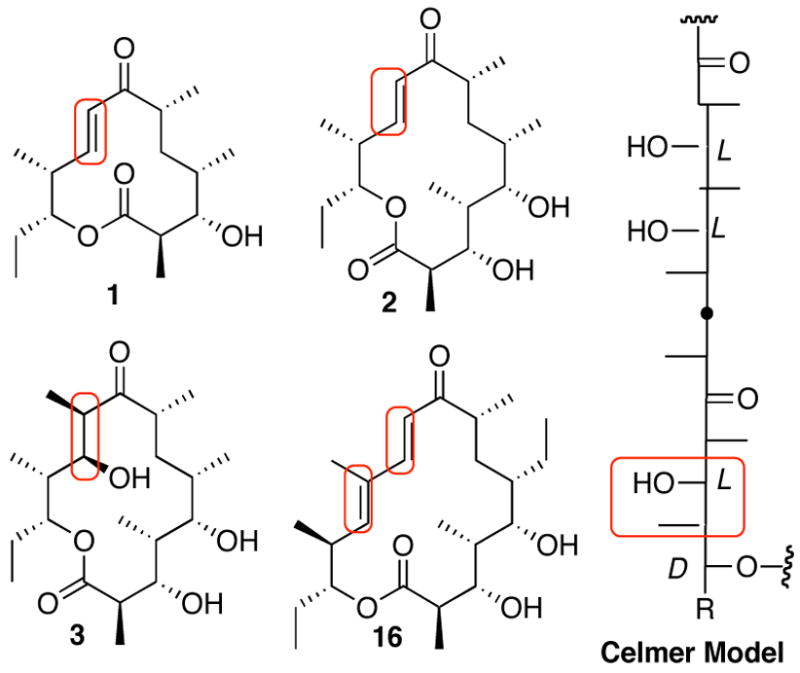
Celmer model of macrolide structure and stereochemistry, illustrating homologous sites of dehydration
The well-known Celmer model of macrolide structure and stereochemistry, based on the alignment of the Fischer projections of classical macrolides, illustrates the structural and mechanistic correlation between pairs of macrolides carrying hydroxyl groups or double bonds at aligned positions (Figure 9).24 Thus by comparison of the structure of tylactone (16) with that of 6-deoxyerythronolide B (3) it can be seen that the 16-membered macrolide aglycone 16 possesses a trisubstituted (E)-12,13-double bond where the 14-membered 6-dEB (3) carries an L-methyl and a D-hydroxyl group at the corresponding 10- and 11-positions. The corresponding (E)-10,11-double bond of narbonolide (2), as well as the homologous disubstituted (E)-8,9-double bond of 10-deoxymethynolide (1) and the (E)-10,11-double bond of tylactone (16) can all be adapted to this structural alignment model by assuming an intervening dehydration reaction. Notably, however, simple dehydration of 6-dEB to give an (E)-10,11-double bond would require a net anti-elimination of water, opposite to the established stereochemistry of fatty acid synthase dehydratase-catalyzed reactions.
Stereochemistry of the ketoreductase reaction
We have previously determined that the KR domains of DEBS modules 1, 2, 5, and 6 are all specific for the H-4si hydride (B-face) of the NADPH cofactor,25 in common with the established stereospecificity of fatty acid synthase ketoreductases.26 Although these KR domains share the same cofactor stereospecificity, their substrate specificity is different, with the KR domain of DEBS module 1 reducing its β-ketoacyl-SACP substrate to generate a D-3-hydroxyacyl product while DEBS modules 2, 5, and 6 all generate L-hydroxy groups. We have previously demonstrated that the stereochemistry of reduction is an intrinsic property of each KR domain and is independent of both substrate structure and modular context.27 Both Reid19 and Caffrey20 have independently compared the sequences of some 200 modular KR domains belonging to the superfamily of short-chain dehydrogenases/reductases (SDRs). These investigators both noted an interesting correlation between the formation of D-hydroxy groups in the final polyketide product and the presence of a conserved Asp residue in the responsible KR domain. By contrast, KR domains that generate L-hydroxy groups lack this conserved aspartate. Caffrey has also pointed out that this aspartate is part of a conserved Leu-Asp-Asp motif in such D-specific ketoreductases.20 Intriguingly, this conserved Asp residue is also present in modular KR domains of unknown stereochemical specificity that are paired with a DH domain, such as the KR domains of PICS module 2 and tylactone synthase modules 2 and 3. Such modules typically generate polyketide chain elongation intermediates with a trans (E) double bond. The clear inference from this bioinformatic analysis is that the cryptic products of such KR domains will all have the D-hydroxy configuration. In fact, the KR domain of PICS module 2 contains the conserved Asp residue as part of a Leu-Ser-Asp motif.
Our finding that PICS module 2+TE and the inactive dehydratase mutant, PICS module 2(DH0)+TE, both catalyze the formation of the D-3-hydroxy triketide lactone 10 establishes that the ketoreductase generates exclusively the D-3-hydroxytriketide acylthioester intermediate. These observations, which constitute the first experimental determination of the stereochemistry of a modular PKS KR domain that is paired with a functional DH domain, support the above-described configurational predictions that were based purely on bioinformatic analysis. This finding is therefore of considerable theoretical as well as practical importance. One clear implication is that genetic engineering of PKS modules by the introduction of DH domains is likely to succeed in generating the derived dehyration product only if the paired KR domain naturally produces a D-hydroxyacylthioester intermediate to serve as substrate for the heterologous DH domain. Indeed, insertion of rapamycin DH domains into DEBS modules 2, 5 or 6 was able to produce the corresponding unsaturated polyketide product only when the complete KR-DH pair derived from rapamycin module 4 was swapped for the native DEBS KR domain.28
PICS module 2+TE
Unsaturated polyketide intermediates have previously been generated in situ as part of intact cell experiments with engineered combinations of PKS modules.5,28–30 In earlier in vitro studies, we have also tested a wide variety of unsaturated triketide-SNAC analogues as surrogate substrates for DEBS modules 2 and 3.13b Recently we reported that a hybrid fusion protein consisting of DEBS module 1 and PICS module 2 generated the decarboxylated tetraketide 17, presumably by the intermodular transfer of the acyl group of the enzyme-bound triketide intermediate, (4S,5R)-4-methyl-5-hydroxy-2-heptenoyl-SACP2, to PICS module 3+TE, followed by an additional round of chain elongation (Scheme 5).9d By contrast, recombinant PICS module 3+TE carrying an appropriate N-terminal docking domain did not accept as substrates either diketide-SNAC 4 or the natural enzyme-bound, saturated 3,5-dihydroxy triketide generated by DEBS1 (DEBS modules 1 and 2). [1-14C]Diketide 4 also failed to acylate PICS module 3+TE, suggesting that PICS module 3 is highly specific for the natural unsaturated triketide product of the upstream PICS module 2.
Scheme 5.
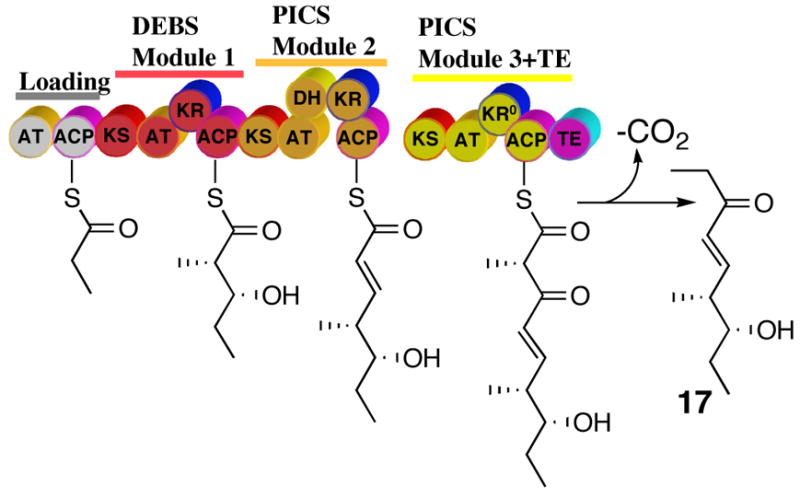
In vivo generation and processing of the unsaturated triketide by a hybrid DEBS/PICS trimodular PKS containing PICS modules 2 and 3.
The expression and analysis of PICS module 2+TE reported herein is the first in vitro study of the biochemistry and mechanism of a single DH-containing PKS module that catalyzes a chain elongation with formation of a 2,3-unsaturated acylthioester product. Fusion of the PICS TE domain to the C-terminus of PICS module 2 allows the release of the expected chain elongation product, unsaturated triketide acid 5, whose structure and stereochemistry was rigorously confirmed by direct comparison of its NMR and mass spectra to those of a synthetic standard. Unexpectedly, the D-3-hydroxy-triketide lactone 10 was also a major product of the incubation (Scheme 3). The formation of 10 is the consequence of an anomalous kinetic competition between the normal DH-catalyzed dehydration of the D-3-hydroxy-triketide acylthioester intermediate and TE-catalyzed cyclization of this same substrate. This adventitious reaction is not an issue when PICS module 2 operates in its normal functional context in which the only competition would be between the reactions catalyzed by the individual KR and DH domains of PICS module 2 (Figure 2) and premature transfer to the downstream PICS module 3. In fact, such abortive transfer to PICS module 3 would be kinetically disfavored in light of the apparent preference of this downstream module for unsaturated substrates.9d
Active sites of KR and DH domains
Introduction of strategic mutations into the DH and KR domains of PICS module 2+TE has allowed the trapping and release of the corresponding 3-hydroxyacyl and 3-ketoacyl triketide intermediates (Figure 2, Scheme 3). The dehydration reaction can be suppressed by inactivation of the PICS DH2 domain. Extensive sequence alignments have identified a conserved HXXXGXXXXP motif among both fatty acid synthase and PKS dehydratases.18 The crystal structure of Pseudomonas aeroginosa (3R)-hydroxyacyl-ACP dehydratase (FabZ) confirms the presence of the homologous conserved His49 residue at the active site as part of the known His/Glu catalytic dyad.31 By alignment of sequence of the DH domain of PICS module 2 with known PKS DH sequences we identified the conserved active site His residue as H3611 (Figure 6A). The importance of this amino acid for DH function is supported by the observation that the corresponding PICS module 2(DH0)+TE-H3611F mutant has an inactive DH domain, but is fully catalytically functional for generation of the D-3-hydroxy triketide lactone 10 (Scheme 3, Table 1).
Leadlay and coworkers have reported that an analogous H2409F mutation in the DH domain of DEBS module 4 abolished 6-dEB formation by mutant strains of the erythromycin producer Saccharopolyspora erythraea.18c These researchers were unable, however, to detect any of the expected shunt product, 6,7-dihydro-7-hydroxy-6-dEB. It is conceivable that the abortive 2-methyl-3-hydroxypentaketide intermediate was simply not processed by the KS domain of the downstream DEBS module 5, perhaps because it would have had the anti-2,3-stereochemistry, which is known to be incompatible with DEBS module 5.9b Interestingly, the 3-hydroxyl group generated by DEBS KR4 is predicted to have the D-configuration,19,20 although the configuration of the adjacent methyl group in the 2-methyl-3-hydroxypentaketide intermediate remains unknown.
As previously noted, extensive sequence alignments of a large number of modular PKS KR domains as well as crystallographic analysis of several members of the superfamily of dehydrogenases/reductases have identified a highly conserved triad of active site Lys, Ser, and Tyr residues.19,20 Modeling and crystallographic studies have indicated that the Ser and Tyr residues interact directly with the substrate by binding and activating the target carbonyl group, while the Lys residue exerts a more indirect influence on the reaction through hydrogen bonding to the 2′,3′-hydroxyl groups of the nicotinamide ribose ring and possible reduction of the pKa of the Tyr hydroxyl through ionic interactions.32 The two PICS module 2(KR0)+TE mutants, harboring the S4237A and Y4250F mutations, respectively, produced the expected triketide ketolactone 7 as the exclusive product, while the corresponding K4211Q also generated 7 in addition to the normal reduction and dehydration products 10 and 5 (Scheme 3). These experiments confirm the predicted importance of each of the three active site residues in the function of the KR domain, while elucidating the reactions of the individual domains of PICS module 2. Interestingly, similar results have been obtained by introducing analogous mutations into the KR domain of DEBS module 6 within the complete DEBS PKS, although only the Tyr to Phe mutation completely suppressed the target ketoreductase reaction.19
Kinetic analysis of PICS module 2+TE and mutants
PICS module 2+TE generates the combined triketide products 10 and 5 with a net kcat of 0.6 min−1 that is well within the range observed for several other DEBS and PICS modules examined in vitro.3,9b,9c The observed Km values for both substrates, (2S,3R)-4 and malonyl-CoA are typical of such modules. PICS module 2 shows a strong preference for the natural syn-(2S,3R)-diketide-SNAC 4 as substrate, as illustrated by the 20- and 40-fold greater kcat/Km compared to the anti- and enantio-syn diketide diastereomers 14 and 13, respectively (Table 2). Similar stereochemical preferences have been demonstrated for DEBS modules 2, 3, 5, and 6, as well as PICS module 5, while PICS module 6 shows a 2.5:1 preference for syn-(2R,3S)-13 over syn-(2S,3R)-4, although none of these other modules will process either anti-diketide-SNAC diastereomer.9b,9c,16
Since the KS acylation experiments with labeled (2S,3R)-4 and PICS module 2 were carried out at lower temperature (22 °C instead of 30 °C) and higher glycerol concentration (20% instead of 10%) than the turnover experiments with PICS module 2+TE, the experimentally determined KS acylation rate of 0.3 min−1 and the measured kcat for the overall reaction of 0.6 min−1 cannot be directly compared. Nonetheless, they are of the same order of magnitude, and the respective Ks and Km values for diketide 4 are in good agreement. If one makes the reasonable assumption that the 8 °C difference in incubation temperature corresponds to a ~2-fold effect on the rate constant, the two processes are certainly kinetically compatible within reasonable experimental limits. Notably, AT-catalyzed acylation of the ACP domain is at least 2 orders of magnitude faster than the KS-catalyzed reaction (Scheme 4).
The steady-state kinetic properties of the various DH and KR mutants vary only a minor amount from those of the wild-type PICS module 2+TE, indicating that the overall function of the other domains has not been compromised in any significant way by the single amino acid substitutions that inactivate each of the targeted domains.
Conclusions
The above-described investigation of PICS module 2+TE and its mutants have established for the first time the structure and stereochemistry of the intermediates of the chain elongation cycle that adds an equivalent of acetate and generates a new double bond, while confirming the importance of key active site residues in both the KR and DH domains. These studies provide a framework for the study of additional DH-containing modules, while providing valuable guidelines for the rational engineering of polyketide biosynthesis.
Experimental Section
Materials
Plasmid pKW119 was a gift from Dr. Kenji Watanabe of the Khosla laboratory. [2-14C]Malonyl-CoA was from American Radiolabeled Chemicals. (2S,3R)-[1-14C]-2-methyl-3-hydroxypentanoic acid N-acetylcysteamine thioester ([1-14C]-4, 55.0 mCi/mmol) was prepared by custom synthesis by Amersham Pharmacia Biotech. Diketide-SNAC 4, triketide ketolactone 7, triketide lactone 10, and (4S,5R)-4-methyl-5-hydroxy-2-heptenoic acid (5) were each synthesized as previously described.9b,13,14 Isopropyl-thio-D -galactopyranoside (IPTG) was from Gibco. N,O -Bis(trimethylsilyl)trifluoracetamide (BSTFA) was obtained from Pierce. All other chemicals, including non-labeled CoA derivatives, were from Sigma. PD-columns were from Amersham Pharmacia Biotech and Centriprep-50 concentrators (MWCO 50 kDa) were from Amicon. SDS-PAGE gradient gels (4–15% acrylamide) were from BioRad. Nickel-nitriloacetic acid (Ni-NTA) resin was obtained from Qiagen. Solvents were reagent grade and were further dried when necessary. Analytical thin-layer chromatography was performed on aluminum plates precoated with silica gel (250 μm, Whatman), with detection by UV and/or spraying with anisaldehyde solution. Flash chromatography was carried out on silica gel (60 Å, 32–63 μm) purchased from Sorbent Technologies.
General Methods
Standard recombinant DNA and protein manipulations were carried out according to published procedures.33 PCR and restriction digestions were run in a MiniCycler thermocycler from MJ Research, equipped with a hot-bonnet. PICS module 2+TE mutants were generated with the Stratagene QuikChange II XL Site-Directed Mutagenesis™ kit using the manufacturer’s protocols, modified as indicated. DNA sequencing was performed by the HHMI Biopolymer/Keck Foundation Biotechnology Resource Laboratory at the Yale University School of Medicine, New Haven (CT), using the dideoxy dye terminator method and automated fluorescence sequencing. Protein concentrations determined by the Bradford assay34 using bovine serum albumin (BSA) as standard showed standard deviations ≤5% for samples measured at different times. Phosphorimaging was carried out using a Bio-Rad FX Molecular Imager System and Bio-Rad K-series phosphorimager screens. Liquid scintillation counting was performed on a Beckman LS6500 Liquid Scintillation Counter in 10 ml of Opti-Fluor™ scintillation cocktail. 1H and 13C NMR utilized Bruker Avance AM300 and AM400 spectrometers. High resolution ESI Mass spectra were recorded on an Applied Biosystems QSTAR TOF mass spectrometer. GC-EI-mass spectra were obtained on a Jeol JMS-600H mass spectrometer. Kinetic data were analyzed by direct fitting to the relevant kinetic equations using the KaleidaGraph data analysis software (Synergy Software.)
Construction of expression plasmid pTJZ61 for PICS module 2 + TE
Plasmid DNA from pHL4-65b, the previously described expression plasmid for PICS TE,10 was used as the template for PCR amplification of bp 3145-4038 of the pikAIV gene (GenBank accession No. AF079138) with PfuUltra DNA polymerase. Foward primer with HindIII site :5′-ACTGAGTAAGCTTTCCGGGCCGACACCGGCG-3′ (Tm 70.7°C); reverse primer with XhoI site and stop codon: 5′-AGTGAGTCTCGAGTCACTTGCCCGCCCCCTCGATGCCC -3′ (Tm 72.6°C). The resultant PCR product was purified by gel electrophoresis on 1% agarose (TAE buffer) and the excised DNA was purified with a Qiagen gel extraction kit. The purified DNA was then digested with HindIII and XhoI and ligated into the corresponding sites of pET28a(+). The sequence of the 900-bp insert in the resulting plasmid, pTJZ41, was verified by DNA sequencing. Plasmid pKW119, which harbors PICS module 2 fused to the N-terminal linker region of DEBS module 3 (EryL3), had been constructed by inserting BsaB1/HindIII-digested DNA, corresponding to bp 7820–13464 of PikAI (Genbank accession no. AF079138) into the corresponding sites of pRSG34, a pET21c-derived PKS expression vector containing bp 14–119 of eryA ORF 2 (eryL3, GenBank accession no. M63677) upstream of the BsaB1 site.9a pKW119 was digested with NdeI and HindIII and the 5.8-kb fragment was ligated into the corresponding sites of pTJZ41 to give pTJZ61 encoding (EryL3)-PICS module 2+TE with an appended N-terminal His6-tag. The integrity of the final pTJZ61 expression vector was verified by restriction digests and sequencing around all DNA ligation sites.
Construction of PICS module 2+TE mutants by site-directed mutagenesis
Site-directed mutagenesis was carried out on pTJZ61 using the Stratagene QuikChange II XL Site-Directed Mutagenesis Kit™. Extension times were increased to 30 min in order to generate full-length DNA harboring the desired mutations. The primer pairs used for mutagenesis are listed in Table 3.
Table 3.
PCR primers for site-directed mutagenesis of pTJZ61/PICS module 2+TE.
| mutant | primera |
|---|---|
| DH0
H3611F |
fwd: 5′-CCC GTG GCT CGC GGA CTT CGC CGT CCT GGG GAG CGT CC-3′ |
| rev: 5′-GGA CGC TCC CCA GGA CGG CGA AGT CCG CGA GCC ACG GG -3′ | |
| KR0
S4237A |
fwd: 5′- GCG TTC GTC ATG TTC TCC GCC GCC GCC GCC GTC TTC GG -3′ |
| rev: 5′- CCG AAG ACG GCG GCG GCG GCG GAG AAC ATG ACG AAC GC -3′ | |
| KR0
Y4250F |
fwd: 5′- GCG GGG CAG GGC GCC TTC GCC GCC GCC AAC -3′ |
| rev: 5′- GTT GGC GGC GGC GAA GGC GCC CTGCCC CGC -3′ | |
| KR0
K4211Q |
fwd: 5′- CAC GTA CTG CGG CCC CAG GTC GAC GCC GCG TTC CTC C -3′ |
| rev: 5′- GGA GGA ACG CGG CGT CGA CCT GGG GCC GCA GTA CGT G -3′ |
mutant sequences indicated in bold.
Expression and purification of WT and mutant PICS module 2+TE
Expression plasmids for PICS module 2+TE (pTJZ61) and mutants were transformed by electroporation into electrocompetent cells of E. coli BAP1. The transformed cells were grown in LB/kanamycin medium to OD 0.6 and protein expression was induced by adding 0.1 mM IPTG at 18 °C. After expression for 12 h, cells from 2 L of LB culture were harvested and lysed in 30 mL of lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 1 mM benzamidine, 2 mg/L leupepsin, 2 mg/L pepstatin, 5 mM β-mercaptoethanol, pH 8.0) by French press at 10,000 psi. The resulting cell lysate was centrifuged at 53,000g to remove cell debris and the supernatant was mixed with 10 mL of Ni-NTA resin. After gentle shaking on an ice-bath for 40 min, the slurry was transferred to a column (ID 1.5 cm) and subjected to a gradient elution using the Econo Buffer Delivery System at a buffer flow rate of 2 mL/min. The column was washed with 5 bed volumes of 10 mM imidazole in lysis buffer followed by increasing imidazole concentrations up to 250 mM in 50 min (10 bed volumes buffer). Pure protein fractions were pooled, concentrated, and exchanged using PD10 columns into storage buffer (100 mM phosphate, 1 mM EDTA, 1 mM TCEP, 10% glycerol, pH 7.2). Aliquots were flash-frozen in liquid nitrogen and stored at −80 °C until use.
Preparative-scale incubation of PICS module 2+TE. Formation of triketide acid 5 and triketide lactone 10
Purified PICS module 2+TE (20 μM) was incubated in a total volume of 30 mL of storage/reaction buffer at 30 °C with 19 mM diketide 4 (added as a stock solution in 1 mL of DMSO), NADPH (8 mM), and 0.4 mL of a stock solution of 100 mg (0.116 mmol) of malonyl-CoA in 1 mL of H2O. Additional 0.4-mL and 0.3-mL batches of malonyl-CoA were added after 20 and 40 min. The incubation was continued overnight (18 h), and then quenched with HCl along with added solid NaCl to adjust the pH to 4. The protein precipitate was removed by centrifugation and the aqueous supernatant was extracted with 5 × 100 mL of ethyl acetate. The solvent was removed in vacuo and the crude products were separated by SiO2 flash chromatography and preparative TLC to afford two triketide products in ~5% purified yield, based on substrate diketide (which also undergoes competing TE-catalyzed hydrolysis): Rf 0.56 (CH2Cl2:CH3OH:CH3CO2H 95:5:0.1), (3S,4S,5R)-3,5-dihydroxy-4-methyl-n-heptanoic acid-δ-lactone (10) 1H NMR (400 MHz, CDCl3) δ 4.61 (m, 1H, CHCH2CH3), 4.04 (m, 1H, CHOH), 2.81 (dd, J=18.04, 5.4 Hz, 1H, CHHCO), 2.55 (dd, J=18.31 Hz, 3.01 Hz, 1H, CHHCO), 1.95 (m, 1H, CHCH3), 1.78 (m, 1H, CHHCH3), 1.55 (m, 1H, CHHCH3), 1.00 (t, J=7.41 Hz, 3H, CH2CH3), 0.93 (d, J =7.12 Hz, 3H, CHCH3). Rf 0.30, (4S,5R)-4-methyl-5-hydroxy-2-heptenoic acid (5) 1H NMR (400 MHz, CDCl3) δ 7.07 (m, J=16.11 Hz, 7.08 Hz, 1H, CH=CHCO2), 5.87 (d, J=15.9 Hz, 1H, CH=CHCO2), 3.53 (m, 1H, CHCOH), 2.48 (m, 1H, CHCH3), 1.54 (m, 1H, CH3CHH), 1.40 (m, 1H, CH3CHH), 1.10 (d, J=6.73 Hz, 3H CHCH3), 0.97 (t, J=7.42 Hz, 3H, CH3CH2); 13C-NMR (100 MHz, CDCl3) δ 171.2, 153.8, 120.5, 75.4, 42.3, 27.4, 13.8, 10.2.
Preparative-scale incubation of PICS module 2+TE without NADPH. Formation of triketide ketolactone 7
A preparative scale reaction was carried out using the crude cell-free extract from a 4-L growth of E. coli BAP1/pTJZ61. The supernatant after ultracentrifugation was diluted to 100 mL with lysis buffer, to which was added 160 mg (0.69 mmol) of diketide 4 in 1 mL DMSO and and 0.4 mL of a stock solution of 100 mg (0.116 mmol) of malonyl-CoA in 1 mL of H2O. Additional 0.4- and 0.3-mL batches of malonyl-CoA were added after 20 and 40 min. The incubation was continued overnight at 30 °C and the organic product isolated and purified as above to give (4S,5R)-3-oxo-4-methyl-5-hydroxy-n- hepatanoic acid-δ-lactone (7) in ~5% purified yield based on 4: Rf 0.65 (CH2Cl2:CH3OH:CH3CO2H 95:5:0.1), 1H NMR (400 MHz, CDCl3) δ 4.53 (m, 1H, CHCH3), 3.55 (d, J=19.51 Hz, 1H, CHHCO), 3.38 (d, J=19.51 Hz, 1H, CHHCO), 2.69 (qd, J=7.34, 3.36 Hz, 1H, CHCH2), 1.77–1.60 (m, 2H, CH2CH3), 1.14 (d, J=7.36 Hz, 3H, CH3CH), 1.07 (t, J=7.42 Hz, 3H, CH3CH2); 13C NMR (100 MHz, CDCl3) δ 203.5, 167.5, 80.1, 45.6, 45.3, 24.0, 10.0,9.3; FAB-MS calcd for C8H12O3: 156.08, observed: 157.08 [M+H]+.
Kinetic characterization of WT and mutant PICS module 2+TE
(2S,3R)-Diketide-SNAC 4 (0.5 to 16 mM) and malonyl-CoA (0.05 to 1 mM, 2 mCi/mmol) were incubated at 30 °C with 2 mM NADPH and 1 to 5 μM PICS module 2+TE or mutant in 25 μL of reaction buffer (100 mM sodium phosphate, 1 mM EDTA, 1 mM TCEP, 10% glycerol, 8% (v/v) DMSO, pH 7.2) for 40 min, during which the production of the products had been shown to be linear. The reactions were quenched by addition of 2 μL of 1 M HCl. Solid NaCl was then added the mixtures were extracted immediately with 3 180-μL portions of ethyl acetate. The combined organic extracts were concentrated on a SpeedVac. The residue was redissolved in 30 μL of ethyl acetate and spotted on TLC plates which were developed with dichloromethane containing 5% methanol and 0.1% acetic acid. Under such conditions, the synthetic standards have the following Rf values: triketide ketolactone 7, 0.65; triketide lactone 10, 0.56; triketide acid 5, 0.30. The developed TLC plates were analyzed by phosphorimaging and radioactive spots with the above-mentioned Rf values were quantitated by using the known amounts of [2-14C]malonyl-CoA co-spotted on the TLC plates as standard. Each reaction was repeated at least 3 times using different protein preparations. Negative controls were performed for all assays where enzyme was left out to verify there was no detectable non-enzymatic reaction. The resulting initial velocity data, representing 15-25 turnovers, were fit to the Michaelis-Menten equation to calculate the kinetic parameters kcat and Km. The kinetics for diketide diastereomers 4, 13, and 14 were determined in a similar manner, as well the formation of 7 by PICS module 2+TE in the absence of NADPH.
Acylation of the KS domain of PICS module 2
The acylation of the KS domain of PICS module 2 was monitored by a modification of the previously reported procedures for acylation of DEBS module 2.8 PICS module 2 (14 μM) was incubated with [1-14C]diketide-SNAC 4 (2.85 mCi/mmol, 1 to 8 mM) in reaction buffer (100 mM sodium phosphate buffer, pH 7.2, containing 1 mM EDTA, 1 mM TCEP, 8% DMSO, and 20% (v/v) glycerol, total vol. 60 μL) at 22 °C. Aliquots were withdrawn at periodic intervals from 1 to 20 min and the reactions quenched with 1 mL of ice-cold acetone together with 0.2 mg of BSA. The precipitated protein was collected by centrifugation, and the pellets were washed 4–5 times with portions of cold 50% trichloroacetic acid until there was no detectable 14C activity in the washings. The protein pellet was then dissolved in 200 μL of 8.0 M guanidinium·HCl and the covalently bound radioactivity quantitated by liquid scintillation counting. Control incubations contained all components except PICS module 2+ΔTE protein. Each data point was determined at least three times. Control incubations analyzed by SDS-PAGE and phosphorimaging verified that all the radioactivity was associated with the single protein band corresponding to PICS module 2.
For monitoring the acylation of the ACP domain by malonyl-CoA, reactions were carried out using a KinTek Corp. Model RQF-3 Rapid Chemical Quench Flow apparatus. Sample syringe 1 contained 300 μL of 23 μM PICS module 2 in 100 mL of sodium phosphate buffer, pH 7.2, containing 1 mM EDTA, 1 mM TCEP, and 20% glycerol. Sample syringe 2 contained 300 μL of 200 μM [2-14C]malonyl-CoA (2 mCi/mmol) in the same buffer. The quench syringe contained 50% trichloroacetic acid (TCA). For a typical reaction, 20 μL of protein solution and 20 μL of substrate solution were loaded into the loading loops and reaction was triggered by injection of the two components into the reaction loop. After incubation for a preset time, varying from 50 ms to 5 s, the quench solution was released into the reaction loop with transfer of the quenched mixture to an Eppendorf tube which was immediately flash frozen in liquid nitrogen. For analysis, the thawed quench mixture was mixed with 1 mL of cold acetone and centrifuged at high speed in a bench top centrifuge for 10 min. The protein pellet was collected, washed 4–5 times with cold 50% TCA until no radioactivity was detected in the washes and the protein was then dissolved in 200 μL of guanidinium hydrochloride and the radioactivity assayed by liquid scintillation counting.
Acknowledgments
This research was supported by NIH Grants GM22172 (D.E.C.) and CA66736 (C.K.). We thank Dr. Tun-Li Shen for assistance with the mass spectrometric analyses.
Footnotes
Supporting Information Available: GC-MS analysis of trimethylsilylated triketide acid and triketide lactone. This material is available free of charge via the Internet at http://pubs.acs.org. See any current masthead page for ordering information and Web access instructions.
References
- 1.a) Hori T, Maezawa I, Nagahama N, Suzuki M. J Chem Soc Chem Commun. 1971:304–305. [Google Scholar]; b) Omura S, Takeshima H, Nakagawa A, Miyazawa J. J Antibiot. 1976;29:316–317. doi: 10.7164/antibiotics.29.316. [DOI] [PubMed] [Google Scholar]; c) Lambalot RH, Cane DE. J Antibiot. 1992;45:1981–1982. doi: 10.7164/antibiotics.45.1981. [DOI] [PubMed] [Google Scholar]
- 2.a) Xue Y, Wilson D, Sherman DH. Gene. 2000;245:203–211. doi: 10.1016/s0378-1119(00)00003-2. [DOI] [PubMed] [Google Scholar]; Xue Y, Zhao L, Liu H-w, Sherman DH. Proc Natl Acad Sci USA. 1998;95:12111–12116. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tang L, Fu H, Betlach MC, McDaniel R. Chem Biol. 1999;6:553–558. doi: 10.1016/S1074-5521(99)80087-8. [DOI] [PubMed] [Google Scholar]
- 3.Beck BJ, Yoon YJ, Reynolds KA, Sherman DH. Chem Biol. 2002;9:575–583. doi: 10.1016/s1074-5521(02)00146-1. [DOI] [PubMed] [Google Scholar]; Xue Y, Sherman DH. Nature. 2000;403:571–575. doi: 10.1038/35000624. [DOI] [PubMed] [Google Scholar]; Aldrich CC, Beck BJ, Fecik RA, Sherman DH. J Am Chem Soc. 2005;127:8441–8452. doi: 10.1021/ja042592h. [DOI] [PubMed] [Google Scholar]; Aldrich CC, Venkatraman L, Sherman DH, Fecik RA. J Am Chem Soc. 2005;127:8910–8911. doi: 10.1021/ja0504340. [DOI] [PubMed] [Google Scholar]
- 4.Kao CM, Luo G, Katz L, Cane DE, Khosla C. J Am Chem Soc. 1995;117:9105–9106. [Google Scholar]
- 5.a) Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Ann Rev Biochem. 1999;68:219–253. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]; b) Staunton J, Wilkinson B. In: Comprehensive Natural Products Chemistry, Polyketides and Other Secondary Metabolites Including Fatty Acids and Their Derivatives. Sankawa U, editor. Vol. 1. Elsevier; Oxford: 1999. pp. 495–532. [Google Scholar]
- 6.a) Donadio S, Staver MJ, Mcalpine JB, Swanson SJ, Katz L. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]; b) Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. Nature. 1990;348:176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]; c) Pieper R, Luo GL, Cane DE, Khosla C. Nature. 1995;378:263–266. doi: 10.1038/378263a0. [DOI] [PubMed] [Google Scholar]
- 7.Cane DE, Lambalot RH, Prabhakaran PC, Ott WR. J Am Chem Soc. 1993;115:522–526. [Google Scholar]
- 8.Wu J, Kinoshita K, Khosla C, Cane DE. Biochemistry. 2004;43:16301–16310. doi: 10.1021/bi048147g. [DOI] [PubMed] [Google Scholar]
- 9.a) Gokhale RS, Tsuji SY, Cane DE, Khosla C. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]; b) Wu N, Kudo F, Cane DE, Khosla C. J Am Chem Soc. 2000;122:4847–4852. [Google Scholar]; c) Yin Y, Lu H, Khosla C, Cane DE. J Am Chem Soc. 2003;125:5671–5676. doi: 10.1021/ja034574q. [DOI] [PubMed] [Google Scholar]; d) Watanabe K, Wang CC, Boddy CN, Cane DE, Khosla C. J Biol Chem. 2003;278:42020–42026. doi: 10.1074/jbc.M305339200. [DOI] [PubMed] [Google Scholar]
- 10.Lu H, Tsai SC, Khosla C, Cane DE. Biochemistry. 2002;41:12590–12597. doi: 10.1021/bi026006d. [DOI] [PubMed] [Google Scholar]
- 11.Walsh CT, Gehring AM, Weinreb PH, Quadri LE, Flugel RS. Curr Opin Chem Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 12.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 13.a) Kinoshita K, Williard PG, Khosla C, Cane DE. J Am Chem Soc. 2001;123:2495–2502. doi: 10.1021/ja004139l. [DOI] [PubMed] [Google Scholar]; b) Cane DE, Kudo F, Kinoshita K, Khosla C. Chem Biol. 2002;9:131–142. doi: 10.1016/s1074-5521(02)00089-3. [DOI] [PubMed] [Google Scholar]
- 14.a) Brandänge S, Leijonmarck H. Tetrahedron Lett. 1992;33:3025–3028. [Google Scholar]; b) Hinterding K, Singhanat S, Oberer L. Tetrahedron Lett. 2001;42:8463–8465. [Google Scholar]
- 15.a) Kim SJ, Kang HY, Sherman DH. Synthesis. 2001;12:1790–1793. [Google Scholar]; b) Ranganathan A, Timoney M, Bycroft M, Cortes J, Thomas IP, Wilkinson B, Kellenberger L, Hanefeld U, Galloway IS, Staunton J, Leadlay PF. Chem Biol. 1999;6:731–741. doi: 10.1016/s1074-5521(00)80020-4. [DOI] [PubMed] [Google Scholar]
- 16.Beck BJ, Aldrich CC, Fecik RA, Reynolds KA, Sherman DH. J Am Chem Soc. 2003;125:12551–12557. doi: 10.1021/ja034841s. [DOI] [PubMed] [Google Scholar]
- 17.a) Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. Science. 1994;263:378–380. doi: 10.1126/science.8278811. [DOI] [PubMed] [Google Scholar]; b) Lau J, Fu H, Cane DE, Khosla C. Biochemistry. 1999;38:1643–1651. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]
- 18.a) Donadio S, Katz L. Gene. 1992;111:51–60. doi: 10.1016/0378-1119(92)90602-l. [DOI] [PubMed] [Google Scholar]; b) Bevitt DJ, Cortes J, Haydock SF, Leadlay PF. Eur J Biochem. 1992;204:39–49. doi: 10.1111/j.1432-1033.1992.tb16603.x. [DOI] [PubMed] [Google Scholar]; c) Bevitt DJ, Staunton J, Leadlay PF. Biochem Soc Trans. 1992;21:30S. doi: 10.1042/bst021030s. [DOI] [PubMed] [Google Scholar]; d) Aparicio JF, Molnar I, Schwecke T, Konig A, Haydock SF, Khaw LE, Staunton J, Leadlay PF. Gene. 1996;169:9–16. doi: 10.1016/0378-1119(95)00800-4. [DOI] [PubMed] [Google Scholar]
- 19.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 20.Caffrey P. ChemBioChem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 21.Kitz R, Wilson IB. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 22.Shiomi K, Omura S. In: Macrolide Antibiotics. Chemistry, Biology, and Practice. 2. Omura S, editor. Academic Press; San Diego, CA: 2002. pp. 1–56. [Google Scholar]
- 23.Sedgwick B, Morris C, French SJ. J C S Chem Commun. 1978:193–194. [Google Scholar]
- 24.Celmer WD. J Am Chem Soc. 1965;87:1801–1802. doi: 10.1021/ja01086a038. [DOI] [PubMed] [Google Scholar]; Celmer WD. Pure Appl Chem. 1971;28:413–453. doi: 10.1351/pac197128040413. [DOI] [PubMed] [Google Scholar]; Cane DE, Celmer WD, Westley JW. J Am Chem Soc. 1983;105:3594–3600. [Google Scholar]
- 25.McPherson M, Khosla C, Cane DE. J Am Chem Soc. 1998;120:3267–3268. [Google Scholar]; Yin Y, Gokhale R, Khosla C, Cane DE. Bioorg Med Chem Lett. 2001;11:1477–1479. doi: 10.1016/s0960-894x(00)00529-1. [DOI] [PubMed] [Google Scholar]
- 26.Benner S. In: Topics in Stereochemistry. Eliel EL, Wilen SH, editors. Vol. 19. Wiley and Sons; New York: 1989. pp. 127–207. [Google Scholar]
- 27.Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. J Am Chem Soc. 1998;120:2478–2479. [Google Scholar]
- 28.McDaniel R, Kao CM, Fu H, Hevezi P, Gustafsson C, Betlach M, Ashley G, Cane DE, Khosla C. J Am Chem Soc. 1997;119:4309–4310. [Google Scholar]; Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. J Am Chem Soc. 1997;119:11339–11340. [Google Scholar]; McDaniel R, Kao CM, Hwang SJ, Khosla C. Chem Biol. 1997;4:667–674. doi: 10.1016/s1074-5521(97)90222-2. [DOI] [PubMed] [Google Scholar]; McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Ashley G. Proc Natl Acad Sci USA. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes-Thomas ZA, Stark CB, Bohm IU, Staunton J, Leadlay PF. Angew Chem Int Ed Engl. 2003;42:4475–4478. doi: 10.1002/anie.200351375. [DOI] [PubMed] [Google Scholar]
- 30.Donadio S, McAlpine JB, Sheldon PJ, Jackson M, Katz L. Proc Natl Acad Sci U S A. 1993;90:7119–7123. doi: 10.1073/pnas.90.15.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimber MS, Martin F, Lu Y, Houston S, Vedadi M, Dharamsi A, Fiebig KM, Schmid M, Rock CO. J Biol Chem. 2004;279:52593–52602. doi: 10.1074/jbc.M408105200. [DOI] [PubMed] [Google Scholar]
- 32.a) Fisher M, Kroon JT, Martindale W, Stuitje AR, Slabas AR, Rafferty JB. Structure. 2000;8:339–347. doi: 10.1016/s0969-2126(00)00115-5. [DOI] [PubMed] [Google Scholar]; b) Price AC, Zhang YM, Rock CO, White SW. Biochemistry. 2001;40:12772–12781. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 34.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]