

Abstract
Glioblastoma is the most common astrocytic brain tumor in humans. Current therapies for this malignancy are mostly ineffective. Photodynamic therapy (PDT), an exciting treatment strategy based on activation of a photosensitizer, has not yet been extensively explored for treating glioblastoma. We used 5-aminolevulinic acid (5-ALA) as a photosensitizer for PDT to induce apoptosis in human malignant glioblastoma U87MG cells and to understand the underlying molecular mechanisms. Trypan blue dye exclusion test showed a decrease in cell viability after exposure to increasing doses of 5-ALA for 4 h followed by PDT with a broad spectrum blue light (400–550 nm) at a dose of 18 J/cm2 for 1 h and then incubation at 37°C for 4 h. Following 0.5 and 1 mM 5-ALA-based PDT (5-ALA-PDT), Wright staining and ApopTag assay showed occurrence of apoptosis morphologically and biochemically, respectively. After 5-ALA-PDT, down regulation of nuclear factor kappa B (NFκB) and baculovirus inhibitor-of-apoptosis repeat containing-3 (BIRC-3) protein indicated inhibition of survival signals. Besides, 5-ALA-PDT caused increase in Bax:Bcl-2 ratio and mitochondrial release of cytochrome c and apoptosis-inducing factor (AIF). Activation of calpain, caspase-9, and caspase-3 occurred in course of apoptosis. Calpain and caspase-3 activities cleaved α-spectrin at specific sites generating 145 kD spectrin break down product (SBDP) and 120 kD SBDP, respectively. The results suggested that 5-ALA-PDT induced apoptosis in U87MG cells by suppression of survival signals and activation of proteolytic pathways. Thus, 5-ALA-PDT can be an effective strategy for inducing apoptosis in glioblastoma.
Keywords: 5-ALA-PDT, Apoptosis, Calpain, Caspases, Glioblastoma
1. Introduction
The most common and deadliest brain tumor is glioblastoma that defies all current therapies. New treatment strategies are needed for controlling the growth of glioblastoma. 5-Aminolevulinic acid (5-ALA), a physiological compound and heme precursor, is widely used as a photosensitizer in treating various tumors [1]. Photodynamic therapy (PDT), which is an emerging treatment strategy for various cancers, uses a photosensitizer for irradiation with specific wavelength of visible light [2, 3]. PDT generates highly reactive oxygen species, particularly singlet oxygen (1O2), from the photosensitizer leading to cell death [4, 5]. The 5-ALA-based PDT (5-ALA-PDT) causes mitochondrial and nuclear DNA damage [6]. After 5-ALA-PDT, apoptosis occurs due to mitochondrial release of cytochrome c [5], endoplasmic reticulum stress, decrease in Bcl-2 and Bcl-xL, and activation of caspase-9 and caspase-3 [3]. Besides, 5-ALA-PDT induces mitochondrial release of apoptosis-inducing factor (AIF) for caspase-independent apoptosis [7]. Also, 5-ALA-PDT causes cytoskeletal changes in human adenocarcinoma WiDr and glioblastoma D54MG cells [8]. Hyperthermia enhances the 5-ALA-PDT cytotoxicity in glioblastoma spheroids [9]. However, the efficacy of 5-ALA-PDT has not yet been explored in human glioblastoma U87MG cells.
Here, we examined the ability of 5-ALA-PDT for induction of apoptosis in U87MG cells and explored the mechanisms involved in this process. We found that 5-ALA-PDT suppressed expression of survival factors such as NFκB, BIRC-3, and Bcl-2 and activated calpain and mitochondria-mediated caspase-dependent and caspase-independent pathways for apoptosis in U87MG cells.
2. Materials and methods
2.1. Treatment of U87MG cells
Human glioblastoma U87MG cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were grown at 37°C in 75-cm2 flasks containing 10 ml of 1xRPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (GIBCO-BRL, Grand Island, NY, USA) in a fully-humidified incubator containing 5% CO2 and 95% air. Following trypsinization using a trypsin/EDTA solution, cells were serially passaged to achieve about 80% confluency. Then, cells were incubated in a low serum condition (1% FBS) for 24 h prior to any treatment and maintained in this low serum condition during all treatments. It should be noted that a low concentration (0.5 – 1%) of serum is the most extensively used method to obtain the synchronized cells in the G1 phase of the cell cycle [10]. A freshly prepared 100 mM 5-ALA solution in 1xRPMI 1640 containing 1% FBS was used. Dose-response (0.1 – 2 mM) studies were conducted to determine the suitable doses of 5-ALA for induction of apoptosis [3]. Cells were treated with 0.5 and 1 mM 5-ALA for 4 h followed by PDT with a broad spectrum blue light (400–550 nm) at a dose of 18 J/cm2 for 1 h and incubation at 37°C for 4 h [3]. Control cells were not treated with 5-ALA but exposed to blue light.
2.2. Trypan blue dye exclusion test for cell viability
Following all treatments the viability of attached and detached cell populations was estimated by trypan blue dye exclusion test [11]. Trypan blue dye could not penetrate the intact plasma membrane of viable (white) cells but crossed the derelict plasma membrane of dead (blue) cells.
2.3. Detection of morphological and biochemical features of apoptosis
Cells from each treatment were washed with PBS, pH 7.4, and sedimented onto the microscopic slide, fixed in 95% ethanol before examination of morphology with Wright staining as we reported previously [12]. The morphology of the apoptotic cells included such characteristic features as chromatin condensation, cell-volume shrinkage, and membrane-bound apoptotic bodies. Four randomly selected fields were counted for at least 800 cells. The percentage of apoptotic cells was calculated from three separate experiments. For detection of DNA fragmentation as a biochemical marker of apoptosis, cells on the microscopic slides were subjected to the TdT-mediated dUTP Nick-End Labeling (TUNEL) using the ApopTagR Peroxidase assay kit (Intergen, Purchase, NY, USA). ApopTag positive cells were brown in a pale green background and considered as apoptotic cells. Experiments were conducted in triplicate and percentage of ApopTag-positive cells was determined by counting the brown cells from randomly selected fields under the light microscope [11, 12].
2.4. Western blotting for detection of specific proteins
We used sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting [11, 12] with necessary modifications. Mitochondrial and cytoplasmic fractions were prepared from cells by a standard procedure [13]. Briefly, cells (107) from each treatment were harvested, washed once with ice-cold PBS, and gently lysed for 1 min in 100 μl ice-cold lysis buffer [250 mM sucrose, 1 mM EDTA, 0.05% digitonin, 25 mM Tris-HCl, pH 6.8, 1 mM 1,4-dithio-DL-threitol (DTT), 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, 1 mM benzamidine, and 0.1 mM phenylmethylsulfonyl fluoride (PMSF)], and centrifuged at 12,000×g at 4°C for 3 min to obtain pellet (fraction containing mitochondria) and supernatant (cytosolic extract without mitochondria). Pellets and supernatants were analyzed by Western blotting. Also, total proteins were extracted following homogenization of control or treated cells in ice-cold (4°C) protein homogenization solution (50 mM Tris-HCl, pH 7.4, 320 mM sucrose, 0.1 mM PMSF, and 1 mM EDTA). CoomassieR Plus Protein Assay Reagent (Pierce, Rockford, IL, USA) was used for colorimetric determination of protein concentration at 595 nm. Protein samples were loaded onto the SDS–polyacrylamide gradient (4–20%) gels, resolved by SDS-PAGE [14], and electroblotted to membranes using Towbin transfer buffer [15]. Membranes were blocked for 2 h in Tris-buffered saline (TBS: 20 mM Tris-HCl, pH 7.6, 137 mM NaCl) containing 5% (w/v) non-fat powdered milk and washed three times in TBS containing 0.1% Tween 20 (TBST). Blots were incubated overnight with a monoclonal or polyclonal IgG primary antibody appropriately diluted in TBST containing 1% (w/v) non-fat powdered milk, washed in TBST, and incubated for 45 min with (1:2000) alkaline horseradish peroxidase (HRP)-conjugated anti-mouse IgG or anti-rabbit IgG secondary antibody. Specific protein bands on the blots were detected by alkaline HRP-catalyzed oxidation of luminol in presence of H2O2 using enhanced chemiluminescence system (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and autoradiography with exposure to X-OMAT XAR-2 film (Eastman Kodak, Rochester, NY, USA). Film exposure times were calibrated for analyzing the optical density (OD) of specific protein bands within the linear range [16]. Autoradiograms were scanned to image and digitize the bands using PhotoShop software (Adobe Systems, Seattle, WA, USA). The OD values of bands were estimated by using Quantity One software [12].
2.5. Statistical analysis
Data were expressed as mean + standard error of mean (SEM) of separate experiments (n≥3) and compared by one-way analysis of variance (ANOVA) followed by Fisher’s post hoc test. Changes in Bax and Bcl-2 expression (n≥3) were presented as Bax:Bcl-2 ratios, which were analyzed for statistical significance. Difference between two treatments was considered significant at p≤0.05.
3. Results
3.1. Decrease in cell viability after 5-ALA-PDT
Dose-dependently, 5-ALA-PDT decreased cell viability in U87MG cells (Fig. 1). Compared with control cells, 5-ALA-PDT induced morphological changes in dead cells (Fig. 1A). Residual cell viability was determined by trypan blue dye exclusion test (Fig. 1B). Compared with control cells, 0.5 and 1 mM 5-ALA-PDT significantly (p<0.0001) decreased cell viability (29% and 41%, respectively) in U87MG cells (Fig. 1B).
Fig. 1.
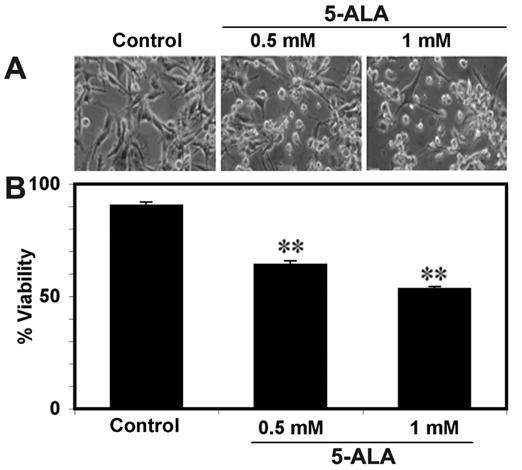
(A) Effect of 5-ALA-PDT on morphology of U87MG cells. (B) Trypan blue dye exclusion test for residual cell viability after 5-ALA-PDT. Bar graph shows residual cell viability. Significant difference from control was indicated by **p<0.001.
3.2. Induction of morphological features of apoptosis after 5-ALA-PDT
Following 5-ALA-PDT, Wright staining showed morphological features of apoptosis in U87MG cells (Fig. 2A). 5-ALA-PDT induced morphological features such as reduction in cell-volume, chromatin condensation, and/or presence of cell membrane blebbing in U87MG cells indicating occurrence of apoptotic cell death, as we reported previously in other studies [10, 11]. Compared with control cells, treatment with 0.5 and 1 mM 5-ALA-PDT caused 26% (p<0.0001) and 36% (p<0.0001) apoptosis, respectively.
Fig. 2.
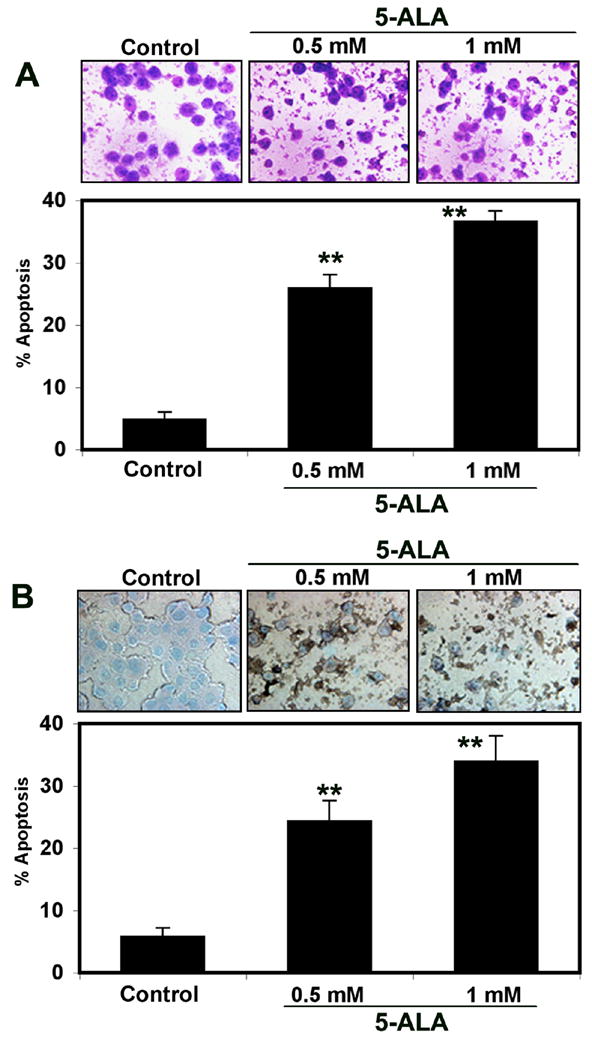
(A) Wright staining for determination of morphological features of apoptosis in U87MG cells following 5-ALA-PDT. Bar graph shows percent apoptosis based on Wright staining. (B) ApopTag assay for determination of morphological as well as biochemical features of apoptosis following 5-ALA-PDT. Bar graph shows percent apoptosis based on ApopTag assay. Significant difference from control was indicated by **p<0.001.
3.3. Induction of DNA fragmentation after 5-ALA-PDT
Induction of DNA fragmentation as a biochemical feature of apoptosis (brown color cell) was examined by ApopTag assay (Fig. 2B). Control cells showed no brown color, confirming almost absence of apoptosis. After 5-ALA-PDT, many cells were apoptotic as indicated by brown staining and also characteristic morphological changes such as cell shrinkage, pyknotic nuclei, cell blebbing, and formation of apoptotic bodies (Fig. 2B). Compared with control cells, treatment with 0.5 and 1 mM 5-ALA-PDT caused 24% (p<0.001) and 34% (p<0.0001) apoptosis, respectively (Fig. 2B). Thus, both Wright staining and ApopTag assay confirmed occurrence of similar amounts of apoptosis in U87MG cells after 5-ALA-PDT.
3.4. Suppression of cell-survival signals after 5-ALA-PDT
In many cancers, upregulation of nuclear factor kappa B (NFκB) exerts anti-apoptotic effect leading to tumor cell-survival, transformation, and resistance to radiation and drug therapies [17]. In contrast, down regulation of NFκB leads to apoptosis [18]. Besides, inhibitor-of-apoptosis proteins (IAPs) exert anti-apoptotic effects by a positive feedback system with NFκB [12]. Currently, there are eight IAPs that are also known as baculovirus inhibitor-of-apoptosis repeat containg (BIRC) proteins [19]. Among these, BIRC-2 and BIRC-3 were identified as possible oncogenes [20]. Therefore, we determined the levels of their expression following 5-ALA-PDT. Western blotting showed the changes in levels NFκB and BIRC-3 expression in U87MG cells after 5-ALA-PDT (Fig. 3A), but no change in BIRC-2 expression (data not shown). Level of β-actin expression remained almost unchanged (Fig. 3A). Both doses of 5-ALA-PDT resulted in a similar decreased expression of NFκB and BIRC-3 in U87MG cells (Fig. 3A).
Fig. 3.
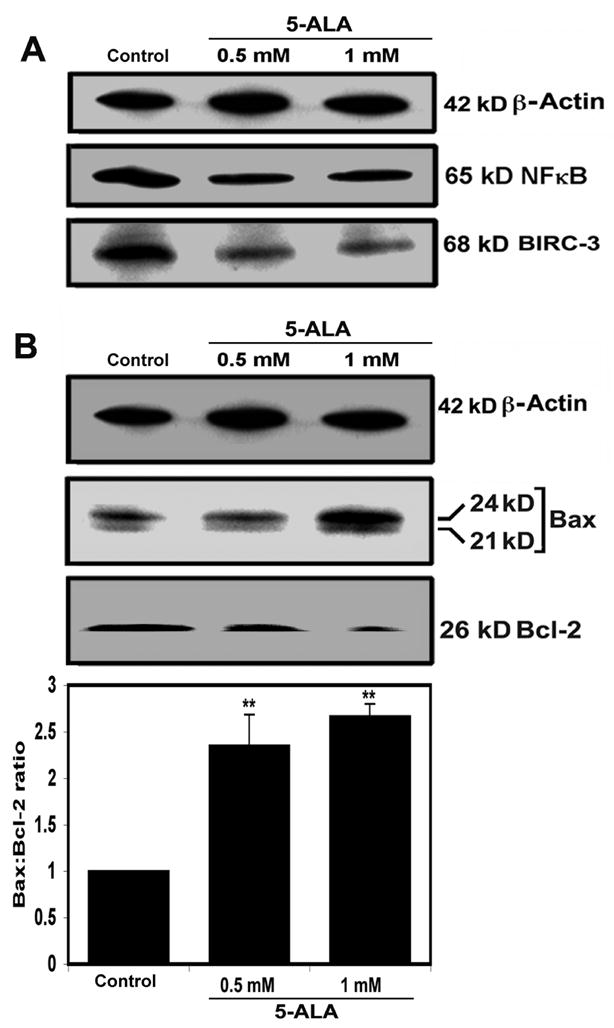
(A) Western blotting showed decreases in expression of 65 kD NFκB and 68 kD BIRC-3 in U87MG cells following 5-ALA-PDT. (B) Western blotting for examining changes in Bax and Bcl-2 expression and determining the Bax:Bcl-2 ratios. Significant difference from control was indicated by **p<0.001.
3.5. Increase in Bax:Bcl-2 ratio after 5-ALA-PDT
Alterations in expression of pro-apoptotic and anti-apoptotic proteins of the Bcl-2 family regulate the commitment of cells to apoptosis. Levels of expression of Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic) were altered so as to increase the Bax:Bcl-2 ratio significantly by 235% (p<0.01) and 267% (p<0.003) after treatment with 0.5 and 1 mM 5-ALA-PDT, respectively, compared with control cells (Fig. 3B). The increase in Bax:Bcl-2 ratio after 5-ALA-PDT indicated a commitment of U87MG cells to apoptosis.
3.6. Mitochondrial release of pro-apoptotic factors after 5-ALA-PDT
Mitochondrial release of several pro-apoptotic molecules such as cytochrome c and AIF can lead to apoptosis via caspase-dependent and caspase-independent pathways, respectively [21–23]. Mitochondrial release of cytochrome c is a well known precondition for formation of apoptosome and activation of caspases for apoptosis [24], as we also reported [12]. After 5-ALA-PDT, cytochrome c level was increased in the cytosolic fraction and decreased in the mitochondrial fraction of U87MG cells (Fig. 4A). Moreover, 5-ALA-PDT markedly increased the cytosolic level of AIF (Fig. 4A), indicating induction of caspase-independent pathway of apoptosis as well. Increased release of both cytochrome c and AIF after 5-ALA-PDT indicated mitochondrial involvement in apoptosis in U87MG cells.
Fig. 4.
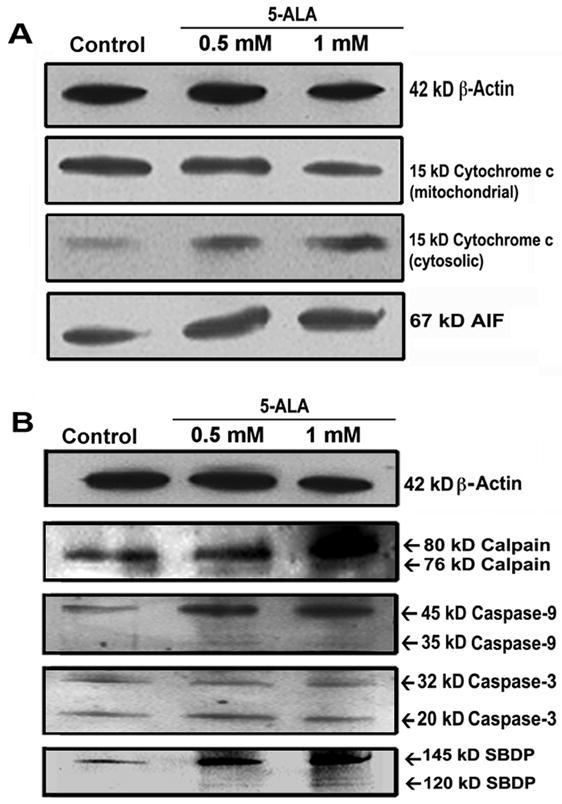
(A) Western blotting to examine mitochondrial release of cytochrome c and AIF in U87MG cells following 5-ALA-PDT. (B) Western blotting showed formation of 76 kD active calpain, 35 kD active caspase-9, and 20 kD active caspase-3, and activities of calpain and caspase-3 in the generation of specific SBDPs.
3.7. Increased activation of calpain, caspase-9, and caspase-3 after 5-ALA-PDT
Calpain is a neutral Ca2+-dependent cysteine protease that plays an important role in apoptosis [11, 12]. Co-operation between calpain and caspase-3 has been demonstrated in apoptotic cells [25]. After 5-ALA-PDT, activation of calpain, caspase-9, and caspase-3 was increased (Fig. 4B). Activation of calpain was detected in the generation of 76 kD active calpain (Fig. 4B). We also observed activation of caspase-9 in the generation of 35 kD active caspase-9 (Fig. 4B) that, in turn, activated caspase-3 with the formation of 20 kD active caspase-3 (Fig. 4B). Activation of caspase-3 is known to cause cleavage of specific substrates at the late phase of apoptosis [26].
3.8. Activation of calpain and caspase-3 cleaved α-spectrin after 5-ALA-PDT
Activation of calpain and caspase-3 can cause cleavage of 270 kD α-spectrin at specific sites to generate 145 kD spectrin breakdown product (SBDP) [27] and 120 kD SBDP, respectively, in apoptotic cells [28]. After 5-ALA-PDT, Western blotting used to assess the calpain and caspase-3 activities in U87MG cells (Fig. 4B). Compared with control cells, 5-ALA-PDT increased the formation of 145 kD SBDP and 120 kD SBDP (Fig. 4B). Our current observation of activation of these proteolytic pathways is in line with our previous reports [11, 12].
4. Discussion
Current investigation showed that 5-ALA-PDT induced apoptosis in human malignant glioblastoma U87MG cells. After 5-ALA-PDT, decrease in cell viability (Fig. 1) occurred due to induction of cell death in U87MG cells (Fig. 2) with characteristic morphological and biochemical features of apoptosis [29, 30], as we also reported previously [12, 31]. Earlier studies showed that 5-ALA-PDT could induce apoptosis in human leukemia Reh cells [7] and K562 cells [32]. However, this is the first report showing that 5-ATAL-PDT is capable of suppressing survival factors and activating caplain and michotochodria-mediated caspase-dependent and caspase-independent pathways for apoptosis in U87MG cells.
After 5-ALA-PDT, we examined the changes in expression of cell survival factors. Our results showed for the first time that 5-ALA-PDT caused down regulation of both NFκB and BIRC-3 in U87MG cells (Fig. 3A) for suppressing survival signals. An elevation in Bax:Bcl-2 ratio (Fig. 3B) was associated with mitochondrial release of cytochrome c (Fig. 4A), suggesting that mitochondria played an important role in apoptosis in U87MG cells following 5-ALA-PDT. Also, mitochondrial release of AIF (Fig. 4A) indicated involvement of caspase-independent pathway of apoptosis as well. So, mitochondria played an essential role in apoptosis in U87MG cells after 5-ALA-PDT.
It has repeatedly been reported that calpain has a significant role in cell death [11, 12, 31, 33]. Calpain can cause Bax translocation to mitochondria to trigger release of cytochrome c for caspase-dependent apoptosis [34] and also AIF for caspase-independent apoptosis [35, 36]. Activation of caspase-9 and caspase-3 in U87MG cells after 5-ALA-PDT (Fig. 4B) suggested an important role of caspase-dependent pathway in apoptosis. A network between calpain and caspase proteolytic systems can work together during apoptosis [25]. Indeed, 5-ALA-PDT activated both calpain and caspase cascades for induction of apoptosis in U87MG cells (Fig. 4B).
Degradation α-spectrin at specific sites can indicate induction of calpain and caspase-3 activities in apoptosis. For example, generation of 145 kD SBDP is specific for calpain activity [27] and 120 kD SBDP is specific for caspase-3 activity [28]. We measured the calpain and caspase-3 activities in the generation of 145 kD SBDP and 120 kD SBDP, respectively (Fig. 4B). Increases in generation of 145 kD SBDP and 120 kD SBDP after 5-ALA-PDT confirmed increases in proteolytic activities of calpain and caspase-3 for apoptosis in U87MG cells. This observation of α-spectrin cleavage is consistent with an earlier study where we demonstrated induction of calpain and caspase-3 activities for α-spectrin cleavage during apoptosis in glioblastoma cells [12].
In conclusion, 5-ALA-PDT induced apoptosis in U87MG cells via suppression of cell survival factors (NFκB, BIRC-3, and Bcl-2) and induction of calpain and mitochondria-mediated caspase-dependent and caspase-independent pathways of apoptosis. Further exploration of 5-ALA-PDT as a treatment strategy in pre-clinical models of glioblastoma may pave the path to combat this malignancy in humans in the future.
Acknowledgments
This investigation was supported in part by the R01 grants (CA-91460 and NS-57811) from the National Institutes of Health (Bethesda, MD, USA) and also by the Spinal Cord Injury Research Fund (SCIRF-0803) from the State of South Carolina.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peng Q, Warloe T, Berg K, Moan J, Kongshaug M, Giercksky KE, Nesland JM. 5-Aminolevulinic acid-based photodynamic therapy: Clinical research and future challenges. Cancer. 1997;79:2282–2308. doi: 10.1002/(sici)1097-0142(19970615)79:12<2282::aid-cncr2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Henderson BW, Dougherty TJ. How does photodynamic therapy work? Photochem Photobiol. 1992;55:145–157. doi: 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- 3.Grebenova D, Kuzelova K, Smetana K, Pluskalova M, Cajthamlova H, Marinov I, Fuchs O, Soucek J, Jarolim P, Hrkal Z. Mitochondrial and endoplasmic reticulum stress-induced apoptotic pathways are activated by 5-aminolevulinic acid-based photodynamic therapy in HL-60 leukemia cells. J Photochem Photobiol B. 2003;69:71–85. doi: 10.1016/s1011-1344(02)00410-4. [DOI] [PubMed] [Google Scholar]
- 4.Wild PJ, Krieg RC, Seidl J, Stoehr R, Reher K, Hofmann C, Louhelainen J, Rosenthal A, Hartmann A, Pilarsky C, Bosserhoff AK, Knuechel R. RNA expression profiling of normal and tumor cells following photodynamic therapy with 5-aminolevulinic acid-induced protoporphyrin IX in vitro. Mol Cancer Ther. 2005;4:516–528. doi: 10.1158/1535-7163.MCT-04-0141. [DOI] [PubMed] [Google Scholar]
- 5.Kriska T, Korytowski W, Girotti AW. Role of mitochondrial cardiolipin peroxidation in apoptotic photokilling of 5-aminolevulinate-treated tumor cells. Arch Biochem Biophys. 2005;433:435–446. doi: 10.1016/j.abb.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 6.Onuki J, Chen Y, Teixeira PC, Schumacher RI, Medeiros MH, Van Houten B, Di Mascio P. Mitochondrial and nuclear DNA damage induced by 5-aminolevulinic acid. Arch Biochem Biophys. 2004;432:178–187. doi: 10.1016/j.abb.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 7.Furre IE, Shahzidi S, Luksiene Z, Moller MT, Borgen E, Morgan J, Tkacz-Stachowska K, Nesland JM, Peng Q. Targeting PBR by hexaminolevulinate-mediated photodynamic therapy induces apoptosis through translocation of apoptosis-inducing factor in human leukemia cells. Cancer Res. 2005;65:11051–11060. doi: 10.1158/0008-5472.CAN-05-0510. [DOI] [PubMed] [Google Scholar]
- 8.Uzdensky A, Kolpakova E, Juzeniene A, Juzenas P, Moan J. The effect of sublethal ALA-PDT on the cytoskeleton and adhesion of cultured human cancer cells. Biochim Biophys Acta. 2005;1722:43–50. doi: 10.1016/j.bbagen.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 9.Hirschberg H, Sun CH, Tromberg BJ, Yeh AT, Madsen SJ. Enhanced cytotoxic effects of 5-aminolevulinic acid-mediated photodynamic therapy by concurrent hyperthermia in glioma spheroids. J Neurooncol. 2004;70:289–299. doi: 10.1007/s11060-004-9161-7. [DOI] [PubMed] [Google Scholar]
- 10.Campbell L, Gumbleton M. Basic aspects of cell growth and cell cycle in culture. In: Lehr CM, editor. Cell Culture Models of Biological Barriers. Taylor and Francis; New York: 2002. pp. 3–19. [Google Scholar]
- 11.Ray SK, Karmakar S, Nowak MW, Banik NL. Inhibition of calpain and caspase-3 prevented apoptosis and preserved electrophysiological properties of voltage-gated and ligand-gated ion channels in rat primary cortical neurons exposed to glutamate. Neuroscience. 2006;139:577–595. doi: 10.1016/j.neuroscience.2005.12.057. [DOI] [PubMed] [Google Scholar]
- 12.Karmakar S, Weinberg MS, Banik NL, Patel SJ, Ray SK. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience. 2006;141:1265–1280. doi: 10.1016/j.neuroscience.2006.04.075. [DOI] [PubMed] [Google Scholar]
- 13.Pique M, Barragan M, Dalmau M, Bellosillo B, Pons G, Gil J. Aspirin induces apoptosis through mitochondrial cytochrome c release. FEBS Lett. 2000;480:193–196. doi: 10.1016/s0014-5793(00)01922-0. [DOI] [PubMed] [Google Scholar]
- 14.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 15.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schumacher PA, Siman RG, Fehlings MG. Pretreatment with calpain inhibitor CEP-4143 inhibits calpain I activation and cytoskeletal degradation, improves neurological function, and enhances axonal survival after traumatic spinal cord injury. J Neurochem. 2000;74:1646–1655. doi: 10.1046/j.1471-4159.2000.0741646.x. [DOI] [PubMed] [Google Scholar]
- 17.Beg AA, Baltimore D. An essential role for NFκB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 18.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. NFκB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 19.MacKenzie A, LaCasse E. Inhibition of IAP’s protection by Diablo/Smac: new therapeutic opportunities? Cell Death Differ. 2000;7:866–867. doi: 10.1038/sj.cdd.4400747. [DOI] [PubMed] [Google Scholar]
- 20.Dai Z, Zhu WG, Morrison CD, Brena RM, Smiraglia DJ, Raval A, Wu YZ, Rush LJ, Ross P, Molina JR, Otterson GA, Plass C. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum Mol Genet. 2003;12:791–801. doi: 10.1093/hmg/ddg083. [DOI] [PubMed] [Google Scholar]
- 21.Earnshaw WC. Apoptosis: A cellular poison cupboard. Nature. 1999;397:387–389. doi: 10.1038/17015. [DOI] [PubMed] [Google Scholar]
- 22.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 23.Jaattela M. Multiple cell death pathways as regulators of tumor initiation and progression. Oncogene. 2004;23:2746–2756. doi: 10.1038/sj.onc.1207513. [DOI] [PubMed] [Google Scholar]
- 24.Kim R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer. 2005;103:1551–1560. doi: 10.1002/cncr.20947. [DOI] [PubMed] [Google Scholar]
- 25.Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem. 2003;278:14162–14167. doi: 10.1074/jbc.M212255200. [DOI] [PubMed] [Google Scholar]
- 26.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 27.Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK. Non-erythroid α-spectrin breakdown by calpain and interleukin 1β-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. Simultaneous degradation of αII- and βII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem. 1998;273:22490–22497. doi: 10.1074/jbc.273.35.22490. [DOI] [PubMed] [Google Scholar]
- 29.Wyllie AH, Beattie GJ, Hargreaves AD. Chromatin changes in apoptosis. Histochem J. 1981;13:681–692. doi: 10.1007/BF01002719. [DOI] [PubMed] [Google Scholar]
- 30.Shi YF, Szalay MG, Paskar L, Sahai BM, Boyer M, Singh B, Green DR. Activation-induced cell death in T cell hybridomas is due to apoptosis. Morphologic aspects and DNA fragmentation. J Immunol. 1990;144:3326–3333. [PubMed] [Google Scholar]
- 31.Sur P, Sribnick EA, Patel SJ, Ray SK, Banik NL. Dexamethasone decreases temozolomide-induced apoptosis in human gliobastoma T98G cells. Glia. 2005;50:160–167. doi: 10.1002/glia.20168. [DOI] [PubMed] [Google Scholar]
- 32.Kuzelova K, Grebenova D, Pluskalova M, Marinov I, Hrkal Z. Early apoptotic features of K562 cell death induced by 5-aminolaevulinic acid-based photodynamic therapy. J Photochem Photobiol B. 2004;73:67–78. doi: 10.1016/j.jphotobiol.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Karmakar S, Banik NL, Patel SJ, Ray SK. Curcumin activated both receptor-mediated and mitochondria-mediated proteolytic pathways for apoptosis in human glioblastoma T98G cells. Neurosci Lett. 2006;407:53–58. doi: 10.1016/j.neulet.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 34.Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 36.Sanges D, Marigo V. Cross-talk between two apoptotic pathways activated by endoplasmic reticulum stress: differential contribution of caspase-12 and AIF. Apoptosis. 2006;11:1629–1641. doi: 10.1007/s10495-006-9006-2. [DOI] [PubMed] [Google Scholar]