

Abstract
We are investigating the use of recombinant streptavidin (rSAv) as a carrier molecule for the short-lived α-particle emitting radionuclides 213Bi (t1/2 = 45.6 min) and 211At (t1/2 = 7.21 h) in cancer therapy. To utilize rSAv as a carrier, it must be modified in a manner that permits rapid chelation or bonding with these short-lived radionuclides, and also modified in a manner that diminishes its natural propensity for localization in kidney. Modification for labeling with 213Bi was accomplished by conjugation of rSAv with the DTPA derivative p-isothiocyanato-benzyl-CHX-A″ (CHX-A″), 3a. Modification for direct labeling with 211At was accomplished by conjugation of rSAv with an isothiocyanatophenyl derivative of a nido-carborane (nCB), 3b, or an isothiocyanatophenyl-dPEG™/decaborate(2-) derivative, 3c. After conjugation of the chelating or bonding moiety, rSAv was further modified by reaction with an excess (50–100 equivalents) of succinic anhydride. Succinylation of the lysine amines has previously been shown to greatly diminish kidney localization. rSAv modified by conjugation with 3a and succinylated radiolabeled rapidly with 213Bi (< 5 min), providing a 72% isolated yield. 211At labeling of modified rSAv was accomplished in aqueous solution using chloramine-T as the oxidant. Astatination of rSAv conjugated with 3b and succinylated occurred very rapidly (<1 min), providing a 50% isolated radiochemical yield. Astatination of rSAv conjugated with 3c and succinylated was also very rapid (<1 min) providing 66–71% isolated radiochemical yields. Astatination of succinylated rSAv, 2a, which did not have conjugated borane cage moieties, resulted in much lower radiolabeling yield (18%). The 213Bi- or 211At-labeled modified rSAv preparations were mixed with the corresponding 125I-labeled rSAv, and dual-label in vivo distributions were obtained in athymic mice. The in vivo data show that 213Bi-labeled succinylated rSAv [213Bi]6a has tissue concentrations similar to 125I-labeled modified rSAv [125I]6b, suggesting that 213Bi is quite stable towards release from the chelate in vivo. In vivo data also indicate that the 211At-labeled rSAv conjugated with 3b or 3c and succinylated are stable to in vivo deastatination, whereas succinylated rSAv lacking a boron cage moiety is subject to some deastatination. The modified rSAv conjugated with nido-carborane derivative 3b has a higher retention in many tissues than rSAv without the carborane conjugated. Interestingly, the rSAv conjugated with 3c, which also contains a m-dPEG12™ moiety, has significantly decreased concentrations in blood and other tissues when compared with direct labeled rSAv, suggesting that it may be a good candidate for further study. In conclusion, rSAv that has been modified with CHX-A″ and succinylated (i.e. 5a) may be useful as a carrier of 213Bi. The encouraging results obtained with the PEGylated decaborate(2-) derivative 3c and succinylated (i.e. 5c) suggests that its further study as a carrier of 211At in pretargeting protocols is warranted.
INTRODUCTION
α-Emitting radionuclides are of high interest for Targeted Radionuclide Therapy (TRT1) of disseminated (micro)metastatic cancer (1–4). The short path length of the α-particle results in deposition of a large amount of energy in a few cell diameters, providing very effective cell killing for single cells or small clumps of cells without affecting the surrounding normal tissues. Two of the most promising α-particle emitting radionuclides for use in TRT are 213Bi and 211At (5). Unfortunately, both of these radionuclides have relatively short half-lives (45.6 min and 7.21 h, respectively) making it imperative that rapid and efficient radiolabeling chemistry be available or developed. The short half-lives of these radionuclides also makes it imperative that the cancer-targeting carrier molecule be able to localize rapidly to the cancer cells in the body. Further, the high cytotoxicity of these radionuclides requires that they remain attached to the carrier molecule during the period they are present in the body.
The cancer-targeting agent is a critical component in TRT. While there are a large number of biological agents to choose from, making the choice of an appropriate cancer-targeting agent to carry highly cytotoxic, short half-lived, α-emitting radionuclides is not easy. For systemic delivery, intact monoclonal antibodies (mAbs) and their F(ab′)2 fragments are questionable for carrying 213Bi or 211At due to their inherent properties (6–8) and unfavorable tumor-targeting pharmacokinetics. Smaller mAb fragments, engineered proteins and peptides are also unfavorable due to their propensity to localize in kidney, although a number of studies to decrease localization have been published (9–13). As an alternative, we have been investigating the use of two different 2-step approachs2 for targeting 213Bi and 211At to cancer cells in vivo termed “pretargeting”. In one form of the 2-step pretargeting approach (14, 15), monoclonal antibodies (mAbs) are conjugated with a biotin derivative and that conjugate is injected to target the cancer cells. At some time later (i.e. 24 h post injection of the mAb-biotin), a radiolabeled recombinant streptavidin (rSAv) is injected to bind with the tumor cell surface-associated mAb-biotin.
Proteins, such as rSAv, can not be labeled with 213Bi or 211At without conjugation of a binding or bonding moiety (BM) first. Furthermore, proteins such as rSAv, which have molecular weights of ~50 kDa, can localized rapidly to the kidney (16), posing the same serious limitations of mAb fragments and peptides. Fortunately, prior studies have reported several methods to decrease the kidney localization of rSAv without significantly affecting the biotin-binding affinity (17–19). Of these options, succinylation of the amines on rSAv is perhaps the most readily achieved, and provides low kidney concentrations. Thus, use of rSAv as a carrier of radionuclides requires two modification steps; the first is to conjugate a binding or bonding moiety and the second is to react all (or most) of the remaining surface lysine amines with a molecule that neutralizes the lysine’s positive charge, such as a succinic anhydride moiety.
A cyclohexyl-DTPA derivative, isothiocyanato-benzyl-CHX-A″ was chosen for conjugation with rSAv for 213Bi labeling. This choice was based on our (20, 21), and other investigators (22–26), prior studies that have demonstrated proteins conjugated with the CHX-A″ moiety are rapidly labeled with 213Bi in high radiochemical yields. The choice for modification of rSAv to attain efficient 211At labeling was not as readily apparent. Unlike radioiodination, direct astatination of proteins can result in an unstable label when injected in vivo (27, 28). The application of p- or m-astatobenzoate NHS esters often used in protein astatinations (29, 30) was considered for labeling rSAv. However, the use of such astatinated benzoate derivatives appeared problematic due to the requirement of modification of rSAv to eliminate kidney localization following the radiolabeling step. Additionally, it was desirable to obtain a simpler and perhaps higher yielding approach for astatination of rSAv. In other studies we have shown that astatination of a mAb Fab’ can be accomplished rapidly in high yield using borane cage moieties (31). Because of the differences noted previously for nido-carborane conjugates and decaborate(2-) conjugates, we felt it was important to evaluate both options in this investigation. The protein-reactive α-emitting radionuclide’s binding (chelating) or bonding moieties used to modify rSAv are shown in Figure 1.
Figure 1.

Structures of protein-reactive (phenylisothiocyanate) compounds used to modify recombinant streptavidin for chelation of 213Bi (3a) or bonding with 211At (3b, 3c). In the structures of the borane cage moieties, the open circles represent boron or B-H atoms and solid (black) circles represent carbon atoms.
The primary goals of this investigation were to determine if modifications made to rSAv for 213Bi- and 211At-labeling altered its in vivo properties and if the rSAv conjugates prepared for study were stable to in vivo release of the radionuclides. A secondary goal was to evaluate the efficiency of radiolabeling with these expensive radionuclides, as that may impact on whether these radionuclides can be successfully applied to cancer therapy. The results obtained in the investigation are reported herein.
EXPERIMENTAL PROCEDURES
General
All chemicals purchased from commercial sources were analytical grade or better and were used without further purification. Succinic anhydride and most other chemicals were obtained from Aldrich (Sigma-Aldrich Corp., Milwaukee, WI). The m-dPEG12™ amine was obtained from Quanta BioDesign, Ltd (Powell, OH). Chloramine-T (ChT) and phosphate buffered saline (PBS) were obtained from Sigma (St. Louis, MO). Avidin was obtained from Pierce (Rockford, IL). Bismuth metal used as the target for preparation of 211At was obtained from Alpha Aesar (Ward Hill, MA) as Puratronic 99.999%. Solvents for HPLC analysis were obtained as HPLC grade and were filtered (0.2 μm) prior to use. Recombinant streptavidin (rSAv) was obtained from Boehringer Mannheim (Indianapolis, IN). Silica gel chromatography was conducted with 70–230 mesh 60 Å silica gel (Aldrich Chemical Co.). Sephadex G-25 desalting columns (PD-10 and NAP-10) were obtained from Amersham Biosciences (Piscataway, NJ). Centricon-30 centrifugation concentrators were obtained from Amicon (Beverly, MA).
Radioactivity
All radioactive materials were handled according to approved protocols at the University of Washington. Na[125I]I was purchased from Perkin-Elmer Life and Analytical Services as high concentration/high specific activity radioiodide in 0.1 N NaOH. [225Ac]Actinium nitrate was purchased from the Department of Energy (Oak Ridge, TN) on a column (actinide resin, 100–200 mesh, EiChrom Technology Inc., Darien, IL) ready for elution of 213Bi in the radiolabeling experiments. 211At was produced by an α-particle irradiation (28 MeV) of natural bismuth, 209Bi(α, 2n)211At, on a Scandatronix MC-50 cyclotron. The 211At activity was distilled from the bismuth target at 650–700°C into a cooled trap as previously described (32). The dry distillation of 211At was conducted in a charcoal filtered glove box (Innovative Technologies, radioisotope glove box). The distilled 211At was trapped in a cooled (−78°C) vial or peek tubing (1 mm ID × 35 cm length) (33) and was rinsed from the vial with CHCl3 or 0.05 N NaOH for use in the labeling experiments.
Radiohalogenation reactions were conducted in a charcoal-filtered Plexiglas enclosure (Biodex Medical Systems Inc., Shirley, NY) housed within a radiochemical fume hood. The radiohalogenation reactions (125I or 211At) were conducted in vials capped with Teflon-coated septa. Reaction vials were vented through a 10 mL charcoal-filled syringe. Additions of reagents and radionuclides to, or removal of materials from, radiohalogenation vials were conducted by passing a syringe needle through the septa.
Measurement of 213Bi, 211At and 125I was accomplished on a Capintec CRC-15R. The calibration numbers provided by Capintec Technical Services were used for measurement of 213Bi and 211At on these instruments. Tissue samples containing these radionuclides were counted in a Wallac Wizard™ gamma counter (PerkinElmer Life and Analytical Sciences, Wellesley, MA). The tissue samples had either 213Bi or 211At, along with 125I. Total radioactivity counts (cpm) for the samples were obtained by counting in all channels of the detector. To determine the counts from each radionuclide in tissues, the tissue samples were allowed to decay for 1–2 day resulting in <0.01% 213Bi being present (<0.01%) or 3–5 days resulting in <0.1% 211At being present. Recounting tissue samples after the decay period provide the 125I counts. The total counts and 125I counts for tissues were transferred to an Excel (Microsoft Corp., Redmond, WA) spreadsheet. The counts for 213Bi or 211At radioactivity were calculated by subtracting the 125I counts from the total cpm for each tissue sample, and the 213Bi and 211At counts were corrected for decay of each sample during the counting process.
Spectral Analyses
1H and 11B NMR spectra for compounds were obtained on either a Bruker AV 300 (300 MHz) or Bruker AV 500 (500 MHz) instrument. 1H chemical shifts are expressed as ppm using tetramethylsilane as an internal standard (δ = 0.0 ppm). 11B chemical shifts were referenced to BF3OEt2 as an external standard. High-resolution mass spectra (HRMS) and low-resolution mass spectra (LRMS) were obtained on a Bruker APEX III 47e Fourier Transform Mass Spectrometer using electrospray ionization. For analysis, the samples were dissolved in 50/50 MeOH/H2O and were introduced by an integral syringe infusion pump (Cole Parmer Series 74900).
Analytical Chromatography
All synthetic reactions were monitored by HPLC. Samples were assessed on a system that contained a Hewlett-Packard quaternary 1050 gradient pump, a variable wavelength UV detector (254 nm), and a Varex ELSD MKIII evaporative light-scattering detector. Analyses of HPLC data were conducted using Hewlett-Packard HPLC ChemStation software. Reversed-phase HPLC chromatography was carried out on an Alltech Altima C-18 column (5 μm, 250 × 4.5 mm) using a gradient solvent system at a flow rate of 1 mL/min. A gradient of MeOH and H2O/0.1% HOAc was used. Starting with 40% MeOH, the initial solvent mixture was held for 2 min, then the gradient was increased to 100% MeOH over the next 10 min, then held at 100% MeOH for 8 min. Retention times (tR) under these conditions for new compounds are provided with the compound experimental.
HPLC separations of the (non-radioactive) modified rSAv were conducted on a Hewlett-Packard isocratic system consisting of a Model 1050 pumping system and a Model 1050 Multiple Wavelength Detector (280 nm). The proteins were evaluated on a Protein Pak 300 SW glass size exclusion column (10 μm, 8 mm × 30 cm, Waters Corporation, Milford, MA) run at 1.0 mL/min eluting with 50 mM sodium phosphate buffer (pH 6.8) which contained 300 mM NaCl, 1 mM EDTA, and 1 mM sodium azide. Analyses of the HPLC data were conducted on Hewlett-Packard HPLC ChemStation software. Retention times (tR) for the succinylated r-SAv was 9.8 min and boron cage-modified rSAv were ~10.5 min using these conditions.
Radiolabeled rSAv
Size-exclusion chromatography was also used to evaluate radiolabeled modified rSAv. HPLC separations of radioiodinated proteins were conducted on a system consisting of a Model 1050 pumping system, a Waters Lambda-Max 481 UV detector, and a Beckman model 170 radioisotope detector. The detectors were connected to a computer through a Hewlett-Packard Model 35900 interface, and data from the chromatograms was obtained on a computer running Hewlett-Packard ChemStation Software. The same size-exclusion column and elution conditions were used for the radiolabeled proteins as unlabeled proteins.
Isoelectric Focusing
IEF were obtained on a Novex PowerEase 500 instrument with the XCell II chamber using Invitrogen (Novex) precast gels, pH 3–10 (1.0 mm, 12 well) running under the standard IEF Program. The protein was stained with GelCode Blue Stain (Pierce). IEF standards were from Serva Electrophoresis GmbH: (pI) 10.7 - cytochrome C; 9.5 - ribonuclease A; 8.3, 8.0, 7.8 - lectin; 7.4, 6.9 - myoglobin; 6.0 - carbonic anhydrase; 5.3, 5.2 - β-lactoglobulin; 4.5 - trypsin inhibitor; 4.2 - glucose oxidase; 3.5 – amyloglucosidase.
Syntheses
The following compounds were prepared as previously described: isothiocyanatobenzyl-CHX-A″ DTPA, 3a (34)3; 3-(3′-(nido-carboranyl)propionyl)amino-1-isothiocyanatobenzene, 3b (35); 5-N-(tert-butyloxycarbonyl)-aminoisophthalate bis-tetrafluorophenyl ester, 7 (36); [Et3NH]B10H9-CO-trioxatridecanediamine, 10 (31).
5-(N-tert-Butyloxycarbonylamido)-3-(m-dPEG12™-amido)-isophthalamic acid 2,3,5,6-tetra-fluorophenyl ester, 9
A solution of m-dPEG12™-amine, 8, (100 mg, 0.18 mmol) in anhydrous DMF (2 mL) was added dropwise over 30 min to a solution of 7 (207 mg, 0.36 mmol) and Et3N (30 μL, 0.36 mmol) in anhydrous DMF (5 mL) at room temperature. After the addition was complete the reaction mixture was stirred at room temperature for an additional 10 min, then the volatile materials were removed under vacuum. The crude residue was purified by silica gel chromatography eluting with 10% MeOH/EtOAc to give 138 mg (80%) of 9 as a colorless oil. 1H NMR (CD3OD, 300 MHz): δ 1.59 (s, 9H), 3.35 (s, 3H), 3.51–3.71 (m, 48H), 7.46–7.58 (m, 1H), 8.23 (t, J = 2.0 Hz, 1H), 8.28 (t, J = 1.6 Hz, 1H), 8.51 (t, J = 1.9 Hz, 1H), 8.72 (t, J = 5.2 Hz, 1H). HRMS (ES+) C44H66N2O17F4Na (M+Na)+; calcd 993.4195; found 993.4193. HPLC: tR = 14.2 min.
5-(N-tert-Butyloxycarbonylamido)-3-(m-dPEG12™-amido)-1-((13′-nonahydro-closo-decaborate(2-))-4,7,10-trioxatridecanediamine)-aminoisophthalate, 11
A solution containing [Et3N]10 (53 mg, 0.113 mmol), 9 (110 mg, 113 μmol), Et3N (32 μL, 0.227 mmol) and anhydrous DMF (5 mL) was stirred at room temperature for 2 h. EtOAc (20 mL) was added and the mixture was stirred for an additional 10 min. The liquid was decanted from the residue. The crude product was redissolved in MeOH/water (1/1) and purified by flash chromatography (Biotage C18 FLASH 25+M column) eluting with a gradient mixture composed of MeOH and 0.1% aqueous HOAc (pH 3.25). The starting gradient mixture of 25% MeOH was held for 2 min, increased to 100% MeOH over the next 10 min, then held at 100% MeOH for 8 min. Isolation yielded 112 mg (78%) of 11 as light-brown tacky solid. 1H NMR (DMSO-d6, 500 MHz): δ 1.17 (t, J = 7.3 Hz, 9H), 1.46 (s, 9H), 1.71–1.81 (m, 4H), 3.04–3.09 (m, 6H), 3.21 (s, 3H), 3.29 (dd, J = 6.6, 12.4 Hz, 2H), 3.39–3.72 (m, 71H), 7.72 (bs, 1H), 7.82 (s, 1H), 7.98 (d, J = 8.0 Hz, 2H), 8.43 (t, J = 5.5 Hz, 1H), 8.47 (t, J = 5.5 Hz, 1H), 9.28 (bs, 1H), 9.58 (s, 1H). 11B NMR (DMSO-d6, 96.29 MHz): δ −5.03 (2B), −15.90 (2B), −24.66 (6B). LRMS (MALDI−) C49H95B10N4O20K (M+K)−; calcd 1208.7; found 1208.8. HPLC: tR = 11.0 min.
5-(Isothiocyanato)-3-(m-dPEG12™-amido)-1-((13′-nonahydro-closo-decaborate(2-))-4,7,10-trioxatridecanediamine)-aminoisophthalate, 3c
A quantity of 11 (100 mg, 0.079 mmol) was dissolved in neat TFA (1 mL), and that solution was stirred at room temperature for 10 min. The excess TFA was removed under a stream of Argon, then the residue was dissolved in a solution of MeOH (3 mL) and Et3N (0.2 mL). After solvents were evaporated on a rotoevaporator, the deprotected compound was dried under vacuum for 2 h, then dissolved in anhydrous DMF (1 mL).
To the DMF solution was added thiocarbonyldiimidazole (28 mg, 0.157 mmol). That solution was stirred at room temperature for 1 h. Et2O was added and the solution was stirred for an additional 30 min. The solvents were decanted and the residue was washed with EtOAc (3 × 15 mL) to afford 91 mg (95%) of 3c. 1H NMR (DMSO-d6, 500 MHz): δ 1.16 (t, J = 7.2 Hz, 9H), 1.71–1.77 (m, 4H), 3.06 (q, J = 7.3 Hz, 6H), 3.16 (s, 1H), 3.21 (s, 2H), 3.30 (dd, J = 6.6, 12.3 Hz, 2H), 3.39–3.74 (m, 71H), 7.40 (s, 2H), 7.93 (d, J = 8.1 Hz, 2H), 8.24 (s, 1H), 8.51 (s, 1H), 8.66 (t, J = 5.2 Hz, 1H), 8.75 (t, J = 5.1 Hz, 1H). 11B NMR (DMSO-d6, 96.29 MHz): δ −5.25 (2B), −16.67 (2B), −25.99 (6B). HPLC: tR = 13.1 min.
Succinylation of rSAv, 2a
Succinylation of rSAv was accomplished as previously described (17). Briefly, a 200 μL aliquot of a 5 mg/mL solution of rSAv (1 mg, 1.9 × 10−2 μmol) was added to 370 μL of 50 mM NaHCO3 buffer (pH 8.5). To this solution was added 100 equivalents (188 μg; 1.88 μmol) of succinic anhydride in 20 μL DMSO. After 30 min at room temperature, the contents were transferred to a Centricon-10 and concentrated to approximately 50 μL. PBS (500μL) was added and the solution was again concentrated to 50 μL. This wash step was repeated 5 times.
Preparation of Succinylated rSAv-CHX-A″, 5a
Three steps were taken to prepare the rSAv conjugate, 5a, such that it could be radiolabeling with 213Bi. The first step involved demetallation of rSAv. The second step involved conjugation of the CHX-A″ chelate 3a, then succinylation of that conjugate. The third step was purification of the modified rSAv under demetallation conditions. The conditions used in each step are described below.
Demetallation of rSAv
The rSAv (5 mg) was dialyzed against 2 liters of metal-free HEPES buffer with a minimum of 5 buffer changes over 3 days at 4°C using a Slide-A-Lyzer® 10K cassette (Pierce, Rockford, IL). All buffers used were prepared with metal-free (18 mega-ohm) water passed over a column of Chelex-100 resin (250 g/12 L). Chelex-100 resin (10 g) was added to each buffer change. The demetallated mAbs were removed from dialysis and placed in acid washed 15 mL conical vials, and care was taken to assure that no metals were introduced.
Conjugation of 3a and Succinylation
To the demetallated rSAv (4.5 mg, 85 nmol) in 1 mL demetallated HEPES buffer, pH 8.5 at room temperature was added 44 μL (1.7 μM) of an isothiocyanatobenzyl-CHX-A″, 3a, solution (22.4 mg/mL in DMSO). The reaction mixture was stirred at room temperature for 18 hours to yield 4a. After that time, 0.43 mg (4.24 μmol) of succinic anhydride was added and the mixture was stirred for another 1 h at room temperature to give 5a (80%, 4.5 mg/mL, 0.8 mL).
Purification and Second Dialysis
After the conjugation reaction was complete, the reaction mixture was placed in a dialysis cassette and dialyzed against 3 × 2 liters of metal-free citrate buffer (50 mM sodium citrate, 150 mM NaCl, 0.05% NaN3 adjusted to pH 5.5) over 2 days; against 4 × 2 liters of 150 mM NaCl for 2 days. The number of CHX-A″ chelates per rSAv molecule in 5a was assessed using the yttrium-arsenazo III complex spectrophotometric assay (37). The modified rSAv was stored at 4°C until used.
Preparation of Succinylated rSAv-nCB, 5b
To 2.5 mg (500 μL at 5 mg/mL in PBS pH 7.2) of rSAv was added 1 mL of 50 mM sodium bicarbonate buffer, pH 8.5, followed by 40 μL of a solution of 3b (4.85 mg/mL in DMSO). This was allowed to react overnight at room temperature. The solution was then passed over a PD-10 column (Pharmacia, Uppsala, Sweden) eluting 1 mL fractions with 0.9% saline. The protein fractions were combined and concentrated using a Centricon-30 (Millipore, Bedford, MA) to give 2.4 mg (400 μL of 6 mg/mL; 96%) of rSAv-nCB, 4b.
To 1.0 mg of rSAv-nCB (167 μL of 6 mg/mL in 0.9% saline solution) was added 334 μL of 50 mM sodium bicarbonate buffer, pH 8.5, followed by 20 μL of a solution of 4.7 mg/mL succinic anhydride in DMSO. After 30 min at room temperature, the solution was passed over a PD-10 column and collected in 0.9% saline. The protein fractions were combined and concentrated in a Centricon-30 to give 0.94 mg (260 μL of 3.6 mg/mL solution; 94%) of succinylated rSAv-nido-carborane, 5b.
Preparation of Succinylated rSAv-B10H9-m-dPEG12™, 5c
To 11 mg (207 nmol) of rSAv in 1 mL of 500 mM sodium borate buffer pH 8.0 was added 137 μL (2.08 μmol) of a solution of 3c (20 mg/mL in DMF). This was allowed to react overnight at room temperature. The solution was then passed over a PD-10 column and collected in PBS pH 7.2. The protein containing fractions were combined and concentrated in a Centricon-30 to give 790 μL of 12.7 mg/mL solution (91%). To 700 μL of this solution (8.9 mg) was added 700 μL of 100 mM sodium bicarbonate pH 8.5 followed by 1.68 mg of succinic anhydride in 30 μL of DMF. After 30 min at room temperature, the solution was concentrated in a Centricon-30 and washed with 4 × 1 mL of PBS pH 7.2. This yielded 7.9 mg (1.1 mL at 7.2 mg/mL; 88%) of succinylated rSAv-B10/dPEG™, 5c.
Preparation of [213Bi]6a
213Bi was obtained from 225Ac (t1/2 = 10 days) by elution of the generator column with 1 M HCl using a dual syringe pump (38). As the elution proceeded, the 213Bi/HCl solution was mixed with water (in a plastic chamber) and run across a MP-50 cation exchange column. The 213Bi was trapped on the ion exchange column and the column was removed from the generator system. The column was then eluted with freshly prepared 0.1N HI, then the pH of the eluant was adjusted to 4.2 – 4.5 using 3M NH3OAc (Ultrex grade).
A 200 μg quantity of demetallated and succinylated rSAv with approx. 1.5 CHX-A″ chelates/molecule in 120 μL PBS was diluted with 100 μL 1 M NaOAc (pH 4), then 300 μL of 213Bi eluant (95 MBq; 2.56 mCi) was added. After 5 min at room temperature the reaction mixture was passed over a PD-10 column and 68 MBq (72%) of [213Bi]6a was obtained. The radiochemical purity of the [213Bi]6a, was analyzed using instant thin layer chromatography (ITLC) on an ITLC-SG strip (Gelman, Ann Arbor, MI). After spotting, the ITLC strips were allowed to air dry, then were placed in a development chamber containing 80% methanol/20% 10 mM DTPA in water as eluant. When the solvent had nearly reached the top of the strip, it was air-dried, cut in sections, and counted in a gamma counter. The percent bound activity was obtained by dividing the activity in the first half of the strip (from point of spotting) divided by the total activity on the ITLC strip (x100).
211At/125I Labeling; Preparation of [211At]2b, [125I]2b, [125I]6b, [211At]6c, [125I]6c, [211At]6d, [125I]6d
The following general procedures were used to obtain the 211At- and 125I-labeled rSAv conjugates.
[125I]6b
To solution of 5a (100 μL of a 4.5 mg/mL in PBS) was added 25 μL of sodium phosphate (500 mM, pH 7.4), then 1 μL of Na[125I]I (195 μCi in 0.1 N NaOH). That addition was followed by 12 μL of a 1 mg/mL solution of ChT in water. After 5 min. at room temperature, the reaction was quenched with 1.2 μL of a 10 mg/mL solution of sodium metabisulfite in water. The reaction mixture was then placed on a NAP-10 column (Amersham Pharmacia Biotech) and eluted with PBS. The protein fractions were combined to yield 166 μCi (85%) of [125I]6b.
[125I]2b and [125I]6c
To 600 μg succinylated rSAv-nCB, 5b, (1.7 mg/mL in PBS) or succinylated rSAv, 2a, (12.6 mg/mL in PBS) was added 25 μL of sodium phosphate 500 mM, pH 7.4. This was followed by 1.5 μL (310 μCi) of Na[125I]I in 0.1 N NaOH and 38 μL of a solution of 1 mg/mL ChT in water. After 30 sec at room temperature, 3.8 μL of a solution of 10 mg/mL sodium metabisulfite in water was added to quench the reaction. The mixture was then passed over a NAP-10 column and collected in 0.9% saline. The protein fractions were combined. This procedure gave radiochemical yields of 54% for [125I]2b and 68% for [125I]6c.
[211At]2b and [211At]6c
To 600 μg succinylated rSAv-nido-carborane, 5b, (1.7 mg/mL in PBS) or succinylated rSAv, 2a, (12.6 mg/mL in PBS) was added 100 μL of sodium phosphate 500 mM, pH 7.4. This was followed by 40 μL (360–410 μCi) of Na[211At]At solution at pH 7 and 38 μL of a solution of 1 mg/mL ChT in water. After 30 sec at room temperature, 3.8 μL of a solution of 10 mg/mL sodium metabisulfite in water was added to quench the reaction. The mixture was then passed over a NAP-10 column (Pharmacia, Uppsala, Sweden) and collected in 0.9% saline. The protein fractions were combined. This procedure gave radiochemical yields of 18% for [211At]2b, and 50% for [211At]6c.
[211At]rSAv-B10, [211At]6d
To 750 μg, 5c, (2.97.2 mg/mL in PBS) was added 100 μL sodium phosphate 500 mM, pH 7.4. This was followed by addition of 400–500 μL (600–660 μCi) of Na[211At]At solution in 0.05 N NaOH. To this solution was added 75 μL of a solution of 1 mg/mL ChT in water. After 1 min at room temperature, the reaction was quenched with 75 μL of 1 mg/mL sodium metabisulfite in water. The mixture was then passed over a NAP-10 column and collected in 0.9% saline. The protein fractions were combined to give radiochemical yields of 66 and 71% (2 radiolabeling reactions) for [211At]6d.
[125I]rSAv-B10, [125I]6d
To 750 μg, 5c, (7.2 mg/mL in PBS) was added 25 μL sodium phosphate 500 mM, pH 7.4. This was followed by addition of 2 μL (800–980 μCi) of Na[125I]I solution in 0.1N NaOH. To this solution was added 20 μL of a solution of 1 mg/mL ChT in water. After 30 seconds at room temperature, the reaction was quenched with 20 μL of 1 mg/mL sodium metabisulfite in water. The mixture was then passed over a NAP-10 column and collected in 0.9% saline. The protein fractions were combined to give radiochemical yields of 81 and 86% (2 radiolabeling reactions) for [125I]6d.
Biodistribution Studies
The animal studies were approved by the University of Washington’s Institutional Animal Care and Use Committee prior to being conducted. Male athymic mice (BALB/c nu/nu), 4–5 weeks of age were obtained from Simonsen Laboratories (Gilroy, CA). The mice were housed for a minimum of 1 week in the animal facility prior to the study. In dual label experiments, the 213Bi- or 211At-labeled modified rSAv was mixed with the appropriate (matched) 125I-labeled modified rSAv. That mixture was diluted in phosphate buffered saline (PBS) to prepare quantities of ~100 μL for administration. In each set of experiments, the dual-labeled succinylated rSAv was injected into 15 athymic mice via the lateral tail vein. The actual amount of injectate each animal received was determined by weighing the administering syringe before and after injection. Groups of five mice were sacrificed by cervical dislocation at 45, 90 and 180 min for samples containing 213Bi, or 1, 4, and 24 h post injection for samples containing 211At. Blood samples were obtained by cardiac puncture immediately before sacrifice. Urine samples were obtained by syringe bladder tap at the time the tissues were excised. The tissues were excised, blotted free of blood, weighed, and counted. Excised tissues are listed in Tables S1–S4 (Supporting Information). The amount of radioactivity and quantity of rSAv, as well as the average animal weights in the biodistributions, are provided in the Table legends. Calculation of the percent injected dose (%ID) and percent injected dose per gram (% ID/g) in the tissues was obtained by measuring standards containing 10 μL of the injected dose. The 213Bi and 211At counts were adjusted for decay from the time of counting standards. Total blood volume was estimated to be 6.0% of body weight (39) for the calculations. Statistical analysis of the data was conducted using the paired Student’s t test. Differences were considered statistically significant when the p < 0.05.
RESULTS
Reagents for Conjugation
Reagents containing bonding or chelating moieties that were used for modification of rSAv were synthesized. The protein-reactive isothiocyanatobenzyl-CHX-A″ DTPA derivative, 3a, was obtained in a multi-step synthesis as previously reported (34)3. The isothiocyanatophenyl-nido-carborane derivative, 3b, was also obtained in a multi-step synthesis as previously described (31). The protein-reactive decaborate(2-) derivative, 3c, was obtained as shown in Scheme 1. In that synthesis, reaction of an excess of 5-N-(t-Boc)-aminoisophthalate bis-tetrafluorophenyl (TFP) ester, 7, (36) with m-dPEG12™ amine, 8, and Et3N in DMF for 30 minutes at room temperature provided 5-N-(t-Boc)-1-(m-dPEG12™ amino)-aminoisophthalate TFP ester, 9, in 80% yield after purification. Reaction of 9 with 2-(4,7,10-trioxatridecane-13-amine-1-amido)-undecahydro-closo-decaborate(2-), 10, and Et3N in DMF for 2h at room temperature gave a 78% yield of 11. Deprotection of the aniline in neat TFA for 10 min at room temperature yielded the free amine, which was converted to the isothiocyanate derivative 3c in 95% yield by reaction with thiocarbonyldiimidazole in DMF for 1 h at room temperature.
Scheme 1.

Synthetic pathway for preparation of protein-reactive phenylisothiocyanato-undecahydro-decaborate(2-) derivative, 3c. In the structures of the decaborate(2-) moiety, the open circles represent boron or B-H atoms.
(a) 8, DMF, Et3N, rt, 40 min, 80%; (b) 10, Et3N, rt, 2 h, 78%; (c) TFA, rt, 10 min; (d) TCDI, rt, 1h, 95%
Modification of rSAv
The reactions used to modify rSAv are shown in Scheme 2. In all examples studied, rSAv was modified by succinylation of surface amines to decrease kidney localization. For comparison, succinylated rSAv, 2a, was prepared without a binding or bonding moiety (BM). That preparation was accomplished by reaction of rSAv with 50–1004 molar equivalents of succinic anhydride in NaHCO3 buffer, pH 8.5 at room temperature for 30 min. Good protein recovery yields (e.g. 85–95%) are generally obtained in this reaction. In the preparation of the other modified rSAv, succinylation was conducted after conjugation of 3a, 3b, or 3c.
Scheme 2.

Chemical steps for modification of rSAv and radiolabeling with 213Bi, 211At or 125I.
(a) 3a, 3b or 3c, HEPES, pH 8.6, rt, 18h; (b) succinic anhydride, DMSO, NaHCO3, pH 8.5, 30 min, rt;
(c) BiCl3, pH 4–5, NaOAc, HOAc; (d) Na[211At]At or Na[125I]I, ChT, H2O, HOAc, rt, 30 sec;
Modification of rSAv for binding (chelating) 213Bi or bonding 211At was conducted by reaction with 10 molar equivalents of the protein-reactive reagents 3a, 3b and 3c (Figure 2) in HEPES buffer, pH 8.5, at room temperature for 18 h to provide 4a, 4b and 4c. After that time, the modified rSAv species were not isolated, but rather 50–1004 equivalents of succinic anhydride were added, and the reaction was allowed to proceed for an additional 30 min. Prior to, and after, conjugation of the modified DTPA derivative, 3a, the rSAv was demetallated using Chelex-100 resin. The recovery of modified rSAv species, 5a, 5b and 5c varied with the protein-reactive reagent used. Size-exclusion HPLC of the modified rSAv showed very little change from unmodified rSAv. For rSAv modified with 3a (CHX-A″), the number of CHX-A″ moieties conjugated was determined by the spectrophotometric assay using yttrium-arsenazo III complex (37). Using this method it was determined that there were ~1.5 chelates/rSAv in the preparation used for the animals study (i.e. Table S1, supporting information). Quantification of the number of 3b (nido-CB moieties) and 3c (dPEG™/B10 moieties) conjugated with rSAv was not readily accomplished. However, changes in isoelectric points (pI) brought about by conjugation were evaluated by IEF analyses. An example of an IEF gel is shown in Figure 2. IEF gels showed that rSAv had protein bands in the range of pH 7.4–7.8. The protein bands (pI) shifted to pH 7.0–5.2 when conjugated with the mono-anion 3b. The protein bands (pI) shifted further to pH 5.2-4.3 when rSAv-3b (4b) was succinylated. Greater shifts in the pI were noted (5.9-4.7) when rSAv was conjugated with the di-anionic 3c. Succinylation of rSAv-3c (4c) shifted the pI even further (4.1-3.7). The fact that none of the unmodified rSAv is present after conjugation of 3b or 3c indicates that all rSAv tetramers have at least 1 of these moieties conjugated. In fact, it is likely that each tetramer has more than 2 conjugates or some of the unmodified rSAv would be present.
Figure 2.
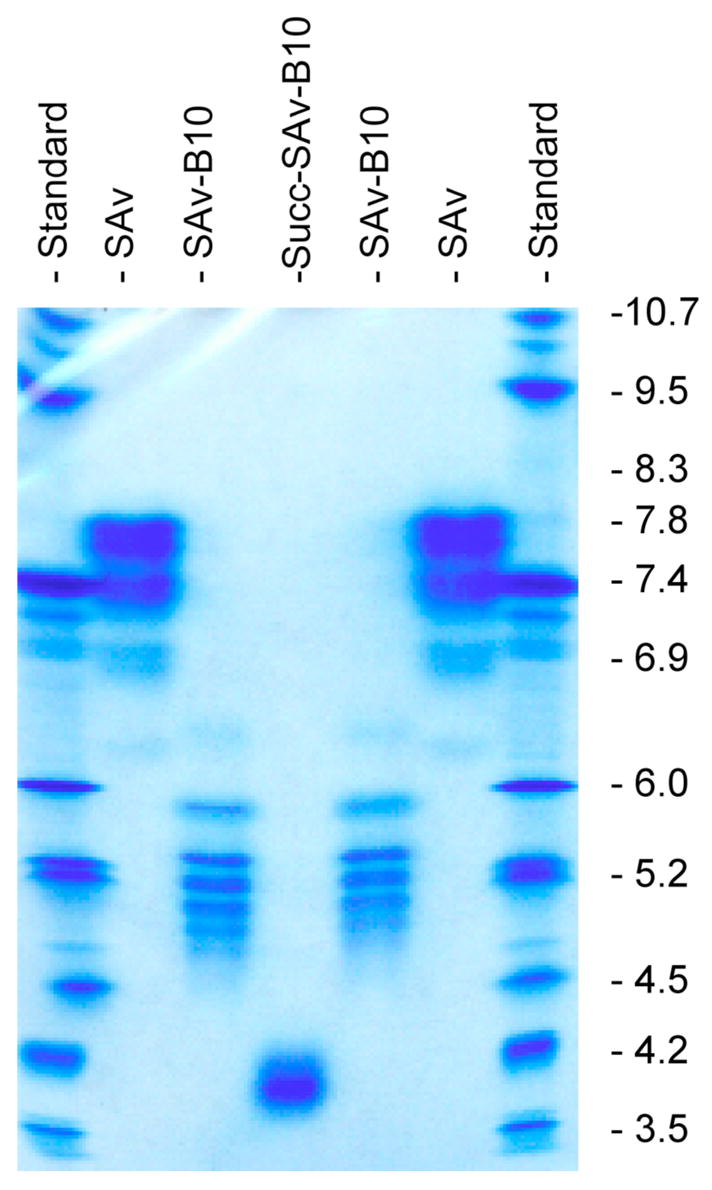
Digitized image of an IEF gel showing changes in pI when rSAv is modified with the decaborate(2-) derivative 3c to prepare conjugate 4c, and after further modification with succinic anhydride to give 5c. Lanes 1 & 7: IEF standards (Serva Electrophoresis GmbH, Broad pI; 3–10) run in IEF standards run in the direction of application (bottom to top) and opposite direction to application of sample (e.g. top to bottom). Lanes 2 & 6: unmodified rSAv. Lane 3 & 5: r-SAv conjugated with 3c. Lane 4: r-SAv conjugated with 3c and further modified by reaction with an excess of succinic anhydride.
Radiolabeled modified rSAv, Preparation of 2b, 6a, 6b, 6c
The modified rSAv was radiolabeled with either 213Bi or 211At to assess radiochemical yields and in vivo stability. rSAv conjugates 2a, 5a, 5b and 5c were also radioiodinated with 125I. Radioiodination of rSAv was conducted to provide a comparison with the astatination yields and to provide an internal control for comparisons in vivo. The 225Ac/213Bi generator system previously described (38) was used to obtain the 213Bi for labeling. The succinylated rSAv-CHX-A″, 5a, was reacted in 1M NaOAc buffer, pH 4 for 5 min. Purification was accomplished by size-exclusion chromatography (NAP-10 column) to give a 72% isolated yield of [213Bi]6a. Dry distillation of an irradiated bismuth target provided 211At for labeling. Astatination was very rapid (<1 min) using chloramine-T in phosphate buffered saline (PBS), pH 7.4. After purification by size-exclusion chromatography (NAP-10), isolated yields of 18% for [211At] 2b and 66% - 86% for [211At]6b and [211At]6c were obtained.
Biodistribution Studies
Four biodistribution studies were conducted in athymic mice. In each study, the 213Bi or 211At-labeled rSAv was mixed with a radioiodinated version of the same modified rSAv and injected into three sets of 5 mice each. Because the rSAv is very resistant to protease degradation, direct radioiodination can be used to provide a standard that closely reflects the distribution of the modified rSAv (16). In the first biodistribution study, [213Bi]6a and [125I]6b were coinjected and tissue samples were obtained at 45 min, 90 min and 180 min (~1, 2 and 4 half-lives) post injection (pi). The data obtained at 90 and 180 min are plotted in Figure 3, panel A, and the data obtained at all three time points are provide in Table S1 (Supporting Information). The plotted data show that the tissue concentrations of the two radionuclides are very similar at the two time points. Analysis of the concentrations of radionuclides in mice by the paired Student’s t test indicated that the only tissue with a significant difference in concentrations of 213Bi and 125I was kidney at the 180 min time point.
Figure 3.
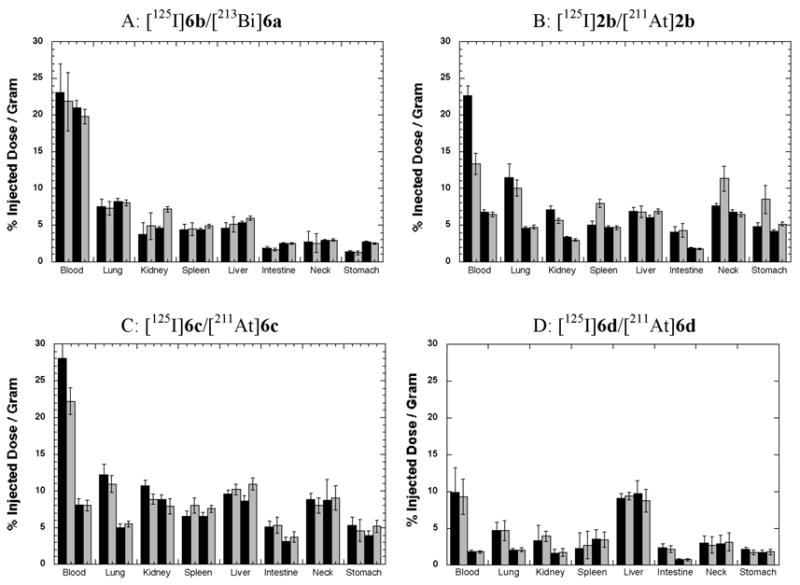
Bar graphs of selected biodistribution data obtained from co-injections (125I- and α-emitter 213Bi-or 211At-labeled) modified rSAv. 125I-labeled modified rSAv data are plotted as black bars in each panel. 213Bi or 211At biodistribution data are plotted as grey bars. Panel A shows tissue distributions of 213Bi-labeled succinylated rSAv-CHX-A″, [213Bi]6a, in comparison with 125I-labeled succinylated rSAv-CHX-A″, [125I]6b, at 1.5 h (1st pair of bars) and 3 h (2nd pair of bars) pi. Panel B shows tissue distributions of 211At- and 125I-labeled succinylated rSAv that had no boron cage conjugated, [211At]2b and [125I]2b, at 4 (1st pair of bars) and 24 h (2nd pair of bars) pi. Panel C shows tissue distributions of 211At- and 125I-labeled succinylated rSAv that had nido-carborane cages conjugated, [211At]6c and [125I]6c, at 4 and 24 h pi. Panel D shows tissue distributions of 211At- and 125I-labeled succinylated rSAv that had decaborate(2-) cages conjugated, [211At]6d and [125I]6d, at 4 and 24 h post injection. Tables (S1–S4) of biodistribution data are provided in Supporting Information.
In a second biodistribution, 211At-labeled succinylated rSAv, [211At]2b, and 125I-labeled succinylated rSAv, [125I]2b, were coinjected and tissue samples were obtained at 1, 4 and 24 h (~0.14, 0.6, and 3.3 half-lives) pi. The data obtained at 4 and 24 are plotted in Figure 3, panel B, and the data obtained at all three time points are provided in Table S2 (Supporting Information). The plotted data show differences in concentrations of radionuclides in blood, kidney, spleen, neck and stomach. When analyzed by the Student’s t test, several differences were found to be significant. At the 1 and 4 h time points, the concentration of radionuclides in blood, spleen, neck and stomach were significantly different. Differences in concentrations of 211At and radioiodine in lung, spleen, neck and stomach is an indication that there was free [211At]astatide in vivo (40). It is interesting to note that at 24 h pi, there is no difference between the radionuclide concentrations in any tissue except muscle (Table S2).
In the third biodistribution, [211At]6c and [125I]6c were coinjected and tissue samples were obtained at 1, 4, and 24 h pi. The data obtained at 4 and 24 are plotted in Figure 3, panel C, and the data obtained at all three time points are provided in Table S3 (Supporting Information). By inspection of Figure 3C it can be seen that, with the exception of concentrations of radionuclides in blood at the 1h time point, the concentrations of the two radionuclides in tissues are quite similar at the two time points plotted. A small but significant difference in the radionuclide concentrations was found in the stomach at 1 h pi, but the lack of significant differences in concentrations in other tissues makes it likely that very little free [211At]astatide was present. At the 4 h time point, significant differences in radionuclide concentrations were seen in blood, muscle, kidney and spleen. Again, lack of significant differences in lung, stomach and neck makes it unlikely that the differences were reflective of astatide release from the rSAv conjugates.
In a fourth biodistribution, [211At]6d and [125I]6d were coinjected and tissue samples were obtained at 1, 4, and 24 h pi. The data obtained at 4 and 24 are plotted in Figure 3, panel D, and the data obtained at all three time points are provided in Table S4 (Supporting Information). From the plotted data it is apparent that concentrations of 211At and 125I are essentially the same in all tissues, indicating a high degree of stability towards deastatination. When subjected to the paired Student’s t test, a small, but significant difference was noted for the radionuclide concentration in only one tissue (spleen) examined, and only at 4 h pi. Because there were no differences observed in the lung, stomach or neck, it seems unlikely that the observed difference was due to free astatide. Interestingly, the concentration of both radionuclides (125I and 211At) was considerably lower in blood and other tissues than observed with the other radiolabeled rSAv conjugates. Indeed, the blood and tissue concentrations of [125I]6d are less than half that obtained with the other labeling methods. While that is the case, the liver concentrations were found to be similar to that observed for the nido-carborane conjugate, [125I]6c. Those concentrations were 3–5 %ID/gram higher than found for the rSAv conjugated with CHX-A″, [125I]6b, or when there was no bonding/binding moiety conjugated, [125I]2b.
DISCUSSION
There are only a few α-emitting radionuclides that are considered reasonable candidates for TRT applications. Of those, 213Bi and 211At are particularly attractive due to the fact that they do not decay to α-particle emitting daughter radionuclides. While they are very different in many aspects, choosing between 213Bi and 211At for TRT is difficult as each radionuclide has its own strengths and weaknesses. For example, 213Bi is available from a generator system, 225Ac/213Bi (41), which could potentially allow widespread use in clinics and treatment centers. Further, the short half-life of 213Bi may allow repeated (fractionated) treatments in an outpatient setting while posing minimal danger to the patients’ family and health care providers. However, the half-life of 213Bi may be problematic as targeting approaches may not be rapid enough in some tissues to adequately target cancer cells before decay occurs. The longer half-life of 211At (~9.4x) may circumvent this shortcoming of 213Bi. Additionally, at present it is difficult to obtain clinical quantities of 225Ac (for 213Bi production) whereas 211At can be prepared in quantities sufficient for clinical studies (42). On the other hand, treatment with an 211At-labeled molecule may require being within a short distance from the cyclotron used for its production. Rather than argue the merits of either 213Bi or 211At, our approach has been to investigate both. The strengths and weaknesses of each radionuclide will likely dictate usefulness for a specific therapy application.
A key issue with the use of the short half-lived α-emitting radionuclides 213Bi and 211At is employing an appropriate cancer-targeting approach. Several preclinical therapy studies using intact monoclonal antibodies or their fragments have shown promising results with 213Bi (43–46) and 211At (47, 48). However, we believe that another antibody-based approach, termed pretargeting (49) may be the most promising for the short half-lived α-emitting radionuclides. That belief is based primarily on results from studies using antibody-rSAv chemical conjugates or fusion proteins in conjunction with biotin labeled with β-emitting radionuclides (50–53). The belief is also supported by results obtained in a study using 213Bi in a pretargeting protocol for therapy of adult T-cell leukemia in a murine model (54). In that study pretargeted 213Bi provided an effective therapy whereas 90Y did not.
In the majority of pretargeting studies, including those cited above, the radionuclide is carried by biotin. But, there is an alternative approach where a biotinylated mAb is administered first (pretargeted) and radiolabeled rSAv is administered later (49, 55–58). In this investigation, initial steps of rSAv toward evaluating the potential for this alternate pretargeting approach to target short half-lived α-emitting radionuclides have been taken. We hypothesize that using the streptavidin as a carrier for treating minimal residual disease with α-emitting radionuclides may have advantages. Its longer serum half-life should allow higher concentrations in blood and tissues, perhaps allowing a lower quantity of expensive radionuclides to be administered. While recombinant streptavidin, a ~53 kDa protein, will not penetrate solid tumors as rapidly as radiolabeled biotin, with minimal residual disease such as micrometastatic lesions, tissue penetration may not be a problem. Further, in contrast to significant normal tissue irradiation from β-emitting radionuclides, such as 90Y, the short path length of the α-particle should cause minimal normal tissue damage and toxicity when decaying in blood. Data from bone marrow conditioning studies have provided some evidence that supports this hypothesis (20). Although the use of succinylated rSAv as a carrier of radionuclide in pretargeting protocols may limit second administration of the radioactive material due to its immunogenicity, we believe that the potential for efficient targeting of α-emitting radionuclides to metastatic disease warrants investigation.
The primary goals of this investigation were to evaluate methods for modifying radiolabeling rSAv with 213Bi and 211At and determine the effect that modification had on tissue distribution and pharmacokinetics in the mouse model. An important factor in the studies was normal tissue localization of the radiolabeled rSAv. Fortunately, prior investigations (17, 18) had shown that the natural propensity for kidney localization of rSAv (i.e. 20+ %ID/g; (16)) could be dramatically decreased by succinylation. The results of these studies demonstrate that even in the case where succinylation of rSAv is conducted after modifying with binding or bonding moieties, this modification greatly diminishes sequestration in the kidney.
Another important aspect of the studies was to determine whether the α-emitting radionuclides remained stably attached to the rSAv in vivo. While quantification of released bismuth is not readily achieved, a reasonable assessment of the stability of 213Bi-labeled rSAv can be obtained by comparing coinjected 213Bi- and 125I-labeled succinylated rSAv localized in kidney. Free bismuth localizes to kidney (59, 60), thus, if excess 213Bi activity is found in kidney it is a good indicator that 213Bi has been released from the labeled rSAv. The DTPA derivative isothiocyanatobenzyl-CHX-A″, 3a, was chosen for conjugation with rSAv for chelation of 213Bi. Studies have shown that 111In and 90Y can be chelated rapidly on bioconjugates containing this group, and the resultant labeled molecules have been shown to be stable to in vivo demetallation (61–63). In our prior studies, 3a was conjugated with mAbs against antigens on canine hematopoietic cells, then radiolabeled with 213Bi and evaluated in dogs (20, 21, 38). In the studies, kidney enzymes were monitored for several months and no damage to that tissue was noted, likely indicating that 213Bi was not released in vivo.
In this investigation, rSAv was modified with CHX-A″, succinylated and radiolabeled with 213Bi. The data plotted in Figure 3, panel A (Table S1 in Supporting Information) show that the distributions of the radionuclides are very similar. Indeed, the only significant difference between the 213Bi and 125I concentrations (by Student’s t test) is in the kidney at 3 h pi. From the data it appears that there is a small amount of 213Bi released in vivo. However, the important consideration is that release is relatively slow and the magnitude of difference is quite small. At the 3 h time point, 4 half-lives of 213Bi have passed so ~94% of the radioactivity has decayed. An alternative labeling method is to conjugate the DOTA chelation group to rSAv for binding 213Bi (64). This may result in essentially no release of 213Bi in vivo, but it is much more difficult to obtain the radiolabeled rSAv, as elevated temperatures (e.g. 37–45°C) are required for up to an hour to achieve reasonable labeling yields. Thus, it appears that the CHX-A″ is the appropriate chelation group for executing 213Bi-labeled rSAv studies.
Instability of 211At-labeled proteins and small molecules has presented a serious problem with its application to cancer therapy. We considered using m-[211At]astatobenzoate NHS ester (29) and p[211At]astatobenzoate NHS ester (32) for labeling rSAv with 211At. Either of these derivatives would be expected to be stable to in vivo deastatination because rSAv is very resistant to protease degradation. However, in our experience, 40–60% labeling yields in the two-step labeling procedure are the best that can be (consistently) obtained with those reagents. Obtaining a high efficiency of radionuclide labeling is very important with 211At as well. Rather than use the benzoate labeling reagents, we chose to evaluate anionic borane cage pendant groups already under investigation in this laboratory (31). With conjugates of boron cage moieties, direct astatination yields of up to 81% had been obtained. Unfortunately, studies of 211At-labeled benzoate and nido-carborane derivatives (35) did not provide encouraging results regarding in vivo stability. However, due to the protease resistance of rSAv, we chose to investigate a nido-carborane conjugate 3b as a pendant group for direct astatination. The nido-carborane derivative 3b had been previously evaluated in conjugates of a mAb Fab’ fragment. We chose to investigate a decaborate(2-) derivative as well since its use with Fab’ gave higher 211At labeling yields and appeared to impart less perturbation of the in vivo properties of the protein.
For this investigation, we could not readily adapt either of the two decaborate(2-) derivatives previously prepared for use with 211At (31). One decaborate(2-) derivative was conjugated by reaction with sulfhydryl groups while the other contained a biotin moiety. Since rSAv does not have disulfide bonds to reduce for reaction with maleimide derivatives (and we did not want to use reagents to produce sulfhydryl groups on rSAv) the sulfhydryl-reactive reagent did not appear to be a good candidate. Similarly, use of a biotin-containing decaborate(2-) was not appropriate for conjugation with rSAv. Therefore, a new reagent containing a decaborate(2-) moiety and a methoxyl-terminated discrete-length (dPEG™) polyethylene glycol moiety was prepared. The rationale for the use of dPEG™ moiety was that such a group might keep the dianionic decaborate moiety from interacting with serum proteins. Further, other investigators have shown that PEGylation of avidin (Av) (albeit with larger PEG moieties) can improve their in vivo properties for pretargeting applications (65, 66).
Before labeling the rSAv conjugated with 3b or 3c, direct labeling of succinylated rSAv, 2a, was evaluated. Astatination studies of a mAb Fab’ fragment resulted in none (<1%) of the astatinated protein being recovered after labeling using ChT and reducing with sodium metabisulfite (31). Other investigators obtained similar results (e.g. <5% bound) when ChT was used as oxidant (67–69). Therefore, it was interesting that an 18% isolated radiochemical yield was obtained with directly labeled succinylated rSAv in this investigation. The reason for the difference in radiolabeling yields is not known. However, it may be due to the presence of disulfides in the Fab’ molecule, or lack of them in the rSAv molecule. Investigators have shown that electrochemical oxidation and oxidation with H2O2 can provide higher yields of astatinated proteins (68), however, studies indicate that the 211At is bound with sulfhydryl groups rather than tyrosine moieties (69, 70). An important point is that rSAv has no disulfides or cysteine or methionine residues (71), so the labeling can not be associated with sulfhydryl or thioether groups. Streptavidin has six tyrosine residues per subunit. Although one tyrosine residue is buried in the biotin-binding pocket, five are on each subunit giving a total of twenty tyrosine residues per molecule that might be astatinated. In related studies, 211At-labeling of succinylated avidin, which has only one tyrosine per subunit (in biotin binding pocket), gave low radiochemical yields (unreported results)5. The combined results are suggestive that electrophilic 211At can react with tyrosine residues in proteins when no disulfides are present. Early astatine labeling studies demonstrated that direct labeling of tyrosine was possible (72).
The three 211At-labeled rSAv preparations ([211At]2b, [211At]6c, [211At]6d) have noticeably different distributions. The tissue distribution of radioiodinated succinylated rSAv, [125I]2b, correlates well with data obtained in a previous study (16). If that iodinated derivative is compared with directly labeled [211At]2b, it is apparent that there is some instability of the astatinated rSAv. On the other hand when the boron cage moieties 3b and 3c are conjugated to rSAv, in vivo deastatination is not detected. A noticeable difference in tissue (e.g. kidney, spleen, liver) accumulation and retention is observed for the nido-carborane conjugated rSAv relative to succinylated [125I]rSAv. In contrast to this, the astatinated decaborate(2-) rSAv conjugate [211At]6d appears to be cleared from the blood much more rapidly than the succinylated native protein. It is not known why this conjugate has such a rapid blood clearance. Clearance is more rapid from all tissues except liver, which has concentrations similar to native protein at the times studied. One possible explanation for the rapid clearance is that the m-dPEG™12 moiety destabilizes the rSAv tetramer by inserting between the rSAv monomers. Higher molecular weight fusion proteins made of antibody single chain constructs and rSAv have also been noted to have much more rapid clearance than rSAv or chemical conjugates of rSAv and antibodies (51). A plausible explanation for the observed fast clearance in that case is that the fused four scFv destabilize the rSAv tetramer.
Summary
This investigation has demonstrated that after appropriate modification, rSAv can be labeled with either 213Bi or 211At in good radiochemical yield. Most importantly, the data obtained from biodistributions conducted in athymic mice have shown that the 213Bi-labeled succinylated rSAv has nearly the same distribution and pharmacokinetics as the non-metallated rSAv conjugate. While there is slight (but significant) difference in the distributions after 4 half-lives (3 h) of 213Bi, at that time there is less than 10% of the activity remaining and the magnitude of specific kidney localization appears small. From the data we conclude that the 213Bi-labeled succinylated CHX-A″-rSAv has adequate stability to be used in pretargeting protocols. The biodistribution data obtained for succinylated rSAv directly labeled with 211At, i.e. [211At]2b, indicate that this labeling approach is not adequate for pretargeting studies due to low radiochemical yields and low in vivo stability. Higher in vivo stability was obtained with the nido-carborane conjugated rSAv, [211At]6c, but that functional moiety appears to increase blood and tissue retention such that it is not an ideal conjugate either. The data obtained from the in vivo studies with the PEGylated decaborate(2-)-conjugated rSAv, [211At]6d, suggests that this method of labeling not only provides a stable label, but also provides very good radiochemical yields and modifies the in vivo pharmacokinetics in what appears to be a favorable manner. Therefore, additional studies with the PEGylated decaborate(2-) conjugate are planned to evaluate its use in pretargeting protocols.
Supplementary Material
Numerical data for the four biodistributions conducted in this investigation are provided in Tables S1–S4. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Kent Buhler, Janna Quinn, Nicole Lavoie, Holly Nguyen and Dr. Robert Vessella for assistance with the animal studies conducted in this investigation, and Dr. Ruedi Risler for assistance with production of 211At. We are particularly grateful to the Department of Defense (DAMD 17-98-1-8258) and National Cancer Institute, NIH (5 RO1 CA113431) for financial support of the studies. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank Quanta BioDesign (http://www.quantabiodesign.com) for the gift of m-dPEG™12 amine used in the studies.
Footnotes
ABREVIATIONS: Av, avidin; BM, binding moiety or bonding moiety; ChT, chloramine-T; cpm, counts per minute; decaborate(2-), undecahydrodecaborate, [B10H9]2--R; DTPA, diethylenetetraaminepentaacetic acid; EDTA, ethylenediaminetetraacetic acid; IEF, isoelectric focusing; iv, intravenous; mAb, monoclonal antibody; %ID/g, percent injected dose/gram; nCB, nido-carborane; PBS, phosphate-buffered saline; pi, post injection; rSAv, recombinant streptavidin; rt, room temperature, SAv, streptavidin
The term 2-step is often not accurate as a “clearing” step may be used to rid the blood of remaining mAb-biotin or mAb-SAv conjugates, in effect making it a 3-step approach.
This reagent is available commercially from Macrocyclics, Dallas, Tx (www.macrocyclics.com).
The initial studies were conducted with 50 equivalents succinic anhydride, but later studies used 100 equivalents to be certain that as many amines are capped as possible.
Astatination of succinylated avidin and succinylated avidin conjugated with the nido-carborane moiety, 3b, was also conducted to show the difference in yields. Avidin has only 1 tyrosine residue per subunit, making it less likely to be astatinated. Astatination of succinylated avidin gave a radiochemical yield of 5% whereas astatination of succinylated avidin conjugated with 3b gave a 52% radiochemical yield.
LITERATURE CITED
- 1.Mulford DA, Scheinberg DA, Jurcic JG. The promise of targeted α-particle therapy. J Nucl Med. 2005;46(Suppl 1):199S–204S. [PubMed] [Google Scholar]
- 2.Couturier O, Supiot S, Degraef-Mougin M, Faivre-Chauvet A, Carlier T, Chatal JF, Davodeau F, Cherel M. Cancer radioimmunotherapy with alpha-emitting nuclides. Eur J Nucl Med Mol Imaging. 2005;32:601–14. doi: 10.1007/s00259-005-1803-2. [DOI] [PubMed] [Google Scholar]
- 3.Imam SK. Advancements in cancer therapy with alpha-emitters: a review. Int J Rad Oncol Biol Phys. 2001;51:271–8. doi: 10.1016/s0360-3016(01)01585-1. [DOI] [PubMed] [Google Scholar]
- 4.Zalutsky MR, Vaidyanathan G. Astatine-211-labeled radiotherapeutics: an emerging approach to targeted alpha-particle radiotherapy. Curr Pharm Des. 2000;6:1433–55. doi: 10.2174/1381612003399275. [DOI] [PubMed] [Google Scholar]
- 5.Feinendegen LE, McClure JJ. Alpha-Emitters for Medical Therapy - Workshop of the United States Department of Energy. Radiation Res. 1997;148:195–201. [Google Scholar]
- 6.Goldenberg DM. Targeted therapy of cancer with radiolabeled antibodies. J Nucl Med. 2002;43:693–713. [PubMed] [Google Scholar]
- 7.Melo e Silva MC, Patricio L, Gano L, Sa e Melo ML, Inohae E, Mataka S, Thiemann T. Synthesis and biological evaluation of two new radiolabelled estrogens: [125I](E)-3-methoxy-17α-iodovinylestra-1,3,5(10),6-tetraen-17β-ol and [125I](Z)-3-methoxy-17α-iodovinylestra-1,3,5(10),6-tetraen-17β-ol. Appl Radiat Isot. 2001;54:227–239. doi: 10.1016/s0969-8043(99)00258-4. [DOI] [PubMed] [Google Scholar]
- 8.Stinchcomb TG, Roeske JC. Analytic microdosimetry for radioimmunotherapeutic alpha emitters. Med Phys. 1992;19:1385–1393. doi: 10.1118/1.596770. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Yazaki PJ, Anderson AL, Crow D, Colcher D, Wu AM, Williams LE, Wong JY, Raubitschek A, Shively JE. Improved biodistribution and radioimmunoimaging with poly(ethylene glycol)-DOTA-conjugated anti-CEA diabody. Bioconjugate Chem. 2006;17:68–76. doi: 10.1021/bc0502614. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi H, Le N, Kim IS, Kim MK, Pie JE, Drumm D, Paik DS, Waldmann TA, Paik CH, Carrasquillo JA. The pharmacokinetic characteristics of glycolated humanized anti-Tac Fabs are determined by their isoelectric points. Cancer Res. 1999;59:422–30. [PubMed] [Google Scholar]
- 11.Fu PP, Unruh LE, Miller DW, Huang LW, Yang DTC. Synthesis of 3-Aryl-3,4-dihydroisocoumarins. J Org Chem. 1985;50:1259–1261. [Google Scholar]
- 12.Fujioka Y, Satake S, Uehara T, Mukai T, Akizawa H, Ogawa K, Saji H, Endo K, Arano Y. In vitro system to estimate renal brush border enzyme-mediated cleavage of Peptide linkages for designing radiolabeled antibody fragments of low renal radioactivity levels. Bioconjugate Chem. 2005;16:1610–6. doi: 10.1021/bc050211z. [DOI] [PubMed] [Google Scholar]
- 13.Behe M, Becker W, Gotthardt M, Angerstein C, Behr TM. Improved kinetic stability of DTPA-dGlu as compared with conventional monofunctional DTPA in chelating indium and yttrium: preclinical and initial clinical evaluation of radiometal labelled minigastrin derivatives. Eur J Nucl Med Mol Imag. 2003;30:1140–1146. doi: 10.1007/s00259-003-1178-1. [DOI] [PubMed] [Google Scholar]
- 14.Goodwin DA, Meares CF. Pretargeting: General Principles. Cancer (Suppl) 1997;80:2675–2680. doi: 10.1002/(sici)1097-0142(19971215)80:12+<2675::aid-cncr45>3.3.co;2-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhu H, Jain RK, Baxter LT. Tumor Pretargeting for Radioimmunodetection and Radioimmunotherapy. J Nucl Med. 1998;39:65–76. [PubMed] [Google Scholar]
- 16.Wilbur DS, Stayton PS, To R, Buhler KR, Klumb LA, Hamlin DK, Stray JE, Vessella RL. Streptavidin in Antibody Pretargeting. Comparison of a Recombinant Streptavidin with Two Streptavidin Mutant Proteins and Two Commercially Available Streptavidin Proteins. Bioconjugate Chem. 1998;9:100–107. doi: 10.1021/bc970152s. [DOI] [PubMed] [Google Scholar]
- 17.Wilbur DS, Hamlin DK, Buhler KR, Pathare PM, Vessella RL, Stayton PS, To R. Streptavidin in antibody pretargeting. 2. Evaluation of methods for decreasing localization of streptavidin to kidney while retaining its tumor binding capacity. Bioconjugate Chem. 1998;9:322–330. doi: 10.1021/bc970182v. [DOI] [PubMed] [Google Scholar]
- 18.Wilbur DS, Hamlin DK, Meyer DL, Mallett RW, Quinn J, Vessella RL, Press OW. Streptavidin in Antibody Pretargeting. 3. Comparison of Biotin Binding and Tissue Localization of 1,2-Cyclohexanedione and Succinic Anhydride Modified Recombinant Streptavidin. Bioconjugate Chem. 2002;13:611–620. doi: 10.1021/bc015574n. [DOI] [PubMed] [Google Scholar]
- 19.Wilbur DS, Hamlin DK, Sanderson J, Lin Y. Streptavidin in antibody pretargeting. 4. Site-directed mutation provides evidence that both arginine and lysine residues are involved in kidney localization. Bioconjugate Chem. 2004;15:1454–63. doi: 10.1021/bc049869n. [DOI] [PubMed] [Google Scholar]
- 20.Sandmaier BM, Bethge WA, Wilbur DS, Hamlin DK, Santos EB, Brechbiel MW, Fisher DR, Storb R. Bismuth 213-labeled anti-CD45 radioimmunoconjugate to condition dogs for nonmyeloablative allogeneic marrow grafts. Blood. 2002;100:318–26. doi: 10.1182/blood-2001-12-0322. [DOI] [PubMed] [Google Scholar]
- 21.Bethge WA, Wilbur DS, Storb R, Hamlin DK, Santos EB, Brechbiel MW, Fisher DR, Sandmaier BM. Selective T-cell ablation with bismuth-213-labeled anti-TCRαβ as nonmyeloablative conditioning for allogeneic canine marrow transplantation. Blood. 2003;101:5068–75. doi: 10.1182/blood-2002-12-3867. [DOI] [PubMed] [Google Scholar]
- 22.Camera L, Kinuya S, Garmestani K, Wu C, Brechbiel MW, Pai LH, McMurry TJ, Gansow OA, Pastan I, Paik CH, Carrasquillo JA. Evaluation of the Serum Stability and In Vivo Biodistribution of CHX-DTPA and Other Ligands for Yttrium Labeling of Monoclonal Antibodies. J Nucl Med. 1994;35:882–889. [PubMed] [Google Scholar]
- 23.Milenic DE, Roselli M, Mirzadeh S, Pippin CG, Gansow OA, Colcher D, Brechbiel MW, Schlom J. In vivo evaluation of bismuth-labeled monoclonal antibody comparing DTPA-Derived bifunctional chelates. Cancer Biother Radiopharm. 2001;16:133–146. doi: 10.1089/108497801300189227. [DOI] [PubMed] [Google Scholar]
- 24.Adams GP, Shaller CC, Chappell LL, Wu C, Horak EM, Simmons HH, Litwin S, Marks JD, Weiner LM, Brechbiel MW. Delivery of the a-emitting radioisotope bismuth-213 to solid tumors via single-chain Fv and diabody molecules. Nucl Med Biol. 2000;27:339–346. doi: 10.1016/s0969-8051(00)00103-7. [DOI] [PubMed] [Google Scholar]
- 25.Nikula TK, McDevitt MR, Finn RD, Wu C, Kozak RW, Garmestani K, Brechbiel MW, Curcio MJ, Pippin CG, Tiffany-Jones L, Geerlings MW, Sr, Apostolidis C, Molinet R, Geerlings MW, Jr, Gansow OA, Scheinberg DA. Alpha-emitting bismuth cyclohexylbenzyl DTPA constructs of recombinant humanized anti-CD33 antibodies: pharmacokinetics, bioactivity, toxicity and chemistry. J Nucl Med. 1999;40:166–76. [PubMed] [Google Scholar]
- 26.McDevitt MR, Finn RD, Ma D, Larson SM, Scheinberg DA. Preparation of alpha-emitting 213Bi-labeled antibody constructs for clinical use. J Nucl Med. 1999;40:1722–7. [PubMed] [Google Scholar]
- 27.Vaughan ATM, Fremlin JH. The Preparation of Astatine Labelled Proteins Using an Electrophilic Reaction. Int J Nucl Med Biol. 1978;5:229–230. doi: 10.1016/0047-0740(78)90145-6. [DOI] [PubMed] [Google Scholar]
- 28.Vaughan ATM, Bateman WJ, Fisher DR. The In Vivo Fate of a 211At labelled Monoclonal Antibody with Known Specificity in a Murine System. Int J Rad Oncol Biol Phy. 1982;8:1943–1946. doi: 10.1016/0360-3016(82)90453-9. [DOI] [PubMed] [Google Scholar]
- 29.Zalutsky MR, Narula AS. Astatination of Proteins using an N-Succinimidyl Tri-n-Butylstannyl Benzoate Intermediate. Appl Radiat Isot. 1988;39:227–232. doi: 10.1016/0883-2889(88)90176-1. [DOI] [PubMed] [Google Scholar]
- 30.Hadley SW, Wilbur DS, Gray MA, Atcher RW. Astatine-211 labeling of an antimelanoma antibody and its Fab fragment using N-succinimidyl p-[211At]astatobenzoate: comparisons in vivo with the p-[125I]iodobenzoyl conjugate. Bioconjugate Chem. 1991;2:171–9. doi: 10.1021/bc00009a006. [DOI] [PubMed] [Google Scholar]
- 31.Wilbur DS, Chyan MK, Hamlin DK, Vessella RL, Wedge TJ, Hawthorne MF. Reagents for Astatination of Biomolecules. 2. Conjugation of Anionic Boron Cage Pendant Groups to a Protein Provides a Method for Direct Labeling that is Stable to In Vivo Deastatination. Bioconjugate Chem. 2007;18:1226–1240. doi: 10.1021/bc060345s. [DOI] [PubMed] [Google Scholar]
- 32.Wilbur DS, Vessella RL, Stray JE, Goffe DK, Blouke KA, Atcher RW. Preparation and evaluation of para-[211At]astatobenzoyl labeled anti-renal cell carcinoma antibody A6H F(ab′)2. In vivo distribution comparison with para-[125I]iodobenzoyl labeled A6H F(ab′)2. Nucl Med Biol. 1993;20:917–27. doi: 10.1016/0969-8051(93)90092-9. [DOI] [PubMed] [Google Scholar]
- 33.Lindegren S, Back T, Jensen HJ. Dry-distillation of astatine-211 from irradiated bismuth targets: a time-saving procedure with high recovery yields. Appl Radiat Isot. 2001;55:157–160. doi: 10.1016/s0969-8043(01)00044-6. [DOI] [PubMed] [Google Scholar]
- 34.Brechbiel MW, Gansow OA. Synthesis of C-Functionalized trans-Cyclohexyldiethylenetriaminepenta-acetic Acids for Labelling of Monoclonal Antibodies with the Bismuth-212 α-Particle Emitter. J Chem Soc Perkin Trans. 1992;1:1173–1178. [Google Scholar]
- 35.Wilbur DS, Chyan MK, Hamlin DK, Kegley BB, Risler R, Pathare PM, Quinn J, Vessella RL, Foulon C, Zalutsky M, Wedge TJ, Hawthorne MF. Reagents for astatination of biomolecules: comparison of the in vivo distribution and stability of some radioiodinated/astatinated benzamidyl and nido-carboranyl compounds. Bioconjugate Chem. 2004;15:203–23. doi: 10.1021/bc034175k. [DOI] [PubMed] [Google Scholar]
- 36.Hamblett KJ, Kegley BB, Hamlin DK, Chyan MK, Hyre DE, Press OW, Wilbur DS, Stayton PS. A streptavidin-biotin binding system that minimizes blocking by endogenous biotin. Bioconjugate Chem. 2002;13:588–98. doi: 10.1021/bc010087t. [DOI] [PubMed] [Google Scholar]
- 37.Pippin CG, Parker TA, McMurry TJ, Brechbiel MW. Spectrophotometric Method for the Determination of a Bifunctional DTPA Ligand in DTPA-Monoclonal Antibody Conjugates. Bioconjugate Chem. 1992;3:342–345. doi: 10.1021/bc00016a014. [DOI] [PubMed] [Google Scholar]
- 38.Bethge WA, Wilbur DS, Storb R, Hamlin DK, Santos EB, Brechbiel MW, Sandmaier BM. Radioimmunotherapy with Bismuth-213 as Conditioning for Nonmyeloablative Allogeneic Hematopoietic Cell Transplantation in Dogs: A Dose Deescalation Study. Transplantation. 2004;78:352–359. doi: 10.1097/01.tp.0000128853.62545.b2. [DOI] [PubMed] [Google Scholar]
- 39.Kaplan HM, Brewer NR, Blair WH. The Mouse in Biomedical Research. In: Foster HL, Small JD, Fox JG, editors. The Mouse in Biomedical Research. Academic Press; New York: 1983. pp. 248–292. [Google Scholar]
- 40.Hamilton JG, Asling CW, Garrison WM, Scott KG. The Accumulation, Metabolism and Biological Effects of Astatine in Rats and Monkeys. Univ Cal Publ Pharmacol. 1953;2:283–343. [PubMed] [Google Scholar]
- 41.Bray LA, Tingey JM, DesChane JR, Egorov OB, Tenforde TS, Wilbur DS, Hamlin DK, Pathare PM. Development of a Unique Bismuth (Bi-213) Automated Generator for Use in Cancer Therapy. Ind Eng Chem Res. 2000;39:3189–3194. [Google Scholar]
- 42.Zalutsky MR, Zhao XG, Alston KL, Bigner D. High-level production of •-particle-emitting 211At and preparation of 211At-labeled antibodies for clinical use. J Nucl Med. 2001;42:1508–1515. [PubMed] [Google Scholar]
- 43.Qu CF, Song EY, Li Y, Rizvi SM, Raja C, Smith R, Morgenstern A, Apostolidis C, Allen BJ. Pre-clinical study of 213Bi labeled PAI2 for the control of micrometastatic pancreatic cancer. Clin Exp Metastasis. 2005;22:575–86. doi: 10.1007/s10585-005-5788-9. [DOI] [PubMed] [Google Scholar]
- 44.Milenic D, Garmestani K, Dadachova E, Chappell L, Albert P, Hill D, Schlom J, Brechbiel M. Radioimmunotherapy of human colon carcinoma xenografts using a 213Bi-labeled domain-deleted humanized monoclonal antibody. Cancer Biother Radiopharm. 2004;19:135–47. doi: 10.1089/108497804323071904. [DOI] [PubMed] [Google Scholar]
- 45.McDevitt MR, Barendswaard E, Ma D, Lai L, Curcio MJ, Sgouros G, Ballangrud AM, Yang WH, Finn RD, Pellegrini V, Geerlings MW, Jr, Lee M, Brechbiel MW, Bander NH, Cordon-Cardo C, Scheinberg DA. An α-particle emitting antibody ([213Bi]J591) for radioimmunotherapy of prostate cancer. Cancer Res. 2000;60:6095–6100. [PubMed] [Google Scholar]
- 46.Behr TM, Behe M, Stabin MG, Wehrmann E, Apostolidis C, Molinet R, Strutz F, Fayyazi A, Wieland E, Gratz S, Koch L, Goldenberg DM, Becker W. High-linear energy transfer (LET) alpha versus low-LET beta emitters in radioimmunotherapy of solid tumors: therapeutic efficacy and dose-limiting toxicity of 213Bi- versus 90Y-labeled CO17-1A Fab’ fragments in a human colonic cancer model. Cancer Res. 1999;59:2635–43. [PubMed] [Google Scholar]
- 47.Zhang M, Yao Z, Zhang Z, Garmestani K, Talanov VS, Plascjak PS, Yu S, Kim HS, Goldman CK, Paik CH, Brechbiel MW, Carrasquillo JA, Waldmann TA. The Anti-CD25 Monoclonal Antibody 7G7/B6, Armed with the α-Emitter 211At, Provides Effective Radioimmunotherapy for a Murine Model of Leukemia. Cancer Res. 2006;66:8227–32. doi: 10.1158/0008-5472.CAN-06-1189. [DOI] [PubMed] [Google Scholar]
- 48.Elgqvist J, Andersson H, Back T, Hultborn R, Jensen H, Karlsson B, Lindegren S, Palm S, Warnhammar E, Jacobsson L. Therapeutic efficacy and tumor dose estimations in radioimmunotherapy of intraperitoneally growing OVCAR-3 cells in nude mice with (211)At-labeled monoclonal antibody MX35. J Nucl Med. 2005;46:1907–15. [PubMed] [Google Scholar]
- 49.Paganelli G, Malcovati M, Fazio F. Monoclonal Antibody pretargetting techniques for tumour localization: the avidin-biotin system. Nucl Med Commun. 1991;12:211–234. doi: 10.1097/00006231-199103000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Pagel JM, Hedin N, Subbiah K, Meyer D, Mallet R, Axworthy D, Theodore LJ, Wilbur DS, Matthews DC, Press OW. Comparison of anti-CD20 and anti-CD45 antibodies for conventional and pretargeted radioimmunotherapy of B-cell lymphomas. Blood. 2003;101:2340–8. doi: 10.1182/blood-2002-03-0874. [DOI] [PubMed] [Google Scholar]
- 51.Pagel JM, Lin Y, Hedin N, Pantelias A, Axworthy D, Stone D, Hamlin DK, Wilbur DS, Press OW. Comparison of a tetravalent single-chain antibody-streptavidin fusion protein and an antibody-streptavidin chemical conjugate for pretargeted anti-CD20 radioimmunotherapy of B-cell lymphomas. Blood. 2006;108:328–36. doi: 10.1182/blood-2005-11-4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Press OW, Corcoran M, Subbiah K, Hamlin DK, Wilbur DS, Johnson T, Theodore L, Yau E, Mallett R, Meyer DL, Axworthy D. A comparative evaluation of conventional and pretargeted radioimmunotherapy of CD20-expressing lymphoma xenografts. Blood. 2001;98:2535–2543. doi: 10.1182/blood.v98.8.2535. [DOI] [PubMed] [Google Scholar]
- 53.Axworthy DB, Reno JM, Hylarides MD, Mallett RW, Theodore LJ, Gustavson LM, Su F, Hobson LJ, Beaumier PL, Fritzberg AR. Cure of human carcinoma xenografts by a single dose of pretargeted yttrium-90 with negligible toxicity. Proc Natl Acad Sci U S A. 2000;97:1802–7. doi: 10.1073/pnas.97.4.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang M, Yao Z, Garmestani K, Axworthy DB, Zhang Z, Mallett RW, Theodore LJ, Goldman CK, Brechbiel MW, Carrasquillo JA, Waldmann TA. Pretargeting radioimmunotherapy of a murine model of adult T-cell leukemia with the a-emitting radionuclide, bismuth 213. Blood. 2002;100:208–216. doi: 10.1182/blood-2002-01-0107. [DOI] [PubMed] [Google Scholar]
- 55.Zhang M, Yao Z, Saga T, Sakahara H, Nakamoto Y, Sato N, Nakada H, Yamshina I, Konishi J. Improved Intratumoral Penetration of Radiolabeled Streptavidin in Intraperitoneal Tumors Pretargeted with Biotinylated Antibody. J Nucl Med. 1998;39:30–33. [PubMed] [Google Scholar]
- 56.Maraveyas A, RowlinsonBusza G, Murray S, Epenetos AA. Improving tumour targeting and decreasing normal tissue uptake by optimizing the stoichiometry of a two-step biotinylated-antibody/streptavidin-based targeting strategy: Studies in a nude mouse xenograft model. Int J Cancer. 1998;78:610–617. doi: 10.1002/(sici)1097-0215(19981123)78:5<610::aid-ijc14>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 57.Saga T, Weinstein JN, Jeong JM, Heya T, Lee JT, Le N, Paik CH, Sung C, Neumann RD. Two-Step Targeting of Experimental Lung Metastases with Biotinylated Antibody and Radiolabeled Streptavidin. Cancer Res. 1994;54:2160–2165. [PubMed] [Google Scholar]
- 58.Paganelli G, Pervez S, Siccardi AG, Rowlinson G, Deleide G, Chiolerio F, Malcovati M, Scassellati GA, Epenetos AA. Intraperitoneal Radio-Localization of Tumors Pre-Targeted by Biotinylated Monoclonal Antibodies. Int J Cancer. 1990;45:1184–1189. doi: 10.1002/ijc.2910450632. [DOI] [PubMed] [Google Scholar]
- 59.Hassfjell S, Brechbiel MW. The development of the alpha-particle emitting radionuclides 212Bi and 213Bi, and their decay chain related radionuclides, for therapeutic applications. Chem Rev. 2001;101:2019–36. doi: 10.1021/cr000118y. [DOI] [PubMed] [Google Scholar]
- 60.Zidenberg-Cherr S, Parks NJ, Keen CL. Tissue and subcellular distribution of bismuth radiotracer in the rat: considerations of cytotoxicity and microdosimetry for bismuth radiopharmaceuticals. Radiat Res. 1987;111:119–29. [PubMed] [Google Scholar]
- 61.Kobayashi H, Wu CC, Yoo TM, Sun BF, Drumm D, Pastan I, Paik CH, Gansow OA, Carrasquillo JA, Brechbiel MW. Evaluation of the in vivo biodistribution of yttrium-labeled isomers of CHX-DTPA-conjugated monoclonal antibodies. J Nucl Med. 1998;39:829–836. [PubMed] [Google Scholar]
- 62.Roselli M, Milenic DE, Brechbiel MW, Mirzadeh S, Pippin CG, Gansow OA, Colcher D, Schlom J. In Vivo Comparison of CHX-DTPA Ligand Isomers in Athymic Mice Bearing Carcinoma Xenografts. Cancer Biother Radiopharm. 1999;14:209–20. doi: 10.1089/cbr.1999.14.209. [DOI] [PubMed] [Google Scholar]
- 63.Chen JQ, Strand SE, Brechbiel MW, Gansow OA, Sjoegren HO. Combination of biotinylation and indium-111 labeling with chelate SCN-Bz-CHX-A-DTPA for chimeric BR96: Biodistribution and pharmacokinetic studies in colon carcinoma isografted rats. Tumor Targeting. 1996;2:66–75. [Google Scholar]
- 64.Junghans RP, Dobbs D, Brechbiel MW, Mirzadeh S, Raubitschek AA, Gansow OA, Waldmann TA. Pharmacokinetics and Bioactivity of 1,4,7,10-tetra-azacylododecane N, N′, N″N‴-tetraacetic acid (DOTA)-Bismuth-conjugated Anti-Tac Antibody for α-Emitter (212Bi) Therapy. Cancer Res. 1993;53:5683–5689. [PubMed] [Google Scholar]
- 65.Caliceti P, Chinol M, Roldo M, Veronese FM, Semenzato A, Salmaso S, Paganelli G. Poly(ethylene glycol)-avidin bioconjugates: suitable candidates for tumor pretargeting. J Controlled Release. 2002;83:97–108. doi: 10.1016/s0168-3659(02)00199-2. [DOI] [PubMed] [Google Scholar]
- 66.Salmaso S, Semenzato A, Bersania S, Chinol M, Paganelli G, Caliceti P. Preparation and characterization of active site protected poly(ethyleneglycol)-avidin bioconjugates. Biochim Biophys Acta. 2005;1726:57–66. doi: 10.1016/j.bbagen.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 67.Smit JA, Myburgh JA, Neirinckx RD. Specific Inactivation of Sensitized Lymphocytes in Vitro Using Antigens Labelled with Astatine-211. Clin Exp Immunol. 1973;14:107–116. [PMC free article] [PubMed] [Google Scholar]
- 68.Aaij C, Tschroots WRJM, Lindner L, Feltkamp TEW. The Preparation of Astatine Labelled Proteins. Int J Appl Rad Isot. 1975;26:25–30. doi: 10.1016/0020-708x(75)90006-x. [DOI] [PubMed] [Google Scholar]
- 69.Visser GWM, Diemer EL, Kaspersen FM. The Nature of the Astatine-Protein Bond. Int J Appl Rad Isot. 1981;32:905–912. doi: 10.1016/0020-708x(81)90078-8. [DOI] [PubMed] [Google Scholar]
- 70.Visser GWM, Diemer EL, Kaspersen FM. The Preparation and Stability of Astatotyrosine and Astato-iodotyrosine. Int J Appl Rad Isot. 1979;30:749–752. [Google Scholar]
- 71.Savage MD, Mattson G, Desai S, Nielander GW, Morgensen S, Conklin EJ. Avidin-Biotin Chemistry: A Handbook. Pierce Chemical Company; Rockford, IL: 1992. [Google Scholar]
- 72.Vaughan ATM, Fremlin JH. The Preparation of Astatotyrosine. Int J Appl Rad Isot. 1977;28:595–598. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Numerical data for the four biodistributions conducted in this investigation are provided in Tables S1–S4. This information is available free of charge via the Internet at http://pubs.acs.org.