

Abstract
Store-operated channels (SOCs) mediate Ca2+ entry signals in response to endoplasmic reticulum (ER) Ca2+ depletion in most cells. STIM1 senses decreased ER luminal Ca2+ through its EF-hand Ca2+-binding motif and aggregates in near-plasma membrane (PM) ER junctions to activate PM Orai1, the functional SOC. STIM1 is also present in the PM, although its role there is unknown. STIM1-mediated coupling was examined using the stable EF20 HEK293 cell line expressing the STIM1-D76A/E87A EF-hand mutant (STIM1EF) deficient in Ca2+ binding. EF20 cells were viable despite constitutive Ca2+ entry, allowing study of SOC activation without depleting ER Ca2+. STIM1EF was exclusively in stable near-PM junctions, 3.5-fold larger than formed with STIM1WT. STIMEF-expressing cells had normal ER Ca2+ levels but substantially reduced ER Ca2+ leak. Expression of antiapoptotic Bcl-2 proteins (BCl-2, MCL-1, BCL-XL) were increased 2-fold in EF20 cells, probably reflecting survival of EF20 cells but not accounting for decreased ER Ca2+ leak. Surface biotinylation and streptavidin pull-down of cells expressing STIM1WT or STIM1EF revealed strong PM interactions of both proteins. Although surface expression of STIM1WT was clearly detectable, STIM1EF was undetectable at the cell surface. Thus, the Ca2+ binding-defective STIM1EF mutant exists exclusively in aggregates within near-PM junctions but, unlike STIM1WT, is not trafficked to the PM. Although not inserted in the PM, external application of a monoclonal anti-N-terminal STIM1 antibody blocked constitutive STIMEF-mediated Ca2+ entry, but only in cells expressing endogenous STIM1WT and not in DT40 STIM1 knock-out cells devoid of STIMWT. This suggests that PM-STIM1 may play a regulatory role in SOC activation.
Ca2+ signals control a vast array of cellular functions ranging from short term responses, such as contraction and secretion, to longer term regulation of cell growth and proliferation (1, 2). Receptor-induced Ca2+ signals involve two closely coupled components. The initial phase is a rapid, inositol 1,4,5-triphosphate-mediated release of Ca2+ from ER4 stores. The depletion of stores triggers Ca2+ entry across the plasma membrane through store-operated channels (SOCs) (3-5). The activation of SOCs is crucial to mediating longer term cytosolic Ca2+ signals and for replenishing intracellular stores (4, 5).
Recent studies have identified the type 1A transmembrane protein, STIM1, as a key mediator of SOC activation (6-9). The protein is believed to function as the sensor of Ca2+ within stores (6, 7). Located in the ER membrane, STIM1 contains an intraluminal N-terminal EF-hand Ca2+ binding domain, which is shown to have low affinity for Ca2+ with a Kd of ∼0.4 mm (10). Upon depletion of Ca2+ from stores, the STIM1 protein in the ER undergoes a profound change from a diffuse labeling pattern across the ER to an aggregated or “punctate” appearance (6, 7). The time course of this translocation of STIM1 correlates well with the activation of SOCs (7, 11). The aggregated STIM1 appears to be within ER that is closely associated with the plasma membrane (7, 11, 12). It was reported that STIM1 may become inserted into the PM following store depletion and hence activate SOCs (9). However, other studies have not confirmed this insertional model (7, 11, 13, 14). Despite this, there is good evidence that STIM1 translocates within the ER into junctional domains that are juxtaposed with the PM and close enough (within 10-25 nm) that direct interactions may occur with the plasma membrane (7, 11, 12). Further screens of gene products involved in the store-operated Ca2+ entry pathway revealed the role of the Orai1 (or CRACM1) protein (15-17). Orai1 is a tetraspanning membrane protein localized to the plasma membrane and appears to be the functional store-operated channel moiety (18-20). Orai1 coexpressed with STIM1 reconstitutes massive levels of the highly Ca2+-selective Ca2+ release-activated Ca2+ current (14, 17, 21, 22), the hallmark of store-operated channel function (3-5). Indeed, mutation of the Orai1 protein at key acidic residues alters the ion selectivity of the channel (18-20), indicating that Orai1 is the pore-forming entity in SOCs. Orai and STIM appear to co-migrate, possibly with other coupling proteins, to form junctional ER-PM coupling domains that are the functional sites of store-operated Ca2+ entry (11, 12).
Although Orai1 appears to be exclusively located in the PM, STIM1 is not exclusive to the ER. Indeed, STIM1 was originally discovered through a novel screening strategy to identify extracellularly expressed PM proteins in stromal cells promoting the proliferation of pre-B cells in culture (23). STIM1 was characterized as a plasma membrane protein involved in controlling cell growth (24-27). It was estimated in myeloid cell lines that as much as 25% of total STIM1 was expressed on the cell surface with its N terminus accessible to the cell exterior (26), although clearly the expression of STIM1 in the ER predominates over plasma membrane expression (13, 26). In the current study, we have addressed the question of the location, translocation, and function of STIM1 by generating stable cell lines expressing STIM1 with mutated acidic residues to prevent Ca2+ binding in the EF-hand domain. Earlier studies revealed that such mutants of STIM1 could constitutively activate store-operated Ca2+ entry, mimicking the store-depleted condition (7-9). Examination of the function of the EF-hand-mutated STIM1 molecule is particularly important in these cells, since the activation process for SOCs can be studied under conditions in which ER Ca2+ stores are not depleted, thus bypassing the severe stress effects of depleting Ca2+ from within the ER (1, 28). The establishment of cell lines with constitutively active STIM1 was remarkable, since the prolonged entry of Ca2+ would have been predicted to be highly damaging to the cells. Our results reveal that important compensatory Ca2+ homeostatic mechanisms probably counteract the increased entry, and we document that one such compensation is a turn-off of the powerful but enigmatic ER Ca2+ leak pathway. We establish that, unlike wild type STIM1, the STIM1 EF-hand mutant is exclusively expressed in the ER and can activate plasma membrane Orai1 without becoming inserted into the plasma membrane. Although not inserted in the membrane during store depletion, the STIM1 mutant undergoes a strong interaction with the plasma membrane. In contrast to the EF-hand mutant, wild type STIM1 is clearly expressed in the plasma membrane as well as ER. Although plasma membrane STIM1 may not be essential for Orai1 channel activation, the results indicate that it may play a role in regulating the activation of store-operated channels.
EXPERIMENTAL PROCEDURES
DNA Constructs and Transfections—Wild type (WT) human STIM1 and STIM2 were subcloned into pIRESneo (Clontech, Palo Alto, CA) as previously described (25). Human STIM1-D76A/E87A mutations were introduced using the QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by sequencing. Orai1 knockdown was achieved using stealth small interfering RNA (Invitrogen) targeting position 901 (UGGUGCCCUUCGGCCUGAUCUUUAU) or 1381 (CCUCCUCCUGUCCUGUCCGUCUCAA). A scrambled RNA sequence (UGGCCUCCUGGCCGUUAUCUGUUAU) was used as a control. DNA constructs and RNA sequences were introduced by electroporation using the Gene Pulser II electroporation system (Bio-Rad) at 350 V, 960 microfarads, and infinite resistance, followed by 48 h in culture.
Development of Stable Cell Lines—Human embryonic kidney 293 (HEK293) cells were maintained as previously described (13). HEK293 stable cell lines were generated by electroporation of the above described human STIM2 or STIM1 (WT or D76A/E87A) constructs, followed by selection with G418 and cloning as previously described (12). Positive clones were selected based upon STIM1 expression and the level of store-operated Ca2+ entry.
Intracellular Ca2+ Measurements—Ratiometric imaging of intracellular Ca2+ using fura-2 was as previously described (29). Cells grown on coverslips were placed in cation-safe solution (107 mm NaCl, 7.2 mm KCl, 1.2 mm MgCl2, 11.5 mm glucose, 20 mm Hepes-NaOH, pH 7.2) and loaded with fura-2/AM (2 μm) for 30 min at 20 °C. Cells were washed, and dye was allowed to de-esterify for a minimum of 30 min at 20 °C. Approximately 95% of the dye was confined to the cytoplasm, as determined by the signal remaining after saponin permeabilization (30). Ca2+ measurements were made using an InCyt dual wavelength fluorescence imaging system (Intracellular Imaging Inc.). Fluorescence emission at 505 nm was monitored with excitation at 340 and 380 nm; intracellular Ca2+ measurements are shown as 340/380 nm ratios obtained from groups (35-45 cells each) of single cells. Resting intracellular free Ca2+ concentration was determined based on ratiometric measurements of cells maintained in 1 mm Ca2+ and calculated according to the following formula of Grynkiewcz et al. (31),
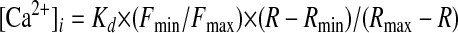 |
(Eq.1) |
where R represents the ratio of the fluorescence intensities measured at 340 and 380 nm during the experiments, and F is the fluorescence intensity measured at 505 nm. Rmin, Rmax, Fmin, and Fmax were determined from in situ calibration of unlysed cells using 40 μm ionomycin in the absence (Rmin and Fmin; 10 mm EGTA) and presence (Rmax and Fmax) of Ca2+. Kd (135 nm) is the dissociation constant for fura-2 at room temperature. Measurements shown as means ± S.E. of traces from ∼30-40 individual cells and are representative of a minimum of three and in most cases a larger number of independent experiments.
Luminal ER Ca2+ Measurements—CFP (436Ex/480Em) and FRETraw (436Ex/535Em) were imaged at 5 mHz in HEK293 cells transfected with the ER-targeted cameleon, D1ER (32), using a Leica DMI 6000B fluorescence microscope controlled by Slide-Book 4.2 software (Olympus). Two-channel corrected FRET was calculated based on the formula,
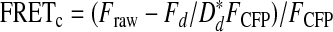 |
(Eq.2) |
where FRETc represents the corrected total amount of energy transfer, Fraw represents measured fluorescence measured through the CFP/YFP ET filter cube, FCFP represents measured CFP fluorescence, and Fd/Dd represents measured bleed-through of CFP through the YFP filter (0.592248). In order to determine absolute Ca2+ concentration, maximum FRETc (Rmax) was measured via the addition of 10 mm Ca2+ and 1 mm Mg2+/ATP to digitonin (25 μm)-permeabilized cells, whereas minimum FRETc was measured upon the subsequent addition of 3 μm Br-A23187 with 5 mm EGTA (33). ER Ca2+ content was then determined based on the following formula of Palmer and Tsien (33),
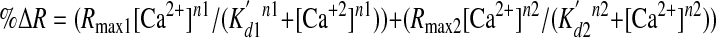 |
(Eq.3) |
in which %ΔR = (R - Rmin)/Rmax - Rmin)*100, K′d1 = 0.58, K′d2 = 56.46, Rmax1 = 28, Rmax2 = 72, n1 = 1.18, and n2 = 1.67. Immunoprecipitation and Cell Lysis—Cells were lysed in Nonidet P-40 lysis buffer (1% (w/v) Nonidet P-40, 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, with protease inhibitors), cleared by centrifugation, and normalized for protein content. For immunoprecipitation, lysates (200 μg) were mixed with 50 μl of a 50% protein G slurry (Calbiochem) that was precoated for 1 h with the indicated antibodies (5 μl/sample). The samples were incubated for 2 h at 4 °C with rotation and were then washed three times with lysis buffer. After the final wash, beads were resuspended in gel-loading buffer and boiled for 5 min.
Surface Biotinylation—Biotinylation studies were performed on HEK293 cells using NHS-LC-Biotin (1 mg/ml; Pierce) for 30 min. Unbound biotin was quenched with Tris (75 mm) prior to lysis. Biotinylated proteins were either run on gels as whole lysates (15 μg/lane) or separated from unbiotinylated proteins using streptavidin-coated beads (100 μg/lane). STIM1 and STIM2 were detected by Western analysis using an anti-STIM1 monoclonal antibody that cross-reacts with STIM2 (BD Biosciences) (25). The samples were incubated for 2 h at 4 °C with rotation and then washed three times with lysis buffer, resuspended in gel-loading buffer, and boiled for 5 min before loading on gels.
Western Blot and Flow Cytometry—For Western blots, proteins were resolved on 6% SDS-polyacrylamide gels, transferred to nitrocellulose paper, and analyzed with the indicated antibodies, as previously described (29). For flow cytometry, HEK293 cell lines were labeled with anti-STIM1 (BD Biosciences), followed by phycoerythrin-conjugated anti-mouse IgG (Invitrogen). Cells were analyzed on a FACScan (BD Biosciences), using Cellquest software (BD Biosciences). Results are histograms based upon 10,000 events/sample. Each experiment was repeated at least three times.
Immunocytochemistry—HEK293 cells were transfected as described and plated onto 10-mm circular coverslips in OPTI medium supplemented with 10% fetal bovine serum, as previously described (13). Fixation and permeabilization were achieved by incubation (10 min) with 3.7% formaldehyde followed by permeabilization in the following buffer: 100 mm PIPES, 1 mm EGTA, 0.1% Triton X-100, 4% polyethylene glycol 8000, pH 6.9, with KOH. After blocking in bovine serum albumin (0.5%), cells were blocked with BSA and labeled with affinity-purified rabbit anti-STIM1 CT (1:50 dilution), followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:50 dilution; Invitrogen). For nonpermeabilized cells, labeling with antibodies was undertaken on live cells, followed by fixation with formaldehyde. Fixed and labeled cells were mounted onto slides in Cytoseal 60 (VWR) and examined with a Zeiss LSM410 confocal laser-scanning microscope, using a ×63/numerical aperture 1.4 objective.
TIRF Microscopy—HEK293 cells were transfected with YFP-STIM1-WT or YFP-STIM1-D76A and plated onto 25-mm circular coverslips in OPTI medium supplemented with 10% fetal bovine serum, as previously described (13). On the day of the experiment, cells were transferred to cation-safe solution. The images were recorded on a high resolution Hamamatsu ORCA-ER camera using a through-the-lens ×60 TIRF objective in a Nikon Eclipse TE2000U microscope while exciting with a 488-nm argon ion laser set to TIRF mode. Store depletion was achieved with the addition of thapsigargin (2 μm) while recording. Image analysis of puncta was undertaken using Image J and SlideBook 4.2 (Olympus). Background was subtracted from the raw image, and noise (less than 10 consecutive pixels) was filtered out. Images were imported into SlideBook, and the size and number of puncta were measured using the mask function. The density of puncta was obtained by dividing the number of puncta by the size of the cell. Relative cell size was measured from cell profile using Image J.
Quantitative PCR—Real time quantitative PCR was performed in a model 7300 sequence detector (Applied Biosystems) using Superscript II Platinum SYBR Green one-step quantitative reverse transcription-PCR kits, and the 7300 System SDS Software. Reaction mixtures contained reverse transcriptase and TaqDNA polymerase in a single enzyme mix with SYBR Green I fluorescent dye, ROX as reference dye, 30 ng of total RNA, and gene-specific primers. Both cDNA synthesis and PCR were performed in a single tube. Total RNA was isolated using TRIzol reagent (34). Primers (forward and reverse) designed using Primer Express 2.0 software (Applied Biosystems) were as follows: Bcl-2, TGCGGCCTCTGTTTGATTTC and GGGCCAAACTGAGCAGAGTCT; BCL-XL, CCATGGCAGCAGTAAAGCAA and CCGGTACCGCAGTTCAAACT; MCL-1, GCATCGAACCATTAGCAGAAAGT and GCCAGTCCCGTTTTGTCCTT; BIM, GCCCAGCACCCATGAGTT and GCCTGGCAAGGAGGACTTG; glyceraldehyde-3-phosphate dehydrogenase, ATGGAAATCCCATCACCATCTT and CGCCCCACTTGATTTTGG. The standard program included one cycle of 30 min at 50 °C for synthesis of cDNA, and amplification included 40 cycles of two steps, each comprising heating to 95 °C and heating to 60 °C, each 0.15 min and 1 min, respectively. Fluorescent product was detected at the last step of each cycle. To verify purity of products, polyacrylamide DNA gels were run using a DNA standard marker. For each RNA set, a control was run with total RNA to check for any DNA contamination. To determine the relative quantitation of gene expression, the comparative threshold cycle method (ΔCt) was used (35). The final mRNA levels were normalized according to their Ct values from the standard curves and expressed in relation to respective glyceraldehyde-3-phosphate dehydrogenase level.
Materials—Peptide affinity-purified specific antibodies to STIM1-NT and STIM1-CT (used for immunocytochemistry and immunoprecipitation) were produced by Chiron Technologies (Clayton, Victoria, Australia), as previously described (25). The anti-STIM1 antibody used for Western blots was from BD Biosciences (San Diego, CA). Horseradish peroxidase-conjugated goat anti-rabbit and rabbit anti-mouse antibodies and Texas Red-conjugated donkey anti-sheep antibodies were from Jackson Laboratories. AlexaFluor488-conjugated goat anti-rabbit IgG was from Invitrogen. Thapsigargin was from EMD Biosciences (San Diego, CA). Carbachol and G418 were from Sigma. Fura-2/AM was from Molecular Probes, Inc. (Eugene, OR).
RESULTS AND DISCUSSION
Generation of Viable, Stable STIM1 EF-hand Mutant-expressing Cells—The STIM1 protein plays an essential role in the activation of store-operated Ca2+ entry (6-9). It is believed to function as the sensor of luminal Ca2+ and probably transduces the store depletion signal to the plasma membrane through translocation within the ER into areas closely juxtaposed with the plasma membrane (7, 7-9, 36). As described above, the protein is expressed not only in the ER but also in the plasma membrane (23-27), although its role in this membrane is not yet clear. Within the ER membrane, STIM1 has a luminal facing Ca2+-sensing EF-hand motif. Upon Ca2+ store depletion, Ca2+ dissociation from the EF-hand results in a conformational change in the STIM1 molecule (10), leading to aggregation and translocation toward the plasma membrane (7, 10-12, 36, 37). Expression of EF-hand-mutated STIM1 results in constitutive SOC activation without emptying stores (7-9, 13), allowing examination of the coupling process to activate SOCs without the stress effects of ER Ca2+ store depletion (1, 28). To study this constitutive SOC coupling process, we sought to develop HEK293 cell lines stably expressing the E76A/D87A double mutant of STIM1 with a defective EF-hand Ca2+ binding site (STIM1EF). STIM1EF-expressing clonal cell lines were generated by transfecting the STIM1-E76A/D87A mutant within the pIRES-neo vector, selection in G418 medium, and cloning procedures previously described (13). A number of clones of cells expressing high levels of the mutant STIM1 protein were identified by Western analysis using an N-terminal STIM1 antibody (Fig. 1A). Expression levels of STIM1EF protein were similar to those of a control cell line expressing wild type STIM1 (Fig. 1A). Ca2+ entry in three separate clonal lines expressing STIM1EF mutant was compared with that in control-transfected HEK293 cells or in HEK293 cells expressing similar amounts of wild type STIM1 protein (Fig. 1B). Each of the cell lines expressing the STIM1-D76A/E87A construct exhibited constitutive Ca2+ entry, with clone EF20 showing the largest and most rapid entry. In contrast, control empty vector-expressing cells or cells expressing wild type STIM1 displayed almost no constitutive Ca2+ entry (Fig. 1B).
FIGURE 1.

Characterization of STIM1 EF-hand mutant and wild type clonal cell lines. HEK 293 cells were transfected with the STIM1 EF hand mutant (D76A/E87A), selected with G418, and cloned as described under “Experimental Procedures.” A, Western analysis of STIM1 EF-hand mutant-expressing cell clones (EF4, EF20, and EF21) in comparison with expression of wild type STIM1-expressing clone (STIM1) and a clone expressing only the pIRES vector control (Vector). B, constitutive Ca2+ entry in STIM1 EF-hand mutant clones and control cells shown in A was assessed by the addition of 1 mm Ca2+ to cells incubated in Ca2+-free solution for 10 min. Results are shown as means ± S.E. of traces from 30-40 individual cells. C-F, immunocytochemical localization of wild type and STIM1-D76A/E87A EF-hand mutant in clonal expressing HEK293 cell lines. C, confocal images of WT STIM1 distribution in a stably expressing cell line. D-F, confocal images of the distribution of the STIM1 EF-hand mutant in stably expressing clones, EF4, EF20, and EF21, as shown. Cells were fixed, permeabilized, blocked, and hybridized with rabbit anti-STIM1-CT antibody and goat anti-rabbit secondary antibody, as described under “Experimental Procedures.”
The successful generation of cells stably expressing the constitutively active STIM1 EF-hand mutant was surprising. It would have been expected that the substantial constitutive entry of Ca2+ would be detrimental to cell survival. We measured resting Ca2+ levels in the EF20 mutant cell line, which were ∼130 nm under normal conditions of extracellular Ca2+ (data not shown). This was significantly higher than with control cells, in which resting Ca2+ levels were in the 20-50 nm range. In the EF20 line, readdition of Ca2+ following treatment with nominally Ca2+-free medium resulted in a transient increase in Ca2+ well above the resting level, returning within a few minutes to close to resting levels (Fig. 1B). This overshoot in Ca2+ is typical of that seen in cells with fully depleted Ca2+ stores (3, 4, 30, 38). Although large increases in Ca2+ were observed in all lines expressing the EF-hand mutant, the return to lower levels was not necessarily as rapid (e.g. clones EF4 and EF21). In the EF21 cell line, the rate of Ca2+ entry was also slower; the reason for this is unclear and may reflect an adaptive response to constitutive Ca2+ entry. We examined whether the levels of Ca2+ pumps (both the SERCA pump in the ER and PMCA in the plasma membrane), might have been altered to compensate for increased constitutive Ca2+ entry. Western analysis of both Ca2+ pump proteins indicated that there was no alteration in the expression of either pump in the clonal lines (data not shown). This does not rule out the possibility that existing pumps in the mutant lines could be redistributed closer to the sites of Ca2+ entry and mediate more efficient Ca2+ extrusion.
STIM1 EF-hand Mutant-expressing Cells Have Permanently Junctional STIM1—Using immunocytochemistry, we examined the cellular distribution of both wild type STIM1 and the STIM1EF mutant using a STIM1 N-terminal antibody. As shown in Fig. 1C, wild type STIM1-expressing HEK293 cells revealed a labeling pattern consistent with distribution in the ER and probably also the PM. In contrast to this, the three HEK293 cell lines expressing the STIM1EF mutant showed a totally aggregated and punctal distribution of the protein (Fig. 1, D-F). This is consistent with previous studies in which the distribution of EF-hand-mutated STIM1 mimicked the distribution of wild type STIM1 following depletion of stores (7, 13). Thus, the stable EF-hand mutant-expressing cells are growing continuously with what appears to be entirely redistributed STIM1 protein. Significantly, in the EF-hand mutant expressing cells, there was no obvious appearance of the protein within the plasma membrane (Fig. 1, D-F) in contrast to the distribution of wild type STIM1 (Fig. 1C). In resting cells, wild type STIM1 is normally widely distributed over the ER (7). If the redistribution of STIM1 into puncta in the EF-hand mutant-expressing cells represented a corresponding redistribution of the entire ER, this would indicate a gross alteration of ER structure and function. The fact that the cells are relatively normal in appearance and in growth properties would argue this is not the case. Indeed, it appears that the store emptying-induced redistribution of STIM1 is through protein translocation within the ER into specific local regions close to the plasma membrane, without alteration of the overall distribution of ER markers (7, 39).
Large, Stable ER-PM Junctions Exist in STIM1EF-expressing Cells—Earlier studies indicated that upon store depletion, STIM1 within the ER undergoes translocation into areas closely juxtaposed with the plasma membrane (7, 9). Using HEK293 cells transiently expressing YFP-labeled STIM1, we undertook TIRF microscopy to assess the presence of STIM1 within the region ∼100 nm from the PM (Fig. 2). Under store-replete conditions, TIRF analysis revealed only a diffuse presence of YFP-labeled STIM1 within the TIRF field (Fig. 2, A and C). Following store depletion, there was a clear increase in the appearance of YFP-STIM1 within the TIRF field (Fig. 2, B and D). The label appears in clearly defined areas, consistent with the movement of STIM1 into puncta close to the plasma membrane. The time course for the relocation of STIM1 closely follows the time of store-emptying, substantial redistribution occurring within 100 s (Fig. 2B). It seems that puncta increase in size with time subsequent to store emptying. Thus, at 100 s, the puncta are relatively small (Fig. 2B), whereas they have considerably enlarged at 240 s (Fig. 2D). In HEK293 cells expressing the YFP-STIM1-D76A mutant, the STIM1 protein is constitutively within near-surface puncta (Fig. 2F) as compared with cells expressing wild type YFP-STIM1 (Fig. 2E). Consistently, the size of punctal areas of EF-hand mutant STIM1 in the TIRF field were even larger than with wild-type STIM1 following store depletion, appearing as structures ∼1 μm in diameter (Fig. 2F). From image analysis, the size of the punctal areas in the STIM1 EF-hand mutant-expressing cells was more than 3-fold greater than the puncta seen in thapsigargin-treated control cells (Fig. 2G), whereas there was little change in the punctal density across the whole cell (Fig. 2H). It seems that the punctal structures may represent substantial areas of interaction between internal membranes and the plasma membrane. Thus, the mutant EF-hand-expressing cells may exist with a substantially altered architecture. Wu et al. (39) revealed that some of the punctal domains into which STIM1 is translocated may be preexisting interactional domains, although de novo interacting domains are also formed following store depletion. The results here suggest that the domains may become more extensive in size with time. Although we cannot tell what proportion of the interactions preexist in normal cells, certainly the mutant EF-hand-expressing cells exist with extensive and perhaps permanent areas of ER-PM membrane interaction.
FIGURE 2.

TIRF microscopy of the distribution of YFP-labeled wild type and EF-hand mutant STIM1. A-D, HEK293 cells expressing either YFP-labeled WT STIM1 or YFP-labeled EF-hand mutant STIM1-D76A were examined by TIRF microscopy either immediately before (pre-TG) or at the times shown after the addition of 2 μm thapsigargin (TG). E and F, comparison of HEK293 cells transiently expressing either YFP-labeled WT STIM1 or YFP-labeled STIM1-D76A under identical conditions in store-replete HEK293 cells. G and H, statistical analysis of the relative size of puncta (G) and the overall density of puncta (H) in the images of either STIM1 EF-hand mutant-expressing cells (EF STIM1; from F) or control cells 240 s after thapsigargin (control after TG; from D). Details of the image analysis are given under “Experimental Procedures.”
The Ca2+ Leak Pathway Is Deactivated in EF-20 Cells—The fact that substantial ER-PM junctional areas exist in the STIM1EF mutant-expressing cells suggested that functional coupling between Ca2+ stores and the PM might be altered. We therefore examined the extent of releasability of Ca2+ stores in EF20 cells in response to SERCA pump blockade, PLC-coupled receptor activation, or Ca2+ ionophore application. In control cells, application of the SERCA blocker, thapsigargin, resulted in a substantial release of Ca2+ from stores (Fig. 3A). Surprisingly, the thapsigargin-induced release of Ca2+ in EF20 cells was ∼80% smaller (Fig. 3D). In these experiments, Ca2+ was transiently added before thapsigargin; only in the EF20 cells was there a substantial entry of Ca2+ due to constitutively active SOCs. The small Ca2+ release in EF20 cells suggested a large reduction in stored Ca2+ in these cells. This was unexpected, especially since the prior induction of Ca2+ entry would have been expected to increase rather than decrease store filling. We considered that the prior Ca2+ entry seen in Fig. 3D might somehow have induced store depletion. However, the thapsigargin-induced Ca2+ release response was similarly reduced without the prior addition of extracellular Ca2+ (data not shown). To determine whether Ca2+ stores were indeed depleted in EF20 cells, we compared the extent of Ca2+ release induced by either the PLC-coupled muscarinic agonist, carbachol (Fig. 3, B and E) or the ionophore, ionomycin (Fig. 3, C and F). The addition of carbachol activated rapid InsP3-mediated Ca2+ release, the rate and extent of which were almost identical in control and EF20 cells. Similarly, the rate and extent of ionomycin-induced Ca2+ release were almost the same in the two cell types. With both carbachol and ionomycin, the release of Ca2+ was not affected by the prior transient addition of Ca2+ (data not shown).
FIGURE 3.

EF20 cells expressing the STIM1-D76A/E87A EF-hand mutant have a substantially reduced thapsigargin-induced leak of Ca2+ from ER. Ca2+ release from stores was induced in either control-transfected HEK293 cells (A-C) or EF20 cells expressing the STIM1EF-mutant (D and E). Cytoplasmic Ca2+ release from stores was monitored with fura-2 in response to 2 μm thapsigargin (TG) after prior transient addition of 1 mm external Ca2+ (A and D) or after the addition of either 10 μm carbachol (B and E) or 2 μm ionomycin (C and F). Results are shown as means ± S.E. of traces from 15-30 individual cells. G and H, ER luminal Ca2+ in pIRES control cells (G) and EF20 cells (H) was measured either in the absence or presence of 1 mm extracellularly Ca2+. Cells were transiently transfected with the ER-specific cameleon, D1ER, and ER luminal Ca2+ was measured by two-channel FRET as described under “Experimental Procedures.”
The Ca2+ release results with InsP3 and ionomycin suggested that loading of Ca2+ within the ER was not altered in EF20 cells. However, it was important to assess this directly by measuring Ca2+ levels within the ER lumen. We transfected EF20 cells and pIRES control cells with the ER-targeted cameleon, D1ER (32), and measured ER luminal Ca2+, as described by Palmer and Tsien (33) (see “Experimental Procedures”). As shown in Fig. 3 (G and H), the levels of Ca2+ in pIRES control cells and EF20 cells were very similar. We measured Ca2+ levels both in the absence and presence of extracellular Ca2+ to determine whether the constitutive entry of Ca2+ mediated by the STIM1EF influenced luminal ER Ca2+ levels. For control cells, ER Ca2+ in nominally free external Ca2+ was 124.9 ± 9.0 μm in control cells and 122.9 ± 5.6 μm in the presence of 1 mm external Ca2+. In EF20 cells, luminal ER Ca2+ levels were 131.5 ± 12.2 and 173 ± 17.3 μm in the absence and presence of extracellular Ca2+, respectively. These results indicate that the size of Ca2+ stores is effectively the same in the two cell types. Therefore, the change in releasability following Ca2+ pump blockade by thapsigargin suggests that the presence of the STIM1EF mutant causes reduced leak of Ca2+ from ER stores.
The leak of Ca2+ that follows thapsigargin-induced pump blockade has been defined as an important “enigma of signaling” (40). Several candidate proteins have been reported to mediate ER Ca2+ leak, including the antiapoptotic Bcl-2 protein (41), the ribosome-translocon protein complex (42, 43), the InsP3 receptor (44), and recently, the presenilin 1 and presenilin 2 proteins (45). Since complete elimination of InsP3 receptors in DT40 B cells has no effect on leak (46), the InsP3 receptor is an unlikely leak mediator. Our results indicate that permanent rearrangement of the STIM1 protein occurring in the EF20 cells has substantially deactivated the leak pathway. Normally, rearrangement of STIM1 into junctions and leak of Ca2+ from stores both occur following treatment of cells with thapsigargin; thus, aggregation of STIM1 per se does not cause leak turn-off. Instead, the results suggest that the leak pathway becomes deactivated with the permanent rearrangement of STIM1 into junctional aggregates in the EF20 cells. Leak pathway down-regulation might be expected as a compensatory mechanism to control increased Ca2+ resulting from permanent coupling to activated SOCs. Indeed, the observation that EF20 cells maintain only modestly elevated resting cytosolic Ca2+ levels despite constitutive Ca2+ entry may reflect turn-off of the leak pathway. Thus, by monitoring the expression and distribution of candidate leak proteins, the EF20 cells may provide a useful system to identify the leak-mediating machinery.
Expression of Antiapoptotic Bcl-2 Family Members in EF-20 Cells—We also considered that survival of the EF20 cells despite constitutive Ca2+ entry might be related to expression of the antiapoptotic Bcl-2 family of proteins (47). Indeed, the antiapoptotic proteins Bcl-2 (41) and Bcl-XL (48) have been shown to increase ER Ca2+ leak, whereas proapoptotic Bcl-2 family members counteract this activity (48). Based on this, we quantified the level of expression of three antiapoptotic Bcl-2 family members (Bcl-2, Bcl-XL, and MCL-1) and one proapoptotic Bcl-2 family member (BIM) by quantitative PCR (Fig. 4). If changes in Bcl-2 family member expression accounted for the decrease in ER Ca2+ permeability observed in EF-20 cells, then expression of the antiapoptotic members of the Bcl-2 family might be expected to decrease and/or expression of proapoptotic Bcl-2 family members would be expected to go up. In contrast, we observed that expression levels of all three of the antiapoptotic Bcl-2 family members examined increased by ∼2-fold, whereas there was no observable change in the level of expression of BIM. Hence, we cannot account for decreased ER Ca2+ permeability by changes in the level of expression of Bcl-2 family members. However, the observed increase in the expression of antiapoptotic proteins may provide some insight into how EF-20 cells are able to survive and proliferate despite constitutively activated Ca2+ channels.
FIGURE 4.

Expression of antiapoptotic Bcl-2 family members in EF-20 cells. The relative expression levels of several antiapoptotic (left) and proapoptotic (right) Bcl-2 family members was measured from total RNA extracted from EF20 and pIRES control cells maintained under optimal conditions by quantitative PCR. Primers were designed to amplify 50-100-bp sections of each gene using Primer Express 3.0 software (Applied Biosystems). Total mRNA content is expressed as arbitrary units normalized to glyceraldehyde-3-phosphate dehydrogenase (×1000). Results are shown as means ± S.E. (n = 3). Statistical significance was determined based on paired Student's t test (*, p < 0.05).
Surface Biotinylation of the EF-hand-mutated STIM1—STIM1 was first identified as a cell surface protein; indeed, it was originally discovered through a novel screening strategy to identify external PM proteins on stromal cells (23). STIM1 is expressed more within the ER than in the plasma membrane (13, 26), and its luminal Ca2+-sensing role in the ER appears well defined. However, it has been suggested that STIM1 in the plasma membrane may also play a role in the activation of SOCs (8, 9). Therefore, we sought to assess whether the stably expressed EF-hand mutant had a similar PM presence and role. We assessed the presence of the mutated STIM1 protein in the PM by undertaking surface biotinylation and streptavidin pull-down. Two lines of HEK293 cells expressing the E76A/D87A double mutant were treated with biotin to label surface proteins. After binding to streptavidin beads, biotinylated proteins were assessed by Western analysis using an N-terminal antibody that interacts with both STIM1 and its close homologue STIM2. As shown in Fig. 5A, the whole lysate from cells reveals the total extent of expression of the STIM1EF mutant protein in the EF20 and EF21 cell lines, as compared with cells stably expressing either wild type STIM1 or wild type STIM2. After pull-down with streptavidin, both wild type STIM1 and STIM1EF are present in the extract from beads, whereas STIM2 was almost undetectable in streptavidin pull-downs (Fig. 5B). The inference from this study would be that both wild type STIM1 and the STIM1EF are present at the plasma membrane surface, where they can become biotinylated. Certainly, the lack of STIM2 in the pull-downs is consistent with our previous observations that STIM2 is not expressed in the plasma membrane (13) despite the similarity in its structure with STIM1.
FIGURE 5.

Biotinylation and streptavidin pull-down of STIM1-WT, STIM1-D76A/E87A-, and STIM2-expressing clonal HEK293 cell lines. Details of biotinylation, streptavidin pull-downs, and detection of STIM1 and STIM2 using a monoclonal antibody cross-reacting with STIM1 and STIM2 are described under “Experimental Procedures.” A, Western analysis of whole lysates of biotinylated samples from STIM1-D76A/E87A mutant cell clones EF21 (lane 1), EF20 (lane 2), or stable lines expressing WT STIM1 (lane 3), WT STIM2 (lane 4), or empty pIRES vector (lane 5). B, Western analysis of streptavidin pull-downs of samples described in A.
The STIM1EF Mutant Is Tightly Associated with but Not Inserted in the Plasma Membrane—We sought to confirm the plasma membrane insertion of the EF-hand mutant of STIM1 by undertaking immunodetection of the protein at the cell surface by both flow cytometry and immunofluorescence. Indeed, we considered this important, since in the immunocytochemical staining in Fig. 1, it appeared as if there was little surface expression of the STIM1EF mutant protein, in contrast with the biotinylation data. We subjected the stably expressing EF20 cell line to analysis by flow cytometry after labeling with the STIM1 N-terminal antibody recognizing the extracellular domain of STIM1 expressed in the PM. As shown in Fig. 6A, we were unable to detect significant surface expression of the D76A/E87A STIM1 mutant. The detection peak was exactly aligned with that seen using the control pIRES vector-expressing cell line (Fig. 6A). Indeed, there was no significant difference (p > 0.05) in the labeling of these cells and those not overexpressing STIM1 (Fig. 6B). In contrast, stable wild type STIM1-expressing cells had clearly identifiable cell surface STIM1 (Fig. 6B).
FIGURE 6.

The STIM1-D76A/E87A mutant does not get inserted into the plasma membrane. A and B, flow-cytometric comparison of surface expression of STIM1-WT and the STIM1-D76A/E87A in clonal HEK 293 cell lines. Cells were stained with a primary mouse antibody against extracellular N-terminal STIM1, the phycoerythrin-conjugated goat anti-mouse secondary antibody. A, fluorescence-activated cell sorting histograms for cells stably transfected with either STIM1-WT (blue), STIM1-D76A/E87A (red), or empty pIRES vector (black). B, statistical analysis of three separate fluorescence-activated cell sorting analyses for STIM1-WT (blue), STIM1-D76A/E87A (red), or empty pIRES vector (black), showing mean ± S.E. in comparison with vector control. C-F, surface and intracellular immunocytochemical staining to compare expression of STIM1-WT and STIM1-D76A/E87A EF-hand mutant. C-E, nonpermeabilized cells expressing either pIRES-vector, STIM1-WT, or STIM1-D76A/E87A were first stained with a primary sheep antibody directed against the extracellular N-terminal domain of STIM1, followed by a fluorescent goat anti-sheep secondary antibody, and then fixed. F-H, cells from the same three stable lines were fixed, permeabilized, and stained with the same sheep anti-STIM1 N-terminal antibody and fluorescent goat anti-sheep secondary antibody. Details are described under “Experimental Procedures.”
These results were consistent with immunofluorescence data examining the direct surface labeling of STIM1 and comparing this with intracellularly expressed STIM1 by permeabilizing the cells (Fig. 6, C-H). In these experiments, we performed confocal imaging comparing permeabilized and nonpermeabilized cells using a sheep STIM1 N-terminal antibody reactive with the extracellular domain of STIM1. Intact cells expressing wild type STIM1 showed clear labeling on their surface (Fig. 6D). In contrast, no significant surface labeling could be detected using the EF20 cells expressing the D76A/E87A mutated STIM1 (Fig. 6E). However, after permeabilization of cells, the immunoreactivity of STIM1 in wild type STIM1-expressing cells (Fig. 6G) and EF20 cells (Fig. 6H) were very similar. From this, it is clear that there is little difference in the total expression or in situ labeling efficiency of wild type STIM1 as opposed to the STIM1-D76A/E87A mutant, consistent with the Western data in Fig. 1A. Therefore, we can be confident that the STIMEF mutant is starkly different from wild type STIM1 in not getting to the plasma membrane. Whereas the STIMEF mutant is not expressed in the plasma membrane, the surface labeling of wild type STIM1 had a punctal appearance (Fig. 6D), suggesting that it is not uniformly distributed in the plasma membrane.
The flow-cytometric and immunocytochemical results together indicate that the EF-hand mutation prevents the entry of STIM1 into the PM. Several previous studies on expression of STIM1 had concluded that the protein does not get into the plasma membrane (7, 11, 14, 49). However, each of these studies utilized N-terminally tagged derivatives of STIM1, either GFP, YFP, HA, or horseradish peroxidase (7, 11, 14, 49). In their recent study, Hauser and Tsien (50) provided direct evidence that these larger tags on the N terminus prevented any expression of STIM1 in the PM. Using the smaller His6-tagged N-terminal derivative of STIM1, they showed that the STIM1 protein was clearly observable in the plasma membrane. A significant conclusion in the present study is that the untagged but constitutively active STIM1EF mutant also does not reach the plasma membrane. This provides yet further evidence against the “insertional” model for the STIM1-mediated activation of SOCs. Thus, it was suggested that STIM1 becomes inserted into the plasma membrane, and the insertion may be necessary for activation of SOCs (9). If the STIM1EF mutant is sufficient for activating SOCs, then it is reasonable to conclude that the process of insertion is unlikely to be part of the SOC activation process. Indeed, it is interesting that the STIM1EF mutant, which exists constitutively within puncta very close to the plasma membrane, never actually finds its way into the plasma membrane. Probably, its predominant existence within large aggregates in the ER prevents STIM1 from trafficking to the plasma membrane. In contrast, the wild type protein gets expressed in significant quantities within the plasma membrane, indicating that its trafficking and insertion in the plasma membrane is independent of its entry into puncta. This is an important point and emphasizes that insertion into the PM is independent of the close association that occurs during store emptying.
The flow cytometry and immunofluorescence labeling studies conclusively revealed the absence of EF-hand-mutated STIM1 in the plasma membrane. In apparent contradiction to this, the biotinylation data (Fig. 5) indicated that the EF-hand-mutated STIM1 protein was present at the cell surface. The explanation for this divergence is important. The biotinylation experiments do not distinguish between actual biotinylated surface proteins and nonsurface proteins that may exist in tight membrane-associated complexes with biotinylated proteins. Thus, the streptavidin pull-down contains not only directly biotinylated proteins but also proteins that are tightly associated with the biotinylated proteins. Hence, we may conclude that the EF-hand-mutated protein exists in tight association with a cell surface component.
Wild-type STIM1 and the STIMEF Mutant Can Still Cross-react—It appears clear that interactions between STIM1 molecules are important in store-induced activation of SOCs (7, 9, 10, 13). Clearly, homomeric interactions between STIM1 molecules within the ER membrane play an important role in the initial response to store depletion (10). We examined whether the exclusively ER-located STIM1 EF-hand mutant was able to undergo interactions with wild type STIM1. Thus, whereas the mutant is in the Ca2+-dissociated configuration and hence is constitutively aggregated and active, native STIM1 in the same cells is in the inactive, nonaggregated Ca2+-bound configuration. Therefore, we assessed whether the two proteins could interact under store-replete conditions. As shown in Fig. 7, there is a clear interaction between wild type STIM1 and the D76A/E87A STIM1EF mutant. In this study, vector control cells (pIRES), WT STIM1-expressing cells, or STIMEF mutant-expressing EF20 cells were transiently transfected with YFP-STIM1. Pull-down of untagged STIM1 was then examined using anti-STIM1 antibodies after immunoprecipitation with a rabbit anti-GFP antibody. Although no STIM1 was pulled down by the GFP antibody without expression of YFP-STIM1 in EF20 cells (lanes 1 and 5), similar amounts of STIM1 were pulled down in STIM1-WT-expressing and EF20 cells after YFP-STIM1 transfection (lanes 7 and 8). Hence, the EF-hand mutant STIM1 protein is able to interact with wild type STIM1 equally well, since wild type STIM1 is able to undergo interaction with itself. Thus, the aggregation process and translocation of the EF-hand mutant into junctions does not prevent nonaggregated wild type STIM1 from interacting with it. This suggests that native STIM1 is not excluded from ER-PM junctions and probably is diffusing in and out of junctional areas.
FIGURE 7.

Coimmunoprecipitation reveals that wild type YFP-STIM1 interacts with the STIM1-D76A/E87A mutant. Stable HEK293 cell lines expressing either empty vector (pIRES), WT STIM1, or STIM1-D76A/E87A were either transiently transfected with YFP-STIM1 or not, as shown. Whole cell lysates (A) or samples that had been pulled down with an anti-GFP antibody (B) were analyzed by Western blot using an anti-N-terminal STIM1 antibody. Immunoprecipitation methods were as described under “Experimental Procedures.”
Role of Orai1 and Plasma Membrane STIM1 in SOC Activation by the STIMEF Mutant—Last, we examined what components in the plasma membrane may functionally interact with the exclusively ER-located STIM1 EF-hand mutant. Initially, we assessed whether the constitutive Ca2+ entry induced by the STIM1 EF-hand mutant was mediated by the Orai1 protein, now functionally identified as the Ca2+ release-activated Ca2+ channel itself (18-20). Clearly, this was the case, since treatment of the EF20 cells with Orai1 small interfering RNA substantially reduced the entry of Ca2+ (Fig. 8A). Also, by immunoprecipitation, we were able to detect interactions between the EF-hand STIM1 mutant and CFP-labeled Orai1 similar to the interaction between wild type STIM1 and Orai1 (data not shown). Thus, the constitutively active STIM1 EF-hand mutant is coupling to Orai1 channels and mimicking the store-emptied condition. However, since the STIM1-EF hand mutant is excluded from the PM, these observations indicate that Orai1 can interact with STIM1 moieties that are exclusively localized to the ER.
FIGURE 8.

Effects of RNA interference for Orai1 and extracellular application of N-terminal STIM1 antibody on constitutive Ca2+ entry in EF20 cells and DT40 cells expressing STIM1-D76A/E87A. A, the EF20 HEK293 cells were transfected with Orai1 small interfering RNA (gray) or control small interfering RNA (black) for 48 h before fura-2 loading. Analysis of cytosolic free Ca2+ was undertaken in Ca2+-free medium, with 1 mm Ca2+ added as shown (arrow). B, EF20 cells were pretreated for 30 min either with (gray) or without (black) 20 μg/ml mouse monoclonal antibody against the extracellular STIM1 N terminus (residues 25-139). C and D, constitutive Ca2+ entry was examined in either wild type DT40 B cells (C) or DT40 cells deficient in the STIM1 gene (D). In each case, cells were transiently transfected (12 h) with the STIM1-D76A EF-hand mutant. Cells were pretreated for 30 min either with (gray) or without (black) 20 μg/ml mouse monoclonal antibody against the STIM1 N terminus (residues 25-139). Ca2+ entry was examined after the addition of 1 mm external Ca2+ as shown (bar).
The second question we addressed was the possibility of interactions between the ER-specific STIM1 EF-hand mutant and wild type STIM1 in the plasma membrane. It has been hypothesized that intermembrane association between ER and plasma membrane STIM1 molecules is involved in the coupling process to activate store-operated channels (8, 9, 13, 51). Although wild-type STIM1 and the constitutively active EF-hand mutant are able to interact, the data in Fig. 7 do not distinguish between whether the interaction occurs between adjacent molecules in the same membrane or between STIM1 molecules in different membranes. We addressed this question by examining the effects of a specific N-terminal STIM1 monoclonal antibody, which we revealed has a substantial inhibitory effect on store-operated Ca2+ entry when applied to the outside of cells (8). As shown in Fig. 8B, the antibody also strongly suppressed the constitutive Ca2+ entry mediated by the D76A/E87A STIM1 mutant in EF20 cells, although in these cells we know that none of the mutant STIM1 is in the plasma membrane. The inhibitory action of the externally applied STIM1 N-terminal antibody indicates that wild type STIM1 in the plasma membrane is exerting an effect on the constitutive activation of Orai1 by the EF-hand STIM1 mutant.
To examine further the possible role of plasma membrane STIM1 in mediating the inhibitory action of the STIM1 antibody, we examined the actions of the antibody using the DT40 chicken B cell STIM1 knock-out cell line described in previous studies (49), which is devoid of endogenous STIM1. We compared the effect of the N-terminal STIM1 antibody on constitutive Ca2+ entry resulting from expression of the D76A STIM1 EF-hand mutant in either wild type DT40 cells or STIM1-k/o DT40 cells (Fig. 8, C and D). The results reveal that in wild type DT40 cells expressing the EF-hand mutant (Fig. 8C), the constitutive Ca2+ entry was almost completely blocked by the antibody. In contrast, in the STIM1-k/o cells expressing the STIM1 EF-hand mutant, the constitutive Ca2+ entry was not affected by application of the same antibody. Thus, in cells expressing only the EF-hand mutant of STIM1, which our experiments reveal does not get to the PM, the antibody has no effect. This provides yet further evidence that the inhibitory action of the externally applied STIM1 antibody is mediated through its interaction with STIM1 inserted in the plasma membrane.
Conclusions—Considering the biotinylation data indicating that the STIM1EF mutant undergoes strong interaction with the PM and considering also that the STIM1EF mutant can undergo interactions with native STIM1, the results may indicate that STIM1 molecules in the ER and plasma membrane undergo transmembrane interactions. We had previously suggested that the inhibitory action of the antibody might reflect a role of surface STIM1 in the activation of SOCs (8). Indeed, as shown in Fig. 9A, one model we considered was that activation of SOCs was actually mediated through plasma membrane STIM1 (8). The recent report using DT40 chicken B cells in which STIM1 expression was knocked out (49) revealed that expression of N-terminal GFP-tagged STIM1, known to be excluded from the plasma membrane (50), was still able to mediate activation of SOCs. From this it was concluded that, at least in DT40 cells, there was no obligatory role for plasma membrane STIM1. Although our data do not prove or disprove an obligatory role of surface STIM1, they do indicate that the plasma membrane STIM1 can influence Ca2+ channel activation mediated by ER STIM1. One possibility is that plasma membrane STIM1 may bind to and exert control over the function of Orai1. However, although STIM1 immunoprecipitated Orai1 in whole cell lysates, we could not detect an interaction of surface STIM1 with Orai1 by applying the N-terminal STIM1 antibody to intact cells. Another possibility is that transmembrane STIM1-STIM1 interactions may exert control over the STIM1-mediated coupling to activate plasma membrane Orai1, as depicted in Fig. 9B. Under this scenario, the effect of the antibody may be to aggregate surface STIM1 and, through trans-membrane interactions with ER STIM1, prevent the latter from being able to effectively interact with and activate Orai1. Thus, we noted that surface labeling of STIM1 with extracellular N-terminal antibody has a distinctly punctal appearance (see Fig. 6D). Recent work of Hauser et al. (50) visualizing surface STIM1 with an N-terminal antibody revealed a similar aggregation of surface STIM1 and was suggested to result from antibody cross-linking.
FIGURE 9.

Possible scenarios for the involvement of plasma membrane STIM1 in the activation of Orai1 by ER-STIM1. A, the possibility that STIM1 in the PM plays an obligatory coupling role in activation of Orai by ER-STIM1 is now discounted. B, STIM1 aggregated in the ER is the primary direct activator of PM Orai1 channels; however, STIM1 in the PM exerts a modulatory role on this channel activation by interacting with aggregated STIM1 in the ER.
Overall, the current study provides insight into the nature of STIM1-mediated SOC activation. The findings establish that plasma membrane Orai1 can be activated by STIM1 that remains within the ER and is not inserted into the PM. The biotinylation data indicate that despite not getting into the plasma membrane, the constitutively active EF-hand mutant of STIM1 undergoes a strong interaction with the plasma membrane. The work also reveals that wild type STIM1 clearly exists in the plasma membrane as well as ER. Whereas plasma membrane STIM1 does not appear to be required for the activation of Orai1 channels, it may play a role in regulating the activation SOCs, possibly by interacting with Orai1 in the plasma membrane or with STIM1 in the ER. Future study of the nature of the interactions of STIM1 with components in the plasma membrane will probably throw light on the coupling process between ER and PM that leads to activation of the channels.
This work was supported in whole or in part, by National Institutes of Health Grants HL55426 (to D. L. G.) AI058173 (to D. L. G.). This work was also supported by the American Heart Association (to J. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ER, endoplasmic reticulum; PM, plasma membrane; SOC, store-operated channel; WT, wild type; HEK, human embryonic kidney; PIPES, 1,4-piperazinediethanesulfonic acid; TIRF, total internal reflection fluorescence; InsP3, inositol 1,4,5-trisphosphate; GFP, green fluorescent protein; CFP, cyan fluorescent protein.
References
- 1.Berridge, M. J., Lipp, P., and Bootman, M. D. (2000) Nat. Rev. Mol. Cell. Biol. 1 11-21 [DOI] [PubMed] [Google Scholar]
- 2.Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003) Nat. Rev. Mol. Cell. Biol. 4 517-529 [DOI] [PubMed] [Google Scholar]
- 3.Parekh, A. B., and Penner, R. (1997) Physiol. Rev. 77 901-930 [DOI] [PubMed] [Google Scholar]
- 4.Venkatachalam, K., van Rossum, D. B., Patterson, R. L., Ma, H. T., and Gill, D. L. (2002) Nat. Cell Biol. 4 E263-E272 [DOI] [PubMed] [Google Scholar]
- 5.Parekh, A. B., and Putney, J. W., Jr. (2005) Physiol. Rev. 85 757-810 [DOI] [PubMed] [Google Scholar]
- 6.Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., Safrina, O., Kozak, J. A., Wagner, S. L., Cahalan, M. D., Velicelebi, G., and Stauderman, K. A. (2005) J. Cell Biol. 169 435-445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E., Jr., and Meyer, T. (2005) Curr. Biol. 15 1235-1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spassova, M. A., Soboloff, J., He, L. P., Xu, W., Dziadek, M. A., and Gill, D. L. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 4040-4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang, S. L., Yu, Y., Roos, J., Kozak, J. A., Deerinck, T. J., Ellisman, M. H., Stauderman, K. A., and Cahalan, M. D. (2005) Nature 437 902-905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stathopulos, P. B., Li, G. Y., Plevin, M. J., Ames, J. B., and Ikura, M. (2006) J. Biol. Chem. 281 35855-35862 [DOI] [PubMed] [Google Scholar]
- 11.Wu, M. M., Buchanan, J., Luik, R. M., and Lewis, R. S. (2006) J. Cell Biol. 174 803-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varnai, P., Toth, B., Toth, D., Hunyady, L., and Balla, T. (2007) J. Biol. Chem. 282 29678-29690 [DOI] [PubMed] [Google Scholar]
- 13.Soboloff, J., Spassova, M. A., Hewavitharana, T., He, L. P., Xu, W., Johnstone, L. S., Dziadek, M. A., and Gill, D. L. (2006) Curr. Biol. 16 1465-1470 [DOI] [PubMed] [Google Scholar]
- 14.Mercer, J. C., Dehaven, W. I., Smyth, J. T., Wedel, B., Boyles, R. R., Bird, G. S., and Putney, J. W., Jr. (2006) J. Biol. Chem. 281 24979-24990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., Hogan, P. G., Lewis, R. S., Daly, M., and Rao, A. (2006) Nature 441 179-185 [DOI] [PubMed] [Google Scholar]
- 16.Vig, M., Peinelt, C., Beck, A., Koomoa, D. L., Rabah, D., Koblan-Huberson, M., Kraft, S., Turner, H., Fleig, A., Penner, R., and Kinet, J. P. (2006) Science 312 1220-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang, S. L., Yeromin, A. V., Zhang, X. H., Yu, Y., Safrina, O., Penna, A., Roos, J., Stauderman, K. A., and Cahalan, M. D. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 9357-9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeromin, A. V., Zhang, S. L., Jiang, W., Yu, Y., Safrina, O., and Cahalan, M. D. (2006) Nature 443 226-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., and Hogan, P. G. (2006) Nature 443 230-233 [DOI] [PubMed] [Google Scholar]
- 20.Vig, M., Beck, A., Billingsley, J. M., Lis, A., Parvez, S., Peinelt, C., Koomoa, D. L., Soboloff, J., Gill, D. L., Fleig, A., Kinet, J. P., and Penner, R. (2006) Curr. Biol. 16 2073-2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peinelt, C., Vig, M., Koomoa, D. L., Beck, A., Nadler, M. J., Koblan-Huberson, M., Lis, A., Fleig, A., Penner, R., and Kinet, J. P. (2006) Nat. Cell Biol. 8 771-773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soboloff, J., Spassova, M. A., Tang, X. D., Hewavitharana, T., Xu, W., and Gill, D. L. (2006) J. Biol. Chem. 281 20661-20665 [DOI] [PubMed] [Google Scholar]
- 23.Oritani, K., and Kincade, P. W. (1996) J. Cell Biol. 134 771-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams, R. T., Senior, P. V., Van Stekelenburg, L., Layton, J. E., Smith, P. J., and Dziadek, M. A. (2002) Biochim. Biophys. Acta 1596 131-137 [DOI] [PubMed] [Google Scholar]
- 25.Williams, R. T., Manji, S. S., Parker, N. J., Hancock, M. S., Van Stekelenburg, L., Eid, J. P., Senior, P. V., Kazenwadel, J. S., Shandala, T., Saint, R., Smith, P. J., and Dziadek, M. A. (2001) Biochem. J. 357 673-685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manji, S. S., Parker, N. J., Williams, R. T., Van Stekelenburg, L., Pearson, R. B., Dziadek, M. A., and Smith, P. J. (2000) Biochim. Biophys. Acta 1481 147-155 [DOI] [PubMed] [Google Scholar]
- 27.Dziadek, M. A., and Johnstone, L. S. (2007) Cell Calcium 42 123-132 [DOI] [PubMed] [Google Scholar]
- 28.Berridge, M. J. (2002) Cell Calcium 32 235-249 [DOI] [PubMed] [Google Scholar]
- 29.Soboloff, J., Spassova, M. A., Xu, W., He, L. P., Cuesta, N., and Gill, D. L. (2005) J. Biol. Chem. 280 39786-39794 [DOI] [PubMed] [Google Scholar]
- 30.Ma, H.-T., Patterson, R. L., van Rossum, D. B., Birnbaumer, L., Mikoshiba, K., and Gill, D. L. (2000) Science 287 1647-1651 [DOI] [PubMed] [Google Scholar]
- 31.Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985) J. Biol. Chem. 261 3440-3450 [PubMed] [Google Scholar]
- 32.Palmer, A. E., Jin, C., Reed, J. C., and Tsien, R. Y. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 17404-17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer, A. E., and Tsien, R. Y. (2006) Nat. Protoc. 1 1057-1065 [DOI] [PubMed] [Google Scholar]
- 34.Chomczynski, P. (1993) BioTechniques 15 532-537 [PubMed] [Google Scholar]
- 35.Livak, K. J., and Schmittgen, T. D. (2001) Methods 25 402-408 [DOI] [PubMed] [Google Scholar]
- 36.Soboloff, J., Spassova, M. A., Dziadek, M. A., and Gill, D. L. (2006) Biochim. Biophys. Acta 1763 1161-1168 [DOI] [PubMed] [Google Scholar]
- 37.Luik, R. M., Wu, M. M., Buchanan, J., and Lewis, R. S. (2006) J. Cell Biol. 174 815-825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma, H. T., Venkatachalam, K., Parys, J. B., and Gill, D. L. (2002) J. Biol. Chem. 277 6915-6922 [DOI] [PubMed] [Google Scholar]
- 39.Chu, X., Cheung, J. Y., Barber, D. L., Birnbaumer, L., Rothblum, L. I., Conrad, K., Abrasonis, V., Chan, Y. M., Stahl, R., Carey, D. J., and Miller, B. A. (2002) J. Biol. Chem. 277 34375-34382 [DOI] [PubMed] [Google Scholar]
- 40.Camello, C., Lomax, R., Petersen, O. H., and Tepikin, A. V. (2002) Cell Calcium 32 355-361 [DOI] [PubMed] [Google Scholar]
- 41.Pinton, P., Ferrari, D., Magalhaes, P., Schulze-Osthoff, K., Di Virgilio, F., Pozzan, T., and Rizzuto, R. (2000) J. Cell Biol. 148 857-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lomax, R. B., Camello, C., Van Coppenolle, F., Petersen, O. H., and Tepikin, A. V. (2002) J. Biol. Chem. 277 26479-26485 [DOI] [PubMed] [Google Scholar]
- 43.Van Coppenolle, F., Vanden Abeele, F., Slomianny, C., Flourakis, M., Hesketh, J., Dewailly, E., and Prevarskaya, N. (2004) J. Cell Sci. 117 4135-4142 [DOI] [PubMed] [Google Scholar]
- 44.Oakes, S. A., Scorrano, L., Opferman, J. T., Bassik, M. C., Nishino, M., Pozzan, T., and Korsmeyer, S. J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 105-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tu, H., Nelson, O., Bezprozvanny, A., Wang, Z., Lee, S. F., Hao, Y. H., Serneels, L., De Strooper, B., Yu, G., and Bezprozvanny, I. (2006) Cell 126 981-993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma, H.-T., Venkatachalam, K., Li, H. S., Montell, C., Kurosaki, T., Patterson, R. L., and Gill, D. L. (2001) J. Biol. Chem. 276 18888-18896 [DOI] [PubMed] [Google Scholar]
- 47.Youle, R. J., and Strasser, A. (2008) Nat. Rev. Mol. Cell. Biol. 9 47-59 [DOI] [PubMed] [Google Scholar]
- 48.White, C., Li, C., Yang, J., Petrenko, N. B., Madesh, M., Thompson, C. B., and Foskett, J. K. (2005) Nat. Cell Biol. 7 1021-1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baba, Y., Hayashi, K., Fujii, Y., Mizushima, A., Watarai, H., Wakamori, M., Numaga, T., Mori, Y., Iino, M., Hikida, M., and Kurosaki, T. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 16704-16709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hauser, C. T., and Tsien, R. Y. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 3693-3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchant, J. S. (2005) Curr. Biol. 15 R493-R495 [DOI] [PubMed] [Google Scholar]