

Abstract
Many cytosolic proteins are recruited to the plasma membrane (PM) during cell signaling and other cellular processes. Recent reports have indicated that phosphatidylserine (PS), phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2), and phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3) that are present in the PM play important roles for their specific PM recruitment. To systematically analyze how these lipids mediate PM targeting of cellular proteins, we performed biophysical, computational, and cell studies of the Ca2+-dependent C2 domain of protein kinase Cα (PKCα) that is known to bind PS and phosphoinositides. In vitro membrane binding measurements by surface plasmon resonance analysis show that PKCα-C2 nonspecifically binds phosphoinositides, including PtdIns(4,5)P2 and PtdIns(3,4,5)P3, but that PS and Ca2+ binding is prerequisite for productive phosphoinositide binding. PtdIns(4,5)P2 or PtdIns(3,4,5)P3 augments the Ca2+- and PS-dependent membrane binding of PKCα-C2 by slowing its membrane dissociation. Molecular dynamics simulations also support that Ca2+-dependent PS binding is essential for membrane interactions of PKCα-C2. PtdIns(4,5)P2 alone cannot drive the membrane attachment of the domain but further stabilizes the Ca2+- and PS-dependent membrane binding. When the fluorescence protein-tagged PKCα-C2 was expressed in NIH-3T3 cells, mutations of phosphoinositide-binding residues or depletion of PtdIns(4,5)P2 and/or PtdIns(3,4,5)P3 from PM did not significantly affect the PM association of the domain but accelerated its dissociation from PM. Also, local synthesis of PtdIns(4,5)P2 or PtdIns(3,4,5)P3 at the PM slowed membrane dissociation of PKCα-C2. Collectively, these studies show that PtdIns(4,5)P2 and PtdIns(3,4,5)P3 augment the Ca2+- and PS-dependent membrane binding of PKCα-C2 by elongating the membrane residence of the domain but cannot drive the PM recruitment of PKCα-C2. These studies also suggest that effective PM recruitment of many cellular proteins may require synergistic actions of PS and phosphoinositides.
One of the hallmarks of cell signaling proteins is their reversible recruitment to the plasma membrane (PM)5 in response to receptor activation (1–4). Although some of these proteins are known to translocate to the PM through protein-protein interactions, a large number of proteins are recruited to the PM by directly interacting with lipids present in the PM (4). The inner leaflet of the PM of mammalian cells is known to be rich in anionic phospholipids, PS (about 20 mol %) (5) and inositol phospholipids, including PtdIns(4,5)P2 (about 1 mol %) (6–8). These anionic lipids have been shown to play key roles in PM recruitment of a wide variety of cytosolic proteins. A series of in vitro membrane binding and cellular translocation studies of various proteins, including protein kinase C (PKC) (9) and sphingosine kinase (10), as well as their isolated lipid binding domains (11), have indicated that PS-selective proteins are targeted to the PM through direct interactions with PS in the PM. More recently, two independent studies have tried to determine the relative contribution of PS and phosphoinositides (PtdInsPs) to specific PM targeting of cytosolic proteins by means of elegant lipid depletion and sensing methods; however, these studies have produced conflicting results. On one hand, Heo et al. (12) reported that the PM targeting of small G proteins with polybasic clusters requires the presence of both PtdIns(4,5)P2 and PtdIns(3,4,5)P3 and that the interaction with PS does not provide a driving force for their membrane targeting. On the other hand, Yeung et al. (13) reported that PS binding is important for PM targeting of cytosolic proteins with polybasic motifs, including small G proteins. It has been reported that monovalent PS and multivalent PtdInsPs can generate distinct electrostatic fields (7). However, there is little mechanistic information as to how differently PS and PtdInsPs contribute to the PM targeting of cytosolic proteins. The present study was designed to provide such mechanistic insight through extensive computational, biophysical, and cell studies of the C2 domain of PKCα that has been reported to bind both PS (11, 14) and PtdIns(4,5)P2 (15, 16) with high affinity and be recruited to the PM when expressed in mammalian cells (11, 15–18).
C2 (protein kinase C conserved 2) domains are small (about 120 amino acids) modular lipid-binding domains that have been identified in various signaling and membrane trafficking proteins, including PKCs (19–22). High-resolution crystal structures determined for the isolated C2 domains of various proteins have shown that the domains have a common fold of 8-stranded anti-parallel β-sandwich connected by variable loops. Many C2 domains bind to the membranes in a Ca2+-dependent manner via the three calcium-binding loops (CBL) that are located at one end of the β-sandwich (see Fig. 2B). Ca2+ binding can drive the association of different C2 domains to different membranes (23). Also, most C2 domains contain a cationic patch in the concave face of the β-sandwich, known as the β-groove, which may interact with anionic lipids (22) (see Fig. 2B).
FIGURE 2.

The structure of PKCα C2 domain. A, initial
configuration of PKCα-C2 and lipids for the system,
 .
The crystal structure of PKCα-C2 (PDB code 1DSY) complexed with two
Ca2+ ions and a PS molecule is shown in schematic representation.
POPC molecules are in yellow, POPS in light blue, and
PtdIns(4,5)P2 in magenta. Calcium ions are shown as
red spheres. For clarity, hydrogen atoms on lipids are not shown and
protein loops are smoothened. B, CBL and cationic β-groove
residues. CBL residues are shown in blue and β-groove residues
in magenta. The PS head groups as occurring in the crystal structure
are shown in stick representation. C, Cα trace of the protein
with the first two principal axes (PA). The first PA is shown with a
solid straight line, whereas the second one is displayed by a
dashed straight line. CBLs are highlighted by a thicker Cα
trace. The floor of the box is parallel to the membrane surface.
.
The crystal structure of PKCα-C2 (PDB code 1DSY) complexed with two
Ca2+ ions and a PS molecule is shown in schematic representation.
POPC molecules are in yellow, POPS in light blue, and
PtdIns(4,5)P2 in magenta. Calcium ions are shown as
red spheres. For clarity, hydrogen atoms on lipids are not shown and
protein loops are smoothened. B, CBL and cationic β-groove
residues. CBL residues are shown in blue and β-groove residues
in magenta. The PS head groups as occurring in the crystal structure
are shown in stick representation. C, Cα trace of the protein
with the first two principal axes (PA). The first PA is shown with a
solid straight line, whereas the second one is displayed by a
dashed straight line. CBLs are highlighted by a thicker Cα
trace. The floor of the box is parallel to the membrane surface.
The PKCα C2 domain is known to bind two Ca2+ ions and a PS molecule in its CBLs (see Fig. 2, A and B) (14). Subsequent studies have shown that Ca2+ and PS are necessary and sufficient for specific targeting of PKCα-C2 to the PM (11). However, recent studies have suggested that binding of PtdIns(4,5)P2 to the cationic β-groove of PKCα-C2 is critical for the PM translocation (15–18). Because PKCα-C2 is a prototype PS-specific protein that is recruited to the PM, these findings challenge the notion that PS binding can drive the PM targeting of cellular proteins. This controversy as well as a wealth of structural and functional information make PKCα-C2 an excellent model to systematically investigate the differential roles of PS and PtdInsPs in the PM targeting of cellular proteins. In this study, we performed in vitro membrane binding and cellular translocation measurements as well as atomistic level molecular dynamics (MD) simulations of PKCα-C2 and mutants under various conditions. Results from these studies provide new insight into the differential roles of PS, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in membrane targeting of this C2 domain, which also bears implication for the PM targeting of other C2 domains and other lipid-binding domains and proteins.
EXPERIMENTAL PROCEDURES
Materials—1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-phosphoinositol (POPI), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS) were from Avanti Polar Lipids (Alabaster, AL). All PtdInsPs were purchased from Cayman (Ann Arbor, MI). Lyn11-rapamycin-binding protein (FRB) (Lyn11-FRB), cyan fluorescence protein (CFP)-FK505-binding protein (FKBP)-Inp54p (CF-Inp), CFP-FKBP-phosphatidylinositol-4-phosphate 5-kinase (CF-PIPK), CFP-FKBP-PtdInsP 3-kinase p85 subunit (CF-iSH), and yellow fluorescence protein-phospholipase Cδ-pleckstrin homology (PH) domain (YFP-PLCδ1-PH) constructs were generous gifts of Dr. Tobias Meyer. The mKate-C vector was a generous gift of Dr. Akihiro Kusumi. Rapamycin and LY294002 (PtdInsP 3-kinase Inhibitor) were purchased from Cell Signaling Technology (Danvers, MA). Ionomycin was from Invitrogen. PKCα-C2 domain and its N189A and K209A/K211A mutants were prepared as described (11). These genes were subcloned between EcoRI and KpnI sites of either the mKate-C or the mCerulean-C1 vector to yield C2 domains with a C-terminal red fluorescence protein (RFP) tag or CFP, respectively. The fluorescence protein-tagged PH domain constructs of phospholipase C (PLC) δ and Btk were prepared as described (24). Recombinant PKCα-C2 proteins with C-terminal His6 tags were expressed in Escherichia coli and purified as described (25).
Surface Plasmon Resonance (SPR) Measurements of the PKCα C2 Domain—All SPR measurements were performed at 23 °C using a lipid-coated L1 chip in the BIACORE X system as described previously (26). Briefly, after washing the sensor chip surface with the running buffer (20 mm HEPES, pH 7.4, containing 0.16 m KCl), POPC/POPE/PtdInsP (77:20:3) and POPC/POPE (80:20) vesicles were injected at 5 μl/min to the active surface and the control surface, respectively, to give the same resonance unit values. The level of lipid coating for both surfaces was kept at the minimum that is necessary for preventing the nonspecific adsorption to the sensor chips. This low surface coverage minimized the mass transport effect and kept the total protein concentration (P0) above the total concentration of protein binding sites on vesicles (M0) (27). The control surface was also coated with 40 μl of bovine serum albumin (0.1 mg/ml in the running buffer) at a flow rate of 5 μl/min before the injection of C2 domains to minimize nonspecific adsorption of C2 domains to the control surface. All SPR measurements were done at the flow rate of 5 μl/min to allow sufficient time for the R values of the association phase to reach near-equilibrium values (Req) (5). After sensorgrams were obtained for 5 or more different concentrations of each protein within a 10-fold range of Kd, each of the sensorgrams was corrected for refractive index change by subtracting the control surface response from it. Assuming a Langmuir-type binding between the protein (P) and protein binding sites (M) on vesicles (i.e. P + M ↔ PM) (27), Req values were then plotted versus P0, and the Kd value was determined by a nonlinear least-squares analysis of the binding isotherm using equation, Req = Rmax/(1 + Kd/P0) (27). Each data set was repeated three or more times to calculate average and standard deviation values. For kinetic SPR measurements, the flow rate was maintained at 30 μl/min for both association and dissociation phases.
Cell Culture and Transfection—NIH-3T3 cells were seeded into 8 wells of a sterile Nunc Lak-TeKII™ chambered cover glass plate, which was filled with 400 μl of Dulbecco's modified Eagle's medium and 10% (v/v) fetal bovine serum and incubated at 37 °C with 5% CO2 for 24 h. For transfection cells were incubated for 3 h with various constructs (0.5 μg of total DNA/well) in the presence of the Lipofectamine 2000 reagent in Opti-MEM (Invitrogen). Co-transfection of cells with Lyn11-FRB, CF-Inp, and YFP-PLCδ1-PH was performed with three corresponding plasmids in a 4:4:1 ratio to balance the emission intensities of fluorophores. The same protocol was used for the co-transfection of Lyn11-FRB, CF-Inp (or other constructs; CF-iSH and CF-PIPK), and PKCα-C2-mKate (or its mutants). Cells were then incubated in Dulbecco's modified Eagle's medium with 10% fetal bovine serum overnight. Typically, 50–80% of cells were transfected with all three cDNAs. 10–30% of these cells underwent cell death presumably due to the cytotoxicity of overexpressed Inp54p. Thus, cells with the moderate expression level of CF-Inp were selected for measurements.
Live Cell Confocal Imaging—Immediately before imaging, the induction media was removed and the cells were washed and then overlaid with 400 μl of HBSS buffer (4.2 mm HEPES, pH 7.4, containing 0.7 mm MgCl2, 1.3 mm CaCl2, 138 mm NaCl, 4.7 mm KCl, 0.4 mm KH2PO4, 0.3 K2HPO4, 5.6 mm d-glucose). Live cell dual-color imaging was performed on a four-channel Zeiss LSM 510 laser scanning confocal microscope. All measurements were carried out with a ×63, 1.2 numerical aperture water immersion objective, at a minimal laser power and exposure time to reduce photobleaching. Proper filter sets for CFP/YFP and CFP/RFP duel-color images had been used to minimize fluorescence overlaps. Images were taken every 15 s at room temperature during the translocation assay. After initial imaging the translocation of CF-Inp, CF-PIPK, or CF-iSH was monitored in the presence of 5 μm rapamycin (0.05% dimethyl sulfoxide) and/or 50 μm LY294002 (0.0025% dimethyl sulfoxide). The PM translocation of PKCα-C2-RFP and its mutants were then triggered by adding 100 μm ATP (final concentration). The membrane translocation of the N189A mutant was induced by adding 10 μm ionomycin (final concentration). Images were analyzed using the analysis tool provided in the Zeiss biophysical software package. Intensities of fluorescence proteins were determined in selected areas in the cytosol and PM and averaged (n > 10) at each time frame. The fluorescence intensity ratio, which indicates the relative abundance of fluorescence proteins at PM versus cytosol, was then calculated as PM/(PM + cytosol) and plotted as a function of time.
System Model—A POPC patch of dimensions 80 Å ×
80 Å (in xy plane) was created using the “membrane”
plug-in in VMD (28) that left
a margin of at least 15 Å on each side after the protein was placed in
the vicinity of the upper layer (called the positive layer). For
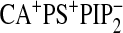 ,
10 POPC molecules in the middle of the upper layer were replaced with 10 POPS
molecules ensuring that the protein had enough POPS molecules in which to
interact. Similarly, for simulations involving both PS and
PtdIns(4,5)P2 (i.e.
,
10 POPC molecules in the middle of the upper layer were replaced with 10 POPS
molecules ensuring that the protein had enough POPS molecules in which to
interact. Similarly, for simulations involving both PS and
PtdIns(4,5)P2 (i.e.
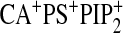 ),
10 POPC molecules were removed, and 8 POPS and 2 PtdIns(4,5)P2
molecules were introduced, whereas only 2 POPC molecules were replaced with 2
PtdIns(4,5)P2 molecules for
),
10 POPC molecules were removed, and 8 POPS and 2 PtdIns(4,5)P2
molecules were introduced, whereas only 2 POPC molecules were replaced with 2
PtdIns(4,5)P2 molecules for
 .
PtdIns(4,5)P2 was introduced in the middle of the bilayer falling
directly below the β3–4 hairpin of the protein where it has been
proposed to interact with the protein
(16,
29)
(Fig. 2A). The
proportion of POPS and PtdIns(4,5)P2 molecules in the membrane
patch used in this study are also biologically relevant
(7,
30). Additionally, under our
experimental conditions, the Ca2+ equilibration with the bilayer is
not necessary as Ca2+ is pre-coordinated in the Ca2+
binding site of the C2 domain.
.
PtdIns(4,5)P2 was introduced in the middle of the bilayer falling
directly below the β3–4 hairpin of the protein where it has been
proposed to interact with the protein
(16,
29)
(Fig. 2A). The
proportion of POPS and PtdIns(4,5)P2 molecules in the membrane
patch used in this study are also biologically relevant
(7,
30). Additionally, under our
experimental conditions, the Ca2+ equilibration with the bilayer is
not necessary as Ca2+ is pre-coordinated in the Ca2+
binding site of the C2 domain.
Force-Field Parameters—Non-standard lipids such as POPS and PtdIns(4,5)P2 together with POPC were parameterized using the Antechamber tool (31) from Amber 7 (32). Antechamber assigns atomic and bond types based on electronic and structural properties of the connected atoms, such as atomic hybridization type, electron donor, or acceptor capability, atomic number, number of connected atoms, number of attached hydrogen atoms, atomic property, which refers to ring properties, aromatic properties, and subtle chemical environments. Generalized amber force-field (33) parameters were used and AM1-bond charge correction (AM1-BCC) (34, 35) was used for generation of high-quality atomic charges. Initial structures of POPS and PtdIns(4,5)P2 were taken from previous works (36, 37). NAMD 2.5 (38) was used for carrying out all molecular dynamics simulations. The parameters used in this study are available at the membrane targeting domain resource, MeTaDor (39). Due to a high heterogeneity because of the presence of two or three different kinds of lipids and unavailability of experimental data about the area per lipid for POPS and PtdIns(4,5)P2, simulations of the bilayer were performed with NTP ensemble with the T = 300 K. The Langevin piston method (40) was used to impose a constant pressure p = 1 atm. The PME method was used for computation of the electrostatic forces (41, 42) with grid spacing below 1.0 Å. The van der Waals interactions were cut off at 12 Å using a switching function starting at 10 Å. No bonds were restrained including the hydrogen bonds and a time step of 1 fs was used in all simulations.
Statistical Ensemble—Periodic boundary conditions were applied in the xy directions to simulate an infinite planar layer and in the z direction to simulate a multilayer system. Due to the negative charge moiety of POPS and PtdIns(4,5)P2, the appropriate number of Na+ ions was added to neutralize the system. The initial translation and rotation of the additional lipid molecules (POPS and/or PtdIns(4,5)P2) with respect to the membrane were optimized to avoid large steric clashes. The system was then subjected to 10,000 steps of conjugate-gradient energy minimization to remove remaining steric overlaps. This was followed by 100 ps of dynamical run.
Docking of Protein on the Membrane Surface—The starting relative orientation of the protein with respect to the membrane was its binding orientation as proposed by theory and validated by experiments (14, 43). In simulations with POPS, phosphorus atoms from one of the POPS heads was made to overlap with that solved with the crystal structure of the domain (Fig. 2a) (14). The penetration of protein into the membrane was also carefully emulated by translation of the protein along the z axis. Additional water was added on the top of the protein-lipid system to create a layer of 15 Å above the protein and appropriate counterions were added. Poor contacts were removed by minimizing the energy of the system by 10,000 steps of conjugate-gradient minimization followed by a dynamic run of 1 to 10 ns.
Computing Observables from MD Simulation—Various properties were calculated for observing the subsequent events compared in different cases. To monitor the movement of the protein, a “membrane surface” was defined as the surface layer in the xy plane with z coordinate equal to the average of z coordinates of phosphorus atoms present in the positive layer of that system. The movement of the protein with respect to this membrane was observed as the change in the difference of the z coordinate of the center of mass of the protein and that of the membrane surface. The initial value of this difference was translated to a value of zero such that the positive value of this distance would indicate the movement of the protein away from the layer as compared with the initial configuration and a negative value would mean its movement toward the membrane.
A structure-function analysis and the crystal structure of PKCα-C2 (11, 14, 44) suggested that PS selectivity derive from the specific recognition of PS by a C2 domain-bound calcium ion and several residues located in the calcium binding loops. In particular, mutation studies on PKCα showed that Asn189, Arg216, Arg249, and Thr251 in the calcium binding loops are involved in PS binding (45). Trp245, Trp247, and Arg249 have also been implicated in membrane binding (25). We monitored the movement of these six residues (Fig. 3) with respect to the membrane by calculating the distance of their Cα atoms from the membrane surface.
FIGURE 3.

Calculation of displacement of PKCα-C2 from the membrane by MD
simulations. Displacement of the CBL and cationic β-groove residues
with respect to the upper membrane layer was determined for four different
systems; A, with Ca2+ and POPC
(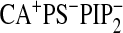 );
B, with Ca2+, POPC, and PtdIns(4,5)P2
(
);
B, with Ca2+, POPC, and PtdIns(4,5)P2
( );
C, with Ca2+, POPC, and POPS
(
);
C, with Ca2+, POPC, and POPS
( );
and (D) with Ca2+, POPC, POPS, and
PtdIns(4,5)P2
(
);
and (D) with Ca2+, POPC, POPS, and
PtdIns(4,5)P2
( ).
Negative value indicates displacement toward and positive value indicates
displacement away from the bilayer. Overlays of initial (green) and
final (red) structures are also shown for the four systems. For
overlays, the membrane plane is parallel to the floor of the box and
solid and dashed lines indicate the first and the second PA,
respectively. E, the number of hydrogen bonds between the protein and
the two PtdIns(4,5)P2 molecules in three systems,
).
Negative value indicates displacement toward and positive value indicates
displacement away from the bilayer. Overlays of initial (green) and
final (red) structures are also shown for the four systems. For
overlays, the membrane plane is parallel to the floor of the box and
solid and dashed lines indicate the first and the second PA,
respectively. E, the number of hydrogen bonds between the protein and
the two PtdIns(4,5)P2 molecules in three systems,
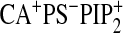 ,
,
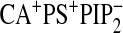 ,
and
,
and
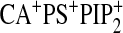 .
PtdIns(4,5)P2 adds about 6 extra hydrogen bonds between the protein
and the bilayer, increasing the average number of the hydrogen bonds from 11
to 17.
.
PtdIns(4,5)P2 adds about 6 extra hydrogen bonds between the protein
and the bilayer, increasing the average number of the hydrogen bonds from 11
to 17.
Tilting of the protein with respect to the membrane surface was investigated in two ways. First, to monitor the inclination in the yz plane (around x axis, Fig. 3), four cationic residues on the β-groove were chosen. The relative movement of these residues with respect to CBL residues would illustrate the rotation of the protein in the yz plane. Second, the first two principal axes (PAs) of the protein were calculated. Because of the shape of the protein, its first PA passes CBLs (Fig. 2C) and its movement is around the x axis in the plane of the paper. The second PA is perpendicular to the first one and elicits the tilting of the protein in the xz plane (roughly around the y axis, perpendicular to the plane of the paper). The two PAs from the initial and final configuration of the protein were overlaid to demonstrate the overall change.
MD Simulation Time—Simulation time for all the control simulations in this study was 1 to 5 ns. On the other hand, the subject simulation was run for a longer time of 5 to 10 ns to ensure that the stability of the subject system was not short-lived but was inherent and persistent. These simulation times are similar to those in many recent and previous studies where simulations of 1–10 ns have been reported while studying different kinds of events related to bilayers such as binding of peptides (37, 46), interactions with small molecules such as cholesterol and trehalose (47–49), lipid reorientation (50), and proton transport on the bilayer surface (51). It should also be noted that a recent study (52) showed significant changes in membrane curvature in response to domain binding were observed within a time scale of 15–20 ns. Thus, a 1–10-ns time scale should be appropriate for studying an event of membrane dissociation or maintaining a membrane-binding orientation, which is expected to be on a shorter time scale than a domain binding the bilayer.
RESULTS
In Vitro Membrane Binding Measurements of PKCα-C2 and Mutants—Both structural (14) and functional (11) studies have firmly established that PKCα-C2 can interact specifically with PS. In contrast, despite several reports indicating that PtdIns(4,5)P2 binds to the β-groove of PKCα-C2 and thereby drives its PM translocation in the cell (15–18), high specificity and affinity of PKCα-C2 for PtdIns(4,5)P2 has not been unambiguously demonstrated. We therefore rigorously determined the lipid specificity and affinity of PKCα-C2 by SPR analysis using vesicles of various lipid compositions coated on the sensor chip (Fig. 1A). First, we measured the binding of PKCα-C2 to various lipid vesicles in the absence of Ca2+. Under this condition, PKCα-C2 bound all anionic lipid vesicles tested with near micromolar affinity (Table 1). Modest membrane affinity and lack of PtdInsP selectivity dispute the notion that PtdIns(4,5)P2 itself provides specificity and driving force for PM recruitment of PKCα-C2.
FIGURE 1.

Kinetic and equilibrium membrane binding measurements of the PKCα C2 domain and a mutant by SPR analysis. A, equilibrium binding of PKCα-C2 to the sensor chip coated with POPC/POPS (80:20) (○), POPC/PtdIns(4,5)P2 (98:2) (▪), POPC/PtdIns(3,4,5)P3 (98:2) (Δ), and POPC/POPS/PtdIns(4,5)P2 (78:20:2) (•) vesicles. PKCα-C2 was injected at 5 μl/min at varying concentrations over each lipid surface and Req values were measured. Binding isotherms were then generated from the Req (average of triplicate measurements) versus the PKCα-C2 concentration. Solid lines represent theoretical curves constructed from Rmax and Kd values determined by nonlinear least-squares analysis of the isotherm using an equation: Req = Rmax/(1 + Kd/P0). B, kinetics of binding of PKCα-C2 (50 nm) to the sensor chip coated with POPC/POPS (80:20), POPC/PtdIns(4,5)P2 (98:2), and POPC/POPS/PtdIns(4,5)P2 (78:20:2). The flow rate was maintained at 15 μl/min for both association and dissociation phases. 10 mm HEPES buffer, pH 7.4, with 0.16 m KCl and 10 μm CaCl2 was used for all measurements.
TABLE 1.
Membrane affinity of the PKCα C2 domain and mutants determined by SPR analysis Values represent the mean ± S.D. from three determinations. All measurements were performed in 10 mm HEPES, pH 7.4, containing 0.16 m KCl.
|
Protein |
Kd |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| POPC/POPS (80:20) | POPC/POPI (80:20) | POPC/PtdIns(4,5)P2 (98:2) | POPC/PtdIns(3,4)P2a(98:2) | POPC/PtdIns(3,4,5)P3 (98:2) | POPC/PtdIns(3)P (98:2) | POPC/PtdIns(4)P (98:2) | POPC/PtdIns(5)P (98:2) | POPC/POPS/PtdIns(4,5)P2 (78:20:2) | POPC/POPS/PtdIns(3,4,5)P3 (78:20:2) | |
| nm | ||||||||||
| WT-Ca2+b | 910 ± 70 | 1200 ± 100 | 790 ± 80 | 800 ± 60 | 900 ± 200 | NMc | NM | NM | 780 ± 70 | 710 ± 50 |
| WTd | 71 ± 8 | 720 ± 60 | 460 ± 40 | 480 ± 30 | 390 ± 30 | 680 ± 50 | 540 ± 30 | 580 ± 40 | 18 ± 3 | 14 ± 2 |
| N189Ad | 340 ± 30 | 730 ± 40 | 470 ± 30 | 470 ± 40 | NMc | NM | NM | NM | 380 ± 50 | NM |
| K209A/K211Ad | 92 ± 9 | 840 ± 50 | 540 ± 70 | 560 ± 60 | NM | NM | NM | NM | 68 ± 8 | NM |
| D248Nb,d | 900 ± 70 | 1100 ± 200 | 920 ± 70 | 940 ± 50 | NM | NM | NM | NM | 880 ± 60 | NM |
PtdIns(3)P, phosphatidylinositol 3-phosphate; PtdIns(4)P, phosphatidylinositol 3-phosphate; PtdIns(5)P, phosphatidylinositol 5-phosphate; PtdIns(3,4)P2, phosphatidylinositol 3,4-bisphosphate
0.1 mm EGTA
NM, not measured
10 μm Ca2+
When we measured the membrane binding of PKCα-C2 in the presence of 10 μm Ca2+, it demonstrated pronounced PS selectivity; i.e. 10-fold higher affinity for POPC/POPS (80:20) than for POPC/POPI (80:20), as reported previously (11). However, even with 10 μm Ca2+, PKCα-C2 showed no PtdIns(4,5)P2 selectivity and it exhibited only modest affinity for PtdIns(4,5)P2-containing vesicles. That is, PKCα-C2 had ∼0.5 μm affinity for POPC vesicles containing 2 mol % of each of 6 PtdInsPs tested (Table 1). Interestingly, when PS is present in the vesicles (i.e. POPC/POPS (80:20) vesicles), addition of 2 mol % of PtdIns(4,5)P2 (i.e. POPC/POPS/PtdIns(4,5)P2 (78:20:2)) or PtdIns(3,4,5)P3 (i.e. POPC/POPS/PtdIns(3,4,5)P3 (78:20:2)) caused the 4–5-fold increase in affinity (see Fig. 1A and Table 1). The comparison of the kinetic sensorgrams (Fig. 1B) of PKCα-C2 for POPC/POPS (80:20) and POPC/POPS/PtdIns(4,5)P2 (78:20:2) vesicles indicates that the increase in affinity by PtdIns(4,5)P2 or PtdIns(3,4,5)P3 is largely due to slower membrane dissociation. Rate constants for association and dissociation could not be accurately determined in this study because the observed kinetics did not conform to either a one-step (i.e. p + M → PM) or a two-step (i.e. p + M → PM → P*M) 1:1 binding model. However, comparison of the half-lives of dissociation (see Fig. 1B) suggested that PKCα-C2 dissociated 4–5 times more slowly from POPC/POPS/PtdIns(4,5)P2 (78:20:2) than from POPC/POPS (80:20). These results thus indicate that Ca2+ and PS binding is essential for the membrane binding of PKCα-C2 and that when Ca2+ and PS are present, PtdInsPs, including PtdIns(4,5)P2 and PtdIns(3,4,5)P3, can significantly augment its membrane binding by slowing the membrane dissociation.
To further elucidate the role of Ca2+, PS, and PtdIns(4,5)P2 (or PtdIns(3,4,5)P3) in membrane binding of PKCα-C2, we measured the effects of mutating residues that are involved in binding to Ca2+ (Asp248), PS (Asn189), and PtdInsPs (Lys209 and Lys211), respectively, on membrane affinity (see Table 1). All mutants were bacterially expressed as well as the wild type, suggesting that mutations did not have deleterious effects on protein structure and stability. In agreement with its PS-binding role, the mutation of Asn189 (N189A) not only reduced the affinity of PKCα-C2 for POPC/POPS (80:20) vesicles by 5-fold but also greatly reduced its PS selectivity: i.e. N189A showed only 2-fold higher affinity for POPC/POPS (80:20) than for POPC/POPI (80:20) and had similar affinity for POPC/POPS/PtdIns(4,5)P2 (78:20:2) and POPC/PtdIns(4,5)P2 (98:2) vesicles. Similarly, D248N with much reduced Ca2+ affinity, hence lower PS affinity (notice that PS is coordinated by Ca2+; see Fig. 2B), showed lower membrane affinity and much reduced PS selectivity. It had 13-fold lower affinity than wild type for POPC/POPS (80:20) vesicles, whereas showing little selectivity between POPC/POPS (80:20) and POPC/POPI (80:20) vesicles. Most importantly, unlike the wild type whose affinity was augmented by PtdIns(4,5)P2 (or PtdIns(3,4,5)P3) addition in the presence of Ca2+ and PS, neither N189A nor D248N showed enhanced affinity for POPC/POPS/PtdIns(4,5)P2 (78:20:2) over POPC/POPS (80:20) vesicles. These data underscore that Ca2+-dependent PS binding is prerequisite for effective PtdInsP binding of PKCα-C2.
Consistent with their location in the β-groove, the double mutation of Lys209 and Lys211 (K209A/K211A) did not have a significant effect on the affinity of PKCα-C2 for POPC/POPS (80:20) vesicles but caused 4-fold reduction in affinity for POPC/POPS/PtdIns(4,5)P2 (78:20:2) vesicles (see Table 1). As a result, K209A/K211A had only 40% higher affinity for POPC/POPS/PtdIns(4,5)P2 than for POPC/POPS (80:20). Intriguingly, in the absence of PS (e.g. for POPC/PtdIns(4,5)P2 (98:2)) the mutation did not reduce the affinity of PKCα-C2, again underscoring that PS binding is prerequisite of efficient PtdIns(4,5)P2 binding of the β-groove residues. Collectively, these in vitro membrane binding results strongly support the essential role of Ca2+ and PS binding in the membrane binding of PKCα-C2 and augmenting role of PtdInsPs, including PtdIns(4,5)P2 and PtdIns(3,4,5)P3, in the presence of Ca2+ and PS.
MD Simulations—To gain further insight into different roles
of Ca2+, PS, and PtdIns(4,5)P2 (or
PtdIns(3,4,5)P3) in membrane binding of PKCα-C2, we performed
and analyzed MD simulations with four different combinations of
Ca2+, PS, and PtdIns(4,5)P2 on all-atoms scheme with
explicit solvent: (i) with Ca2+ and POPC
(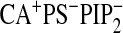 );
(ii) with Ca2+, POPC, and POPS
(
);
(ii) with Ca2+, POPC, and POPS
( );
(iii) with Ca2+, POPC, and PtdIns(4,5)P2
(
);
(iii) with Ca2+, POPC, and PtdIns(4,5)P2
( );
and (iv) with Ca2+, POPC, POPS, and PtdIns(4,5)P2
(
);
and (iv) with Ca2+, POPC, POPS, and PtdIns(4,5)P2
( ).
Our approach is to simulate either the dissociation or the maintenance of the
binding orientation by the domain under different conditions beginning with
the experimentally determined binding orientation with the Ca2+
ions intact (see Fig.
2A). This is distinct from simulating the association and
the subsequent dissociation of the domain beginning from an unbound state
and/or in response to lipid or Ca2+ fluctuations near the bilayer.
Simulation was run 5 to 10 ns to ensure that the stability of the subject
system was not short-lived but was inherent and persistent.
).
Our approach is to simulate either the dissociation or the maintenance of the
binding orientation by the domain under different conditions beginning with
the experimentally determined binding orientation with the Ca2+
ions intact (see Fig.
2A). This is distinct from simulating the association and
the subsequent dissociation of the domain beginning from an unbound state
and/or in response to lipid or Ca2+ fluctuations near the bilayer.
Simulation was run 5 to 10 ns to ensure that the stability of the subject
system was not short-lived but was inherent and persistent.
We first determined the displacement of selected residues in the CBL and the cationic β-groove of PKCα-C2 from the membrane surface as the simulation proceeded for the four systems. CBL residues include those involved in PS (Asn189, Arg216, and Thr251) and membrane binding (Trp245, Trp247, and Arg249) (11, 14, 45) whereas cationic β-groove residues are Lys197, Lys199, Lys209, and Lys211 (see Fig. 2B for CBL and β-groove residues). A larger positive displacement value would indicate a weaker interaction and a negative value would suggest a tighter interaction.
When Ca2+ is present in the CBL but POPS and
PtdIns(4,5)P2 are absent in the bilayer (i.e.
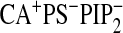 ),
CBL residues displayed a large displacement of up to 5 Å away from the
membrane (Fig. 3A).
Cationic β-groove residues also moved away from the membrane, although
the degree of displacement was lower (about 2 Å) than that for CBL
residues. The addition of PtdIns(4,5)P2 to this system
(
),
CBL residues displayed a large displacement of up to 5 Å away from the
membrane (Fig. 3A).
Cationic β-groove residues also moved away from the membrane, although
the degree of displacement was lower (about 2 Å) than that for CBL
residues. The addition of PtdIns(4,5)P2 to this system
(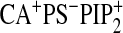 )
did not appreciably change the displacement pattern of either CBL or
β-groove residues, as shown in Fig.
3B, underscoring that PtdIns(4,5)P2 alone
contributes little to the membrane binding without PS. This notion is also
supported by the finding that the PtdIns(4,5)P2-binding
β-groove (15) moved away
from the bilayer for both
)
did not appreciably change the displacement pattern of either CBL or
β-groove residues, as shown in Fig.
3B, underscoring that PtdIns(4,5)P2 alone
contributes little to the membrane binding without PS. This notion is also
supported by the finding that the PtdIns(4,5)P2-binding
β-groove (15) moved away
from the bilayer for both
 and
and
 ,
which is evident in the overlays of the PA of the protein from the initial and
final frames (Fig. 3, A and
B).
,
which is evident in the overlays of the PA of the protein from the initial and
final frames (Fig. 3, A and
B).
When Ca2+ was coordinated to the CBL and POPS was present in the
bilayer
(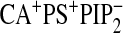 ),
CBL residues fluctuated around their initial binding position with average
displacements within 1 Å on either side
(Fig. 3C). Also,
β-groove residues showed mostly ≤1 Å displacement from the
membrane. As expected from the fact that PtdIns(4,5)P2 does not
interact with CBL, the addition of PtdIns(4,5)P2 to this system
(
),
CBL residues fluctuated around their initial binding position with average
displacements within 1 Å on either side
(Fig. 3C). Also,
β-groove residues showed mostly ≤1 Å displacement from the
membrane. As expected from the fact that PtdIns(4,5)P2 does not
interact with CBL, the addition of PtdIns(4,5)P2 to this system
( )
did not affect the CBL movement (Fig.
3D). However, it had a definite, albeit small, effect on
the positioning of β-groove residues, which fluctuated less in the
presence of PtdIns(4,5)P2 than in its absence and moved closer to
the membrane after 5 ns. This supports that PtdIns(4,5)P2
stabilizes the interaction of the β-groove region with the membrane in
the presence of Ca2+ and PS.
)
did not affect the CBL movement (Fig.
3D). However, it had a definite, albeit small, effect on
the positioning of β-groove residues, which fluctuated less in the
presence of PtdIns(4,5)P2 than in its absence and moved closer to
the membrane after 5 ns. This supports that PtdIns(4,5)P2
stabilizes the interaction of the β-groove region with the membrane in
the presence of Ca2+ and PS.
To further investigate the specific role of PtdIns(4,5)P2 in
augmenting the interaction of the domain with the bilayer, we calculated the
number of hydrogen bonds between the PKCα-C2 and the bilayer for three
systems,
 ,
,
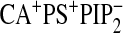 ,
and
,
and
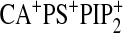 .
The angle and distance criterions for a hydrogen bond to exist between an
acceptor and donor were set to be 60° and 2.8 Å, respectively. As
shown in Fig. 3E, the
addition of PtdIns(4,5)P2 to the PS-containing membrane increased
the average number of hydrogen bonds between the protein and the bilayer from
∼11 to ∼17, indicating that PtdIns(4,5)P2 confers an
additional 6 hydrogen bonds. Specifically, these hydrogen bonds occur between
two PtdIns(4,5)P2 molecules and five cationic residues
(Lys197, Lys199, Lys209, Lys211,
and Arg249) in the β-grove of the domain (data not shown). In
the absence of PS, the number of hydrogen bonds drops to 5 within the first 2
ns even when PtdIns(4,5)P2 is present, which is consistent with the
finding that the domain moves away from the membrane during the same time
(Fig. 3B) and that
PtdIns(4,5)P2 alone cannot stabilize membrane interactions of
PKCα-C2. These calculations indicate that PtdIns(4,5)P2
strengthens Ca2+- and PS-dependent membrane binding of
PKCα-C2 by changing the membrane-bound orientation of PKCα-C2 and
allowing extra stabilizing interactions.
.
The angle and distance criterions for a hydrogen bond to exist between an
acceptor and donor were set to be 60° and 2.8 Å, respectively. As
shown in Fig. 3E, the
addition of PtdIns(4,5)P2 to the PS-containing membrane increased
the average number of hydrogen bonds between the protein and the bilayer from
∼11 to ∼17, indicating that PtdIns(4,5)P2 confers an
additional 6 hydrogen bonds. Specifically, these hydrogen bonds occur between
two PtdIns(4,5)P2 molecules and five cationic residues
(Lys197, Lys199, Lys209, Lys211,
and Arg249) in the β-grove of the domain (data not shown). In
the absence of PS, the number of hydrogen bonds drops to 5 within the first 2
ns even when PtdIns(4,5)P2 is present, which is consistent with the
finding that the domain moves away from the membrane during the same time
(Fig. 3B) and that
PtdIns(4,5)P2 alone cannot stabilize membrane interactions of
PKCα-C2. These calculations indicate that PtdIns(4,5)P2
strengthens Ca2+- and PS-dependent membrane binding of
PKCα-C2 by changing the membrane-bound orientation of PKCα-C2 and
allowing extra stabilizing interactions.
Cellular Translocation of PKCα-C2 and Mutants—To prove the physiological significance of our computational and in vitro binding studies that show the essential role of Ca2+ and PS binding and the ausmenting role of PtdInsPs in membrane targeting of PKCα-C2, we measured membrane recruitment of PKCα-C2 and mutants in NIH-3T3 cells under different conditions. Specifically, we measured the effects of (i) mutating PS- and PtdInsP-binding residues of PKCα-C2, and (ii) depleting PtdIns(4,5)P2 and PtdIns(3,4,5)P3 from PM (or increasing their local concentrations) on the membrane recruitment of PKCα-C2.
The PtdIns(4,5)P2 depletion was performed according to a reported procedure using the Lyn11-FRB and CF-Inp constructs (12, 53), whereas PtdIns(3,4,5)P3 depletion was performed with PtdInsP 3-kinase inhibitor LY294002 (12). In the former system, rapamycin mediates the heterodimerization of the PM-bound Lyn11-FRB and CFP-FK505-binding protein that is fused to a yeast inositol polyphosphate 5-phosphatase (Inp54p), resulting in PtdIns(4,5)P2 hydrolysis in PM. As reported, the addition of 5 μm rapamycin to NIH-3T3 cells co-transfected with Lyn11-FRB, CF-Inp, and PLCδ-PH-YFP resulted in fast and irreversible translocation of CF-Inp to PM (completed within 1 min), which was synchronized with the dissociation of a PtdIns(4,5)P2 marker PLCδ-PH-YFP from PM (Fig. 4A). This finding shows that PM translocation of CF-Inp causes spontaneous and quantitative removal of PtdIns(4,5)P2 from PM under our experimental conditions. We also tested the PtdIns(3,4,5)P3 depletion by LY294002 using a PtdIns(3,4,5)P3 sensor, Bruton tyrosine kinase (Btk)-PH-CFP (24). Even at resting cells, Btk-PH-CFP showed a considerable degree of PM prelocalization (Fig. 4B), implying the presence of a detectable level of PtdIns(3,4,5)P3 at PM under our experimental conditions. Because PtdIns(3,4,5)P3 is generally thought to be produced in small amounts and transiently in response to cell activation, this may imply that Btk-PH is localized to PM through protein-protein interactions. However, a recent report indicated that a significant level of PtdIns(3,4,5)P3 is present at PM in cultured cells (12). To circumvent ambiguity, we added 50 ng/ml platelet-derived growth factor to NIH-3T3 cells to enhance the PtdIns(3,4,5)P3 level at PM as reported previously (24). This led to further PM recruitment of Btk-PH-CFP, which was in turn reversed within 4 min by treatment with 50 μm LY294002 (Fig. 4B). These control experiments thus established the robustness of PtdIns(4,5)P2 and PtdIns(3,4,5)P3 depletion methods under our experimental conditions.
FIGURE 4.


Membrane translocation of PKCα-C2 and mutants under different conditions. A, PtdIns(4,5)P2 depletion control. Addition of rapamycin induces rapid PM translocation of CF-lnp (cyan), which is synchronized with the dissociation of a PtdIns(4,5)P2 sensor, PLCδ-PH-YFP (yellow), from the PM. B, PtdIns(3,4,5)P3 depletion control. Addition of LY294002 induces the dissociation of a PtdIns(3,4,5)P2 sensor, Btk-PH-CFP (cyan), within 4 min. Btk-PH-CFP is prelocalized at the PM to a significant degree in the resting cells; however, to demonstrate the effect of LY294002, PtdIns(3,4,5)P2 formation at PM was induced by platelet-derived growth factor (PDGF). C, the PM translocation of PKCα-C2-RFP (red) was induced by ATP without PtdIns(4,5)P2 or PtdIns(3,4,5)P3 depletion. D, the PM translocation of PKCα-C2-RFP (red) was induced by ATP after PtdIns(4,5)P2 depletion. E, the PM translocation of PKCα-C2-RFP (red) was induced by ATP after PtdIns(3,4,5)P3 depletion. F, the PM translocation of PKCα-C2-RFP (red) was induced by ATP after sequential PtdIns(3,4,5)P3 and PtdIns(4,5)P2 depletion. G, the PM translocation of PKCα-C2-RFP (red) was induced by ATP after PtdIns(3,4,5)P3 induction. H, the PM translocation of PKCα-C2-RFP (red) was induced by ATP after PtdIns(4,5)P2 induction. I, the PM translocation of co-transfected PKCα-C2-WT-CFP (cyan) and K209A/K221A-RFP (red) was induced by ATP without PtdIns(4,5)P2 or PtdIns(3,4,5)P3 depletion. J, the membrane translocation of PKCα-C2-N189A-RFP (red) was induced by adding 10 μm ionomycin. All measurements except G were performed with NIH-3T3 cells co-transfected with Lyn11-FRB and CF-lnp. Single cells that represent the behaviors of the majority of the cell population under different conditions were selected for illustration. Images were taken every 15 s. Time values after individual treatments are separately shown. Decreased fluorescence intensities at later time points are due to photobleaching of fluorescence proteins. Bars indicate 10 μm.
In our cell studies, we induced the membrane translocation of PKCα-C2 by ATP stimulation that is known to enhance the intracellular Ca2+ in NIH-3T3 cells through the activation of PLCβ. The cell populations expressing similar levels of PKCα-C2 domains were selected by visual inspection of RFP fluorescence intensity and used for translocation measurements. A minimum of quadruple measurements were performed under each condition with >10 cells monitored for each measurement. Typically, >70% of the cell population showed similar behaviors with respect to membrane translocation of PKCα-C2.
When NIH-3T3 cells co-transfected with Lyn11-FRB, CF-Inp, and PKCα-C2-RFP were treated with 100 μm ATP, PKCα-C2 completed the translocation to PM within 2 min and largely stayed bound to PM even after 10 min (see Fig. 4C and Fig. 5A). When the same cells were treated sequentially with rapamycin (3 min during and after which extensive PtdIns(4,5)P2 depletion is expected as seen from the PM localization of CF-Inp in Fig. 4D) and ATP, PKCα-C2 still rapidly translocated to PM. In this case, however, PKCα-C2 returned to the cytosol much faster than PKCα-C2 without PtdIns(4,5)P2 depletion; the PM recruitment of PKCα-C2 was almost completely reversed within 10 min (see Fig. 5A). A similar effect was seen with PtdIns(3,4,5)P3 depletion by LY294002 (Fig. 4E). After treatment with 50 μm LY294002 for 6 min, ATP addition triggered fast PM translocation of PKCα-C2, which then returned to the cytosol faster than PKCα-C2 without PtdIns(3,4,5)P3 depletion. As expected from a lower level of PtdIns(3,4,5)P3 than PtdIns(4,5)P2 at PM, PtdIns(3,4,5)P3 depletion showed a lesser effect than the PtdIns(4,5)P2 depletion (see Fig. 5A). We then performed the double depletion of PtdIns(3,4,5)P3 and PtdIns(4,5)P2 by sequentially treating the cells with LY294002, rapamycin, and ATP (see Fig. 4F). Again, PKCα-C2 rapidly translocated to the PM and also rapidly dissociated from PM. Qualitatively, PtdIns(3,4,5)P3 and PtdIns(4,5)P2 depletion seems to have an additive effect (Fig. 5A).
FIGURE 5.

Effects of PtdInsP depletion and induction and the mutation of
PtdInsP-binding site on kinetics of PM translocation of PKCα-C2.
A, the RFP intensity ratio (=PM/(PM + cytosol)) of PKCα-C2-RFP,
which indicates the relative abundance of the protein at PM versus
cytosol, is plotted as a function of time. The plots are shown for no lipid
depletion (•, Fig.
4C), PtdIns(4,5)P2 depletion (○,
Fig. 4D;
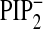 ), PtdIns(3,4,5)P3
depletion (Δ, Fig.
4E;
), PtdIns(3,4,5)P3
depletion (Δ, Fig.
4E; 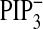 ), and
PtdIns(4,5)P2 + PtdIns(3,4,5)P3 depletion (□;
Fig. 4F;
), and
PtdIns(4,5)P2 + PtdIns(3,4,5)P3 depletion (□;
Fig. 4F;
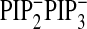 ).
B, the RFP intensity ratio as a function of time is shown for no
lipid induction (•, Fig.
4C), PtdIns(4,5)P2 induction (Δ,
Fig. 4H,
).
B, the RFP intensity ratio as a function of time is shown for no
lipid induction (•, Fig.
4C), PtdIns(4,5)P2 induction (Δ,
Fig. 4H,
 ), and PtdIns(3,4,5)P3
induction (○, Fig.
4G,
), and PtdIns(3,4,5)P3
induction (○, Fig.
4G, 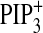 ).
C, the fluorescence intensity ratios of PKCα-C2-RFP (•)
and K209A/K211A (○) co-transfected into NIH-3T3 cells
(Fig. 4I) are plotted
as a function of time. No lipid depletion was performed for this
measurement.
).
C, the fluorescence intensity ratios of PKCα-C2-RFP (•)
and K209A/K211A (○) co-transfected into NIH-3T3 cells
(Fig. 4I) are plotted
as a function of time. No lipid depletion was performed for this
measurement.
We also measured the effect of increasing local PtdIns(3,4,5)P3 and PtdIns(4,5)P2 concentrations on the membrane translocation of PKCα-C2. The PtdIns(4,5)P2 induction was performed using the Lyn11-FRB and CF-PIPK constructs, whereas the PtdIns(3,4,5)P3 increase with Lyn11-FRB and CF-iSH constructs (12, 53). In these systems, rapamycin mediates the PM recruitment of phosphatidylinositol-4-phosphate 5-kinase and the p85 subunit of PtdInsP 3-kinase, respectively, through heterodimerization with Lyn11-FRB, resulting in the local synthesis of PtdIns(4,5)P2 and PtdIns(3,4,5)P3, respectively, in PM. As shown in Fig. 4, G and H (also in Fig. 5B), induced local synthesis of PtdIns(4,5)P2 or PtdIns(3,4,5)P3 considerably elongated the PM residence of PKCα-C2. As a result, PKCα-C2 exhibited only a minor degree of membrane dissociation even after 10 min.
We then monitored the membrane translocation of PKCα-C2-CFP and its K209A/K211A-RFP co-transfected into NIH3T3 cells. When these cells were stimulated with ATP, both proteins translocated to the PM with comparably high speed but K209A/K211A returned to the cytosol much faster than the wild type (see Figs. 4I and 5C). The same trend was seen when cells were co-transfected with PKCα-C2-RFP and K209A/K211A-CFP (data not shown). Qualitatively, K209A/K211A dissociated as fast as the wild type with the PtdIns(3,4,5)P3 and PtdIns(4,5)P2 double depletion, which is consistent with the PtdInsP binding role of the β-groove residues.
Lastly, we measured membrane translocation of N189A. Consistent with its much reduced affinity for POPC/POPS/PtdIns(4,5)P2 (78:20:2) vesicles (see Table 1), this mutant showed much slower translocation to the membrane in response to ATP stimulation in NIH-3T3 cells, which entailed the use of a stronger stimulus. When cells were treated with 10 μm ionomycin, N189A rapidly translocated to membranes but it no longer exhibited exclusive PM localization (Fig. 4J). In fact, this mutant, which was also distributed in the nucleus before stimulation, was recruited to nuclear membranes and cytosolic vesicular structures. This data again underscores the essential role of PS in specific PM translocation of PKCα-C2. The unique subcellular localization pattern is not an artifact due to ionomycin stimulation because PKCα-C2 wild type shows the same localization patterns in response to ATP and ionomycin, respectively (data not shown). Also, PtdIns(4,5)P2 depletion with co-transfection with Lyn11-FRB and CF-lnp had no effect on the subcellular localization of N189A (data not shown). Collectively, these cell data confirm that Ca2+ and PS binding drives the PM targeting of PKCα-C2, whereas PtdInsP binding augments the PM targeting by elongating its membrane residence.
DISCUSSION
The present study systematically investigates the differential roles of PS, PtdIns(4,5)P2, and PtdIns(3,4,5)P3, in membrane recruitment of PKCα-C2. This protein was selected for the study for three reasons. First, PKCα-C2 is a prototype Ca2+-dependent PM-targeting protein, and its membrane recruitment can be triggered by Ca2+ increase, unlike small G proteins that are pre-localized to PM when expressed in mammalian cells. This enables one to gain more mechanistic information through the kinetic analysis of membrane association and dissociation of the protein (11). Second, PKCα-C2 has well defined and spatially separate binding sites for PS and PtdInsPs, respectively. Thus, it allows systematic and unambiguous modulation of PS and PtdInsP binding through site-specific mutation. Third, there has been controversy as to whether PS or PtdIns(4,5)P2 binding is the major determinant of the specific PM targeting of PKCα-C2, which in itself warrants a systematic and rigorous analysis of the membrane binding of the domain. Collectively, PKCα-C2 serves as an excellent model for systematic structure-function and mechanistic studies to determine the differential roles of PS and PtdInsPs in the PM targeting of cytosolic proteins.
Our in vitro membrane binding studies by SPR analysis clarify key misconceptions about the lipid affinity and specificity of PKCα-C2. As demonstrated previously (11), PKCα-C2 has pronounced PS specificity and high affinity for PS-containing vesicles. However, it shows no specificity for any PtdInsP, PtdIns(4,5)P2 in particular, and only modest affinity for vesicles containing POPC and PtdInsPs. PtdInsPs, including PtdIns(4,5)P2 and PtdIns(3,4,5)P3, can augment the Ca2+-dependent membrane binding of PKCα-C2 only in the presence of PS in the membrane. This affinity increase is mainly due to slower membrane dissociation. The essential role of PS and the augmenting function of PtdInsPs are also supported by the observed effects of the mutations of PS- and PtdInsP-binding sites, respectively, on the membrane binding properties of PKCα-C2. The mutations of PS and Ca2+ ligands, Asn189 and Asp248, respectively, greatly reduce the affinity for PS-containing vesicles with or without PtdIns(4,5)P2. In contrast, the mutation of PtdIns(4,5)P2 ligands, Lys209 and Lys211, only shows a significant effect when PS is present in the PtdIns(4,5)P2-containing vesicles.
Our computational work provides further mechanistic insight while supporting the conclusion from the membrane binding studies. Systematic simulations with different permutation of lipids show that PS is the lipid determinant of the stable membrane association of the C2 domain. When PS (or Ca2+) is absent from the system, the domain has a weak affinity for the membrane and drifts away. The presence of PtdIns(4,5)P2 alone in the bilayer does not lead to the formation of a stable PKCα-C2-membrane complex and the domain displays a rotation, which causes the PtdIns(4,5)P2-binding β-groove region of the domain to drift away from the membrane. When both PS and Ca2+ are present, the domain maintains the binding orientation and does not show any rotation. This retention of the binding configuration occurs consistently over a longer time scale of 10 ns and does not occur temporarily due to environmental effects. Under this condition, the addition of PtdIns(4,5)P2 further stabilizes the PKCα-C2-membrane complex due in part to additional hydrogen bonds between PtdIns(4,5)P2 and β-groove residues. Our MD simulations also suggest that these extra interactions are achieved through the molecular movement that positions the β-groove residues closer to PtdIns(4,5)P2 in the membrane. The formation of additional hydrogen bonds account for the elongated membrane residence of PKCα-C2 on the PtdIns(4,5)P2-containing membranes because hydrophobic interactions and short-range specific interactions, such as hydrogen bond, have been shown to slow the membrane dissociation of proteins (3).
Cellular translocation behaviors of PKCα-C2 and mutants under different conditions further verify the notion that Ca2+-dependent PS binding drives the PM targeting of PKCα-C2, whereas PtdInsPs largely function in keeping the domain longer at the membrane. Depletion of PtdIns(4,5)P2 and/or PtdIns(3,4,5)P3 from PM significantly accelerates the dissociation of PKCα-C2 after it translocates to PM in response to ATP stimulation in NIH-3T3 cells. A similar effect is seen with the mutation of PtdInsP-binding β-groove residues. Conversely, induction of local synthesis of PtdIns(4,5)P2 or PtdIns(3,4,5)P3 at PM considerably slows the dissociation of PKCα-C2 from PM. Although it has been thought that PtdIns(3,4,5)P3 is a second messenger that is formed transiently upon activation of PtdInsP 3-kinases, no quantitative information about the cellular levels of PtdIns(3,4,5)P3 has been reported. Recently, Heo et al. (12) reported that the PM targeting of small G proteins was abrogated only when both PtdIns(4,5)P2 and PtdIns(3,4,5)P3 are removed, suggesting the presence of a significant level of PtdIns(3,4,5)P3 in the PM of cultured mammalian cells. Our results are consistent with the finding. PtdIns(3,4,5)P3 depletion, separately or in combination with PtdIns(4,5)P2 depletion, clearly accelerates the PM dissociation of PKCα-C2. Given that PKCα-C2 has similar affinity for PtdIns(4,5)P2 and PtdIns(3,4,5)P3, it is estimated from Fig. 5A that the level of PtdIns(3,4,5)P3 can be as high as a half of that of PtdIns(4,5)P2 in the PM under our experimental conditions. The finding that the association of PKCα-C2 to PM is not significantly affected by PtdInsP depletion and the mutation of PtdInsP-binding site, respectively, underscores that Ca2+-dependent PS binding is the driving force of PM targeting. This notion is also supported by the loss of specific PM targeting by the mutation of a PS-binding residue, Asn189, in the present study as well as the N189F/T250Y mutation in the previous study (16).
Although our results clearly dispute the notion that PtdIns(4,5)P2 binding is the driving force for the membrane targeting of PKCα-C2, it is important to point out how other studies reached a different conclusion. First of all, it should be noted that in earlier reports supporting the critical role of PtdIns(4,5)P2 in the membrane binding of PKCα-C2, PS is always included in the vesicles used for membrane binding measurements (16). Also, Corbin et al. (18) suggested that PKCα-C2 could associate with PtdIns(4,5)P2 at low Ca2+ levels prior to PS association. Indeed, it has been hypothesized for some C2 domains that Ca2+-independent interactions between the cationic β-groove and PtdIns(4,5)P2 in the PM keep the protein in a juxtamembrane location, which promotes rapid membrane interactions in response to a rise in intracellular Ca2+ (20). Our measurements also show that PKCα-C2 has modest (micromolar) affinity for POPC/PtdIns(4,5)P2 (98:2) vesicles at low Ca2+ concentrations. However, this affinity does not compare favorably with those of other PtdIns(4,5)P2-binding domains, such as Epsin ENTH domain (54) and PLCδ PH domain, determined under similar conditions. Thus, PKCα-C2 may not compete with these proteins for the available PtdIns(4,5)P2 pool at PM.
Results described herein provide new insight into the mechanism of cellular PM translocation of PKCα-C2. In response to a rise in intracellular Ca2+, PKCα-C2 would coordinate the Ca2+ ions and bind PS molecules abundant in the cytoplasmic face of the PM (11). The membrane-bound domain would then find the molecular orientation that optimizes the interactions of its cationic β-groove residues with PtdIns(4,5)P2 and PtdIns(3,4,5)P3 in the PM, which in turn elongates its membrane residence and increases the affinity of PKCα-C2 for the PM. This synergistic action of PS and PtdInsPs should confer the high PM targeting specificity on PKCα-C2.
It should be noted that not all PM-targeting domains and proteins have high PS specificity. For those proteins with PS specificity, the principle learned from this study can be applied to explaining their membrane targeting behaviors: i.e. the membrane binding is driven by PS binding and can be synergistically augmented by PtdIns(4,5)P2 or other minor lipids in the PM. For signaling proteins with PtdInsP specificity, including those proteins with PtdIns(4,5)P2-specific PH, PX, ENTH, and ANTH domains, PtdInsP binding should obviously play a primary role in PM targeting and PS might contribute to some degree through nonspecific electrostatic interactions. A situation is less clear for those signaling proteins with little lipid specificity, as two recent studies yielded conflicting results as to whether PS or PtdInsP binding is the main determinant of PM targeting of cytosolic proteins with polybasic motifs (12, 13). Undoubtedly, further studies are needed to fully understand the PM targeting mechanisms of diverse signaling proteins. Nevertheless, the present study demonstrates that a combination of computational and experimental approaches can provide valuable mechanistic insight into this important aspect of cell regulation.
This work was supported, in whole or in part, by National Institutes of Health Grants P01AI060915 (to H. L.) and GM52598, GM68849, and GM76581 (to W. C.). This work was also supported by an Indiana University Biomedical Research Grant, American Heart Association Grant SDG 0735350N, and American Cancer Society Grant IRG-84-002-22) (to R. V. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PM, plasma membrane; Btk, Bruton tyrosine kinase; CBL, calcium binding loop; CF-Inp, CFP-FK505-binding protein-Inp54p; CF-iSH, CFP-FKBP-PtdInsP 3-kinase p85 subunit; CF-PIPK, CFP-FKBP-phosphatidylinositol-4-phosphate 5-kinase; CFP, cyan fluorescence protein; Lyn11-FRB, Lyn11-rapamycin-binding protein; MD, molecular dynamics; PA, principal axis; PH, pleckstrin homology; PtdInsP, phosphoinositide; PtdIns(4,5)P2, phosphatidylinositol (4,5)-bisphosphate; PtdIns(3,4,5)P3, phosphatidylinositol (3,4,5)-trisphosphate; PLC, phospholipase C; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPI, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoinositol; POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine; RFP, red fluorescence protein; PS, phosphatidylserine; SPR, surface plasmon resonance; Inp54p, inositol polyphosphate 5-phosphatase; PKC, protein kinase C.
References
- 1.Teruel, M. N., and Meyer, T. (2000) Cell 103 181-184 [DOI] [PubMed] [Google Scholar]
- 2.Hurley, J. H., and Meyer, T. (2001) Curr. Opin. Cell Biol. 13 146-152 [DOI] [PubMed] [Google Scholar]
- 3.Cho, W., and Stahelin, R. V. (2005) Annu. Rev. Biophys. Biomol. Struct. 34 119-151 [DOI] [PubMed] [Google Scholar]
- 4.Cho, W. (2006) Sci. STKE 2006, pe7. [DOI] [PubMed]
- 5.Vance, J. E., and Steenbergen, R. (2005) Prog. Lipid Res. 44 207-234 [DOI] [PubMed] [Google Scholar]
- 6.Di Paolo, G., Pellegrini, L., Letinic, K., Cestra, G., Zoncu, R., Voronov, S., Chang, S., Guo, J., Wenk, M. R., and De Camilli, P. (2002) Nature 420 85-89 [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin, S., and Murray, D. (2005) Nature 438 605-611 [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin, S., Wang, J., Gambhir, A., and Murray, D. (2002) Annu. Rev. Biophys. Biomol. Struct. 31 151-175 [DOI] [PubMed] [Google Scholar]
- 9.Stahelin, R. V., Digman, M. A., Medkova, M., Ananthanarayanan, B., Rafter, J. D., Melowic, H. R., and Cho, W. (2004) J. Biol. Chem. 279 29501-29512 [DOI] [PubMed] [Google Scholar]
- 10.Stahelin, R. V., Hwang, J. H., Kim, J. H., Park, Z. Y., Johnson, K. R., Obeid, L. M., and Cho, W. (2005) J. Biol. Chem. 280 43030-43038 [DOI] [PubMed] [Google Scholar]
- 11.Stahelin, R. V., Rafter, J. D., Das, S., and Cho, W. (2003) J. Biol. Chem. 278 12452-12460 [DOI] [PubMed] [Google Scholar]
- 12.Heo, W. D., Inoue, T., Park, W. S., Kim, M. L., Park, B. O., Wandless, T. J., and Meyer, T. (2006) Science 314 1458-1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeung, T., Gilbert, G. E., Shi, J., Silvius, J., Kapus, A., and Grinstein, S. (2008) Science 319 210-213 [DOI] [PubMed] [Google Scholar]
- 14.Verdaguer, N., Corbalan-Garcia, S., Ochoa, W. F., Fita, I., and Gomez-Fernandez, J. C. (1999) EMBO J. 18 6329-6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Bautista, S., Marin-Vicente, C., Gomez-Fernandez, J. C., and Corbalan-Garcia, S. (2006) J. Mol. Biol. 362 901-914 [DOI] [PubMed] [Google Scholar]
- 16.Evans, J. H., Murray, D., Leslie, C. C., and Falke, J. J. (2006) Mol. Biol. Cell 17 56-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerrero-Valero, M., Marin-Vicente, C., Gomez-Fernandez, J. C., and Corbalan-Garcia, S. (2007) J. Mol. Biol. 371 608-621 [DOI] [PubMed] [Google Scholar]
- 18.Corbin, J. A., Evans, J. H., Landgraf, K. E., and Falke, J. J. (2007) Biochemistry 46 4322-4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho, W. (2001) J. Biol. Chem. 276 32407-32410 [DOI] [PubMed] [Google Scholar]
- 20.Bai, J., and Chapman, E. R. (2004) Trends Biochem. Sci. 29 143-151 [DOI] [PubMed] [Google Scholar]
- 21.Malmberg, N. J., and Falke, J. J. (2005) Annu. Rev. Biophys. Biomol. Struct. 34 71-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho, W., and Stahelin, R. V. (2006) Biochim. Biophys. Acta 1761 838-849 [DOI] [PubMed] [Google Scholar]
- 23.Murray, D., and Honig, B. (2002) Mol. Cell 9 145-154 [DOI] [PubMed] [Google Scholar]
- 24.Manna, D., Albanese, A., Park, W. S., and Cho, W. (2007) J. Biol. Chem. 282 32093-32105 [DOI] [PubMed] [Google Scholar]
- 25.Medkova, M., and Cho, W. (1998) J. Biol. Chem. 273 17544-17552 [DOI] [PubMed] [Google Scholar]
- 26.Stahelin, R. V., and Cho, W. (2001) Biochemistry 40 4672-4678 [DOI] [PubMed] [Google Scholar]
- 27.Cho, W., Bittova, L., and Stahelin, R. V. (2001) Anal. Biochem. 296 153-161 [DOI] [PubMed] [Google Scholar]
- 28.Humphrey, W., Dalke, A., and Schulten, K. (1996) J. Mol. Graph. 14 33-38, 27-38 [DOI] [PubMed] [Google Scholar]
- 29.Corbalan-Garcia, S., Garcia-Garcia, J., Rodriguez-Alfaro, J. A., and Gomez-Fernandez, J. C. (2003) J. Biol. Chem. 278 4972-4980 [DOI] [PubMed] [Google Scholar]
- 30.Hauser, H., and Poupart, G. (1992) in The Structure of Biological Membranes, (Yeagle, P., ed) pp. 3-71, CRC Press, Boca Raton, FL
- 31.Wang, J., Wang, W., Kollman, P. A., and Case, D. A. (2006) J. Mol. Graph. Model 25 247-260 [DOI] [PubMed] [Google Scholar]
- 32.Case, D. A., Cheatham, T. E., 3rd, Darden, T., Gohlke, H., Luo, R., Merz, K. M., Jr., Onufriev, A., Simmerling, C., Wang, B., and Woods, R. J. (2005) J. Comput. Chem. 26 1668-1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004) J. Comput. Chem. 25 1157-1174 [DOI] [PubMed] [Google Scholar]
- 34.Jakalian, A., Bush, B. L., Jack, D. B., and Bayly, C. I. (2000) J. Comput. Chem. 21 132-146 [DOI] [PubMed] [Google Scholar]
- 35.Jakalian, A., Jack, D. B., and Bayly, C. I. (2002) J. Comput. Chem. 23 1623-1641 [DOI] [PubMed] [Google Scholar]
- 36.Mukhopadhyay, P., Monticelli, L., and Tieleman, D. P. (2004) Biophys. J. 86 1601-1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liepina, I., Czaplewski, C., Janmey, P., and Liwo, A. (2003) Biopolymers 71 49-70 [DOI] [PubMed] [Google Scholar]
- 38.Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., Chipot, C., Skeel, R. D., Kale, L., and Schulten, K. (2005) J. Comput. Chem. 26 1781-1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhardwaj, N., Stahelin, R. V., Zhao, G., Cho, W., and Lu, H. (2007) Bioinformatics 23 3110-3112 [DOI] [PubMed] [Google Scholar]
- 40.Feller, S. E., Zheng, Y. H., Pastor, R. W., and Brooks, B. R. (1995) J. Comp. Phys. 103 4613-4621 [Google Scholar]
- 41.Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995) J. Chem. Phys. 103 8577-8593 [Google Scholar]
- 42.Darden, T., York, D., and Pedersen, L. (1993) J. Chem. Phys. 98 10089-10092 [Google Scholar]
- 43.Kohout, S. C., Corbalan-Garcia, S., Gomez-Fernandez, J. C., and Falke, J. J. (2003) Biochemistry 42 1254-1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stahelin, R. V., and Cho, W. (2001) Biochem. J. 359 679-685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conesa-Zamora, P., Lopez-Andreo, M. J., Gomez-Fernandez, J. C., and Corbalan-Garcia, S. (2001) Biochemistry 40 13898-13905 [DOI] [PubMed] [Google Scholar]
- 46.Berneche, S., Nina, M., and Roux, B. (1998) Biophys. J. 75 1603-1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mihailescu, D., and Smith, J. C. (2000) Biophys. J. 79 1718-1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smondyrev, A. M., and Berkowitz, M. L. (2000) Biophys. J. 78 1672-1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Villarreal, M. A., Diaz, S. B., Disalvo, E. A., and Montich, G. G. (2004) Langmuir 20 7844-7851 [DOI] [PubMed] [Google Scholar]
- 50.Kasson, P. M., and Pande, V. S. (2004) Biophys. J. 86 3744-3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smondyrev, A. M., and Voth, G. A. (2002) Biophys. J. 82 1460-1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blood, P. D., and Voth, G. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 15068-15072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inoue, T., Heo, W. D., Grimley, J. S., Wandless, T. J., and Meyer, T. (2005) Nat. Methods 2 415-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stahelin, R. V., Long, F., Peter, B. J., Murray, D., De Camilli, P., McMahon, H. T., and Cho, W. (2003) J. Biol. Chem. 278 28993-28999 [DOI] [PubMed] [Google Scholar]