

Abstract
Fractionation of two Fijian specimens of the sponge Corticium sp., led to the isolation of the known active alkaloid steroid plakinamine A and two new halogenated cyclic peptides, corticiamide A (1) and cyclocinamide B (2). Structural elucidation of 1 and 2 was achieved by an extensive combination of high field NMR and HRFT MS/MS experiments, and the absolute stereochemistry of 2 was determined by acid hydrolysis and Marfey’s analysis. Corticiamide A (1) and cyclocinamide B (2) represent the first peptides to be described from the genus Corticium.
The sponge genus Corticium is widespread in warm temperate to tropical waters of the world’s oceans, yet compounds isolated from the genus are far from prevalent within the chemical literature. To date, only two main structural motifs have been reported: the polycyclic meridene1 and a number of steroidal alkaloids, including members of the plakinamine family2–6 and two unusual steroids containing a seven membered B ring.7 Almost all reported Corticium derived compounds have exhibited in vitro bioactivity. For example, meridene is a potent antifungal compound,1 plakinamine derivatives exhibit significant in vitro cytotoxic activity6 and slight HIV activity,5 and the lokysterolamines have been reported to be cytotoxic, antimicrobial and antifungal agents.2 Recently, an additional class of steroidal alkaloids from Corticium simplex has been described that shows anti-proliferative activity as well as antiangiogenic activity.8 Fractionation of Corticium sp. collected from Fiji in the years 2001–2004 inclusive, led to the isolation of plakinamine A and two new cyclic peptides, corticiamide A (1) and cyclocinamide B (2). Structural elucidation of the two new peptides was achieved by extensive use of NMR and FTMS/MS experiments, and the absolute stereochemistry of cyclocinamide B (2) was determined by Marfey’s analysis. Corticiamide A (1) and cyclocinamide B (2) are the first peptides, and the first halogenated compounds, to be isolated from a species of Corticium.
Results and Discussion
The crude methanol extract of the 2002 collection of the Fijian Corticium sp. was partitioned between EtOAc and H2O, and the bioactive EtOAc soluble material was separated on HP20 resin resulting in 5 fractions. Fraction 3 was the most active and was separated by HPLC yielding 0.8 mg of corticiamide A (1) as a pale yellow oil. Monoisotopic peaks for the doubly charged molecular ions [M+2H]2+ and [M+H+Na]2+ were detected at m/z 948.32495 and m/z 959.31633 in positive mode nanoelectrospray FTMS corresponding to a neutral monoisotopic mass of 1894.63617. On the basis of accurate mass measurements of the molecular and fragment ions, a molecular formula of C80H112Br2N20O22S was determined for the neutral molecule.
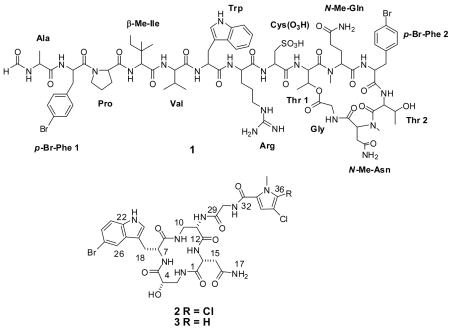
Interpretation of 1D and 2D NMR spectra (1H, TOCSY, gHSQC and gHMBC) resulted in the identification of 14 partial structures. Eight standard amino acid residues were identified as Ala, Val, two Thr, Pro, Trp, Gly, and Arg. In addition, a formamide moiety was determined by the presence of a signal in the HSQC spectrum for a methine with shifts at δH 7.89 and δC 161.2, respectively. The presence of two ortho-coupled proton signals at δH 7.25 and δH 7.40 in the 1H NMR spectrum suggested a para substituted phenyl ring. Correlations in the HMBC spectrum from the ortho-coupled aromatic protons to two quaternary carbons (δC 120.2 and δC 137.6) suggested a p-Br substitution. TOCSY correlations from an NH (δH 7.94) to a methine at δH 4.73 and a diastereotopic methylene (δH 2.73, δH 3.01; δC 38.8), in combination with an HMBC correlation from this methylene to aromatic carbons (δC 132.5 and δC 137.6) established this residue as p-Br-Phe. A second p-Br-Phe residue was identified in a similar manner. The cysteic acid residue was proposed based on the downfield 13C chemical shifts (δC 51.6 and δC 53.0, respectively) of an observed NH-CH-CH2 spin system and was confirmed by MS studies (vide infra). The β-Me-Ile residue was determined by HMBC correlations from an sp3 hybridized quaternary carbon (δC 37.5) to three methyl groups, one of which had an upfield 13C shift at δC 8.7, a methylene, and an NH-CH. An N-Me-Gln was identified starting from an HMBC correlation between an N-methyl (δH 2.93, δC 30.5) and a methine (δC 54.9) that was part of a -CH-CH2-CH2- TOCSY spin system. An HMBC correlation from the γ-methylene to the δ-carbonyl (δC 174.0) provided additional support for the N-Me-Gln. An HMBC correlation from a second N-methyl (δH 2.60, δC 29.2) to a methine (δC 56.7) that was part of a -CH2-CH2- spin system was consistent with an N-Me-Asn. HMBC correlations from the methylene protons (δH 2.34, δH 2.84) to the γ-carbonyl (δC 171.5) confirmed the presence of N-Me-Asn. The linear sequence of (1) was deduced from inter-residue ROESY correlations (see Figure 1) in conjunction with MS experiments.
Figure 1.

Planar structure of corticiamide A (1). Bold bonds represent TOCSY correlations, arrows selected HMBC correlations, and lines selected NOE’s.
Attempts to confirm the structure using ESI FTMS/MS experiments were conducted concurrently with the NMR based structure elucidation. FTMS/MS experiments using Sustained Off-Resonance Irradiation with Collision Induced Dissociation (SORI-CID) were crucial to solving the planar structure of 1 as it enabled accurate mass measurements of MS/MS daughter ions, resulting in the assignment of specific molecular formulas for many fragments.9 The SORI-CID mass spectrum of the isolated [M+2H]2+ at m/z 948 is shown in Figure 2. The majority of the fragmentation occurred in the linear region of the peptide, and the most abundant fragment ion (m/z 7862+) was produced through cleavage of the amide bond on the N-terminal side of the Pro residue. Cleavage N-terminal to a Pro residue has been widely observed and documented as the major fragmentation site for many peptides,10 and the fragmentation pattern observed provides support for the assignment of a Pro at residue 3. The neutral loss between m/z 948 ([M+2H]2+) and m/z 7862+ could be assigned the elemental composition C13H13BrN2O3 corresponding to HCO-Ala-pBr-Phe. The Pro residue could be unambiguously assigned by the observation of the b3 ion. Detection of the b4 and b5 ions, together with their complementary y10″ and y9″ ions, resulted in the sequence assignment of a β-MeIle-Val fragment after the Pro residue. Unique elemental compositions were determined based on accurate mass measurements for these relatively small b3, b4, and b5 fragment ions and were supported by an observed isotopic distribution characteristic of the presence of one Br atom. The sequence was easily extended to Trp and Arg based upon the y-series fragment ions. Unique elemental compositions could not be determined for these relative large y-series fragment ions, but the mass difference between adjacent y-series fragment ions was easily assigned a unique elemental composition based upon accurate mass measurements.
Figure 2.

HR SORI CID mass spectral analysis of major fragments for corticiamide A (1).
The peptide sequence could not be followed after Arg using fragmentation of the molecular ion, as neither b8 nor its complementary y6″ ion (cleavage of CysO3H-Thr) was detected in the positive mode FTMS SORI-CID mass spectrum. Therefore, the abundant doubly charged y12″ ion, m/z 7862+, was isolated and fragmented using SORI-CID. Although the sequence immediately after Arg could not be determined, due again to the lack of fragment ions from the cleavage of CysO3H-Thr, the connection of Gly-NMeAsn-Thr-pBrPhe-NMeGln was established by detection of the following series of ions: m/z 143.08176 (C6H11N2O21+), 368.06100 (C15H19BrN3O31+), 469.10886 (C19H26BrN4O51+), 597.16707 (C24H34BrN6O71+), and 654.18813 (C26H37BrN7O81+). Once again, unique elemental compositions were determined for each ion based upon observed accurate mass measurements for these relatively small fragment ions and supported by the detected isotopic distribution (four of them are characteristic of one Br atom). Complementary ions to the m/z 469 and 597 ions were also detected at m/z 1103.53108 (C48H75N14O14S1+) and 975.47237 (C43H67N12O12S1+), respectively, further supporting the proposed sequence connection. Detailed examination of the SORI-CID mass spectrum for the isolated m/z 7862+ ion indirectly suggested the presence of a CysO3H-Thr fragment. Firstly, the elemental composition for the difference between the two major fragment ions m/z 666 and 975 could be assigned as C9H15N3O7S (Expt, 309.06370, Calcd 309.06307, Δ = 0.63 mmu) corresponding to CysO3H-Thr-Gly, and secondly, the observed m/z 818 ion could be explained as the loss of SO3H2 from the m/z 900 ion, which itself could be explained as resulting from the m/z 975 ion by the loss of Gly and H2O.
Assignment of Cys(O3H) adjacent to Arg was made unambiguously by the detection of the y6″1− ion in the SORI-CID mass spectrum of the isolated singly charged deprotonated molecular ion at m/z 18931−. The difference between m/z 7531− (y6″) and m/z 9041− (y7″) was accurately measured, resulting in a unique elemental composition corresponding to Cys(O3H). The peptide sequence in the linear portion of 1, determined based on FTMS/MS of the doubly charged protonated [M+2H]2+ molecular ion, was identical to that based on the FTMS/MS of the singly charged deprotonated [M−H]1− molecular ion, providing additional support for the sequence assignment.
Both MS and NMR data supported the linear sequence of 1. However, the molecular formula assigned demanded 34 units of unsaturation and only 33 were accounted for above, which implied the presence of an additional ring. The chemical shift for the β-proton of Thr-1 was shifted downfield to δH 5.22, typical of a threonine involved in an ester linkage,11–15 suggesting the site of cyclization. Observation of an HMBC correlation from the β-proton of Thr-1 to the Gly amide carbon (δC 169.0) confirmed that 1 was a cyclic depsipeptide.
Corticiamide A (1) is a member of a family of structurally related peptides that include the discodermins,16–18 halicylindramides,19,20 polydiscamide A21 and microspinosamide A.22 However, corticiamide A (1) is the only member of the family to contain a p-Br-Phe at residue 11, and no other examples are known to contain an N-Me-Asn. Microspinosamide A and polydiscamide A contain the unusual β-Me-Ile at residue 6, whereas the same amino acid is found at residue 5 in 1. Based upon these differences, the trivial name corticiamide A is proposed for 1. All previously reported compounds in this class have a mixture of D and L amino acids. Unfortunately, due to the small amount of 1 isolated, the absolute stereochemistry of the molecule was not determined nor was the bioactivity evaluated.
The crude methanol extract of a 2001 collection of Corticium was partitioned (see Experimental), and the CHCl3 soluble portion was separated using C18 flash chromatography (H2O to CH3OH gradient) resulting in thirteen fractions. While preparing fraction 10 for preliminary 1H NMR analysis, a portion of the material was not soluble in CD3OD and further investigation revealed that this waxy white substance was soluble in DMSO. The 1H and 13C NMR spectra of the DMSO soluble material indicated that the compound was of high purity, and a full structural elucidation was undertaken. The combination of six carbonyl signals in the range δ 169.4 to δ 173.5 in the 13C spectrum (Table 2), typical of amide carbonyls suggested that compound (2) was most likely a peptide.
Table 2.
NMR data for cyclocinamide B (2) in DMSO-d6 (400 MHz).
| position | δC | δH (J in Hz) | COSY | TOCSY | HMBCa |
|---|---|---|---|---|---|
| OH | - | 6.00 d (4.0) | - | 3 | 5 |
| 1 | 171.1 | - | - | - | - |
| 2 | - | 7.05 br t (5.0) | 4 | 4, 3 | 1 |
| 3 | 70.4 | 4.04 m | 4, OH | 4, 2, OH | - |
| 4 | 43.5 | 3.35 m, 3.48 m | 2, 3 | 3, 2, OH | - |
| 5 | 171.6 | - | - | - | - |
| 6 | - | 7.87 d (9.9) | 7 | 7, 18 | 7, 5 |
| 7 | 54.0 | 4.52 m | 6, 18 | 18, 6 | 18, 19, 8, 5 |
| 8 | 173.5 | - | - | - | - |
| 9 | - | 7.98 t (6.0) | 10 | 11, 10 | 8 |
| 10 | 40.8 | 3.34 s | 28, 11 | 11, 9, 28 | - |
| 11 | 55.2 | 4.27 m | 10, 28 | 10, 9, 28 | 29 |
| 12 | 169.4 | - | - | - | - |
| 13 | - | 7.89 d (9.2) | 14 | 14, 15 | 12 |
| 14 | 50.0 | 4.56 m | 13, 15 | 15, 13 | 15, 16, 12 |
| 15 | 37.1 | 2.26 dd (4.8, 15.6) | 11 | 11, 13 | 14, 16, 1 |
| 15′ | 2.45 dd (7.4, 15.6) | 11 | - | 14, 16, 1 | |
| 16 | 172.6 | - | - | - | - |
| 17 | - | 6.80 brs | 17′ | 17′ | 16 |
| 17′ | 7.28 brs | 17 | 17 | - | |
| 18 | 28.5 | 2.95 | 7 | 7, 6 | 7, 8, 20, 19, 28 |
| 19 | 110.2 | - | - | - | - |
| 20 | 126.0 | 7.15 d (1.7) | 18, 21 | 21 | 19 |
| 21 | - | 11.1 br d (1.7) | 20 | 20 | (19, 28) |
| 22 | 135.5 | - | - | - | - |
| 23 | 114.1 | 7.28 d (8.4) | 26, 24 | 26, 24 | 28, 25 |
| 24 | 124.1 | 7.14 dd (1.8, 8.4) | 26, 23 | 26, 23 | 28, 26, 25, 22 |
| 25 | 111.8 | - | - | - | - |
| 26 | 121.4 | 7.63 d (1.8) | 24,23 | 23 | 19,25, 24, 22 |
| 28 | 129.8 | - | - | - | - |
| 28 | - | 8.09 d (7.6) | 11 | 111, 10 | 29 |
| 29 | 169.7 | - | - | - | - |
| 30 | 42.9 | 3.75 m, 3.83 m | 31 | 31 | 29, 32 |
| 31 | - | 8.57 t (5.8) | 30 | 30 | 34, 32 |
| 32 | 161.0 | - | - | - | - |
| 33 | 125.2 | - | - | - | - |
| 34 | 112.2 | 7.02 s | - | - | 33, 36, 37, 32 |
| 35 | 107.9 | - | - | - | - |
| 36 | 119.2 | - | - | - | - |
| 37 | 34.1 | 3.76 s | - | - | 33, 34, 36 |
HMBC correlations from proton(s) to the stated carbons.
The mass spectrum obtained with ESI-FTMS indicated that the compound contained multiple halogens and SORI-CID experiments on the isolated monoisotopic molecular ion as well as the daughter ion m/z 535 established the molecular formula of 2 as C29H32N9O8BrCl2 using the top-down, bottom-up approach (expt. 784.10084, calc. 784.10070).9 Analysis of 1D and 2D NMR data (1H, 13C, COSY, TOCSY, gHSQC-TOCSY, gHSQC and gHMBC) led to the identification of six partial structures. A singlet at δH 11.1 and an upfield quaternary carbon at 111.8 suggested a 5-Br tryptophan residue (5-Br-Trp). Comparison with published data23–25 confirmed the assignment of the 5-Br-Trp. HSQC-TOCSY correlations from the methine carbon at δC 70.4 (δH 4.04) to an NH (δH 7.05), a diastereotopic methylene (δH 3.35, δH 3.48), and a hydroxyl (δH 6.00) indicated the presence of isoserine (iSer). TOCSY correlations were used to establish a spin system incorporating NH-CH2-CH-NH resulting in the assignment of a 1,2-diaminopropionic acid (DAP) residue. Using these spin systems for a database search resulted in cyclocinamide A (3). A comparison of NMR data in conjunction with the MS data indicated that cyclocinamide B (2) contained a dichloropyrrole instead of the monochloropyrrole found in cyclocinamide A (3). HMBC correlations from the aromatic methine singlet at δ 7.02 to the high-field carbonyl (δ 161.0), and two quaternary carbons (δ 125.2 and δ 119.2), established the aromatic methine at position 3 of the pyrrole, and the upfield 13C chemical shift (δ 112.2) suggested an adjacent Cl substitution. Thus, the second Cl was located at position 5 on the pyrrole, leading to the elucidation of an N-Me-2-carbonyl-4,5-dichloropyrrole moiety.
Since cyclocinamide A (3), which was isolated from Psammocinia sp.,26 is so similar to 2, the trivial name cyclocinamide B is proposed for 2. The published 1H NMR spectrum of cyclocinamide A (3) and cyclocinamide B (2) are almost identical, further supporting the proposed structure for cyclocinamide B (2). The only major difference is the iSer OH, which Clark et al. assigned at δ 3.27. However, in cyclocinamide B (2) correlations in the HSQC-TOCSY and HMBC spectra clearly indicate that the doublet at 6.00 can be δH assigned to this hydroxyl. Most likely, the shift may vary and is dependent on the sample preparation (H2O content, pH, etc.).
As a result of limited material Clark et al.26 were only able to determine the absolute stereochemistry at two of the four stereocenters of cyclocinamide A (3). Using a combination of acid hydrolysis and chiral TLC they assigned S stereochemistry for the Br-Trp and Arg residues and suggested that, based upon biosynthetic considerations, the stereochemistry of the DAP and iSer residues should also be S. Although, synthesis of 4R, 11R cyclocinamide A resulted in a product with a similar 1H NMR, the spectrum was not identical to the natural product.27
Cyclocinamide B (2) was subjected to acid hydrolysis as described by Clark et al.26 However, these conditions proved too harsh and resulted in complete decomposition. Complete hydrolysis was achieved at 110 °C in four hours with 6 N HCl in the presence of 0.1% phenol (to preserve the 5-Br-Trp residue). The hydrolysate was subjected to derivatization with Nα-(2,4-dinitro-5-fluorophenyl)-L-alaninamide (L-FDAA) and subsequent analysis by HPLC (Marfey’s analysis).28,29 Reference compounds were prepared with commercially available amino acids utilizing both pure L-amino acids and DL- mixtures. In each case the L-amino acid derivatives eluted before the D isomers. The results of the Marfey’s analysis unambiguously proved the absolute stereochemistry for three of the four amino acids: D-Asp, L-DAP, and D-5-Br-Trp. Unfortunately, the retention times of L-FDAA-D-iSer and L-FDAA-Gly overlapped. To circumvent this problem, LCMS analysis of the Nα-(2,4-dinitro-5-fluorophenyl)-L-valinamide (L-FDVA) derivatized amino acids was undertaken. The greater lipophilicity of L-FDVA vs. L-FDAA allowed for longer retention times and better separation. A coinjection with an L-FDVA-DL-iSer reference and the L-FDVA derivatized hydrolysate of 2 showed an enhancement of L-iSer over D-iSer and confirmed the presence of L-iSer in cyclocinamide B (2).
Both corticiamide A (1) and cyclocinamide B (2) have close structural resemblances to peptides isolated from sponges that are only distantly related, taxonomically, to Corticium. Thus, it seems likely that these two cyclic peptides may be produced by microorganisms associated with the Fijian Corticium sp.
Peptides related to corticiamide A (1), namely the discodermins, halicylindramides, polydiscamide A and microspinosamide A are all known to be cytotoxic in the low μM range and to inhibit the growth of bacteria and fungi.17–22 In addition, microspinosamide A has been shown to inhibit the cytopathic effect of HIV-1 in CEM-SS cells.22 Interestingly, the cyclic nature of these peptides is crucial to their bioactivity, with linear versions exhibiting a loss of activity of at least one order of magnitude. It has been noted that this class of peptides exhibits a high degree of amphiphilic character and that this may lead to observed increases in the membrane permeability of cells and tissues treated with discodermin A.30 Cyclocinamide A (3) was reported to be highly cytotoxic in vitro and exhibited selective cytoxicity toward solid tumors in the soft agar colony formation disk diffusion assay; however, cyclocinamide B (2) had no cytotoxicity against HCT-116 cells.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco DIP-370 polarimeter. UV spectra were obtained using a Perkin Elmer UV/VIS spectrometer Lambda2. IR spectra were recorded on a JASCO FTIR-420 spectrophotometer. NMR data for 1 were collected using either a Varian INOVA 500 (1H 500 MHz, 13C 125 MHz) NMR spectrometer with a 3 mm Nalorac MDBG probe or a Varian INOVA 600 (1H 600 MHz, 13C 150 MHz) NMR spectrometer equipped with a 5 mm 1H[13C,15N] triple resonance cold probe with a z-axis gradient and utilized residual solvent signals for referencing (δH 1.94, δC 118.3 for CD3CN). NMR data for 2 were obtained using a Varian Mercury 400 (1H 400 MHz, 13C 100 MHz) instrument equipped with a 5 mm Nalorac probe and utilized residual solvent signals for referencing (δH 2.49, δC 39.7 for DMSO-d6). LCMS analyses were conducted with a Micromass Q-Tof micro mass spectrometer and a Waters 2795 HPLC. High resolution mass spectra (HRMS) were obtained using a Bruker (Billerica, MA) APEXII FTICR mass spectrometer equipped with an actively shielded 9.4 Tesla superconducting magnet (Magnex Scientific Ltd., UK), an external Bruker APOLLO ESI source, and a Synrad 50W CO2 CW laser. Nanoelectrospray in both positive and negative modes was employed. Typically, a 5 μL sample was loaded into the nanoelectrospray tip (New Objective, Woburn, MA) and a high voltage, about 1000 V, was applied between the nanoelectrospray tip and the capillary. Mass spectra were internally/externally calibrated using HP tuning mix. For FTMS/MS experiments, precursor ions were isolated using correlated sweep and then dissociated using SORI-CID. Analytical and semi-preparative HPLC was accomplished utilizing a Beckman System Gold 126 solvent module and 168 PDA detector. All commercially available reagents and amino acid standards were purchased and used without additional purification.
Biological Material
Corticium sp. was collected by SCUBA. Bulk material was frozen at −4°C until return to The University of Utah where it was stored at −20 °C until required. All of our Fijian collections of Corticium sp. (Homosclerophorida, Plakinidae) are morphologically indistinguishable. It is a common undescribed species of Corticium with a widespread Indo-Pacific distribution. The sponge is encrusting, contracting considerably after collection; it has a black exterior and slighter lighter interior. Sample FJ01-2-014 was collected in the lower Yasawa group (S 18° 42.041′ E 178° 30.309′), sample FJ02-13-059 was from the Great Astrolabe Reef (S 17° 10.211′ E 177° 17.720′); voucher specimens are maintained at the University of Utah.
Extraction and Isolation
Frozen sponge (70 g wet weight) from the 2002 collection (sample FJ02-13-059) was extracted with MeOH and the solvent removed in vacuo. The crude extract was then partitioned between EtOAc and H2O and the EtOAc extracts were further separated on HP20ss resin (H2O to MeOH in 25% steps, 10 mL fractions). The fraction eluting with 1:1 H2O:MeOH was subjected to multiple rounds of RPHPLC as follows. Isocratic 2:3 MeOH:H2O for 20 min, followed by a gradient to 100% MeOH over 40 min, 3.5 mL/min, Phenomenex phenylhexyl, 10 × 250 mm. The fraction that eluted with 80–85% MeOH contained corticiamide A (1) and was subjected to RPHPLC employing a gradient of 1:1 CH3CN:H2O to 4:1 CH3CN:H2O in 20 min, 3.5 mL/min, Phenomenex C18, 10 × 250 mm. The corticiamide A (1) containing fraction eluted with 52–56% CH3CN, and was subjected to a gradient from 3:7 CH3CN:0.1% aq. TFA to 3:2 CH3CN:0.1% aq. TFA over 20 min, and then to 100% CH3CN in 5 min. Corticiamide A (1, 0.8 mg) eluted at 15 min.
Frozen sponge (100 g wet weight) from the 2001 collection (sample FJ01-2-014) was extracted with MeOH and the solvent removed in vacuo. The crude extract was then dissolved in 100 mL of 10% aqueous MeOH and extracted with hexanes (3 × 100 mL). An additional 35 mL of H2O was added to the aqueous MeOH and extracted with CHCl3 (3 × 100 mL). The CHCl3 fraction was further separated by RP C18 flash chromatography using a H2O:MeOH gradient in 10% steps. Fraction 10 contained 9.6 mg of 2.
Corticiamide A (1)
+5 (c 0.16, MeOH); UV (MeOH) λmax nm (log ε) 201 (4.59), 230 (4.23), 275 (3.5); IR (KRS-5 cell, DMSO) νmax 3465, 2971, 1662, 1250, 1005 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS [M+2H]2+ m/z 948.32495 (calcd 948.32465 for C80H114Br2N20O22S2+), [M+H+Na]2+ m/z 959.31633 (calcd 959.31562 for C80H113Br2N20NaO22S2+).
Table 1.
NMR data for corticiamide A (1) in DMSO-d6 (600 MHz).
| position | δCa | δH (J in Hz) | HMBC | position | δCa | δH (J in Hz) | HMBC |
|---|---|---|---|---|---|---|---|
| Alanine | Cysteic Acid | ||||||
| α | 47.1 | 4.29 m | β, C=O | α | 51.6 | 4.60 m | β, C=O |
| β | 19.2 | 0.91 d (6.0) | α, C=O | β | 53.0 | 2.89 m, 2.95 m | α |
| NH | 8.14 | CHO | NH | 8.34 br s | |||
| C=O | 172.0 | C=O | 171.7 | ||||
| CHO | 161.2 | 7.89 br s | α | Threonine (1) | |||
| p-Bromophenylalanine (1) | α | 53.0 | 4.86 d (8.6) | ||||
| α | 52.1 | 4.67 m | β, γ, C=O | β | 69.2 | 5.22 m | Gly-C=O |
| β | 36.9 | 2.65 m 2.90 m | γ, δ | γ | 17.1 | 1.06 d (5.0) | α, β |
| γ | 137.7 | NH | 7.86 br s | ||||
| δ, δ′ | 132.3 | 7.19 d (8.1) | β, δ′,δ, ζ | C=O | 169.4 | ||
| ε,ε′ | 131.4 | 7.38 d (8.1) | γ, ε′,ε, ζ | N-Methylglutamine | |||
| ζ | 120.2 | α | 54.9 | 4.93 m | β, γ | ||
| NH | 8.39 d (8.7) | Ala-C=O | β | 25.4 | 1.55 m | ||
| C=O | 170.1 | γ | 32.0 | 1.76 m | δC=O | ||
| Proline | δC=O | 174.0 | |||||
| α | 60.0 | 4.53 d (6.6) | α, β, C=O | δNH2 | 6.67 br s, (7.1) | ||
| β | 30.5 | 2.06 m | γ | NMe | 30.5 | 2.93 br s | α, Thr(1)-C=O |
| γ | 25.0 | 1.83 m | δ | C=O | 169.4 | ||
| δ | 47.4 | 3.62 br s | β, γ | p-Bromophenylalanine (2) | |||
| C=O | 171.8 | α | 53.0 | 4.73 m | β, γ C=O | ||
| β-Methylisoleucine | β | 38.8 | 2.73 m, 3.01 m | ||||
| α | 58.9 | 4.40 d (8.2) | β, γ, δ, C=O | γ | 137.6 | ||
| β | 37.5 | δ, δ′ | 132.5 | 7.25 d (8.1) | β, δ, ε, ε, ζ | ||
| γ | 31.8 | 1.19 m | α, β, ε | ε, ε′ | 131.4 | 7.40 d (8.1) | γ, ε′, ζ |
| δ | 23.8 | 0.82 s | ζ | 120.2 | |||
| δ | 23.8 | 0.83 s | α, β, γ, ε | NH | 7.94 br d (8.2) | N-MeGln-C=O | |
| ε | 8.7 | 0.75 t (6.8) | β, γ, δ | C=O | 173.2 | ||
| NH | 7.80 br s | Threonine (2) | |||||
| C=O | 171.2 | α | 54.7 | 4.75 m | C=O, pBrPhe-C=O | ||
| Valine | β | 67.4 | 3.88 m | α | |||
| α | 58.8 | 4.12 br t (6.7) | β, γ, C=O | γ | 20.3 | 1.01 d (5.1) | α, β |
| β | 30.9 | 1.81 m | γ | OH | 4.29 | α, β, γ | |
| γ | 18.5 | 0.57 d (6.0) | α, β, γ | NH | 9.18 d (5.6) | pBrPhe C=O | |
| γ | 19.7 | 0.58 d (6.0) | α, β, γ | C=O | 171.3 | ||
| NH | 7.84 | N-Methylasparagine | |||||
| C=O | 173.4 | α | 56.7 | 5.36 m | C=O, β, γC=O | ||
| Tryptophan | β | 35.1 | 2.34 m, 2.84 m | C=O, γC=O | |||
| α | 61.5 | 4.63 m | γC=O | 171.5 | |||
| β | 26.7 | 2.92 m, 3.11 m | 2, 4 | γNH2 | |||
| ε′ NH | 10.72 br s | 3, 4 | NMe | 29.2 | 2.60 br s | α, β, Thr(2)-C=O | |
| 2 | 125.0 | 7.12 br s | 3, 4, 9 | C=O | 169.2 | ||
| 3 | 110.5 | Glycine | |||||
| 3a | 127.8 | α | 40.5 | 3.39 m, 4.26 m | C=O | ||
| 4 | 119.1 | 7.59 d (8.1) | 3, 7, 9 | NH | 8.18 br d (7.7) | ||
| 5 | 118.8 | 6.95 m | 4, 8 | C=O | 169.0 | ||
| 6 | 121.5 | 7.02 m | 5, 9 | ||||
| 7 | 111.8 | 7.31 d (8.1) | 4, 6 | ||||
| 7a | 136.7 | ||||||
| NH | 8.12 d (7.7) | ||||||
| C=O | 172.0 | ||||||
| Arginine | |||||||
| α | 52.4 | 4.31 m | |||||
| β | 32.1 | 1.59 m | |||||
| γ | 28.6 | 1.28 m | |||||
| δ | 40.7 | 2.93 m | |||||
| εNH | 7.48 br s | φC | |||||
| φC | 155.5 | ||||||
| φNH2 | 6.93 m, 6.95 m | φC, γ | |||||
| NH | 8.00 br s | ||||||
| C=O | 171.4 | ||||||
Assignments are from gHMBC, gHSQC, and TOCSY spectra.
Cyclocinamide B (2)
+9.6 (c 0.033, MeOH); UV (MeOH) λmax nm (log ε) 201 (3.53), 274 (2.43) IR (KRS-5 cell, DMSO) νmax 3436, 2973, 2480, 1661, 1372, 1251, 1030, 602 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z 784.10084 (calcd 784.10070 for C29H32N9O8BrCl21+).
Acid Hydrolysis and FDAA Analysis of Cyclocinamide B (2)
To a thick walled micro vial containing 100 μg (0.13 μmoles) of 2 was added 250 μL of 6 N HCl containing 0.1 % phenol (w/v). The vial was sealed and heated at 110 °C for four hours. The reaction was allowed to cool to room temperature and was concentrated to dryness. The resulting dry residue was dissolved in 100 μL of water, and subsequently 300 μL of 1 M aqueous NaHCO3 was added followed by 250 μL of a 1% solution of L-FDAA in acetone (w/v). This mixture was heated at 50 °C for 1.5 h, cooled to room temperature, and diluted with an equal volume of CH3CN. The L-FDAA derivatives were separated by HPLC (1.0 mL/min, Phenomenex C18, 5 μ, 4.6 × 250 mm) using a linear gradient of 10% CH3CN in 0.1 M NH4OAc (pH=5) to 50% CH3CN in 0.1 M NH4OAc over 60 min. The absolute stereochemistry was determined by comparing retention times of L and D, L amino acid standards derivatized with L-FDAA and the derivatized hydrolysate of 2. HPLC retention times in min for the amino acid standards: L-Asp (10.9), D-Asp (13.6), L-iSer (19.9), D-iSer (20.6), Gly (20.4), L-DAP (39.3), D-DAP (40.3), L-5-BrTrp (39.6), D-5-BrTrp (43.3). HPLC retention times in min for L-FDAA derivatized amino acids of 2: D-Asp (13.9), L-DAP (39.2), D-5-BrTrp (42.5).
FDVA derivatization of the hydrolysate of cyclocinamide B (2)
The hydrolysate of 2 was obtained as described above and was concentrated to dryness and dissolved in 75 μL of H2O. Subsequently, 25 μL of 1 M aqueous NaHCO3 was added followed by the addition of 100 μL of a 1% solution of L-FDVA in acetone (w/v). The reaction was heated at 40 °C for 1 h, and then neutralized with 20 μL of 1 N HCl.
LCMS analysis of the FDVA derivatives
The separation of L and D, L FDVA derivatives utilized a 2 × 50 mm, 3 μ, Phenomenex C18 column at 20 °C. A linear gradient (0.2 mL/min) using 0.1% acetic acid/99% CH3CN to 100% CH3CN over 75 min was used. The retention times in min for the reference D and L-iSer-L-FDVA: L-iSer (32), D-iSer (33). The retention time for L-FDVA derivatives of (2): L-iSer (31.9). Additionally, a coinjection with a 1:4 mixture of DL-iSer-L-FDVA standards to L-FDVA derivatives of (2) showed an enhancement of the peak corresponding to L-FDVA-L-iSer.
HCT-116 cytotoxicity assay
The assay was performed as previously described.31
Supplementary Material
A picture of a representative Fijian Corticium sp. is provided. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 3.
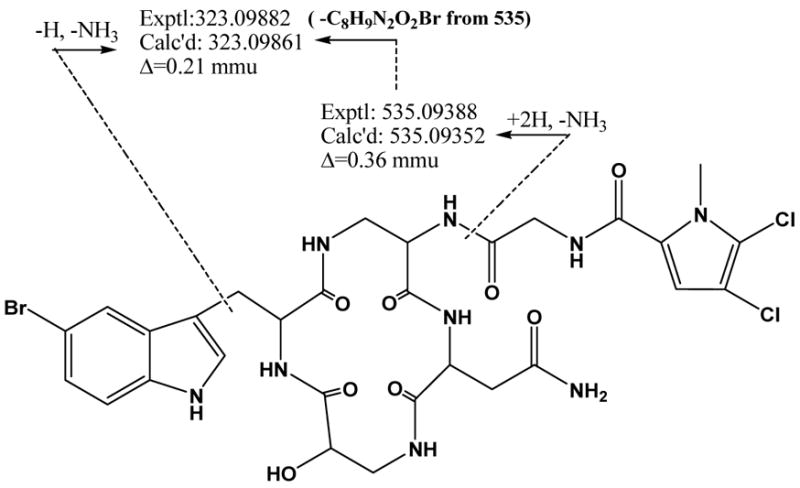
Major MS fragments of cyclocinamide B (2).
Acknowledgments
The authors are grateful to J. J. Skalicky for help in obtaining NMR data and would like to thank Drs. G. T. Carter and F. E. Koehn for their kind support. The authors would like to acknowledge the NIH, RR06262 and RR14768, and NSF grant DBI-0002806 for funding NMR instrumentation in the University of Utah Health Sciences NMR facility. This research was funded by NIH grant CA 067786.
References and Notes
- 1.McCarthy PJ, Pitts TP, Gunawardana GP, Kelly-Borges M, Pomponi S. J Nat Prod. 1992;55:1664–1667. doi: 10.1021/np50089a016. [DOI] [PubMed] [Google Scholar]
- 2.Jurek J, Scheuer PJ. J Nat Prod. 1994;57:1004–1007. doi: 10.1021/np50109a022. [DOI] [PubMed] [Google Scholar]
- 3.De Marino S, Iorizzi M, Zollo F, Roussakis C, Debitus C. Eur J Org Chem. 1999;3:697–701. [Google Scholar]
- 4.Lee HS, Seo Y, Rho JR, Shin J, Paul VJ. J Nat Prod. 2001;64:1474–1476. doi: 10.1021/np0101649. [DOI] [PubMed] [Google Scholar]
- 5.Borbone N, De Marino S, Iorizzi M, Zollo F, Debitus C, Esposito G, Iuvone T. J Nat Prod. 2002;65:1206–1209. doi: 10.1021/np020027r. [DOI] [PubMed] [Google Scholar]
- 6.Ridley CP, Faulkner DJ. J Nat Prod. 2003;66:1536–1536. doi: 10.1021/np0302706. [DOI] [PubMed] [Google Scholar]
- 7.De Marino S, Zollo F, Iorizzi M, Debitus C. Tetrahedron Lett. 1998;39:7611–7614. [Google Scholar]
- 8.Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. J Am Chem Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h. [DOI] [PubMed] [Google Scholar]
- 9.McDonald LA, Barbieri LR, Carter GT, Kruppa G, Feng X, Lotvin JA, Siegel MM. Anal Chem. 2003;75:2730–2739. doi: 10.1021/ac0264731. [DOI] [PubMed] [Google Scholar]
- 10.Breci LA, Tabb DL, Yates JR, III, Wysocki VH. Anal Chem. 2003;75:1963–1971. doi: 10.1021/ac026359i. [DOI] [PubMed] [Google Scholar]
- 11.Nogle LM, Williamson RT, Gerwick WH. J Nat Prod. 2001;64:716–719. doi: 10.1021/np000634j. [DOI] [PubMed] [Google Scholar]
- 12.Hamann MT, Otto CS, Scheuer PJ, Dunbar DC. J Org Chem. 1996;61:6594–6600. doi: 10.1021/jo960877+. [DOI] [PubMed] [Google Scholar]
- 13.Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ. J Nat Prod. 1999;62:655–658. doi: 10.1021/np980553b. [DOI] [PubMed] [Google Scholar]
- 14.Capon RJ, Ford J, Lacey E, Gill JH, Heiland K, Friedel T. J Nat Prod. 2002;65:358–363. doi: 10.1021/np010329d. [DOI] [PubMed] [Google Scholar]
- 15.Matsunaga S, Fusetani N, Konosu S. Tetrahedron Lett. 1984;25:5165–5168. [Google Scholar]
- 16.Ryu G, Matsunaga S, Fusetani N. Tetrahedron Lett. 1994;35:8251–8254. [Google Scholar]
- 17.Ryu G, Matsunaga S, Fusetani N. Tetrahedron. 1994;50:13409–13416. [Google Scholar]
- 18.Li H, Matsunaga S, Fusetani N. J Med Chem. 1995;38:338–343. doi: 10.1021/jm00002a015. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Matsunaga S, Fusetani N. J Nat Prod. 1995;59:163–166. doi: 10.1021/np9600309. [DOI] [PubMed] [Google Scholar]
- 20.Gulavita NK, Gunasekera SP, Pomponi SA, Robinson EV. J Org Chem. 1992;57:1767–1772. [Google Scholar]
- 21.Rashid MA, Gustafson KR, Cartner LK, Shigematsu N, Pannell LK, Boyd MR. J Nat Prod. 2001;64:117–121. doi: 10.1021/np0002379. [DOI] [PubMed] [Google Scholar]
- 22.Capon RJ, Rooney F, Murray LM, Collins E, Sim ATR, Rostas JAP, Butler MS, Carroll AR. J Nat Prod. 1998;61:660–662. doi: 10.1021/np970483t. [DOI] [PubMed] [Google Scholar]
- 23.Djura P, Faulkner DJ. J Org Chem. 1980;45:735–737. [Google Scholar]
- 24.Hernández Franco L, Joffe EB, de K, Puricelli L, Tatian M, Seldes AM, Palermo JA. J Nat Prod. 1998;61:1130–1132. doi: 10.1021/np970493u. [DOI] [PubMed] [Google Scholar]
- 25.Lee NK, Fenical W, Lindquist N. J Nat Prod. 1997;60:697–699. doi: 10.1021/np970042+. [DOI] [PubMed] [Google Scholar]
- 26.Clark WD, Corbett T, Veleriote F, Crews P. J Am Chem Soc. 1997;119:9285–9286. [Google Scholar]
- 27.Grieco PA, Reilly M. Tetrahedron Lett. 1998;39:8925–8928. [Google Scholar]
- 28.Marfey P. Carlsberg Res Commun. 1984;49:591–596. [Google Scholar]
- 29.B’Hymer CJ. Liq Chrom & Rel Technol. 2001;24:3085–3094. [Google Scholar]
- 30.Sato K, Horibe K, Amano K, Mitsui-Saito M, Hori M, Matsunaga S, Fusetani N, Ozaki H, Karaki H. Toxicon. 2001;39:259–264. doi: 10.1016/s0041-0101(00)00123-9. [DOI] [PubMed] [Google Scholar]
- 31.Ratnayake AS, Bugni TS, Feng X, Harper MK, Skalicky JJ, Mohammed KA, Andjelic CD, Barrows LR, Ireland CM. J Nat Prod. 2006;69:1582–1586. doi: 10.1021/np060229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A picture of a representative Fijian Corticium sp. is provided. This material is available free of charge via the Internet at http://pubs.acs.org.