

Abstract
The recognition and catalytic properties of biopolymers derive from an elegant evolutionary mechanism, whereby the genetic material encoding molecules with superior functional attributes survives a selective pressure and is propagated to subsequent generations. This process is routinely mimicked in vitro to generate nucleic-acid or peptide ligands and catalysts. Recent advances in DNA-programmed organic synthesis have raised the possibility that evolutionary strategies could also be used for small-molecule discovery, but the idea remains unproven. Here, using DNA-programmed combinatorial chemistry, a collection of 100 million distinct compounds is synthesized and subjected to selection for binding to the N-terminal SH3 domain of the proto-oncogene Crk. Over six generations, the molecular population converges to a small number of novel SH3 domain ligands. Remarkably, the hits bind with affinities similar to those of peptide SH3 ligands isolated from phage libraries of comparable complexity. The evolutionary approach has the potential to drastically simplify and accelerate small-molecule discovery.
Introduction
The evolutionary approach to in vitro molecular discovery was first introduced forty years ago1, and since then, has become a routine method for isolating functional biopolymers2, 3. The power of the strategy lies its use of nucleic acids to serve as amplifiable blueprints that can be translated into biopolymer sequences, enabling iterative functional selections to be carried out with extremely complex molecular libraries. The rare molecules that are fit to survive an imposed selective pressure eventually overtake the library. The simplicity and efficiency of in vitro evolution contrast with the current methods available for synthetic compound discovery, which rely heavily on robotics and high-throughput screening technologies. The largest small-molecule libraries that have been screened comprehensively are many orders of magnitude smaller than a typical in vitro evolution library, and the infrastructure required to screen them precludes their use by most basic science laboratories.
Applying evolution-based search strategies to synthetic chemical libraries could, in principle, dramatically accelerate small-molecule discovery. However, in vitro selection requires that each molecule be encoded by a genetic blueprint. Recently, we reported a strategy that fulfills this requirement for synthetic compounds by encoding their structure in DNA4–6. Using sequence-directed hybridization5 and DNA-compatible chemical transformations6, collections of single-stranded DNA (ssDNA) molecules are routed through a split-and-pool combinatorial synthesis. The products are covalent small molecule-DNA hybrids (Fig. 1). Because the splitting is based on DNA sequence, the synthesis of each gene product is uniquely programmed by the attached DNA molecule. Consequently, iterated rounds of library selection and amplification are possible. Using this approach, we previously isolated a known antibody epitope out of a library of one-million synthetic peptides4. The experiment required six chemical translation steps for ligand synthesis and three generations for library convergence. A similar control was reported by Gartner and coworkers using DNA-templated synthesis7. In that study, a library of 65 DNA-linked peptide-fumaride macrocycles was synthesized and subjected to one round of affinity purification and amplification, resulting in the enrichment of a DNA sequence encoding a known ligand. Although these efforts clearly illustrate the mechanics of DNA-programmed combinatorial chemistry and selection, they are based on recovering known ligands from “spiked” libraries, and do not demonstrate that the technology will yield new binding motifs from wholly unnatural libraries.
Figure 1.

DNA Display. A library of ssDNA molecules (top) is chemically translated into synthetic compound DNA conjugates. The DNA library is split into sub pools by hybridization of 20 base codons to complimentary oligonucleotide anticodons that are immobilized on separate columns (orange, cyan, pink bars). A distinct chemical transformation is carried out on each sub pool, resulting in the covalent attachment of a chemical building block to the DNA (orange, cyan, pink balls). The library is pooled and then split based on the next coding region (green, brown, yellow bars), and distinct chemical transformations are carried out for each sub pool. The process is iterated until the entire DNA sequence is read. Each codon can exist at only one coding region. The translated library is subjected to selection for a function of interest (binding to the immobilized grey widget), DNA linked to binders is amplified and used as input for the subsequent round of chemical translation. The entire process is repeated until the library converges. Enriched molecules are identified by DNA sequencing and assayed for function as pure compounds.
The utility of blindly screening combinatorial chemistry libraries in search of new classes of ligands has been debated. One prevalent view is that combinatorial chemistry libraries are only useful if the constituent molecules are narrowly focused around a known lead. Biopolymers, however, are viewed quite differently. Nearly all efforts to isolate biopolymer ligands are based on the idea of using blind libraries: those not known a priori to contain a chemical motif that will bind to the target. It is unclear whether these opposite points of view arise because biopolymers are fundamentally different from synthetic compounds (perhaps “privileged”), or because modern combinatorial chemistry libraries are too small to routinely yield potent hits from unfocused libraries. Only experimental data will ultimately resolve this question. Here, we take a step towards an experimental test by applying DNA-programmed synthesis and in vitro selection to a combinatorial chemistry library of 100 million diversity, several orders of magnitude larger than any that has been exhaustively explored to date.
Materials and Methods
Materials
Chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, Missouri, USA), Acros (Geel, Belgium), Novabiochem (La Jolla, California, USA), Chem-Impex International (Wood Dale, Illinois, USA), TCI America (Portland, Oregon, USA) or Fisher Scientific (Hampton, New Hampshire, USA). Microwave assisted coupling was carried out with a Panasonic NN-S954WF 1250 watt microwave.
The N-CrkSH3 peptide ligand (YPPPALPPKRRR) and all oligonucleotides were purchased from the Stanford PAN Facility (Stanford, California, USA). Oligonucleotide coupling experiments utilized an unmodified 10-base oligonucleotide of sequence CGGACTAGAG and a reactive twenty-base oligonucleotide of sequence H2N-X-AGCAGGCGAATTCGTAAGCC, where X represents a C12 (Glen Research, 5′-Amino Modifier C12) or PEG (Glen Research, Spacer Phosphoramidite 18 and 5′-Amino-Modifier 5) linker. All library and selection experiments utilized the PEG linker. Resins, column housings, tubing, pumps, reaction syringes, and all other DNA display materials were identical to those described previously 4–6.
General Methods
HPLC analysis of peptoid synthesis on oligonucleotides was carried out using reverse-phase C18 analytic columns (Microsorb, Varian, Palo Alto, California, USA) and UV detection at 260 nm and 280 nm at 50°C. Linear MeCN gradients in 100 mM TEAA pH 5.5 were used. MALDI-MS analysis of DNA-peptoid conjugates was carried out using an ammonium citrate/HPA matrix at the Stanford PAN facility, typically giving errors of 0.1–0.2%. Conjugates were desalted either with Sephadex G25 (GE Healthcare, Piscataway, NJ, USA) or ZipTip (Millipore, Billerica, MA, USA) prior to MALDI-MS. Solid-phase peptoid synthesis products were purified by reverse-phase C18 semi-preparative HPLC (Zorbax, Agilent, Palo Alto, California, USA) at 50°C with UV detection at 220 nm using linear MeCN gradients in 0.1% TFA. MALDI-MS analysis of peptoids was carried out using α-cyano-4-hydroxycinnamic acid matrix at the Stanford PAN facility.
N-CrkSH3
The N-terminal SH3 domain from c-Crk (residues 134–191) was PCR amplified from a GST-fusion construct generously provided by Wendell Lim (University of California, San Francisco). An N-terminal FLAG-tag and nine amino acid Gly-Ser linker were added by subsequent PCR. The resulting FLAG-Crk construct was cloned into pET24a (NdeI and XhoI), which appended a C-terminal His6 tag. Protein was expressed in BL21 DE3 Escherichia coli, inducing with 1 mM IPTG for 5 hours at 37 °C. Cells were harvested by centrifugation and sonicated. Filtered lysates were passed over a Ni-NTA column in 50 mM Tris-HCl pH 8.0, 300 mM NaCl, washed, and eluted in 100 mM NaH2PO4 pH 3.9, 300 mM NaCl. Pooled fractions were dialyzed into 50 mM Tris pH 8.0 and loaded onto a HiTrap Fast Flow Q-column. After washing, bound protein was eluted with 50 mM Tris pH 8.0, 1 M NaCl and concentrated to approximately 2 mg/ml. Protein was over 90% pure by SDS-PAGE, and verified by MALDI-MS.
Library construction and chemical translation
We assembled the initial DNA library as described5. A fraction of the assembled material was subcloned, and 114 isolates were sequenced to verify sequence degeneracy. New 20-base constant regions (Z0 and Z10) were then added to the ends of the degenerate material via PCR to exclude the possibility of amplifying contaminating DNA from previous library experiments. All library amplification steps used these regions for priming. Single-stranded DNA synthesis, anticodon column synthesis, sequence-directed splitting, DEAE-Sepharose column packing, anticodon-to-DEAE transfer, and endpoint isolation of the library were carried out exactly as described4, 5. One-hundred picomoles of ssDNA was used as starting material for each library round.
Peptoid Coupling
Chemistry columns were washed with 3 mL DEAE bind buffer (10 mM acetic acid, 0.005% Triton-X100), 1 mL water, and 6 mL of methanol. The columns were then incubated (2 × 20 min) with 500 μL of 150 mM DMT-MM and 100 mM ClAcONa in distilled methanol8 in open syringe barrels on a vacuum manifold, and then washed with 3 mL of methanol, 2 mL of 1 M propylamine in methanol, and 3 mL of methanol. Each column was then incubated with 500 μL of the appropriate amine solution (Table S1) and microwaved for 20 seconds at 100% power 6 times over 30 minutes in a closed system between two syringes9. The columns were allowed to cool for 5 minutes after each microwave step. Only 3 microwave steps were used over 30 minutes for methylamine, ethylamine, ethanolamine, tryptamine, ethylenediamine, and 1,3-diaminopropane. After alkylation, the columns were washed with 1 mL of DMSO, and 3 mL of DEAE bind buffer. For oligonucleotide coupling experiments, the columns were then eluted with 2 mL DEAE Elute Buffer (50 mM Tris pH, 1.5 M NaCl, 0.005% Triton X-100), and analyzed by HPLC and MALDI-MS. For library synthesis, a 2 mL PBS wash (and a 2 mL DEAE Elute Buffer wash after the final chemistry step) was carried out just before subsequent hybridization.
Azide containing amines were synthesized exactly as described10. After incorporation into DNA-peptoid conjugates, azide reduction was achieved with 10 mM TCEP, 100 mM Tris-HCl pH 8.0 for 1 hour at room temperature. Reduced products were verified by HPLC and MADLI-MS. During library synthesis, this step was carried out just after duplexing the library.
Proline Coupling
Chemistry columns were washed with 3 mL DEAE bind, 1 mL water, 3 mL of THF, and 6 mL of distilled DCM. 22.5 mg of Fmoc-L-Proline (67 μmoles) was dissolved in 1 mL of DCM containing 300 mM TEA and 150 mM TCA. The solution was carefully added (to avoid vigorous bubbling) to 41.5 mg (158 μmoles) of finely crushed TPP11. The resulting yellow solution was then applied to the chemistry column, and incubated for 30 minutes at room temperature. Following coupling, the column was washed with 6 mL DCM, 3 mL THF, 1 mL water, and 3 mL of DMF. The Fmoc group was removed with 20% piperidine in DMF for 20 minutes, and the column was washed with 3 mL DMF and then 3 mL of DEAE bind buffer. For oligonucleotide coupling experiments, the columns were eluted with 2 mL DEAE Elute Buffer and analyzed by HPLC and MALDI-MS. For library synthesis, a 2 mL PBS wash was carried out just before subsequent hybridization.
Selection
Prior to selection, the ssDNA-peptoid conjugates were duplexed via one-cycle PCR (50μL) containing 20 μM of a single end primer (Z10′), 200 μM of each dNTP, 5 mM MgCl2, 1X Promega Taq reaction buffer, and 5 U of Taq DNA polymerase (95°C for 2.5 min, 58°C for 1 min, 72°C for 15 min). Azide side chains were then reduced, and the library was ethanol precipitated and resuspended in 25 μL of Selection Buffer (20mM HEPES pH 7.5, 50mM NaCl, 0.25 mg/mL BSA, 0.1 mg/mL yeast tRNA). The library was incubated at 4 °C for 1 hour with Pansorbin cells to preclear non-specific binders. After centrifugation, the supernatant was taken (discarding cells) and N-CrkSH3 was added to 2 μM. After a 1 hour 4 °C incubation, 5 μg of ANTI-FLAG M2 antibody (Simga-Aldrich, St. Louis, Missouri, USA) was added. The mixture was incubated for 30 minutes at 4 °C, then mixed with fresh Pansorbin cells (Calbiochem, San Diego, California, USA) for an additional hour at 4 °C. The cells were pelleted and washed three times with 500μL Selection Buffer containing 500 mM NaCl, and once with 500 μL Selection Buffer, all at 4 °C. Bound library members were eluted with 50 μL Selection Buffer containing 200 μM N-CrkSH3 peptide (YPPPALPPKRRR) at room temperature for 1 min. Selected genes were PCR amplified from 10 μL of the elute supernatant. The 50 μL reaction contained 2 μM of each primer (Z0 and Z10′), 200 μM of each dNTP, 1XPhusion HF buffer and Phusion DNA polymerase (98 °C 10 sec, 58 °C 30 sec, 72 °C 15 sec, 25 to 30 cycles). After each round of chemical translation and selection, a fraction of the library was cloned and 96 isolates were sequenced. After the fifth round of selection, 960 isolates were sequenced (MCLab, South San Francisco, CA, USA).
For control selection experiments, the ligand and competitors constructs were generated by PCR using HPLC purified peptide-oligonucleotide conjugates as forward primers6. The constructs were quantified via UV absorbance and ethidium bromide fluorescence using a Typhoon 8600 imager (GE Healthcare, Piscataway, NJ, USA). Selection load and elute amplification products were cleaned up using QIAquick PCR purification kits (Qiagen, Hilden, Germany), digested with BglI and BamHI, analyzed by agarose gel electrophoresis, and quantified via ethidium bromide staining and fluorescence.
Quantitative PCR
Selection inputs and elutes were quantified on a Stratagene (La Jolla, California, USA) Mx3000P quantitative PCR machine using DyNamo HS SYBR Green qPCR reagents (Finnzymes, Espoo, Finland) and 200 nM of primers (Z1 and Z9′). Samples were measured in duplicate and were quantified against a standard curve generated from serial dilutions of dsDNA of known concentration (also measured in duplicate). Dissociation curves indicated a single amplification product for all samples.
Sequence analysis and clustering
DNA sequences were translated computationally into the single letter peptoid code. Only full reads (all 8 coding regions) were considered for further analysis. A similarity matrix for all peptoid 8mers was computed using the Needleman-Wunsch global alignment algorithm12 in MATLAB, with the gap-opening penalty set to 0.5 and no gap extension penalty. Two-dimensional hierarchical clustering was carried out with Cluster and viewed with TreeView (Michael Eisen, University of California, Berkeley).
Solid Phase Peptoid Synthesis
Peptoids were re-synthesized on Fmoc-Rink amide MBHA resin (Novabiochem) using a microwave-assisted protocol9. All amines were used as 1 M solutions in DMSO. Proline residues were incorporated using 10 equivalents of Fmoc-L-proline and HATU and 20 equivalents of DIEA, twice for one hour at 37 °C. The resin was then washed with DMF (8 × 3ml) and the Fmoc group was removed with 20% piperidine for 20 minutes at room temperature. After the final coupling reaction, the resin was washed with anhydrous DMF (5 × 3 ml) and DCM (8 × 3 ml), dried under nitrogen, and cleaved with 95% TFA, 2.5% anisole for 4 hours. After filtration, the crude product was concentrated with a nitrogen stream, HPLC purified, and lyophilized. The purified peptoids were resuspended in water (25 mM stocks) and were analyzed by MALDI-MS.
Binding Assay
Dissociation constants (Kd) were determined by measuring tryptophan fluorescence perturbation as a function of peptoid concentration essentially as described13, 14. The data were fit to the equation:
where Fb = fraction protein bound, P = total protein concentration, and L = total peptoid concentration. N-CrkSH3 concentration was between 0.5 and 1 μM. Measurements were made in triplicate at 22 °C on a FluoroLog-3 spectrofluorometer (HORIBA Jobin Yvon, Edison, New Jersey, USA) with excitation at 295 nm (4 nm band pass) and emission at 340 nm (4 nm band pass). Average Fb values for each peptoid concentration were used to fit the binding curve and extract a Kd value. Errors for Kd values are estimated to be between 5 and 15%. Selection Buffer was used for all assays.
Results
N-substituted glycines, or peptoids (Fig. 2A), comprise an attractive class of molecules for combinatorial discovery15–17. Although structurally similar to peptides, peptoids lack amide hydrogens and are protease resistant, making them potentially more druggable than peptides18, 19. Furthermore, the diversity element in peptoid synthesis is introduced via nucleophilic displacement with a primary amine, of which thousands are available commercially. Extremely large libraries of small peptoids can thus be actualized. Methods for solid-phase peptoid synthesis are well-established, rapid, and highly efficient, making large-scale resynthesis of library hits robust and inexpensive.
Figure 2.

DNA encoded peptoid synthesis. (A) Peptoid structure, where R represents side chains. (B) DNA compatible submonomer peptoid synthesis. Each peptoid residue is constructed by chloroacetylation of a secondary amine followed by nucleophilic displacement with a primary amine. (C) HPLC analysis of submonomer peptoid coupling on DNA. A control 10 mer and a 5′ aminated 20 mer oligonucleotide starting; material were loaded onto DEAE Sepharose microcolumns, and subjected to chloroacetylation (red trace), or to chloroacetylation followed by nucleophilic displacement with propylamine (blue trace). The products were eluted and analyzed by reverse phase HPLC. Each peak was collected and its identity was verified by MALDI-MS. (D) HPLC analysis of polypeptoid synthesis on DNA. A 5′ animated 20 mer oligonucleotide (black trace) served as the starting material for an eight residue peptoid synthesis. The crude peptoid DNA product was analyzed by HPLC (red trace). The major peak (~30% yield) was confirmed as the intended product by MALDI MS (Observed: 7558, Expected: 7556).
Peptoid synthesis is typically carried out in a submonomer format, whereby each residue is constructed in two synthetic steps – bromoacetylation of a secondary amine followed by nucleophilic displacement with a primary amine20. Standard bromoacetylation protocols damage DNA, and had to be modified for DNA-programmed peptoid synthesis. We found that chloroacetylation of oligonucleotides bearing a 5′ amine proceeded efficiently and without detectable DNA damage using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) and sodium chloroacetate (ClAcONa) in methanol. Subsequent nucleophilic displacement to introduce the peptoid side chain was accomplished in high yield with concentrated amine (~1–3 M) in DMSO or water. Taken together, the two-step addition of a peptoid residue to aminated DNA (Fig. 2B, 2C) could be achieved in under 90 minutes. Yields for the addition of each residue ranged from 75 to >95%, depending on the size of the side chain on the secondary amine substrate (Supplementary Fig. 1, Supplementary Table 1). Side-chain hydroxyl and guanidium groups did not require protection, because no significant side reactions of these functional groups were detected. Side chain carboxylic acids and primary amines could be introduced as tert-butyl esters and azides, respectively. Acids were deprotected thermolytically6, and azides were reduced to primary amines using tris(2-carboxyethyl)phosphine (TCEP). DNA-linked peptoids up to eight residues long were synthesized with per residue yields averaging 85–90%, even for sequences containing multiple hindered side chains (Fig. 2D).
We wanted to include cyclic N-substituted glycine monomers (e.g. proline) in our peptoid libraries for added side chain and structural diversity. Although DMT-MM could be used to couple Fmoc-proline to DNA bearing a primary amine, acylation of secondary amines proceeded poorly. More stringent in situ acid chloride formation conditions, using trichloroacetonitrile and triphenylphosphine in dichloromethane, were successful for coupling proline to even the most hindered peptoids tested (Supplementary Fig. 2). With subsequent Fmoc deprotection, total coupling time came to one hour. This acid-free in situ activation strategy may prove to be generally useful for the acylation of hindered amines in a DNA-compatible fashion.
We chose the N-terminal Src homology 3 domain of the c-Crk protein (N-CrkSH3) as our selection target21. SH3 domains are an abundant family of protein fragments that recognize proline-rich peptide sequences22, mediate numerous protein-protein interactions in signaling pathways23, and are regarded as potential drug targets24, 25. Nguyen and coworkers demonstrated that several SH3 domains, including N-CrkSH3, could tolerate one or two proline-to-peptoid substitutions within native peptide ligands13, 14. Furthermore, a well-established, fluorescence-based binding assay exists for N-CrkSH3, facilitating the characterization of potential selection hits13, 14.
Based on previous experiments with DNA-programmed library selections, we pursued an immunoprecipitation-based selection strategy using FLAG-tagged N-CrkSH3, anti-FLAG antibody, and Pansorbin cells, followed by a competitive elution (Fig 3A). When DNA bearing a peptide that binds to N-CrkSH3 was diluted into a solution of DNA bearing a non-binding peptide and subjected to selection, an N-CrkSH3-dependent enrichment of nearly 1,000 fold was observed for the ligand-DNA conjugate (Fig. 3B).
Figure 3.

Selection for N CrkSH3 binding. (A) The selection strategy utilizes an N-CrkSH3 protein (grey) with a prepended FLAG tag (white), as well as an anti FLAG antibody (black Y), and Pansorbin cells (yellow). After incubation of these components with a translated library, the Pansorbin cells are pelleted and washed. Ligand DNA conjugates are eluted by addition of an excess of an N-CrkSH3 binding peptide (black balls). (B) To measure the selection signal lo noise ratio, two peptide DNA conjugates were synthesized. One construct consisted of the known N-CrKSH3 peptide ligand YPPPALPPKRRR linked to a 340 mer dsDNA with a central BgII site (red). The other consisted of the peptide YGGFL linked to a 340 mer dsDNA with a central BamHI site (blue). When the ligand construct was diluted 1 to 1,000 into the non-ligand construct and amplified, BgII cutting was undetectable (lane 1). When this mixture was subjected to a mock selection that lacked N-CrkSH3 and was amplified, BgII cutting was undetectable (lane 2). When subjected to a selection with N-CrkSH3 included and amplified, the BgII containing DNA was enriched nearly 1,000 fold (lane 3).
We designed a 100 million-member 8-mer peptoid library for selection experiments. The DNA-library construct included eight coding regions, with ten mutually exclusive codons at each region, and a 5′ primary amine as a synthetic handle (Fig. 4A). Rather than choosing from thousands of possible side chains at random, we opted to examine26 an N-CrkSH3 peptide co-crystal structure27 to assist our library design, assuming that a peptoid might bind to the target in a fashion similar to a peptide. The local environment of each side chain in the model was assessed qualitatively and used to choose four monomer sets (Fig. 4A). All of the sets, with the exception of the N-terminal set, share at least five side chains, and a great deal of steric and chemical diversity is maintained at each position.
Figure 4.
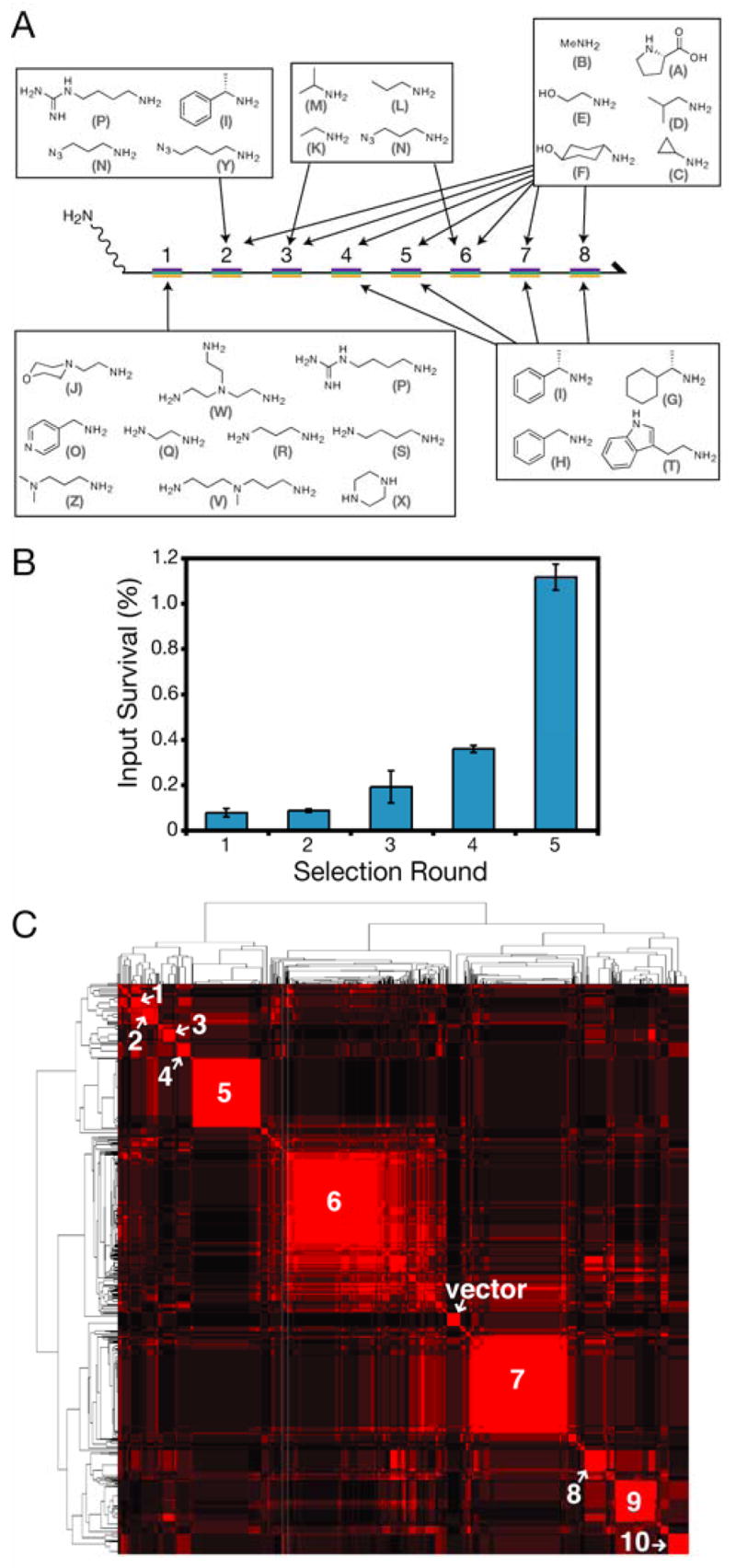
In vitro selection of synthetic ligands that bind to N-CrkSH.3. (A) The library construct consisted of a 340 mer ssDNA molecule with eight coding regions (rainbow bars) and ten potential sequences at each region (108 possible library members). The monomers used at each coding region are indicated. Each monomer is assigned an arbitrary letter code for convenience. The coding regions are numbered according to the N to C sequence of the peptoid product, but are read C to N during chemical translation. (B) The percent of input DNA recovered in the selection eluate is plotted with respect to selection round. DNA concentrations were determined by quantitative PCR. Error bars represent standard deviations of repeat measurements. (C) Clustergram of the sixth generation peptoid sequences. Red indicates sequence similarity. A representative peptoid from each major cluster (numbered in white relief) was synthesized for binding assays. ’Vector’ Indicates sequences derived from the cloning vector, and not from the library.
A fully degenerate DNA library was chemically translated into peptoid-DNA conjugates and subjected to selection for N-CrkSH3 binding. The genetic material of the library members isolated after selection was amplified via polymerase chain reaction (PCR) and used to generate the ssDNA input for the subsequent round of chemical translation. We repeated this process, propagating the library through five rounds of chemical translation and selection. As an assay of bulk library convergence, quantitative PCR was used to measure the fraction of the translated library that survived each round of selection (Fig. 4B). Initially, about 0.1% of the library was recovered, consistent with our signal-to-noise estimates of 1000:1 for the control selection experiments (Fig. 3B). By the third round of the selection, an increase in percent survival was detected. This trend continued, and survival exceeded 1% for the fifth round, suggesting that the library was enriched for ligands. We also assessed the genetic make-up of the library after each selection step by subcloning and sequencing approximately 80 isolates. Clear signs of sequence convergence were apparent by the third library generation, in which roughly one-third of the isolates could be clustered into five sequence groups (the groups included exact matches, single mutants, and double mutants). This trend continued over the next two generations.
We thoroughly characterized the sixth generation affinity-matured library by sequencing 960 clones. Out of the 840 full-sequence reads, we observed only 215 unique sequences, 112 of which occurred at least twice. We computed a similarity matrix for all 840 peptoid 8-mers and clustered the sequences based on their similarity scores, creating a phylogeny of synthetic compounds (Fig. 4C). The data are essentially an evolutionarily-produced structure-activity profile. Over 97% of the sampled sequences had at least five identical residues to a neighbor in the clustergram. A number of dominant sequence families are apparent, representing chemically and functionally related compounds highly enriched from the initial population.
Given the extensive convergence of library sequences, we wanted to directly examine the ability of peptoids encoded by enriched DNA sequences to bind N-CrkSH3. To that end, we used standard solid-phase peptoid synthesis protocols to prepare a representative peptoid from each of the ten prominent sequence clusters. Purified peptoids were assayed for N-CrkSH3 binding via tryptophan fluorescence perturbation (Fig. 5A). Six of the ten peptoids showed significant binding affinity for the SH3 domain (Fig. 5B). Dissociation constants (Kd) fell between 10 μM and 100 μM, values typical for SH3 domain binding interactions with peptide ligands. Crktoid-8 represents the tightest binding wholly unnatural ligand (Kd = 16 μM) for an SH3 domain28. Carrying the library forward additional rounds might have produced tighter binding ligands, but at the expense of ligand diversity. The discovery of several novel ligand families for one naïve target suggests that large synthetic libraries should contain binding motifs for many targets.
Figure 5.

(A) Intrinsic tryplophan fluorescence based binding assay. The fraction of N-CrkSH3 that is bound by ligand is plotted as a function of Crktoid 8 concentration (red points). The data points are averages of three separate experiments, with standard deviations shown, The Kd value was determined by filling the averaged data to a binding curve (black line). (B) Sequences and Kd values of resynthesized peptoids, named according to cluster and sorted by affinity. The numbers above the sequences refer to the coding positions as in Figure 4. Side chains have been categorized as small (yellow), bulky (green), singly positively charged {light blue), and multiply positively charged (dark blue). (C) Chemical structures of the validated ligands sorted by decreasing affinity.
Discussion
Clear patterns appear in the sequences of the validated ligands (Fig. 5B and C). Complete conservation of tris-(2-aminoethyl)-amine as the side chain (W) at the N-terminal peptoid residue suggests that these positive charges play an important role in N-CrkSH3 binding. Ionic interactions between acidic amino acids in SH3-domain loop regions and basic ligand residues flanking the polyproline segment are known to be crucial for the affinity and orientation of peptide binding in a number of SH3 domains27, 29, 30. Our peptoid ligands all possess a sequence of three to four relatively small side chains (methyl, ethyl, prolyl, cyclopropyl, or ethanol) in the central portion of the peptoid chain and bulky substituents at the first and/or second residue. This does not simply reflect the monomers used at each position, as coding regions utilizing the same initial monomer set (e.g. regions 4, 5, 7, and 8) converged differently. The pattern presumably represents the structural features accommodated by the N-CrkSH3 binding site.
The chemical composition of the ligand families also speaks to the robustness of the technology. In controls, the β-branched side chains (residues C, F, G, I, and M; particulary the larger ones F, G, and I) exhibit lower coupling yields (75–90%) than the other residues. With the exception of isopropylamine (M), all of the β-branched residues are well represented in the converged sequence families (Fig 5B and C). Thus, coupling efficiencies do not dominate the outcome of the selection.
Four of the largest sequence clusters do not encode ligands, as determined by a tryptophan fluorescence perturbation binding assay. Possible explanations for this result include 1) DNA contributions to binding, in either a sequence-specific or non-specific manner; 2) selection of minor unintended synthetic products, which are not present in the re-synthesized and purified compound preparations; and 3) selection of ligands that do not induce a perturbation of tryptophn fluorescence. We ruled out sequence-specific DNA binding for the most enriched non-binding sequences (Crktoids 6 and 7) using a control similar to that depicted in Figure 3. Specifically, when Crktoids 6 and 7 were synthesized on heterologous DNA sequences and subjected to selection, the peptoid-DNA conjugates were enriched 20–50 fold over background. Given the different behaviors of the Crktoids, we view small-molecule in vitro selection as being most useful for narrowing the focus of a molecular search problem from a very large library (>108) to a manageable number of molecules, which can then be assayed as purified compounds by a direct screening approach. In this example, we started with a library of 100 million distinct molecules, but performed only ten binding assays to discover six new ligand families.
The Kd values for the peptoid hits are similar to those of natural SH3 ligand peptides31 and those derived from phage display libraries32, despite the fact that the ligand scaffold is unnatural. A comparison of our result to the existing in vitro selection literature is informative. In the well-studied example of selecting linear peptides that bind to streptavidin, phage libraries of 108–109 complexity routinely give ligand Kd values 50 μM, whereas mRNA display33 and ribosome display34 libraries of ~1013 complexity give ligand Kd values in the low nanomolar range. If a similar relationship between affinity and complexity applies to synthetic compound libraries, then compound collections of 1013 complexity should routinely produce nanomolar hits. The systematic comparison between chemical libraries and biopolymer libraries is only now possible: the ability to exhaustively test large populations of synthetic products is a unique capability afforded by chemical evolution technologies. Efforts are currently underway to generate DNA-programmed small-molecule libraries with complexities exceeding 1013.
The DNA-programmed combinatorial chemistry approach has the potential for widespread use because of its simplicity. We applied it here to a compound collection two orders of magnitude more complex than large HTS libraries, and discovered unprecedented peptoid ligands. The molecules that emerged in this work were unknown at the outset. A priority going forward will be to expand the range of novel chemical classes that are amenable to DNA-programmed synthesis. If successful, these efforts should make custom small-molecule reagents as available as antibodies currently are, with applications in chemistry, biology, material science, and medicine.
Supplementary Material
Figures S1–S2, Table S1. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We thank W.A. Lim for providing an N-CrkSH3 construct, P.A. Walker for oligonucleotide synthesis, S. Patel for MALDI-MS analysis, and F.E. Boas, R.S. Fenn, and W.L. Martin for helpful discussion. This work was supported by an NIH Pioneer Award (P.B.H.). S.J.W. was partially supported by the Stanford University Medical Scientist Training Program. R.M.W. was supported by the Gabilan Stanford Graduate Fellowship.
References
- 1.Mills DR, Peterson RL, Spiegelman S. Proc Natl Acad Sci U S A. 1967;58(1):217. doi: 10.1073/pnas.58.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joyce GF. Annu Rev Biochem. 2004;73:791. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- 3.Roberts RW, Ja WW. Curr Opin Struct Biol. 1999;9(4):521. doi: 10.1016/S0959-440X(99)80074-8. [DOI] [PubMed] [Google Scholar]
- 4.Halpin DR, Harbury PB. PLoS Biol. 2004;2(7):E174. doi: 10.1371/journal.pbio.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpin DR, Harbury PB. PLoS Biol. 2004;2(7):E173. doi: 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halpin DR, Lee JA, Wrenn SJ, Harbury PB. PLoS Biol. 2004;2(7):E175. doi: 10.1371/journal.pbio.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305(5690):1601. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunishima M, Kawachi C, Hioki K, Terao K, Tani S. Tetrahedron. 2001;57:1551. [Google Scholar]
- 9.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4(23):4057. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 10.Lee JW, Jun SI, Kim K. Tetrahedron Letters. 2001;42:2709. [Google Scholar]
- 11.Vago I, Greiner I. Tetrahedron Letters. 2002;43:6039. [Google Scholar]
- 12.Needleman SB, Wunsch CD. J Mol Biol. 1970;48(3):443. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen JT, Porter M, Amoui M, Miller WT, Zuckermann RN, Lim WA. Chem, Biol. 2000;7(7):463. doi: 10.1016/s1074-5521(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen JT, Turck CW, Cohen FE, Zuckermann RN, Lim WA. Science. 1998;282(5396):2088. doi: 10.1126/science.282.5396.2088. [DOI] [PubMed] [Google Scholar]
- 15.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, et al. Proc Natl Acad Sci U S A. 1992;89(20):9367. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. J Am Chem Soc. 2003;125(46):13995. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen PE. Pseudo-peptides in drug discovery. Wiley-VCH; Weinheim: 2004. p. 1. [Google Scholar]
- 18.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46(1–3):3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 19.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Drug Development Research. 1995;35(1):20. [Google Scholar]
- 20.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Methods Enzymol. 1996;267:437. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda M, Tanaka S, Nagata S, Kojima A, Kurata T, Shibuya M. Mol Cell Biol. 1992;12(8):3482. doi: 10.1128/mcb.12.8.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren R, Mayer BJ, Cicchetti P, Baltimore D. Science. 1993;259(5098):1157. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 23.Cohen GB, Ren R, Baltimore D. Cell. 1995;80(2):237. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 24.Feller SM, Lewitzky M. Curr Pharm Des. 2006;12(5):529. doi: 10.2174/138161206775474369. [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto S, Hirose M, Hashimoto A, Morishige M, Yamada A, Hosaka H, Akagi K, Ogawa E, Oneyama C, Agatsuma T, Okada M, Kobayashi H, Wada H, Nakano H, Ikegami T, Nakagawa A, Sabe H. Proc Natl Acad Sci U S A. 2006;103(18):7036. doi: 10.1073/pnas.0509166103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The backbone structure of a bound decamer CrkSH3 (PDB accession code 1CKA) peptide ligand was copied and rotated 180 around an axis perpendicular to the helix axis and passing through the C-alpha of the central leucine (residue 5). The rotated structure was aligned with the original structure of the bound peptide and allowed to translate/rotate to minimize the deviation between 1) peptide carbonyl bonds and peptoid carbonyl bonds and 2) peptide C-alpha/C-beta bonds and peptoid amide nitrogen/hydrogen bonds.
- 27.Wu X, Knudsen B, Feller SM, Zheng J, Sali A, Cowburn D, Hanafusa H, Kuriyan J. Structure. 1995;3(2):215. doi: 10.1016/s0969-2126(01)00151-4. [DOI] [PubMed] [Google Scholar]
- 28.Inglis SR, Stojkoski C, Branson KM, Cawthray JF, Fritz D, Wiadrowski E, Pyke SM, Booker GW. J Med Chem. 2004;47(22):5405. doi: 10.1021/jm049533z. [DOI] [PubMed] [Google Scholar]
- 29.Feng S, Chen JK, Yu H, Simon JA, Schreiber SL. Science. 1994;266(5188):1241. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- 30.Lim WA, Richards FM, Fox RO. Nature. 1994;372(6504):375. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- 31.Viguera AR, Arrondo JL, Musacchio A, Saraste M, Serrano L. Biochemistry. 1994;33(36):10925. doi: 10.1021/bi00202a011. [DOI] [PubMed] [Google Scholar]
- 32.Rickles RJ, Botfield MC, Weng Z, Taylor JA, Green OM, Brugge JS, Zoller MJ. Embo J. 1994;13(23):5598. doi: 10.1002/j.1460-2075.1994.tb06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson DS, Keefe AD, Szostak JW. Proc Natl Acad Sci U S A. 2001;98(7):3750. doi: 10.1073/pnas.061028198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamla T, Erdmann VA. J Mol Biol. 2003;329(2):381. doi: 10.1016/s0022-2836(03)00432-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2, Table S1. This material is available free of charge via the internet at http://pubs.acs.org.