

Abstract
In the respiratory chains of aerobic organisms, oxygen reductase members of the heme-copper superfamily couple the reduction of O2 to proton pumping, generating an electrochemical gradient. There are three distinct families of heme-copper oxygen reductases: A-, B- and C-type. The A- and B-type oxygen reductases have an active-site tyrosine that forms a unique crosslinked histidine-tyrosine cofactor. In the C-type oxygen reductases (also called cbb3 oxidases) an analogous active-site tyrosine has recently been predicted by molecular modeling to be located within a different transmembrane helix in comparison to the A- and B-type oxygen reductases. In this work mass spectrometry is used to show that the predicted tyrosine forms a histidine-tyrosine crosslinked cofactor in the active site of the C-type oxygen reductases. This is the first known example of the evolutionary migration of a post-translationally modified active-site residue. It also verifies the presence of a unique cofactor in all three families of proton pumping respiratory oxidases, demonstrating that these enzymes likely share a common reaction mechanism and that the histidine-tyrosine cofactor may be a required component for proton pumping.
Aerobic respiration plays a fundamental role in Earth’s biogeochemical oxygen cycle. It has been estimated that about 75% of the O2 produced by oxygenic photosynthesis is reduced to water via this enzymatically catalyzed process, tightly coupling two of the most widespread metabolisms on earth. Aerobic respiration is also the most exergonic metabolism known and appears to be a requirement for multicellular life. Respiration is performed by a series of integral membrane protein complexes that form electron transfer chains, found within the inner mitochondrial membrane of aerobic eukaryotes and the cytoplasmic membrane of many prokaryotic organisms (1, 2). Mitochondria have a linear electron transfer chain terminating with cytochrome c oxidase, a proton-pumping oxygen reductase which reduces O2 to water. Prokaryotes have more complicated electron transfer chains with branches leading to different terminal electron acceptors (e.g., fumarate, nitrate, Fe3+, O2), allowing for metabolic flexibility when encountering different environments.
Most aerobic prokaryotes utilize respiratory oxidases (i.e., oxygen reductases) that are members of the heme-copper superfamily, which is structurally and catalytically diverse, containing both oxygen reductases and nitric oxide reductases. The mitochondrial cytochrome c oxidase is also a member of the heme-copper superfamily. Heme-copper oxygen reductases catalyze the reduction of O2 to water with the concomitant electrogenic translocation of protons across the membrane, contributing to the generation of a proton electrochemical gradient that can be coupled to energy-requiring cellular processes (1, 2). The oxygen reductases are all multi-subunit protein complexes that span the membrane bilayer. They are classified as A-, B-, or C-type oxygen reductases, based on genomic, phylogenetic and structural analyses(1). All three oxygen reductase families have been shown to pump protons coupled to the reduction of oxygen; however, they differ in biochemical properties such as reaction rate and oxygen affinity. Many prokaryotic genomes encode several heme-copper oxygen reductases which are differentially expressed depending on the environmental conditions.
Subunit I is the core protein in the enzyme complex and is the only subunit shared by all three families of the oxygen reductases. All of the amino acid residues and cofactors necessary for catalysis and proton pumping are within subunit I. The active site of the enzyme is a bimetallic center composed of a copper ion (CuB) and a high-spin heme, together ligated by four conserved histidines (three to CuB and one to the heme Fe). X-ray structures of members of the A- and B-type heme-copper oxygen reductases reveal a unique crosslinked histidine-tyrosine cofactor in the active site between one of the CuB ligands and a tyrosine that is essential for enzyme function (3–5). This tyrosine is postulated to be oxidized to a tyrosyl radical during turnover and to donate a hydrogen atom to facilitate breaking the O-O bond during catalysis (6–8). The crosslink has been verified by mass spectrometry in the B-type oxygen reductase from Thermus thermophilus (9). Figure 1 shows the structure of the crosslinked residues at the active site of the A-type oxygen reductases.
Figure 1.
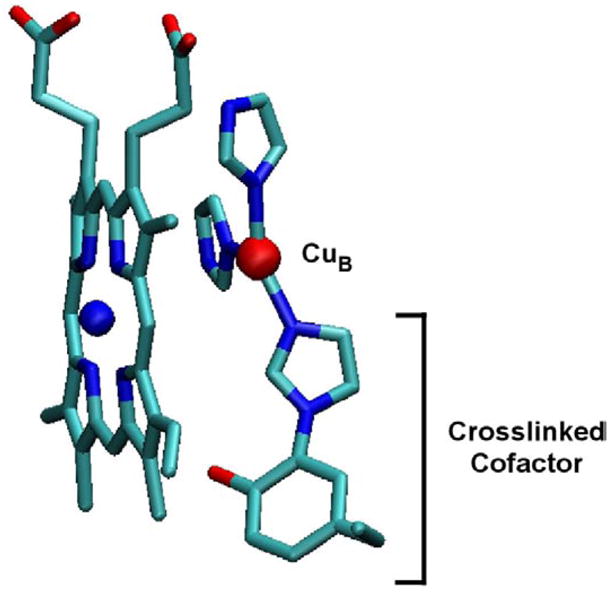
Structure of the active-site cofactor from Rhodobacter sphaeroides A-type heme-copper oxygen reductase. The cofactor is formed by a covalent crosslink between the Nε of a CuB histidine ligand and the Cε of the active site tyrosine and is present throughout the catalytic cycle. The farnesyl tail has been removed from the heme for clarity. This figure was generated using VMD software (45).
Sequence alignments have shown that the active-site tyrosine present in all of the A- and B-type oxygen reductases is absent in the C-type oxygen reductases. Recently, structural models of subunit I for the C-type oxygen reductases from Vibrio cholerae (10) and Rhodobacter sphaeroides (11) were built utilizing the X-ray structures of the A- and B-type oxygen reductases as templates. A surprising result was the prediction that a completely conserved tyrosine (Y255 in V. cholerae) from transmembrane helix VII in the C-type oxygen reductases occupies the same physical position in the active site as the tyrosine located in transmembrane helix VI of the A- and B-type oxygen reductases (10, 11). It was also shown by modeling that it is geometrically feasible for a crosslink to be formed with the equivalent histidine ligand to CuB (H211 in V. cholerae). In the current work, mass spectrometry was used to show that the predicted crosslink is indeed present in subunit I of the V. cholerae C-type oxygen reductase.
MATERIALS AND METHODS
All reagents are from Sigma (St. Louis, MO) unless otherwise noted.
Overexpression of C-type oxygen reductase from Vibrio cholerae
Protein was overexpressed and collected as previously reported (10). Briefly, Vibrio cholerae cells were grown in LB media (USB Corporation) with 100 μg/L ampicillin (Fisher Biotech) and 100 μg/L streptomycin at 37 °C. Gene expression was induced with 0.2% L-(+)-arabinose. The cells were lysed and centrifuged at 40,000 RPM to collect the membranes. Membrane proteins were solubilized by adding 0.5% dodecyl β-D-maltoside (Anatrace). Non-solubilized membranes were removed by centrifuging at 40,000 RPM for 30 minutes.
Purification of oxygen reductase
In order to obtain a preparation sufficiently pure for the mass spectrometry, the enzyme was first purified using immobilized metal affinity chromatography (IMAC) followed by weak anion exchange (WAX) on DEAE-sepharose. IMAC was performed as previously reported (10), using a nickel affinity column (Qiagen, Valencia, CA) in a cold room (4 ºC) at low pressure in 0.05% DDM and eluting the his-tagged protein using a stepped gradient of imidazole. WAX was performed using fast protein liquid chromatography (FPLC) (Amersham Biosciences (now GE Healthcare), Piscataway, NJ) in a cold room using 10 mM ammonium bicarbonate (pH 8.0)/0.05% DDM as solvent A and 1 M ammonium bicarbonate/0.05% DDM as solvent B. Samples were loaded at a percentage of solvent B that was approximately 10% below the expected elution concentration of solvent B for the protein complex as determined by test gradients. A 1 hour gradient to 100% solvent B was then performed and fractions containing the purified protein were combined. After each chromatography step, the sample was concentrated using a centrifugal filter with a mass cutoff of 50 kDa (Millipore, Billerica, MA).
Trypsin digestion of the oxygen reductase and sample prepareation
10 μL purified enzyme (approximately 25 mg/mL) was digested overnight with 20 μg sequencing-grade trypsin (Bio-Rad, Hercules, CA) in 90% 100 mM ammonium bicarbonate (pH 8.0) and 10% acetonitrile at 37 °C. Immediately following trypsin digestion, 50 μL of the sample was applied to a gel filtration spin column with a 6 kDa mass cutoff (Micro Bio-Spin P6, Bio-Rad) to remove low-mass peptides. The spin column was equilibrated four times in 0.05% DDM prior to use. A methanol-chloroform precipitation (12, 13) was then used to separate the remaining peptides from the detergent and soluble peptides. The resulting pellet was resuspended in 500 μL 75% acetic acid and immediately subjected to analysis via mass spectrometry.
Mass spectrometry
Samples were analyzed on a custom-built 8.5 Tesla quadrupole Fourier-transform ion cyclotron resonance mass spectrometer (Q-FTICR MS) (14) using the MIDAS datastation (15). Introduction was performed using electrospray ionization (ESI) from a nanospray robot (Advion BioSciences, Ithaca, NY) at 1.2 kV with a backing gas pressure of 0.5 psi. Broadband scans were taken to identify species of interest for fragmentation followed by quadrupole isolation (2 m/z window) and MS/MS using collisionally activated dissociation (CAD) in the external accumulation octopole (16, 17). In these MS/MS experiments, several CAD acceleration voltages were used in order to generate a wider variety of fragment ions. The transfer time into the ICR cell was also varied in order to compensate for time-of-flight effects.
Data Analysis
Data from the broadband and MS/MS experiments were processed using the THRASH algorithm (18) and analyzed with ProSightPTM (http://prosightptm.scs.uiuc.edu, (19)). The presence of the crosslink required separate fragmentation analysis of each of the two peptides, with the opposite peptide modeled as a single large post-translational modification. C-terminal (B) and N-terminal (Y) type fragment ions (20) were matched at 20 ppm for the crosslinked peptide from the V. cholerae C-type oxygen reductase and 10 ppm for the all other peptides.
RESULTS
Mass spectrometery of a tryptic digest of a C-type oxygen reductase
Mass spectrometry (MS) of a tryptic digest of subunit I from the Vibrio cholerae C-type oxygen reductase was performed to discern the presence of the predicted crosslink. Analysis of the protein sequence predicted that if a crosslink was formed then complete trypsin digestion would result in a peptide containing residues S193-K232 crosslinked to residues L243-K304, but missing the region from residues Q233-R242. This “H-shaped” tryptic peptide would not be present if the crosslink did not exist and was predicted to have a mass equal to that of the two individual peptides, subtracting two daltons for the two protons lost during the formation of the crosslink. A peptide of this expected molecular weight (monoisotopic mass 11478.7 Da) was present in the mass spectrum of the trypsin digest. This crosslinked tryptic fragment was isolated and analyzed by tandem MS (MS/MS) using collisionally activated dissociation (CAD) fragmentation. Figure 2 shows the MS/MS fragment map of the crosslinked peptide. Multiple N- and C-terminal fragment ions were detected from the peptides on either end of the crosslink, demonstrating the existence of a crosslink between the two. In addition to the MS/MS fragment ions containing the crosslinked peptide, several of the MS/MS fragments spanned the crosslinked residue Y255 but did not include the crosslink (Figure 3).
Figure 2.
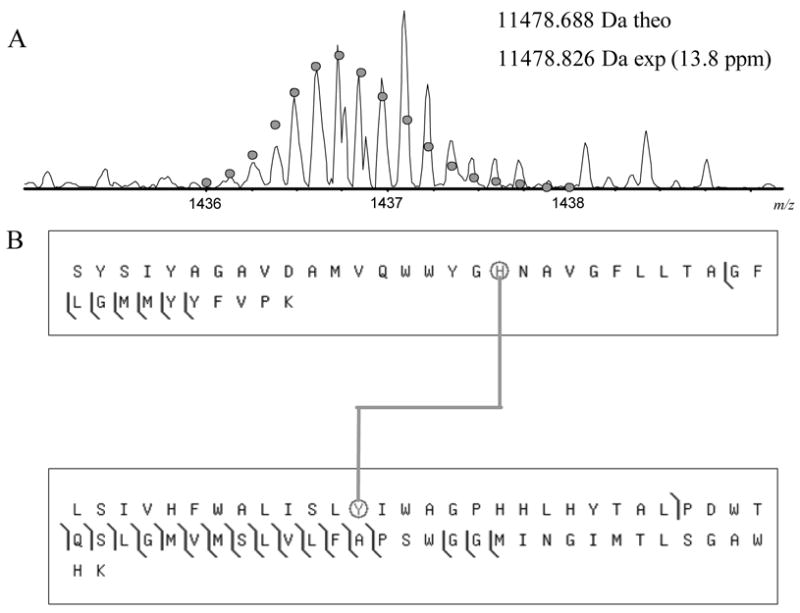
CAD fragmentation map for the crosslinked peptides S193-K232 and L243-K304. (A) The mass spectrum of the crosslinked tryptic peptide. The circles show the theoretical isotope heights for a peptide of the given mass. (B) CAD MS/MS fragmentation results. The crosslinked histidine and tyrosine are circled. Spectra were processed using a modified version of the THRASH algorithm (18) and fragments were matched using ProSight PTM (http://prosightptm.scs.uiuc.edu, (46)) with a match tolerance of 20 ppm.
Figure 3.
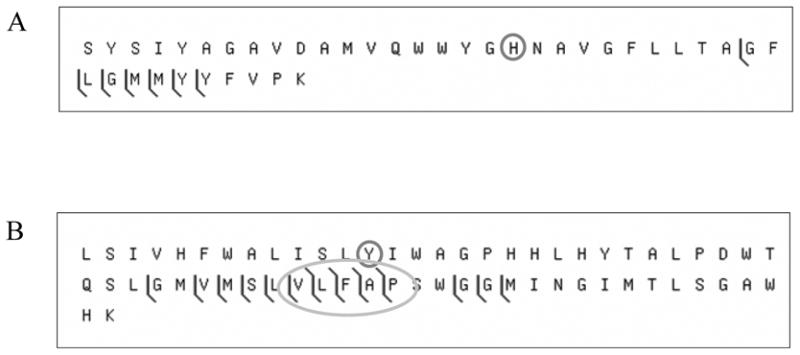
CAD fragmentation of the crosslinked tryptic peptide from the C-type oxygen reductase assuming the absence of the crosslink. The CAD fragmentation dataset used to generate figure 2 was also analyzed as if the crosslink were not present. (A) shows the matches for the tryptic peptide S193-K232 and (B) for L243-K304. The histidine and tyrosine normally involved in the crosslink are circled. The oval highlights several fragments that were found to span the crosslinked residue but do not include the mass of the opposite crosslinked peptide.
Presence of non-crosslinked protein in the digest
The trypsin digest also contained non-crosslinked S193-K232 and L243-K304 peptides and detailed MS/MS fragmentation confirmed their identity (Figures 4 and 5). It is unclear whether the His-Tyr crosslink is normally absent in a portion of the population of the protein, or if the non-crosslinked species is an artifact of the recombinant protein expression or sample preparation. In order to address the lability of the crosslink, the same protocol was used to investigate the crosslink in the A-type oxygen reductase from R. sphaeroides. In the A-type oxygen reductases, the crosslink is between residues that are only 4 amino acids apart on the same transmembrane helix (H284-Y288 in the R. sphaeroides A-type oxygen reductase), whereas there are 44 amino acids between the crosslinked residues in the C-type oxygen reductases, which span two helices (H211-Y255 in the V. cholerae C-type oxygen reductase). The data show definitively that the crosslink is present between His284 and Tyr288 in subunit I of the R. sphaeroides A-type oxygen reductase, and no fragments with the molecular weight expected for the non-crosslinked peptide were detected (Figure 6). In agreement with the latest X-ray structure of the R. sphaeroides A-type oxygen reductase (S. Ferguson-Miller, personal communication), it is concluded that the His-Tyr crosslink is present in the entire population of this A-type oxygen reductase. At this time, it is unclear whether the occupancy of the crosslink in the C-type reductase is less than 100% in vivo or if the non-crosslinked peptides are an artifact of the sample preparation or mass spectrometry. Although the A-type reductase appears to be entirely crosslinked, the His-Tyr bond lability may be higher in the C-type under the analysis conditions.
Figure 4.
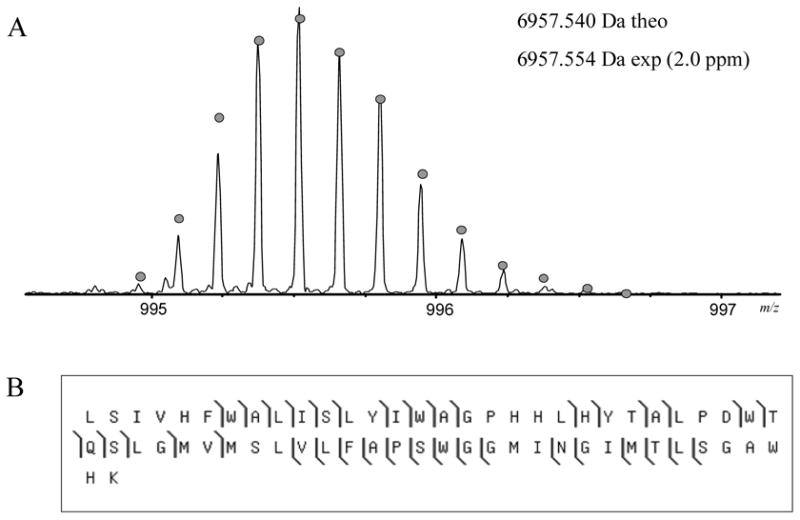
Mass spectrum and fragmentation of the tryptic peptide L243-K304 from the C-type oxygen reductase from V. cholerae. (A) The mass spectrum of the peptide. The circles show the theoretical isotope heights for a peptide of the given mass. (B) CAD MS/MS fragmentation results.
Figure 5.
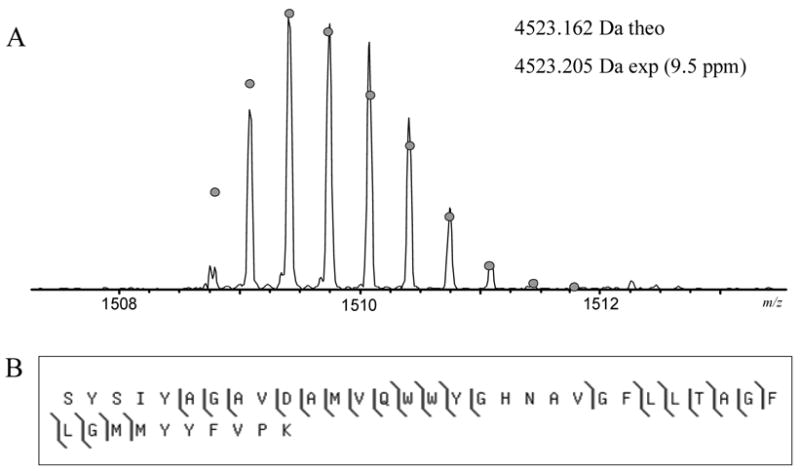
Mass spectrum and fragmentation of the tryptic peptide S193-K232 from the C-type oxygen reductase from V. cholerae. (A) The mass spectrum of the peptide. The circles show the theoretical isotope heights for a peptide of the given mass. (B) CAD MS/MS fragmentation results.
Figure 6.

Mass spectrum and fragmentation of the crosslinked peptide from the A-type oxygen reductase from R. sphaeroides. (A) The mass spectrum of the crosslinked tryptic peptide N258-K307. The circles show the theoretical isotope heights for a peptide of the given mass. The mass spectrum shows that only the crosslinked species is detected in the tryptic digest. (B) CAD MS/MS fragmentation results when analyzed with the crosslink present (the histidine and tyrosine are circled) and (C) without the crosslink. Multiple fragments containing the crosslink are detected whereas no fragments are detected which do not contain it.
DISCUSSION
The presence of an active-site cross-linked cofactor in C-type heme-copper oxygen reductases
The current work demonstrates that a novel crosslinked cofactor is present in all three families of the heme-copper oxygen reductases (Figure 7). This verifies the prediction by molecular modeling (10, 11) of the presence of an active-site tyrosine in the C-type oxygen reductases that is structurally and functionally equivalent to the active-site tyrosine in the A- and B-type oxygen reductases. It is also a unique structural feature which separates the oxygen reductases from other members of the heme-copper superfamily, notably NO reductases. The analysis of the C-type oxidase also revealed the presence of a population of the enzyme without the His-Tyr crosslink, but this is likely an artifact either of the conditions of the expression of the enzyme (e.g., incomplete incorporation of copper) or of the sample preparation. It is clear that collisionally activated fragmenation of the isolated crosslinked peptide during MS/MS analysis can result in scission of the crosslinking bond. However, the non-crosslinked peptide is also apparent in the absence of collisionally activated fragmentation in the mass spectrometer. This suggests that the non-crosslinked enzyme is either present within the trypsin digest or that the crosslink in the C-type enzyme is more labile than that of the A-type either during the electropray process or in the trapping and cooling of the ions in the mass spectrometer. While this manuscript was being revised after initial review, Rauhamaki et al (21) published a paper which demonstrates by MALDI mass spectrometry the presence of the His-Tyr crosslink in the C-type oxygen reductase from R. sphaeroides. The report by Rauhamaki et al (21) does not indicate any non-crosslinked protein, so it is very likely that the crosslink is present in all properly assembled enzyme. It can be concluded from the current work and that of Rauhamaki (21) that the presence of the active-site His-Tyr crosslink is a universal feature of the of the C-type oxygen reductases and, by extension, all heme-copper oxygen reductases.
Figure 7.
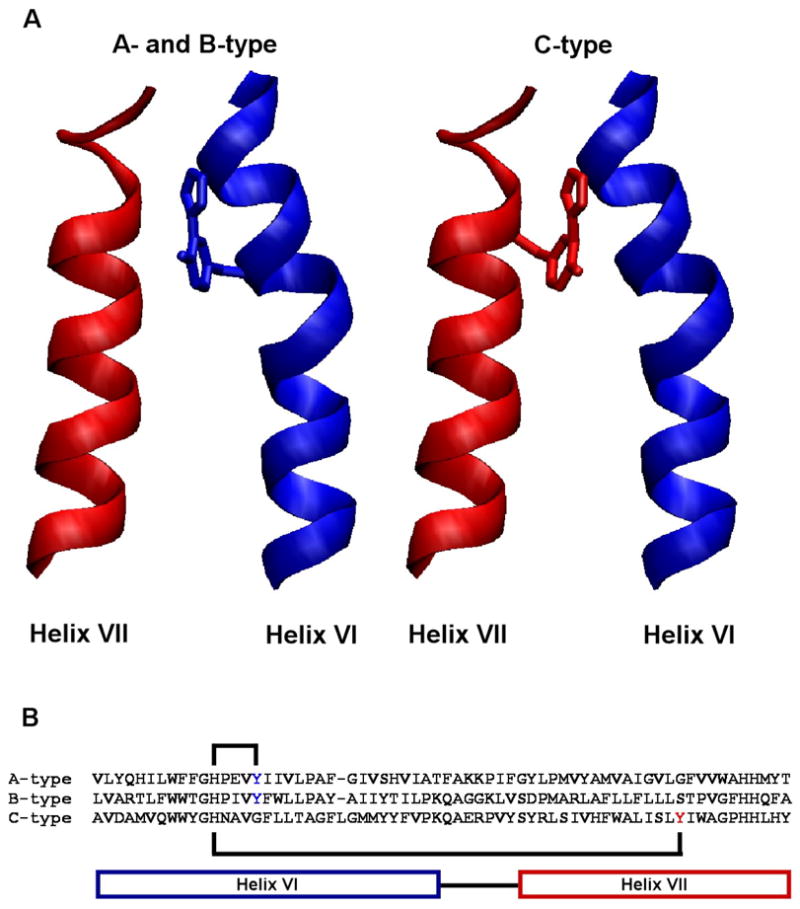
A novel crosslinked cofactor is present in all three heme-copper oxygen reductase families. (A) The active-site tyrosine forming the cofactor originates from helix VI in the A- and B-type oxygen reductases, while in the C-type oxygen reductases it originates from helix VII. This is the first example of the evolutionary migration of a post-translationally modified active-site residue. (B) The crosslink is formed between a histidine and tyrosine within helix VI in the A- and B-type oxygen reductases. In the C-type oxygen reductases the crosslink is formed between a histidine in helix VI and a tyrosine in helix VII, covalently coupling the helices together. This figure was generated using VMD software (45).
A unified catalytic mechanism for all oxygen reductase families
The novel crosslinked cofactor is thought to form as a result of the generation of a tyrosine radical in the active site, presumably upon the initial turnover of the reduced enzyme with O2. Conceivably, this could be a side-reaction and not essential for enzyme function. However, replacement of the active-site tyrosine by a phenylalanine in the R. sphaeroides A-type oxygen reductase resulted not only in an inactive enzyme, but also altered the metal ligation in the active site (22). This suggests that the crosslink is needed to maintain the structure of the active site. Furthermore, work by Uchida et al. (23) demonstrated that substituting d4-Tyr for tyrosine resulted in a large decrease in enzymatic activity of an Escherichia coli A-type oxygen reductase and the spectroscopic properties suggested that the His-Tyr crosslink was not formed. These observations suggest that the crosslink is not simply an irrelevant side-product of the chemistry at the active site, but is essential for the function of the heme-copper oxygen reductases. The active-site tyrosine is proposed to donate both a proton and an electron to facilitate cleavage of the O-O bond (6–8, 24–26). It is clear that an amino acid radical does form during the catalytic cycle of the oxygen reductase (27–31), and the active-site tyrosine is a logical primary electron donor for the chemistry. There is no evidence from rapid-quench electron paramagnetic resonance (EPR) spectroscopy for rapid formation of a tyrosine radical (28, 32), but it would likely be EPR-silent due to proximity to the metals at the active site. Attempts to demonstrate the presence of a radical by Fourier-transform infrared (FTIR) spectroscopy (33, 34) and by iodination of the amino acid radical (6) have provided data consistent with the formation of neutral tyrosyl radical, but these data are acquired over a longer time period allowing for radical migration. Indeed, the strongest argument for the formation of a radical at the active-site tyrosine may be the fact that the His-Tyr crosslink is present. Presumably, the crosslink is a consequence of radical-based chemistry that occurs during the initial turnovers of the enzyme. The data strongly suggest that all heme-copper oxygen reductases utilize the same catalytic mechanism of hydrogen atom donation for oxygen bond scission.
The functional role of the His-Tyr crosslink
The function of the crosslink has been the subject of considerable speculation as well as investigation. Recent studies with model compounds (35–39) as well as computational studies (26, 40) have suggested a possible functional significance for the crosslink. The crosslinked histidine withdraws electrons from the tyrosine, resulting in a lower pKa and a higher midpoint potential of the tyrosine (41). Conversely, the redox state and protonation state of the tyrosine influence the electron donating capacity of the imidazole as a metal ligand, thus controlling the preferred ligand geometry about CuB (35). It has also been suggested that, due to the presence of the crosslinked tyrosine, the histidine ligand to CuB might be labile and move away from the metal during turnover, playing a key role in the proton pump mechanism (40). These studies, in conjunction with the presence of the crosslinked cofactor in all oxygen reductase families, suggests that the cofactor may be a required component for proton pumping. Further work is necessary to elucidate its role.
The evolutionary migration of the post-translationally modified tyrosine
The post-translational modification of active-site amino acid residues to form novel cofactors in situ has been observed in a number of redox active enzymes (42). Some cofactors are produced via chemical modification of amino acid side chains (e.g. oxidation, methylation, hydroxylation) whereas other cofactors are formed by crosslinking two or more amino acids together. These post-translationally crosslinked active-site amino acids can be found in tyrosinase, hemocyanin and catechol oxidase (Cys-His), catalase-peroxidase (Met-Tyr-Trp), galactose oxidase (Tyr-Cys), catalase (His-Tyr-αC) and the A- and B-type heme-copper oxygen reductases (His-Tyr) (see(42)). The evolutionary migration of amino acids within a protein family can be defined as the situation where residues that have the same structural or functional role and which share the same spatial location, derive from different positions within their respective protein sequences. Evolutionary migration has been reported for active-site residues (43, 44), but this is the first report for a post-translationally modified active-site residue. The active-site tyrosine forming the crosslinked cofactor is located within a different transmembrane helix in the C-type oxygen reductases (helix VII) in comparison to the A-type and B-type oxygen reductases (helix VI). It is currently unknown which state (the tyrosine being located in helix VI or helix VII) is ancestral.
Footnotes
This work was supported by grants from the National Institutes of Health GM 067193-04 (to NLK) and HL 16101 (to RBG) and from the National Science Foundation NSF-BES-04-03846 (to TJM).
References
- 1.Pereira MM, Santana M, Teixeira M. A Novel Scenario for the Evoluation of Haem-copper Oxygen Reductases. Biochim Biophys Acta. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB. The Superfamily of Heme-Copper Respiratory Oxidases. J Bacteriol. 1994;176:5587–5600. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 Å Resolution of the Paracoccus denitrificans Two-Subunit Cytochrome c Oxidase Complexed with an Antibody Fv Fragment. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsukihara T, Aoyama H, Yamashita E, Takashi T, Yamaguichi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 5.Soulimane T, Buse G, Bourenkov GP, Bartunik HD, Huber R, Than ME. Structure and Mechanism of the Aberrant ba3-cytochrome c Oxidase fromThermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Oxygen Activation and Reduction in Respiration: Involvement of Redox-Active Tyrosine 244. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 7.Gennis RB. Multiple Proton-conducting Pathways in Cytochrome Oxidase and a Proposed Role for the Active-site Tyrosine. Biochim Biophys Acta. 1998;1365:241–248. [Google Scholar]
- 8.Babcock GT. How Oxygen is Activated and Reduced in Respiration. Proc Natl Acad Sci USA. 1999;96:12971–12973. doi: 10.1073/pnas.96.23.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buse G, Soulimane T, Dewor M, Meyer HE, Blüggel M. Evidence for a Copper-coordinated Histidine-tyrosine Cross-link in the Active Site of Cytochrome Oxidase. Protein Science. 1999;8:985–990. doi: 10.1110/ps.8.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hemp J, Christian C, Barquera B, Gennis RB, Martinez TJ. Helix Switching of a Key Active-Site Residue in the Cytochrome cbb3 Oxidases. Biochemistry. 2005;44:10766–10775. doi: 10.1021/bi050464f. [DOI] [PubMed] [Google Scholar]
- 11.Sharma V, Puustinen A, Wikstrom M, Laakkonen L. Sequence analysis of the cbb3 oxidases and an atomic model for the Rhodobacter sphaeroides enzyme. Biochemistry. 2006;45:5754–65. doi: 10.1021/bi060169a. [DOI] [PubMed] [Google Scholar]
- 12.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–3. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 13.Whitelegge JP, le Coutre J, Lee JC, Engel CK, Prive GG, Faull KF, Kaback HR. Toward the bilayer proteome, electrospray ionization-mass spectrometry of large, intact transmembrane proteins. Proc Natl Acad Sci U S A. 1999;96:10695–8. doi: 10.1073/pnas.96.19.10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patrie SM, Charlebois JP, Whipple D, Kelleher NL, Hendrickson CL, Quinn JP, Marshall AG, Mukhopadhyay B. Construction of a hybrid quadrupole/Fourier transform ion cyclotron resonance mass spectrometer for versatile MS/MS above 10 kDa. J Am Soc Mass Spectrom. 2004;15:1099–108. doi: 10.1016/j.jasms.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 15.Senko MW, Canterbury JD, Guan S, Marshall AG. A high-performance modular data system for Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom. 1996;10:1839–44. doi: 10.1002/(SICI)1097-0231(199611)10:14<1839::AID-RCM718>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.Patrie SM, Ferguson JT, Robinson DE, Whipple D, Rother M, Metcalf WW, Kelleher NL. Top down mass spectrometry of < 60-kDa proteins from Methanosarcina acetivorans using quadrupole FRMS with automated octopole collisionally activated dissociation. Mol Cell Proteomics. 2006;5:14–25. doi: 10.1074/mcp.M500219-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Senko MW, Hendrickson CL, Emmett MR, Shi DH, Marshall AG. External Accumulation of Ions for Enhanced Electrospray Ionization Fourier Transform Ion cyclotron Resonance Mass Spectrometry. J American Society for Mass Spectrometry. 1997;8:970–976. [Google Scholar]
- 18.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectrom. 2000;11:320–32. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 19.LeDuc RD, Taylor GK, Kim YB, Januszyk TE, Bynum LH, Sola JV, Garavelli JS, Kelleher NL. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004;32:W340–5. doi: 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 21.Rauhamaki V, Baumann M, Soliymani R, Puustinen A, Wikstrom M. Identification of a histidine-tyrosine cross-link in the active site of the cbb3-type cytochrome c oxidase from Rhodobacter sphaeroides. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0606254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das TK, Pecoraro C, Tomson FL, Gennis RB, Rousseau DL. The Post-Translational Modification in Cytochrome c Oxidase Is Required To Establish a Functional Environment of the Catalytic Site. Biochemistry. 1998;37:14471–14476. doi: 10.1021/bi981500w. [DOI] [PubMed] [Google Scholar]
- 23.Uchida T, Mogi T, Nakamura H, Kitagawa T. Role of Tyr-288 at the dioxygen reduction site of cytochrome bo studied by stable isotope labeling and resonance raman spectroscopy. J Biol Chem. 2004;279:53613–20. doi: 10.1074/jbc.M409719200. [DOI] [PubMed] [Google Scholar]
- 24.Blomberg MRA, Siegbahn PEM, Babcock GT, Wikström M. O-O Bond Splitting Mechanism in Cytochrome Oxidase. J Inorg Biochem. 2000;80:261–269. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 25.Blomberg MR, Siegbahn PE, Wikström M. Metal-bridging Mechanism for O-O Bond Cleavage in Cytochrome c Oxidase. Inorg Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 26.Bu Y, Cukier RI. Structural character and energetics of tyrosyl radical formation by electron/proton transfers of a covalently linked histidine-tyrosine: a model for cytochrome C oxidase. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2005;109:22013–26. doi: 10.1021/jp053046t. [DOI] [PubMed] [Google Scholar]
- 27.MacMillan F, Kannt A, Behr J, Prisner T, Michel H. Direct Evidence for Tyrosine Radical in the Reaction of Cytochrome c Oxidase with Hydrogen Peroxide. Biochemistry. 1999;38:9179–9184. doi: 10.1021/bi9911987. [DOI] [PubMed] [Google Scholar]
- 28.Wiertz FGM, Richter O-MH, Cherepanov AV, MacMillan F, Ludwig B, de Vries S. An Oxo-ferryl Tryptophan Radical Catalytic Intermediate in Cytochrome c and Quinol Oxidasses Trapped by Microsecond Freeze-hyperquenching (MHQ) FEBS Letters. 2004;575:127–130. doi: 10.1016/j.febslet.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 29.MacMillan F, Budiman K, Angerer H, Michel H. The Role of Tryptophan 272 in the Paracoccus denitrificans Cytochrome c Oxidase. FEBS Lett. 2006;580:1345–9. doi: 10.1016/j.febslet.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 30.Rich PR, Rigby SEJ, Heathcote P. Radicals associated with the Catalytic Intermediates of Bovine Cytochrome c Oxidase. Biochim Biophys Acta. 2002;1554:137–146. doi: 10.1016/s0005-2728(02)00228-1. [DOI] [PubMed] [Google Scholar]
- 31.Budiman K, Kannt A, Lyubenova S, Richter O-MH, Ludwig B, Michel H, MacMillan F. Tyrosine 167: The Origin of the Radical Species Observed in the Reaction of Cytochrome c Oxidase with Hydrogen Peroxide in Paracoccus denitrificans. Biochemistry. 2004;43:11709–11716. doi: 10.1021/bi048898i. [DOI] [PubMed] [Google Scholar]
- 32.Wiertz FGM, de Vries S. Low-temperature Kinetic Measurements of Microsecond Freeze-hyperquench (MHQ) Cytochrome Oxidase Monitored by UV-Visible Spectroscopy with a Newly Designed Cuvette. Biochemical Society Transactions. 2006;34:136–138. doi: 10.1042/BST0340136. [DOI] [PubMed] [Google Scholar]
- 33.Iwaki M, Puustinen A, Wikstrom M, Rich PR. ATR-FTIR spectroscopy and isotope labeling of the PM intermediate of Paracoccus denitrificans cytochrome c oxidase. Biochemistry. 2004;43:14370–8. doi: 10.1021/bi048545j. [DOI] [PubMed] [Google Scholar]
- 34.Nyquist RM, Heitbrink D, Bolwien C, Gennis RB, Heberle J. Direct Observation of Protonation Reactions During the Catalytic Cycle of Cytochrome c Oxidase. Proc Natl Acad Sci USA. 2003;100:8715–8720. doi: 10.1073/pnas.1530408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pesavento RP, Pratt DA, Jeffers J, van der Donk WA. Model studies of the Cu(B) site of cytochrome c oxidase utilizing a Zn(II) complex containing an imidazole-phenol cross-linked ligand. Dalton Trans. 2006:3326–37. doi: 10.1039/b516090a. [DOI] [PubMed] [Google Scholar]
- 36.Pratt DA, Pesavento RP, van der Donk WA. Model studies of the histidine-tyrosine cross-link in cytochrome C oxidase reveal the flexible substituent effect of the imidazole moiety. Org Lett. 2005;7:2735–8. doi: 10.1021/ol050916g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim E, Kamaraj K, Galliker B, Rubie ND, Moenne-Loccoz P, Kaderli S, Zuberbuhler AD, Karlin KD. Dioxygen reactivity of copper and heme-copper complexes possessing an imidazole-phenol cross-link. Inorg Chem. 2005;44:1238–47. doi: 10.1021/ic048907b. [DOI] [PubMed] [Google Scholar]
- 38.Tomson FL, Bailey JA, Gennis RB, Unkefer CJ, Li Z, Silks LA, Martinez RA, Donohoe RJ, Dyer RB, Woodruff WH. Direct Infrared Detection of the Covalently Ring-Linked His-Tyr Structure in the Active Site of the Heme-Copper Oxidases. Biochemistry. 2002;41:14383–14390. doi: 10.1021/bi026370c. [DOI] [PubMed] [Google Scholar]
- 39.Cappuccio JA, Ayala I, Elliott GI, Szundi I, Lewis j, Konopelski JP, Barry BA, Einarsdóttir Ó. Modeling the Active Site of Cytochrome Oxidase: Synthesis and Characterization of a Cross-linked Histidine-Phenol. J Am Chem Soc. 2002;124:1750–1760. doi: 10.1021/ja011852h. [DOI] [PubMed] [Google Scholar]
- 40.Colbran SB, Paddon-Row MN. Could the tyrosine-histidine ligand to CuB in cytochrome c oxidase be coordinatively labile? Implications from a quantum chemical model study of histidine substitutional lability and the effects of the covalent tyrosine-histidine cross-link. J Biol Inorg Chem. 2003;8:855–65. doi: 10.1007/s00775-003-0488-x. [DOI] [PubMed] [Google Scholar]
- 41.McCauley KM, Vrtis JM, Dupont J, van der Donk WA. Insights into the Functional Role of the Tyrosine-Histidine Linkage in Cytochrome c Oxidase. J Am Chem Soc. 2000;122:2403–2404. [Google Scholar]
- 42.Okeley NM, van der Donk WA. Novel cofactors via post-translational modifications of enzyme active sites. Chem Biol. 2000;7:R159–71. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 43.Todd AE, Orengo CA, Thornton JM. Plasticity of enzyme active sites. Trends Biochem Sci. 2002;27:419–26. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- 44.Hasson MS, Schlichting I, Moulai J, Taylor K, Barrett W, Kenyon GL, Babbitt PC, Gerlt JA, Petsko GA, Ringe D. Evolution of an enzyme active site: the structure of a new crystal form of muconate lactonizing enzyme compared with mandelate racemase and enolase. Proc Natl Acad Sci U S A. 1998;95:10396–401. doi: 10.1073/pnas.95.18.10396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 46.Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL. Web and database software for identification of intact proteins using “top down” mass spectrometry. Anal Chem. 2003;75:4081–6. doi: 10.1021/ac0341721. [DOI] [PubMed] [Google Scholar]