

Abstract
A growing number of biological processes have been found to be regulated by the covalent attachment of the ubiquitin-like protein SUMO to key cellular targets. A critical step in the process of analyzing the role of SUMO in regulating the activity of these proteins is the identification of the lysine residues that are targeted by this modification. Unfortunately, current methods aimed at mapping these attachment sites are laborious and often ineffective. We report here the development of a platform that combines the use of different C-terminal SUMO mutants with different protease digestion strategies to enable the rapid and efficient identification of SUMO attachment sites. We successfully apply this approach to several model SUMO substrates as well as to a mixture of SUMO conjugates purified from Saccharomyces cerevisiae. Although we specifically employ this strategy for the identification of SUMO attachment sites in yeast, this general approach can easily be adapted to map the sites of conjugation for other ubiquitin-like proteins from a wide range of organisms.
SYNOPSIS
This manuscript describes the development of a method that utilizes different SUMO mutants in combination with different digestion strategies and mass spectrometry to identify SUMO attachment sites in substrates. The utility of this method is demonstrated through the identification of modification sites in several model SUMO substrates as well as in complex mixtures. This approach can also be adapted to map conjugation sites for other ubiquitin-like proteins in various organisms.
Introduction
The post-translational modification of proteins by ubiquitin and ubiquitin-like proteins (UbLs) has emerged as a critical regulatory mechanism by which the activity of any given protein and its associated biological process can be modulated 1, 2. One such UbL is the small ubiquitin-related modifier, SUMO. The covalent attachment of SUMO, or Smt3p in budding yeast, to lysine residues of intracellular proteins has been shown to regulate a wide range of cellular functions including gene expression, chromatin structure, signal transduction, and the maintenance of genome integrity. Despite its key role in these processes, the exact function of SUMO conjugation in these cases is still poorly understood and remains a topic of active investigation.
Progress in understanding the function of SUMO conjugation had been hindered in part by difficulties in identifying sumoylated substrates that serve as effectors for the SUMO-mediated regulation of biological processes. A number of recent proteomic studies by our group and others have provided significant advances in this area with the identification of hundreds of new putative substrates, many of which have known roles in processes that have been shown to be regulated by SUMO 3-12. That work has set the stage for the in-depth characterization of this plethora of new SUMO targets in order to begin to understand the functional consequences of their sumoylation.
The well-established experimental paradigm for studying the effect of a post-translational modification on the activity of a protein is to identify the site of modification, mutate the site to a chemically similar but non-modifiable amino acid, and then assess the ability of the mutant protein to carry out its biological function. Although this approach has been successfully applied to the study of a number of sumoylated targets, it has also been limited by difficulties in quickly and efficiently identifying SUMO-attachment sites in the target of interest 13-15. In most cases, the mapping of SUMO-attachment sites has been performed by the laborious process of systematically mutating all lysines that conform to a previously identified sequence motif for SUMO attachment that can be directly recognized by the SUMO-conjugating enzyme Ubc9p. While most of the experimentally determined sites of SUMO conjugation conform to this consensus sumoylation motif of ψ-K-X-E/D, where ψ is a hydrophobic residue and X is any amino acid residue, many exceptions have also been identified, further complicating the process of localizing the exact site of modification 13, 14. Alternatively, methods utilizing mass spectrometry to directly identify SUMO modification sites have also been successfully applied 3, 16-18. Unfortunately, these strategies also face many technical challenges that have limited their ability to efficiently detect sites of SUMO conjugation.
In this work, we describe a method for the identification of SUMO attachment sites in cellular proteins. This method utilizes different SUMO mutants in combination with various protease digestion strategies followed by detection using mass spectrometry to directly and specifically map the locations of the modified lysine residues. We successfully demonstrate the utility of this method through the identification of SUMO modification sites in an assortment of previously characterized SUMO substrates as well as complex mixtures. More importantly, this strategy provides a conceptual framework that can be easily adapted to map the conjugation sites for other UbLs from diverse organisms.
Experimental Procedures
Construction of Smt3p Mutants
Smt3p-I96K, Smt3p-I96R, Smt3p-G97K, Smt3p-G97R, and Smt3p-FacX were constructed using polymerase chain reaction (PCR) and primers encoding the appropriate mutations and restriction sites and subsequently cloned into pET28a (Novagen) for expression in E. coli and into the previously described pRS315-PGAL-His6Flag-Smt3p expressing plasmid19. The presence of mutations were confirmed by standard DNA sequencing methods.
In Vitro Sumoylation Assay
His6-Uba2p, His6-Aos1p, His6-Smt3p, and His6-Ubc9p were expressed and purified as previously described 19, 20. His6-Smt3p mutants were expressed in E. coli from pET28a expression vectors and purified using Nickel-nitriloacetic acid (Nickel-NTA) according to the manafacturer’s instructions (Qiagen). LacI, Cdc3p, Cdc11p, and Pol30p were cloned into a variant of pGEX-2T using PCR and primers containing appropriate restriction sites. GST-LacI, GST-Cdc3p, GST-Cdc11p, and GST-Pol30p were expressed in E. coli and purified using GSH-agarose according to the manafacturer’s instructions (GE Healthcare). Small-scale in vitro sumoylation reactions were performed in a total reaction volume of 20 μL and contained 50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 5 mM ATP, 0.5 μg of His6-Uba2 / His6-Aos1p, 1.25 μg of His6-Ubcp9, 2.5 μg of wild-type His6-Smt3p or mutant His6-Smt3p, and 1 μg of GST-tagged substrate, where indicated. These reactions were incubated at 30°C for different periods of time, stopped by the addition of 20 μL of 2X Laemmli buffer, and analyzed by immunoblotting with α-GST antisera (Santa Cruz Biotechnology). Large-scale reactions were performed in 200 μL total volume with all reaction components scaled up accordingly. Large-scale reactions were incubated 12-18 hours at 30°C and stopped by the addition of 10 μL of 0.5M EDTA. The GST-tagged substrate was purified from this reaction using GSH-agarose, digested with the appropriate proteases, and analyzed by mass spectrometry as described below.
Reconstitution of SUMO Conjugation Pathway in E. coli
PCR using primers containing the appropriate restriction sites was used to clone the coding sequences for Uba2p and Aos1p into the pACYC-Duet expression vector (Novagen). UBA2 was cloned into the first multicloning site (MCS1) in-frame with the N-terminal His6 tag while AOS1 was cloned into the second multicloning site (MCS2) in-frame with the C-terminal S-tag. Similarly, UBC9 was cloned into MCS2 of pRSF-Duet in-frame with the C-terminal S-tag while the coding sequence for wild-type Smt3p, Smt3p-I96K, or Smt3p-I96R was cloned into the MCS1 site of the same vector and in-frame with the N-terminal His6 tag. pAYCY-Duet-Aos1p/Uba2p and pRSF-Duet-Ubc9p/Smt3p(wild-type or mutant) were then transformed into BL21(DE3) with a pGEX plasmid that expressed the substrate of interest. A BL21(DE3) carrying all three plasmids was grown to an OD590 ∼ 0.5, induced by the addition of IPTG to a final concentration of 100 μg/mL, and incubated for 12-18 hours at 30°C before harvesting. The GST-tagged was subsequently purified using GSH-agarose (GE Healthcare) according to the manafacturer’s instructions, digested using the appropriate proteolytic digestion strategy, and then analyzed by mass spectrometry.
Purification of Smt3p-conjugates from S. cerevisiae
Smt3p-conjugates were purified from either the previously described yeast strain expressing HisFlag-tagged wild-type Smt3p from a galactose-inducible promoter (GAL10) or derivative strains expressing mutant versions of Smt3p in the same manner 13, 19. Details for the purifications have been described elsewhere 10, 13.
Proteolytic Digestion of Samples
Lys-C and trypsin digestions were performed as previously described 21. Arg-C digestions were performed according to the manafacturer’s instruction at a enzyme:substrate ratio of 1:20 (Roche). Sequential digestions using Lys-C and either Asp-N or Glu-C were performed by digesting the sample in buffer containing 8M Urea and 100 mM Tris-HCl, pH8.5, at an enzyme:substrate ratio of 1:20 for 4 hours at 37°C, diluting the digestion to a final concentration of 2M Urea using 100 mM Tris-HCl, pH 8.5, adding Asp-N or Glu-C to an enzyme:substrate ratio of 1:20 and then incubating for four additional hours at 37°C. Digests were acidified by the addition of formic acid to a final concentration of 5% before analysis by mass spectrometry.
Identification of Smt3p-modification sites using Mass Spectrometry
Peptide digests were analyzed by Multidimensional Protein Identification Technology (MudPIT) using a Thermofinnigan LTQ ion trap mass spectrometer as previously described 10, 22, 23. Data collection for the analysis of sumoylated Pol30p was performed using both data-dependent and data-independent directed MS/MS acquisition strategies. Data-dependent analysis consisted of one full MS scan (m/z range = 400-2000) followed by MS/MS scans of the eight most abundant precursor ions from the full MS scan. The directed MS/MS approach was described in Flick et al. and involved sequentially and cyclically collecting MS/MS from 3 m/z windows centered on the following m/z values: 1910.3, 1273.9, 1724.8, 1150.2, 1317.6, 878.7, 1132.1, 755, and 1087 24. These m/z values correspond to the +2 and +3 ions of LMDIDADFLK(+484)IEELQYDSTLSLPSSEFSK, the +2 and +3 ions of LMDIDADFLK(+114)IEELQYDSTLSLPSSEFSK, the +2 and +3 ions of DLSQLSDSINIMITK(+484)ETIK, the +2 and +3 ions of DLSQLSDSINIMITK(+114)ETIK, and the +2 ion of IEELQYDSTLSLPSSEFSK, respectively. Database searching was performed using the SEQUEST algorithm (version 2.7) and considered the appropriate differential modification masses 25. All database searches were performed using a database consisting of all yeast open reading frames downloaded from the Saccharomyces Genome Database on March 26, 2005. Peptide identifications were filtered using DTASelect 25-27.
Results and Discussion
Mass spectrometry-based approaches for identifying the site of UbL conjugation to a target protein are based on monitoring a diagnostic mass signature corresponding to a specific tag of several amino acids that remains covalently attached to the target lysine after digestion of the modified protein by trypsin. In the case of ubiquitin, for example, digestion of an ubiquitinated protein by trypsin results in a diglycine (-GG) tag from the C-terminus of ubiquitin remaining covalently linked to the target lysine residue of the tryptic peptide spanning the modification site (Figure 1)28, 29. The presence of this diglycine tag on the modified lysine residue leads to a +114 Da increase in the mass of the peptide that can be readily monitored using mass spectrometry. The fragmentation spectra of these modified peptides can be identified using database search algorithms that consider the mass of the modified lysine residue.
Figure 1. Schematic for identification of attachment Sites for ubiquitin and Smt3p.

Digestion of a ubiquitinated protein with trypsin leaves a diagnostic diglycine tag with a mass of 114 Da attached to the modified lysine and that can be monitored by mass spectrometry. Digestion of a protein modified by Smt3p with trypsin leaves a sequence tag of EQIGG which corresponds to a mass increase of 484 Da.
Although this strategy can also be applied to the identification of SUMO-attachment sites, the different amino acid sequence at the C-terminus of SUMO can complicate this approach. For example, in the case of Smt3p in Saccharomyces cerevisiae, digestion of a sumoylated protein with trypsin does not result in a diglycine tag as was the case for ubiquitin. Instead, a longer amino acid sequence (-EQIGG) remains attached to the modified lysine after digestion (Figure 1). This longer amino acid tag results in a mass shift of +484 Da and has been successfully used in several studies to identify the sites of modification in a sumoylated protein 3, 16.
During our previous work analyzing SUMO-conjugates affinity purified from budding yeast, we observed that the identification of the Smt3p attachment sites using the diagnostic +484 Da mass shift present after tryptic digestion was inefficient and resulted in the successful identification of very few sites compared to analogous studies we have performed identifying ubiquitination sites in mixtures of ubiquitin-conjugated proteins. We hypothesized two primary reasons to account for this inefficiency: (1) the longer sequence tag on the modified peptides interfered with their efficient identification and (2) the stoichiometry of sumoylation is often very low (<1%) so that sumoylated proteins are present only in very low quantities in our samples. In this work, we describe the results of several approaches focused on addressing these problems in order to create a platform for the efficient identification of Smt3p-attachment sites.
C-terminal Smt3p Mutants
In order to address the problem of having a long amino acid sequence tag, we generated a series of C-terminal Smt3p mutants that introduced either a tryptic cleavage site or a FactorXa site closer to the C-terminus of Smt3p. The mutants that were constructed and the diagnostic mass signature left after protease digestion are shown in Table 1. These mutants were then examined in several different experimental systems in order to assess whether or not they improved our ability to identify Smt3p attachments compared to wild-type Smt3p.
Table 1.
Smt3p C-terminal Mutants
| Smt3p Mutants | Smt3p after cleavage | Δ Mass |
|---|---|---|
| WT | REQIGG-K → EQIGG-K | +484 |
| I96K | REQKGG-K → GG-K | +114 |
| I96R | REQRGG-K → GG-K | +114 |
| G97K | REQIKG-K → G-K | +57 |
| G97R | REQIRG-K → G-K | +57 |
| Factor Xa | IEGRGG-K → GG-K | +114 |
In Vitro Sumoylation System
We attempted to resolve the problem of having extremely low stoichiometries of sumoylated proteins in our sample by establishing an in vitro system that could generate a large amount of any given sumoylated substrate. In addition to being able to produce large amount of recombinant sumoylated protein, this in vitro system must also be able to transfer the different Smt3p mutants we constructed to substrates and must retain the specificity of in vivo sumoylation events (i.e. acceptor lysine residues that are modified by Smt3p in vivo should also be modified in the in vitro reaction). Although in vitro sumoylation systems have been described in past work, the ability of those systems to meet these latter two requirements has never been extensively examined 30, 31.
To establish a system for in vitro sumoylation, we first expressed and purified from E. coli recombinant versions of the proteins required for SUMO conjugation: His6-Smt3p, His6-Uba2p, His6-Aos1p, and His6-Ubc9p. A reaction containing these proteins as well as ATP was capable of promoting the time-dependent formation of poly-Smt3p chains during incubation at 30°C suggesting that these recombinant proteins possess Smt3p-conjugating activity in vitro (Fig. 2A, lanes 5-7). Importantly, control reactions lacking either Smt3p, the heterodimeric Smt3p-activating enzyme Uba2p/Aos2, the Smt3p-conjugating enzyme Ubc9p, or ATP did not display Smt3p conjugation activity (Fig 2A, lanes 1-4).
Figure 2. Establishment of an in vitro sumoylation system.



(A) An in vitro reaction containing the Smtp3-conjugation machinery is able to catalyze the formation of poly-Smt3p chains. Reaction mixtures containing the indicated components were incubated at 30°C for the indicated times and then analyzed by SDS-PAGE followed by immunoblotting with α-Smt3p antisera to monitor the time-dependent formation of poly-Smt3p chains.
(B) Wild-type Smt3p, Smt3p-I96K, and Smt3p-I96R were effectively incorporated into poly-Smt3p chains in vitro by the Smt3p conjugation machinery. In vitro reactions containing the Smt3p-conjugation machinery and different Smt3p mutant proteins were assayed by immunoblotting for the ability of each mutant to form poly-Smt3p chains in a time-dependent fashion.
(C) Wild-type Smt3p, Smt3p-I96K, and Smt3p-I96R were conjugated to GST-Cdc11p by the Smt3p conjugation machinery. SDS-PAGE followed by immunoblotting with α-GST antisera was performed to examine the ability of different Smt3p mutant proteins to modify the model substrate, GST-Cdc11p, in an in vitro reaction containing the Smt3p-conjugation machinery, the indicated Smt3p mutant proteins, and GST-Cdc11p.
(D) Smt3p is efficiently conjugated in vitro to known substrates. GST-Cdc3p and GST-Pol30p, but not to control proteins, GST or GST-LacI. In vitro Smt3p-conjugation reactions were performed to test the ability of Smt3p to modify GST, GST-LacI, GST-Cdc3p, and GST-Pol30p. SDS-PAGE followed by immunoblotting with α-GST antisera was used to measure the incorporation of Smt3p into the different substrates.
(E) Sumoylation of GST-Pol30p in E. coli strains expressing the Smt3p conjugation machinery in conjunction with either wild-type Smt3p or Smt3p-I96K. Protein extracts were made from an E. coli strain co-expressing GST-Pol30p with His6-Uba2p, S-tagged-Aos1p, S-tagged-Ubc9p, and either His6-Smt3p-WT or His6-Smt3p-I96K as well as a control strain expressing only GST-Pol30p. Samples were analyzed by SDS-PAGE and immunoblotting with α-GST antisera to monitor the ability of Smt3p-WT and Smt3p-I96K to be conjugated to GST-Pol30p during expression in E. coli.
To test the ability of this in vitro sumoylation reaction to utilize the different Smt3p mutants described in Table 1, we next expressed and purified His6-tagged versions of these mutant proteins and assayed their ability to form poly-Smt3p chains in vitro. As shown in Fig 2B, Smt3p-WT, Smt3p-I96K, and Smt3p-I96R were efficiently converted into poly-Smt3p chains while Smt3p-G97K, Smt3p-G97R, and Smt3p-FacX were not effectively utilized in this assay.
In addition to testing whether or not these Smt3p mutants could be incorporated into poly-Smt3p chains, we also wanted to assess their ability to be conjugated to bona fide Smt3p targets. To this end, we expressed and purified GST-Cdc11 from E. coli. Cdc11 is a septin involved in bud growth and cytokinesis during the budding yeast cell cycle and also a well-established target of sumoylation 13. When Cdc11p was added to the in vitro Smt3p reaction, we found that wild-type Smt3p as well as Smt3p-I96K and Smt3p-I96R could be conjugated to Cdc11 while the other mutants failed to do so (Figure 2C). These results confirm that the ability of these mutants to form poly-Smt3p chains as described in Fig. 2B is a good indication of functionality and led us to conclude that Smt3p-I96R and Smt3p-I96K are capable of substituting for wild-type Smt3p in in vitro trans-conjugation reactions while the other mutants cannot be utilized by the Smt3p conjugation machinery.
To further establish the specificity of our in vitro sumoylation system, we examined its ability to sumoylate two other known in vivo substrates of sumoylation, GST-Cdc3p and GST-Pol30p, as well as two negative control proteins, GST and GST-LacI, that would not be expected to be sumoylated 13, 14, 32. Figure 2D shows that Smt3p is actively conjugated to GST-Cdc3p and GST-Pol30p but not to GST and GST-LacI.
Together, the results of these experiments demonstrate that our in vitro Smt3p conjugation system is capable of incorporating wild-type Smt3p as well as the Smtp3-I96K and Smt3p-I96R mutants into several different physiological targets of sumoylation (Cdc11p, Cdc3p, Pol30p) in a specific manner. This ability to generate significant quantities of sumoylated protein overcomes one of the major difficulties encountered in identifying the sites of Smtp3-conjugation in different cellular targets as outlined previously.
In contrast to the ubiquitination machinery, it is important to note that the in vitro conjugation of Smt3p to substrates does not require the presence of SUMO E3 ligases 33, 34. Previous work on the SUMO conjugation machinery that included high-resolution structural studies concluded that the SUMO acceptor site (I/V/L-K-X-D/E) was recognized directly by Ubc9p and the role of SUMO E3 ligases in the process was to increase the effective concentration of the substrate relative to Ubc9p 35, 36. In the in vitro system described here where both the SUMO conjugation machinery and the substrate are present at high concentrations, the addition of yeast SUMO ligases (Siz1p, Siz2p, and Mms21p) did not significantly increase the efficiency of the reaction (data not shown).
One drawback of our in vitro sumoylation system is that the cloning, expression, and purification of each component of the SUMO conjugation machinery including the different mutants as well as the substrates of interest is laborious and time-consuming. As a rapid alternative approach, we also attempted to reconstitute the pathway for yeast Smt3p conjugation in E. coli. This approach is based primarily on a recently published strategy for producing large amounts of sumoylated thymine DNA glycosylase by coexpressing it in E. coli with the rest of the SUMO conjugation machinery 37, 38. We performed the analogous experiment using the pDUET bacterial expression system (Novagen) which facilitates the co-expression of up to eight different proteins using four different plasmids that each contain a unique selection marker and origin of replication as well as two separate T7-based expression cassettes. This system allowed us to simultaneously express Uba2p/Aos1p, Ubc9, wild-type or mutant versions of Smt3p, and a GST or MBP tagged substrate of interest in E. coli. By co-expression of the Smt3p conjugation machinery with the substrate in E. coli, we found that a significant amount of Smt3p could be conjugated to the substrate of interest, which could then be purified directly from bacterial extracts. Large amounts of recombinant protein conjugated to either wild-type Smtp3 or mutant Smt3p can be generated very quickly using this approach and only one protein has to be purified compared to the large number of components that must be purified separately for the in vitro conjugation system. Our implementation of this strategy which utilizes the pDUET expression system potentially offers greater flexibility than the other published approaches using this method in that the expression of additional components such as SUMO E3 ligases can be easily incorporated into the system.
Identification of Smt3p Attachment Sites in Substrates Sumoylated In Vitro
After the establishment of a system for the production of large quantities of sumoylated protein, we next attempted to compare the efficiency with which we could identify the sites of Smt3p modification using either wild-type Smt3p or the Smt3p mutants, I96K and I96R. Recombinant GST-Pol30p modified by either Smt3p-WT or Smt3p-I96K was digested with trypsin and analyzed by LC-MS/MS. Since previous studies had identified K127 and K164 as the two in vivo sites of SUMO modification for Pol30p, we repeatedly collected MS/MS spectra corresponding to the predicted m/z ratio of +2 and +3 versions of the modified tryptic peptides containing each of these sites 14. As described previously in Table 1, modified peptides were predicted to have the mass of the unmodified peptide +484 Da when conjugated to wild-type Smt3p and +114 Da when conjugated to Smt3p-I96K. Spectra collected from these experiments were then analyzed using the SEQUEST algorithm and considered the appropriate mass shifts. The number of spectra identified for each peptide is shown for the K127 site in figure 3A and for the K164 site in figure 3B. In both cases, significantly more spectra were identified when using the Smt3p-I96K mutant than when using wild-type Smt3p. Importantly, the ability of the I96K mutant to increase the efficiency with which these modified peptides can be identified cannot be attributed to differences in the relative quantity of Pol30p-Smt3p-WT and Pol30p-Smt3p-I96K since both immunoblotting (Fig. 2E) and the comparison of spectral abundance for unmodified peptides (Fig. 3C) show the amount of modified Pol30p to be approximately equal between the two samples. We also observed similar results when these two samples were analyzed using data-dependent acquisition strategy instead of the directed MS/MS strategy described above.
Figure 3. Comparison of mass spectrometry-based identification of Smt3p-attachment sites in Pol30p when conjugated to either wild-type Smt3p or Smt3p-I96K.



(A-C) The identification of peptides (+2 and +3 charge states) spanning the (A) K127 and (B) K164 sumoylation sites of GST-Pol30p is more efficient for peptides modified by Smt3p-I96K (+114 Da) than those modified by wild-type Smt3p (+484 Da) while (C) unmodified peptides are identified equally well. GST-Pol30p sumoylated by either Smt3p-WT or Smt3p-I96K was digested by trypsin and analyzed using the directed MS/MS strategy as described in the Experimental Procedures. Spectra were identified using SEQUEST considering appropriate masses for the modified lysine. The number of peptide identifications shown is the average of three independent experiments.
(D) Representative spectra of the sumoylated peptides spanning the K127 site of Pol30p and containing either the -EQIGG tag from Smt3p-WT (left) or the -GG from Smt3p-I96K (right).
(E) Normalized intensities of Pol30p-K127 sumoylated peptides and unmodified control peptides derived from GST-Pol30p conjugated to either Smt3p-WT or Smt3p-I96K. Intensity values are the average of three independent experiments.
(F) Graph showing the ratio of +2 to +3 spectra for sumoylated peptides spanning the K127 site of GST-Pol30 conjugated to either Smt3p-WT or Smt3p-I96K.
After establishing that using the Smt3p-I96K mutant significantly increased our ability to identify Smt3p modification sites, we then focused on trying to understand the basis for this improvement. We first examined the MS/MS spectra of the Pol30p-K127 site modified with wild-type Smt3p or Smtp3-I96K to determine if differences in fragmentation of the two peptides could account for the difference in identification efficiency. Representative spectra from these two peptides are shown in Figure 3D and show no dramatic changes in the fragmentation pattern. Additionally, average Xcorr and ΔCN values were unchanged between the two peptides suggesting that the SEQUEST algorithm could identify both spectra equally well (Supplementary Data). Together, these data suggest that differences in fragmentation do not account for the increased ability to identify Smt3-I96K modified peptides.
We next compared the intensity of the precursor ions for the modified peptides to determine whether intensity differences could potentially explain the improvements in sumoylated peptide identification. Figure 3E shows the normalized intensities of the precursor ions for both the +2 and +3 charge states of the wild-type and I96K modified Pol30p-K127 peptides as well as two other unmodified peptides from Pol30p. We find that the Smt3p-I96K modified peptide for K127 show a 5-10 fold increase in the intensity of the precursor ion compared to the wild-type modified peptide while both unmodified peptides display no significant differences in intensity. This large increase in the intensity of the precursor ions containing the smaller mass signature of the Smt3p mutants is one likely reason behind the increased identification efficiency of the Smt3p-I96K modified peptides.
Although the physical basis for the increased intensity of the mutant-derived precursor ions is not clear, we believe that two possibilities are likely. One possibility is that larger mass signature of the wild-type peptides (-EQIGG) undergoes a higher degree of in-source fragmentation in the mass spectrometer compared to the small mass signature of the mutant peptide (-GG) so that the intensity of the wild-type peptide is distributed over a series of precursor ions corresponding to in-source fragments thus effectively lowering the intensity of the primary precursor ion from which the modified spectra were obtained. A second possibility is that is the smaller size of the mutant-derived peptides shifts the charge envelopes of those peptides down to lower charge states relative to the wild-type peptides. Since most peptide identification algorithms only consider charges states of +3 or less when analyzing MS/MS spectra, a shift in the charge envelope of the mutant-derived peptides to lower charge states would increase the intensity of the +2 and +3 precursor ions from which our peptide identifications are obtained and thereby increase our ability to identify the modified peptide. Evidence supporting the idea that the charge envelope of the Smt3p-I96p modified peptides has been shifted relative to Smt3p-WT modified peptides is shown in Figure 3F. The data in this figure clearly indicate that the ratio of +2 to +3 spectra is much higher for the mutant-derived peptide identifications compared to the wild-type peptide identifications and is consistent with a shift to lower charge states.
Although in-source fragmentation and a shift to a lower charge states represent potential possibilities as to why the Smt3p-I96 mutant peptides are more readily identified than the wild-type peptides, it is important to note that these explanations are largely unsubstantiated and thus speculative in nature and future work characterizing the biophysical properties and fragmentation patterns of these branched-chain peptides is essential to gain a more complete understanding of this process.
Digestion Strategies for Identification of Smt3p Attachment Sites
After establishing the ability of the Smt3p I96K and I96R mutants to improve the identification of modification sites, we next explored the possibility of coupling the use of these mutants to different digestion strategies to (1) enable mapping of Smt3p attachment sites difficult to identify using traditional mass spectrometric approaches and (2) generate SUMO-specific diagnostic mass signatures that can be differentiated from other UbLs.
One example of a SUMO attachment site difficult to identify using mass spectrometry is K412 found near the carboxy terminus of the budding yeast septin Cdc11. As shown in Figure 4A, this region is lysine-rich and the modified peptide predicted to arise from tryptic digestion is a small and unusual branched-chain peptide. The fact that the size of the branch corresponding to the diagnostic wild-type SUMO fragment is nearly as large as the portion of the peptide generating the sequence-specific fragment ions would likely make it difficult to identify using a differential modification search of +484 Da on the modified lysine. Not surprisingly, when we purified large amounts of GST-Cdc11p modified with wild-type Smt3p using our in vitro sumoylation system, digested it with trypsin, and then analyzed it by LC-MS/MS, we were unable to identify any spectra corresponding to this modified peptide. Alternatively, if we were to purify GST-Cdc11p modified with Smt3p-I96R and then digest the modified protein with Arg-C, then a peptide predicted to be more amenable to mass spectrometry and database searching would be generated. We confirmed this prediction by identifying 40 spectra corresponding to this modified peptide from a LC-MS/MS analysis of the sample. This example clearly illustrates how utilizing different combinations of Smt3p mutants and protease digestion strategies can facilitate the identification of Smt3p attachment sites in regions of the protein that might otherwise be difficult to analyze.
Figure 4. Identification of sumoylated peptides using different proteolytic digestion strategies.


(A) Identification of Cdc11p modification site using Smt3p-I96R and Arg-C digestion. GST-Cdc11p modified with either Smt3p-WT or Smt3p-I96R was digested with trypsin or Arg-C, respectively. Peptide digests were then analyzed by MudPIT and spectra corresponding to the peptide spanning the known modification site and containing the appropriate mass tag were identified.
(B) Schematic for digestion strategies that allow discrimination between lysine residues modified by ubiquitin and Smt3p-I96K.
(C) Identification of the K127 SUMO attachment site Pol30p after either Lys-C or Lys-C / Asp-N digestion of Pol30p modified by Smt3p-I96K. GST-Pol30p modified by Smt3p-I96K was digested using the indicated enzymes, analyzed by MudPIT, and peptides corresponding to the site of modification were identified using the SEQUEST algorithm.
(D) Sequence alignment of human ubiquitin and ubiquitin-like modifiers.
One potential disadvantage of using the Smt3p-I96K/R mutants for the identification of modification sites is when the sample being analyzed also contains ubiquitylated proteins. As diagrammed in Figure 4B, the digestion of a protein modified with either ubiquitin or a Smt3p-I96K/R mutant leaves an identical diglycine tag (+114 Da) on the modified lysine after digestion with trypsin making these modified peptides indistinguishable. To circumvent this problem, we have developed a strategy for generating a SUMO-specific mass signature that can be distinguished from ubiquitin by modifying proteins with the Smt3p-I96K and digesting them with either Lys-C or sequentially with Lys-C and a second enzyme such as Glu-C or Asp-N (Figure 4B). In this case, digestion of the modified protein by Lys-C will generate a +114 Da fragment that is specific for SUMO-modified peptides but not for ubiquitinated peptides. Since LysC-only digests often generate peptides that are too large to be easily identified by MS/MS, a second enzyme such as Glu-C or Asp-N can also be added to generate smaller peptides that still retain the diglycine branch. To test this strategy, we examined our ability to identify the K127 SUMO attachment site in Pol30p after its modification by either wild-type Smt3p or Smt3p-I96K and digestion with either Lys-C alone or Lys-C followed by Asp-N. As shown in Figure 4C, we identified six and five spectra corresponding to the predicted SUMO-modified peptides generated by Lys-C digestion (LMDIDADFLK(+114)IEELQYDSTLSLPSSEFSK) and Lys-C / Asp-N digestion (DFLK(+114)IEELQY), respectively. These data clearly demonstrate our ability to identify sumoylated peptides when using alternate digestion strategies aimed at generating SUMO-specific mass tags. To further confirm the specificity of this protease digestion strategy, we also attempted to analyze ubiquitinated Pol30p, but were unable to obtain enough of the modified protein for the study. Nonetheless, our preliminary results attest to the feasibility of this strategy for differentiating between ubiquitinated and sumoylated peptide.
An important point worth highlighting regarding the strategy of combining a set of SUMO mutants with strategies utilizing different proteases is its ability to be a generalized approach for mapping modification sites of other Ubls. This potential can be readily recognized when examining a sequence alignment of the C-termini of several ubiquitin and ubiquitin-like modifiers from humans (Figure 4D). For example, the tryptic digestion of a protein modified by ubiquitin, Nedd8, or ISG15 would result in the same 114 Da diglycine tag being present on the modified peptide and make it impossible to distinguish between these three modifiers. Mutation of the most C-terminal arginine to lysine for one of these modifiers, however, would enable mapping of the modification site using a lys-C based digestion strategy as described above. Human SUMO1 or FAT10 have no tryptic cleavage site in their C-terminus leaving no straightforward method for identifying their attachment sites. Introduction of a tryptic or other protease cleavage site into this C-terminus would enable a mass signature diagnostic of those modifiers to be generated upon digestion with the appropriate enzymes. Indeed, Gocke et al. recently showed that the introduction of a tryptic cleavage site into the C-terminus of SUMO1 permitted the mapping of a SUMO-1 modification site in RanGAP1 18.
Identification of in vivo Smt3p Modification Sites
In addition to their use in in vitro assays, we also tested whether the Smt3p mutants we created could be used to map Smt3p attachment sites in vivo. First, we analyzed the ability of the Smt3p mutants to be incorporated into cellular substrates in vivo. A His6-Flag-tagged version of each mutant on a centromeric plasmid and under the control of a galactose-inducible promoter was expressed in yeast and protein extracts made from these mutant strains were immunoblotted with α-Smt3p antiserum to assess their incorporation into substrates (Figure 5A). Wildtype Smt3p as well as Smt3p-I96K and Smt3p-I96R were efficiently attached to cellular targets while Smt3p-FacX was weakly incorporated and Smt3p-G97K and Smt3p-G97R were unable to modify proteins to any significant extent. Importantly, the pattern of conjugates between the I96K/R mutants and wild-type appear to be quite similar suggesting that the same spectrum of targets are modified. These results are consistent with our in vitro analysis in which only Smt3p-I96K and Smt3p-I96R are efficiently utilized by the SUMO conjugation machinery. These data are also supported by complementation studies in budding yeast where only Smt3p-I96K and Smt3p-I96R could complement a deletion of the SMT3 gene which is essential for viability (data not shown).
Figure 5. Identification of Smt3p attachment sites in vivo using C-terminal Smt3p mutants.

(A) Incorporation of Smt3p-WT, Smt3p-I96K, and Smt3p-I96R into Smt3p-conjugates in vivo. Protein extracts from yeast strains expressing His6-Flag-tagged Smt3p or the indicated mutants were analyzed by SDS-PAGE followed by immunoblotting with α-Smt3p antisera to assess the ability of each Smt3p mutant to be conjugated to in vivo substrates.
(B) Smt3p-WT and Smt3p-I96R are efficiently conjugated to Cdc11p in vivo. His6-Flag-tagged versions of Smt3p-WT or Smt3p-I96R were expressed via a plasmid in a yeast strain expressing TAP-tagged Cdc11p. Smt3p-conjugates were purified using Ni-NTA chromatography and then analyzed by immunoblotting and HRP-conjugated peroxidase-anti-peroxidase (PAP) to detect the presence of TAP-Cdc11p in the mixture of Smt3p-modified proteins.
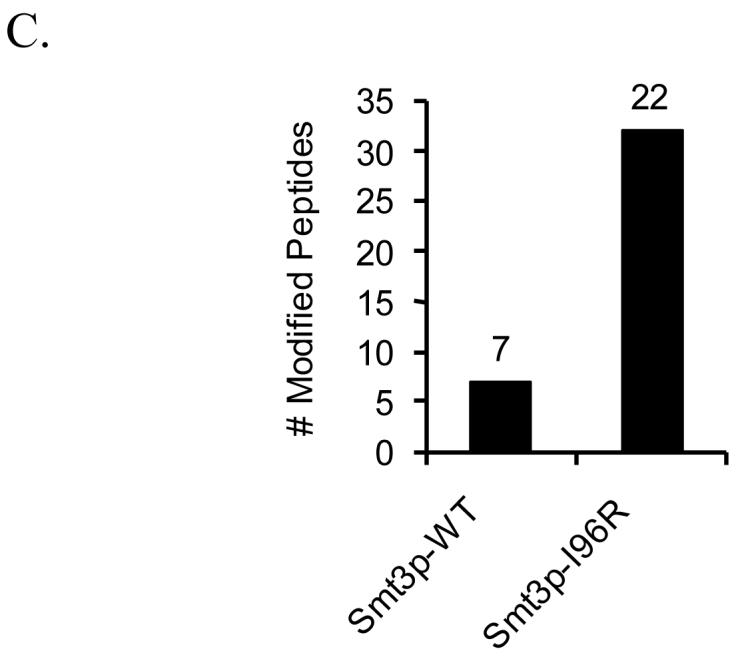
(C) Improved identification of Smt3p-attachment sites in vivo using Smt3p-I96R. Smt3p conjugates were purified from yeast strains containing a chromosomal deletion of the SMT3 locus and expressing His6-Flag-tagged versions of either Smt3p-WT or Smt3p-I96R. Purified samples were analyzed by MudPIT and the number of modified peptides identified from each mixture is plotted here.
Although the pattern of conjugates between wild-type Smt3p and the Smt3p-I96K/R mutants looked similar, we also checked to make sure that these mutants could be effectively incorporated into a known substrate. This was tested by expressing HisFlag-Smt3p-WT and HisFlag-Smt3p-I96R in a yeast strain expressing a TAP-tagged version of Cdc11p. Smt3p-conjugates were purified from these two strains using Ni-NTA chromatography and assayed for the presence of Cdc11p in the pool of Smt3p-conjugated proteins. Figure 5B shows that both wild-type Smt3p and Smt3p-I96R were efficiently conjugated to Cdc11p under these conditions.
Since the Smt3p-I96R mutant could be effectively incorporated into cellular targets of sumoylation, we next attempted to analyze the pool of Smt3p-I96R conjugates using a shotgun proteomics approach (MudPIT) in order to identify Smt3p-modified peptides from the mixture. Briefly, Smt3p-conjugates were purified using Ni-NTA chromatography from strains expressing either wild-type Smt3p or Smt3p-I96R, digested with trypsin, and analyzed by MudPIT. Modified peptides were identified using database search strategies that considered +484 Da for peptides modified by wildtype Smt3p and +114 Da for Smt3p-I96R. The results are shown in Figure 5C where 22 modified peptides were found in the Smt3p-I96R sample while only 7 modified peptides were identified from the wild-type Smt3p sample. A complete listing of these peptides can be found in the supplemental data. It is important to note that several of the sumoylation sites identified in the wild-type sample (-EQIGG) were not identified in the mutant sample (-GG). Although we believe that this likely results from incomplete sampling of these low abundance branched chain peptides in the two samples (a common feature in shotgun proteomic experiments), it could also reflect other, as of yet not understood, differences in the biophysical behavior of these two classes of peptides. Nonetheless, this improved ability to identify Smt3p-I96R-modified peptides compared to wild-type Smt3p-modified peptides is consistent with our in vitro results and suggests that mutant peptides are working in an analogous fashion in vivo. Importantly, this difference cannot be attributed to higher amounts of Smt3p-conjugates in the Smt3p-I96R sample since more unmodified peptides were identified in the wild-type Smt3p sample than in the mutant Smt3p sample (7463 peptides compared to 6370 peptides).
As described earlier, distinguishing sumoylation sites from ubiquitination sites can be difficult when using the Smt3p-I96R mutant since the diagnostic mass signature of 114 Da is identical for SUMO and ubiquitin modified peptides. Although we cannot rule out the possibility that some of the SUMO-modified peptides from the Smt3p-I96R sample are actually ubiquitin-modified peptides, we have taken a number of steps to minimize this possibility. First, the purification specifically enriches for sumoylated proteins so the vast majority of peptides containing this mass signature are likely to come from bona fide SUMO targets and not trace amounts of co-purifying ubiquitinated factors. Second, we only considered a peptide to be sumoylated if it was derived from the list of 271 proteins we have previously shown to be SUMO substrates 10. Finally, those modified peptides bearing the +114 Da mass tag and found in both the wild-type and I96R purifications were likely to be ubiquitinated and filtered out of the final list of sumoylated peptides. Strong evidence that these filtering strategies were successful includes the observation that most of the remaining sumoylated peptides were either previously identified as sites of sumoylation (Pol30p, Cdc3p, Shs1p) or conformed to the known consensus sequence for SUMO attachment.
It is important to note that we also tried to map SUMO modification sites in vivo using the Smt3p-I96K mutant and the LysC-based digestion strategies described earlier. These experiments were unsuccessful, however, due primarily to difficulties in purifying sufficient amounts of Smt3p-I96K conjugated proteins as a result of the lower degree of incorporation of the Smt3p-I96K mutant compared to either wild-type Smtp3 or Smt3p-I96R. As such, future work focusing on improving the conjugation efficiency of this mutant and scaling-up the biochemical purification of SUMO conjugates will be required to improve the utility of this mutant in identifying in vivo sumoylation sites.
Supplementary Material
Acknowledgements
This work was supported by an American Cancer Society Postdoctoral Fellowship to JAW and NIH grants RR11823 (JRY), ES012021 (JRY), GM038328 (SIR) and GM62268 (ESJ).
References
- 1.Melchior F. SUMO--nonclassical ubiquitin. Annu Rev Cell Dev Biol. 2000;16:591–626. doi: 10.1146/annurev.cellbio.16.1.591. [DOI] [PubMed] [Google Scholar]
- 2.Johnson ES. Protein modification by sumo. Annu Rev Biochem. 2004;73:355–82. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 3.Denison C, Rudner AD, Gerber SA, Bakalarski CE, Moazed D, Gygi SP. A proteomic strategy for gaining insights into protein sumoylation in yeast. Mol Cell Proteomics. 2005;4(3):246–54. doi: 10.1074/mcp.M400154-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Gocke CB, Yu H, Kang J. Systematic identification and analysis of mammalian small ubiquitin-like modifier substrates. J Biol Chem. 2005;280(6):5004–12. doi: 10.1074/jbc.M411718200. [DOI] [PubMed] [Google Scholar]
- 5.Hannich JT, Lewis A, Kroetz MB, Li SJ, Heide H, Emili A, Hochstrasser M. Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J Biol Chem. 2005;280(6):4102–10. doi: 10.1074/jbc.M413209200. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Evdokimov E, Shen RF, Chao CC, Tekle E, Wang T, Stadtman ER, Yang DC, Chock PB. Sumoylation of heterogeneous nuclear ribonucleoproteins, zinc finger proteins, and nuclear pore complex proteins: a proteomic analysis. Proc Natl Acad Sci U S A. 2004;101(23):8551–6. doi: 10.1073/pnas.0402889101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panse VG, Hardeland U, Werner T, Kuster B, Hurt E. A proteome-wide approach identifies sumolyated substrate proteins in yeast. J Biol Chem. 2004 doi: 10.1074/jbc.M407950200. [DOI] [PubMed] [Google Scholar]
- 8.Rosas-Acosta G, Russell WK, Deyrieux A, Russell DH, Wilson VG. A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol Cell Proteomics. 2005;4(1):56–72. doi: 10.1074/mcp.M400149-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vertegaal AC, Ogg SC, Jaffray E, Rodriguez MS, Hay RT, Andersen JS, Mann M, Lamond AI. A proteomic study of SUMO-2 target proteins. J Biol Chem. 2004;279(32):33791–8. doi: 10.1074/jbc.M404201200. [DOI] [PubMed] [Google Scholar]
- 10.Wohlschlegel JA, Johnson ES, Reed SI, Yates JR., 3rd Global analysis of protein sumoylation in Saccharomyces cerevisiae. J Biol Chem. 2004;279(44):45662–8. doi: 10.1074/jbc.M409203200. [DOI] [PubMed] [Google Scholar]
- 11.Wykoff DD, O’Shea EK. Identification of sumoylated proteins by systematic immunoprecipitation of the budding yeast proteome. Mol Cell Proteomics. 2005;4(1):73–83. doi: 10.1074/mcp.M400166-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Yu VP, Reed SI. Cks1 is dispensable for survival in Saccharomyces cerevisiae. Cell Cycle. 2004;3(11):1402–4. doi: 10.4161/cc.3.11.1208. [DOI] [PubMed] [Google Scholar]
- 13.Johnson ES, Blobel G. Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J Cell Biol. 1999;147(5):981–94. doi: 10.1083/jcb.147.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419(6903):135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 15.Cardone L, Hirayama J, Giordano F, Tamaru T, Palvimo JJ, Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science. 2005;309(5739):1390–4. doi: 10.1126/science.1110689. [DOI] [PubMed] [Google Scholar]
- 16.Zhou W, Ryan JJ, Zhou H. Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J Biol Chem. 2004;279(31):32262–8. doi: 10.1074/jbc.M404173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper HJ, Tatham MH, Jaffray E, Heath JK, Lam TT, Marshall AG, Hay RT. Fourier transform ion cyclotron resonance mass spectrometry for the analysis of small ubiquitin-like modifier (SUMO) modification: identification of lysines in RanBP2 and SUMO targeted for modification during the E3 autoSUMOylation reaction. Anal Chem. 2005;77(19):6310–9. doi: 10.1021/ac058019d. [DOI] [PubMed] [Google Scholar]
- 18.Knuesel M, Cheung HT, Hamady M, Barthel KK, Liu X. A Method of Mapping Protein Sumoylation Sites by Mass Spectrometry Using a Modified Small Ubiquitin-like Modifier 1 (SUMO-1) and a Computational Program. Mol Cell Proteomics. 2005;4(10):1626–1636. doi: 10.1074/mcp.T500011-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. Embo J. 1997;16(18):5509–19. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson ES, Blobel G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J Biol Chem. 1997;272(43):26799–802. doi: 10.1074/jbc.272.43.26799. [DOI] [PubMed] [Google Scholar]
- 21.McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ, Yates JR., 3rd Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. International Journal of Mass Spectrometry. 2002;219:245–251. [Google Scholar]
- 22.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 23.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–90. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 24.Flick K, Ouni I, Wohlschlegel JA, Capati C, McDonald WH, Yates JR, Kaiser P. Proteolysis-independent regulation of the transcription factor Met4 by a single Lys 48-linked ubiquitin chain. Nat Cell Biol. 2004;6(7):634–41. doi: 10.1038/ncb1143. [DOI] [PubMed] [Google Scholar]
- 25.Eng J, McCormack A, Yates JR., 3rd An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 26.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2(1):43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 27.Tabb DL, McDonald WH, Yates JR., 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1(1):21–6. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng J, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom. 2001;36(10):1083–91. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 29.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21(8):921–6. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 30.Johnson ES, Gupta AA. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 2001;106(6):735–44. doi: 10.1016/s0092-8674(01)00491-3. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi Y, Kahyo T, Toh EA, Yasuda H, Kikuchi Y. Yeast Ull1/Siz1 is a novel SUMO1/Smt3 ligase for septin components and functions as an adaptor between conjugating enzyme and substrates. J Biol Chem. 2001;276(52):48973–7. doi: 10.1074/jbc.M109295200. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi Y, Iwase M, Konishi M, Tanaka M, Toh-e A, Kikuchi Y. Smt3, a SUMO-1 homolog, is conjugated to Cdc3, a component of septin rings at the mother-bud neck in budding yeast. Biochem Biophys Res Commun. 1999;259(3):582–7. doi: 10.1006/bbrc.1999.0821. [DOI] [PubMed] [Google Scholar]
- 33.Desterro JM, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J Biol Chem. 1999;274(15):10618–24. doi: 10.1074/jbc.274.15.10618. [DOI] [PubMed] [Google Scholar]
- 34.Okuma T, Honda R, Ichikawa G, Tsumagari N, Yasuda H. In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem Biophys Res Commun. 1999;254(3):693–8. doi: 10.1006/bbrc.1998.9995. [DOI] [PubMed] [Google Scholar]
- 35.Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108(3):345–56. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 36.Reverter D, Lima CD. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005;435(7042):687–92. doi: 10.1038/nature03588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uchimura Y, Nakamura M, Sugasawa K, Nakao M, Saitoh H. Overproduction of eukaryotic SUMO-1-and SUMO-2-conjugated proteins in Escherichia coli. Anal Biochem. 2004;331(1):204–6. doi: 10.1016/j.ab.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 38.Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature. 2005;435(7044):979–82. doi: 10.1038/nature03634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.