

Abstract
Unsymmetrical cyanine dyes are widely used in biomolecular detection due to their fluorogenic behavior, whereby fluorescence quantum yields can be very low in fluid solution but are significantly enhanced in conformationally restricted environments. Herein we describe a series of fluorinated analogues of the dye thiazole orange that exhibit improved fluorescence quantum yields and photostabilities. In addition, computational studies on these dyes revealed that twisting about the monomethine bridge beyond an interplanar angle of 60° leads to a dark state that decays nonradiatively to the ground state, accounting for the observed fluorogenic behavior. The effects of position and number of fluorine substituents correlates with both observed quantum yield and calculated activation energy for twisting beyond this critical angle.
Introduction
The cyanine class of organic dyes has been studied for over 150 years and continues to be the focus of considerable interest in chemistry, biology and biotechnology.1 Most research and applications have involved symmetrical cyanine dyes, where equal heterocycles are linked by a polymethine bridge. The widely used fluorescent labels Cy3 and Cy5 are examples of symmetrical cyanines, which typically exhibit large molar extinction coefficients (εmax > 105 M-1cm-1) and at least moderate fluorescence quantum yields (φf > 0.1). The development of efficient strategies for synthesizing these dyes in forms that allow facile conjugation to biomolecules2 has spurred their use in a range of applications, including cell microscopy, flow cytometry, “gene-chip” microarrays and single molecule spectroscopy.
The unsymmetrical cyanine dyes3 do not enjoy as long a history as their symmetrical counterparts, but they have been used extensively in sensing applications, particularly in detection of nucleic acids.4 This class of dyes features two different heterocycles linked by a mono- or polymethine bridge. For example, thiazole orange (TO, Chart 1) has benzothiazole and quinoline heterocycles connected by a monomethine group. Different heterocycles and bridge lengths allow tuning of the excitation and emission wavelengths of these dyes throughout the visible spectrum. A variety of substituents can be added to the two heterocyclic nitrogens, allowing further diversification of the dye structure.
Chart 1.

Fluorinated and nonfluorinated cyanine dyes. All dyes are iodide salts, except for S5-F8, for which the counterion is ethyl sulfate.
The most useful property of unsymmetrical cyanine dyes is the environmental sensitivity of their fluorescence quantum yields.5,6 In fluid solutions, these dyes typically exhibit φf < 0.001, i.e. more than 100-fold lower than their symmetrical counterparts. In contrast, viscous solvents such as glycerol promote substantial (>102 fold) enhancements in φf for unsymmetrical dyes. Thus, unsymmetrical cyanines can act as fluorogenic sensors that report on the local viscosity of their environment.
Thiazole orange and many closely related dyes are widely used as stains for nucleic acids, allowing detection of DNA and RNA in gels, flow cytometers or microscopes.4 Most of these dyes bind to nucleic acids by intercalating between base pairs6 (or presumably between individual bases in single stranded nucleic acids). Nucleobases are stacked on one or both faces of the dye in these complexes, effectively raising the viscosity of the dye's local environment and leading to large increases in fluorescence. The strong fluorescence of nucleic acid-bound dye and weak fluorescence of unbound dye result in effective staining for imaging and detection.
In addition to their uses as soluble intercalating probes, unsymmetrical cyanines have been conjugated to a variety of molecules, including peptides,7 proteins,8 DNA9 and DNA analogues such as peptide nucleic acid (PNA).10 In these contexts, the dyes often exhibit substantially greater fluorescence than the free dye.11 Nevertheless, binding of the dye-conjugated probe to a target molecule results in increased fluorescence, with detection sensitivity often extending into the nanomolar concentration range.
We are interested in using fluorogenic cyanine dyes as components in DNA- and RNA-based biosensors. The goal of the research described here was to synthesize and characterize a series of TO analogues that have electron-withdrawing fluoro or trifluoromethyl groups replacing hydrogen atoms on the heterocycles. Fluorination of a symmetrical cyanine dye was previously shown to improve the fluorescence properties of the dye12 and similar effects were expected for the unsymmetrical dyes. While this was observed in some of the dyes, interesting spectral shifts prompted us to perform calculations on the dyes. The calculations show that the shifts may be attributed to the effects of fluorine substitution on the frontier molecular orbitals. The calculations also explore the origin of the observed effects of the dyes' local environment on the quantum yield. Fluorescence quenching in TO has been attributed to intramolecular twisting in the excited state.13 This is consistent with the increase in fluorescence quantum yield observed in environments that rigidify the structure, such as in viscous solvents or upon binding to biomolecules. Ab initio calculations, performed on a small model cyanine, also implicate intramolecular twisting of the dye as the dominant quenching mechanism in unconstrained environments.14 Here, we use semiempirical methods to allow computations on the full dye molecules. These semiempirical calculations are consistent with the ab initio calculations, in that they also find that intramolecular twisting is an efficient quenching pathway. In addition, the semiempirical calculations allow us to study the effects of fluorine substitution on the energetics of this twisting pathway and help explain the observed substituent effects on the fluorescence quantum yield.
Results
Our previous work with symmetrical cyanine dyes such as S5 and S5-F8 (Chart 1) demonstrated that fluorination of the heterocycles can decrease aggregation and improve photostability,12 important parameters for fluorescence labels used in bulk imaging and single molecule measurements. A series of thiazole orange (TO) analogues bearing fluorine substituents on the benzothiazole heterocycle or a trifluoromethyl group on the quinoline heterocycle (Chart 1) was synthesized to investigate the effect of fluorination on unsymmetrical cyanines.
Spectroscopy
UV-vis absorption spectra were recorded for each dye in methanol as well as in the presence of a large excess of double-stranded DNA, where the dye is expected to be intercalated (Figure 1). The absorption maxima for all five dyes shift to the red upon binding to DNA, indicative of the lower dielectric environment of the stacked base pairs (Table 1). In addition, a distinct trend in the maxima is observed in both cases: fluorination of the benzothiazole leads to a progressive blue-shift of the absorption band, whereas addition of a trifluoromethyl group to the quinoline side of the dye causes a red-shift. A similar trend is observed in the emission data. (The low fluorescence of the dyes in methanol necessitated the use of 90% glycerol in water to observe fluorescence in the absence of DNA.)
Figure 1.

UV-vis absorption spectra of TO and fluorinated analogues determined in methanol (left) and in the presence of double-stranded DNA (right). Each spectrum was normalized at its wavelength of maximum absorbance.
Table 1.
Absorption and emission wavelengths (in nm) of thiazole orange (TO) and fluorinated analogues.
| Dye | Absorption in MeOH | Absorption in DNA | Emission in 90% Glycerol | Emission in DNA |
|---|---|---|---|---|
| TO | 502 | 511 | 547 | 528 |
| TO-1F | 500 | 509 | 544 | 529 |
| TO-p2F | 489 | 496 | 534 | 525 |
| TO-4F | 485 | 492 | 530 | 523 |
| TO-CF3 | 516 | 527 | 571 | 549 |
The electronic spectra of thiazole orange and its fluorinated derivatives were modeled to help explain the trends described above. Molecular structures were obtained using the SAM115 Hamiltonian in the AMPAC16 quantum chemistry package. The ground electronic state was optimized using restricted Hartree-Fock (RHF) theory. The excited state was optimized using single configuration interaction (SCI) theory including all excitations between the 50 highest occupied molecular orbitals and 50 lowest unoccupied orbitals. While the optimized ground state is planar, the excited state is highly twisted (as discussed further below). To obtain an estimate of the Stokes' shift in an environment such as DNA that may prevent twisting, the excited state geometries were also optimized under the constraint that the interplanar angle between the quinoline and benzothiazole groups is 0°. These planar excited-state geometries are used below when making spectral predictions for dyes bound to DNA.
The vertical excitation energies predicted by SAM1 (Table 2) do not correlate well with experiment. More reliable predictions of the excitation energy can be obtained from Intermediate Neglect of Differential Overlap (INDO) theory,17 however INDO is only useful after the molecular geometry is known. We therefore adopted a hybrid method in which geometries are obtained from SAM1 while excitation energies are obtained from a direct INDO/SCI18 method that includes excitations between all molecular orbitals. The results in Table 2 and Figure 2 show that this hybrid method leads to good correlation between experiment and theory for both absorption (INDO excitation energy calculated at the SAM1 ground-state geometry) and fluorescence (INDO excitation energy calculation at the SAM1 excited-state geometry). This strong correlation between experiment and theory indicates that the calculations are useful for understanding the sign and relative magnitude of the effects of fluorine substitution. However, the absolute magnitudes of the calculated substituent effects are not as reliable, since a fit of experiment to theory yields a slope of 1.79 for absorption and 1.15 for emission. (The slope should be 1.0 for perfect correlation.)
Table 2.
Results from modeling electronic spectra of TO and fluorinated analogues.
| Experimental
λmax (nm) |
Calculated
λmax(abs) (nm) |
λmax(Fl)
(nm) |
INDO Ground State
(eV) |
|||||
|---|---|---|---|---|---|---|---|---|
| Dye | Abs a | Fl b | SAM1 c | INDO d | INDO e | HOMO f | LUMO g | Δ h |
| TO | 511 | 528 | 476.0 | 476.5 | 489.0 | -10.30 | -4.47 | 5.83 |
| TO-1F | 509 | 529 | 476.1 | 471.4 | 483.3 | -10.44 | -4.57 | 5.87 |
| TO-p2F | 496 | 525 | 472.2 | 468.3 | 479.5 | -10.51 | -4.60 | 5.91 |
| TO-4F | 492 | 523 | 473.2 | 463.0 | 475.7 | -10.72 | -4.79 | 5.93 |
| TO-CF3 | 527 | 549 | 479.1 | 482.5 | 496.4 | -10.47 | -4.69 | 5.78 |
Experimental absorption maximum in DNA
Experimental emission maximum in DNA
Excitation energy from SAM1 for ground state optimized structure
Excitation energy from direct INDO/SCI for SAM1 ground state optimized structure
Excitation energy from direct INDO/SCI for SAM1 excited state optimized planar structure
INDO HOMO energy for SAM1 ground state optimized structure
INDO LUMO energy for SAM1 ground state optimized structure
HOMO-LUMO splitting from INDO for ground state optimized structure.
Figure 2.

Correlation of predicted λmax with experimental λmax, in nm. The dashed line is for absorption and the solid line is for emission.
We next consider whether the substituent effects on the excitation energy can be understood in terms of the HOMO-LUMO gap. Table 2 lists the HOMO and LUMO energies and the HOMO-LUMO gap obtained for the ground state geometries. In Figure 3 the HOMO-LUMO gap is shown to be well correlated with the predicted λmax. This correlation implies that the observed spectral shifts can be understood in terms of the effects of the electron-withdrawing substituents on the HOMO and LUMO orbitals, which are plotted in Figure 4 for the unsubstituted dye TO in a planar geometry. Inductive effects from fluorine substitution may be expected to stabilize a molecular orbital, with the decrease in energy being greater for orbitals that have large populations on the site of substitution. The HOMO is spread evenly between the benzothiazole and quinoline ring systems while the LUMO is mostly on the quinoline. Inductive effects from fluorine substitution on the benzothiazole thereby lower the HOMO energy more than the LUMO energy. This leads to an increase in the HOMO-LUMO gap, in agreement with the hypsochromic shifts seen experimentally. On the other hand, inductive effects from CF3 substitution on the quinoline ring system lower the LUMO more than the HOMO. This leads to a decrease in the HOMO-LUMO gap, in agreement with the bathochromic shift seen experimentally.
Figure 3.

Correlation of the HOMO-LUMO splitting with predicted absorption λmax.
Figure 4.
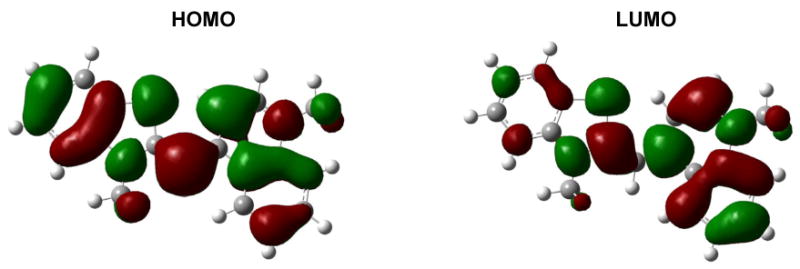
The HOMO and LUMO of TO obtained from SAM1 computations on the ground state structure. The molecules are drawn oriented as in Chart 1.
Fluorogenicity
The effect of viscous solvent or double-stranded DNA on dye fluorescence is shown in Figure 5 for TO-p2F. Table 3 lists the fluorescence quantum yields for the dyes in viscous solution and DNA. In solution, the quantum yields are found to increase with fluorination of the benzothiazole but decrease when a trifluoromethyl group is present on the quinoline. In contrast, the quantum yield decreases for all fluorinated derivatives when bound to DNA. Repeating the experiment at higher DNA concentration does not increase the quantum yields, indicating that the trend shown in Table 3 is not due to weaker binding of the fluorinated dyes to DNA.
Figure 5.

Fluorescence spectra recorded for 1.0 μM TO-p2F in methanol (dashed), 90% glycerol-water (dashed) and double-stranded calf thymus (CT) DNA (solid). Spectra were corrected for differences in absorbance at the excitation wavelength (450 nm).
Table 3.
Fluorescence quantum yields (φf) ecorded in 90% glycerol and DNA.
| Dye | φf (90% Glycerol) | φf (DNA) |
|---|---|---|
| TO | 0.027 | 0.110 |
| TO-1F | 0.029 | 0.091 |
| TO-p2F | 0.038 | 0.092 |
| TO-4F | 0.051 | 0.040 |
| TO-CF3 | 0.017 | 0.030 |
The fluorescence enhancement exhibited by TO and its analogues in viscous solution or DNA is due at least in part to restriction of conformational freedom in the monomethine bridge. As stated above, the excited state geometries of these dyes are highly twisted; attaining such a conformation within the excited state lifetime is hindered by viscous solvent or the geometrical constraints imposed by intercalation into DNA. This prompted additional calculations to better understand the twisting of the dyes in the excited state.
Figure 6 defines two interplanar angles φ1 and φ2 that we will use to characterize the twisting motion. Twisting about φ1 was examined first (this choice will be justified below). Since we do not currently have the ability to perform geometry optimizations within our hybrid computational method, the structures were obtained by optimizing the ground electronic state using SAM1 theory with a constraint applied to φ1. The INDO excitation energies and transition intensities are shown as a function of φ1 in Figure 7 for the unsubstituted dye TO. The lowest singlet excitation (blue circles) drops rapidly above ca. 60° and becomes dark when fully twisted, with the optical intensity shifting to higher energy excited states. This behavior, which is also predicted for the substituted dyes, rationalizes the low quantum yield of these dyes observed in solution. It also helps rationalize the higher quantum yields observed in DNA since insertion into DNA likely hinders this geometric relaxation such that the quantum yield is established by some other mechanism. It is for this reason that, in the above prediction of the Stokes' shift in DNA, a planar constraint was applied while optimizing the excited-state geometry.
Figure 6.

Definition of interplanar angles, φ1 and φ2 used to describe twisting of TO.
Figure 7.

Singlet excitation energies of TO as a function of the interplanar angle φ1, holding φ2 fixed at 0° (angles defined in Figure 6). The radius of the circles is proportional to the predicted absorption intensity. The excitation energies for the S0-S1 transition are shown in blue, while energies for transitions to higher states are shown in purple (S0-S2), violet (S0-S3), magenta (S0-S4) and red (S0-S5).
The drop in the excitation energy as the interplanar angle φ1 exceeds 60° is correlated with a loss of conjugation between the benzothiazole and quinoline rings. This is reflected in the bond orders on the methine bridge. The bond order for the “single” bond (i.e. the bond connecting the bridge to the benzothiazole in the resonance form shown in Chart 1) drops from 1.33 at the planar structure to 0.97 at 90°, while the bond order for the central “double bond” increases from 1.26 to 1.55. The loss in conjugation is also reflected in the molecular orbitals. In the planar geometry, both the HOMO and LUMO are delocalized over the entire dye (Figure 4). However, in the twisted geometry, the HOMO becomes localized to the quinoline ring and the LUMO becomes localized to the benzothiazole ring (Figure 8).
Figure 8.
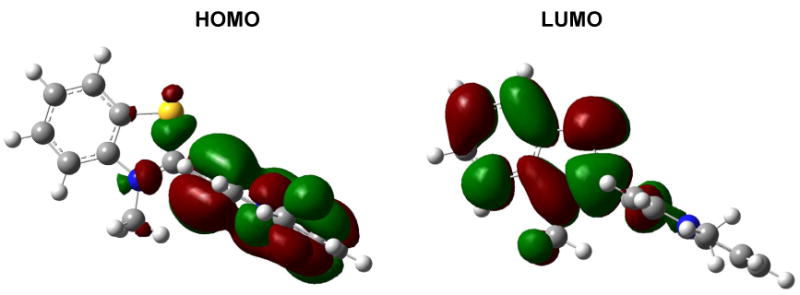
The HOMO and LUMO of TO obtained from SAM1 computations on the 90° twisted structure. The molecules are drawn oriented as in Chart 1.
The drop in excitation energy at 90° can be related to the localization of these frontier orbitals. This requires, however, going beyond just the orbital energies. When electron-electron interactions are taken into account, the excitation energy associated with a HOMO-LUMO promotion, Eexc, is
| (1) |
where ΔE0 is the difference of the HOMO and LUMO orbital energies, J is the Coulomb repulsion integral, and K is the exchange integral, defined as
| (2) |
| (3) |
using chemist's notation.19 The contribution of the various terms to the excitation energy of Eq. 1 is shown as a function of interplanar angle in Fig. 9. The HOMO-LUMO gap does decrease between 0° and 90°, but this accounts for only about 20% of the total drop in excitation energy. Since J has nearly the same value at 0° and 90°, it does not contribute to the difference in excitation energy between the planar and twisted structures. (J does however vary at intermediate angles, in a manner that decreases the barrier between the planar and twisted conformations.) The drop in excitation energy at φ1 > 60° arises primarily from K going to zero in the twisted structure. This is related to the HOMO and LUMO being localized on different ring systems, since the exchange interaction is small between orbitals that occupy different spatial regions.
Figure 9.

Individual contributions of energy terms given in Eq. 1 to the excited state energy of TO as a function of the interplanar angle φ1, holding φ2 fixed at 0°.
We next consider the effects of fluorine substitution on the above twisting mechanism. Figure 10 shows the excited state energy surfaces as a function of φ1 for the series of substituted dyes. The excited-state surface is obtained as the sum of the SAM1 ground state energy and the INDO excitation energy. The potential surfaces have local minima at about 20° and show activated crossing to a twisted structure. Similar barriers have been observed computationally in trimethine cyanine, a small molecule used as a computational model of symmetrical cyanine dyes.14 The substituent effects on the activation barrier are consistent with the trend in quantum yields measured in 90% glycerol: the barrier increases with fluorination of the benzothiazole heterocycle but decreases for the trifluoromethyl-substituted dye. This indicates that activated crossing of this barrier establishes the quantum yield.
Figure 10.

Excited state energy surfaces as a function of φ1 for all dyes. For clarity, the energies are referenced against the excited state energy of the planar structure, which is set to zero. Inset: expanded view of region between 50-70°.
The origin of the barrier in Figure 10 can be understood in terms of the relative importance of the ground state energy, which increases as the molecule deviates from planarity, versus the excitation energy, which decreases as the molecule deviates from planarity. Near φ1 = 0°, the excitation energy decreases more rapidly than the ground-state energy increases, such that the excited-state energy initially decreases with φ1. The increase in the ground-state energy with φ1 dominates between 20° and 60°, leading to a gradual rise from a local minimum near 20° to a maximum at about 60°. Above 60°, the sharp decrease in excitation energy (Figure 7) dominates and the excited-state surface decreases rapidly to a global, twisted minimum.
Above, the excited state surfaces were obtained by optimizing the ground state surface with constraints applied to φ1. In the resulting structures, φ2 adopts a value very near 0°. However, it is possible that the minimum energy path for twisting of the dyes in the excited state involves some twisting about φ2 as well. To explore this, Figure 11 shows a contour plot of the excited state potential energy surface of TO as a function of both φ1 and φ2. The results show that the reaction path is primarily along φ1, with similar results being obtained for the substituted dyes. This supports the above use of φ1 to map out the excited state potential energy surfaces.
Figure 11.
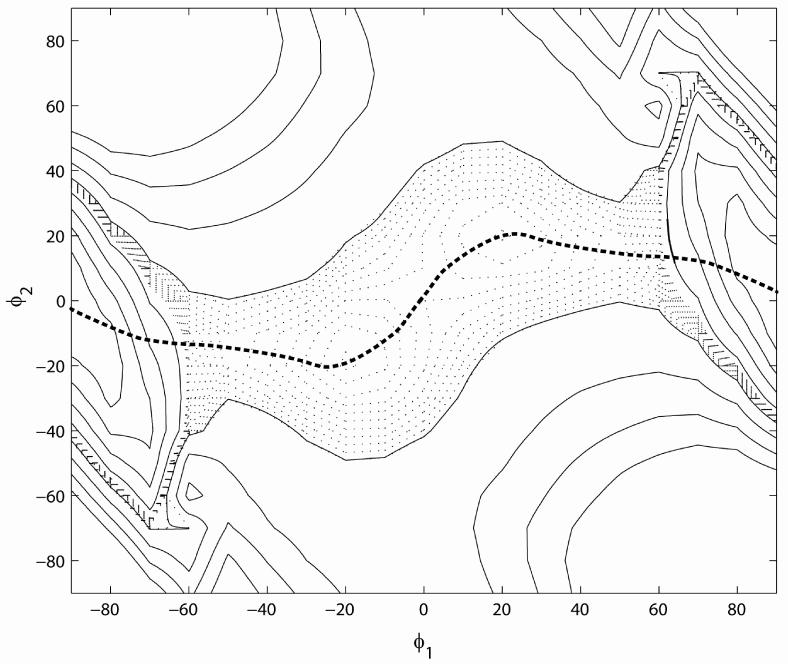
Contour plot of the excited state potential energy surface of TO as a function of φ1 and φ2, obtained from the hybrid method. The dotted contours are from 2.60 to 2.65eV in steps of 0.005eV. The solid contours begin at 2.65 eV with increments of 0.1 eV. The bold dotted line shows the lowest energy path for the twisting motion. This path has a local maximum at (φ1,φ2) = (0,0). Shallow local minima at about φ1 = ±20° are separated from deep minima at φ1 = ±90° by a barrier at ±60° (compare to Figure 10).
To further explore the nature of the twisting motion, we used SAM1 to map out the reaction path between the planar and twisted structures in the excited state. This was done using the chain algorithm in AMPAC with the SAM1 Hamiltonian and the SCI method including all excitations between the 50 highest occupied molecular orbitals and 50 lowest unoccupied orbitals. Table 4 shows the interplanar angles φ1 and φ2 at the predicted local minima and transition states. For all dyes, the results support the above assumption that the reaction path involves mostly φ1. The local minima occur at φ1 of about 20° and φ2 near 0°. The transition states occur at intermediate values of φ1 with φ2 close to 0° (except for TO-CF3 where the local minimum and transition state differ by only about 10° in φ1). The predictions regarding the effects of fluorine substitution on the activation barriers do not agree with those obtained above, and so are not in agreement with the experimental quantum yields seen in solution. This is not too surprising given that, as discussed above, SAM1/SCI does not correctly reproduce the effects of fluorine substitution on the absorption maxima. We therefore take these SAM1/SCI results as giving general support to the assumption that the reaction coordinate for twisting is φ1, and that there is activated crossing to a twisted structure. They also confirm that SAM1/SCI does not give reliable results for the effects of fluorine substitution on these dyes.
Table 4.
Results from SAM1/SCI calculations on the reaction path between planar and twisted structures in the lowest singlet excited state. Shown are the experimental quantum yields, the interplanar angles at both the nearly-planar intermediate (I) and the transition state (TS), along with the activation barrier.
| Dye | φfa | I (φ1,φ2) b | TS (φ1,φ2) c | ΔE (eV) d |
|---|---|---|---|---|
| TO | 0.027 | (16°, -4°) | (65°,4°) | 0.160 |
| TO-1F | 0.029 | (19°, 3°) | (53°,3°) | 0.073 |
| TO-p2F | 0.038 | (13°, 14°) | (70°,1°) | 0.027 |
| TO-4F | 0.051 | (7°, 6°) | (49°,0°) | 0.027 |
| TO-CF3 | 0.017 | (64°, 3°) | (72°,1°) | 0.063 |
Experimental quantum yield φf (90% glycerol),
φ1 and φ2 at the local minima,
φ1 and φ2 at the transition state,
the activation energy for transition from the intermediate to the transition state.
Willets et al20 attributed the fluorogenic behavior of DCDHF fluorophores (Scheme 1) to excited state twisting motions that have some similarities to that implicated here for TO. In both DCDHF and TO, the excited state surface exhibits a planar minimum and a twisted minimum, with the twisted structure decaying nonradiatively. However, in DCDHF, strong couplings between the various interplanar angles (α, φ, and δ in Scheme 1) lead to a more complex situation than the essentially one-dimensional mechanism put forward here for TO. In DCDHF, the ground state structure has (α,φ,δ) = (1°,-39°,0°), and the potential along φ is sufficiently weak that the room temperature population at φ = -90° is about 10%. Excitation is then to a Franck-Condon region near (1°,-39°,0°) which relaxes either to a fluorescent planar configuration at (0,-9,1) or to a nonradiative twisted configuration at (0, -42, 88). The experimentally observed environmental sensitivity of the fluorescence properties of DCDHF is attributed to the effects of the environment on this branching ratio.
Scheme 1.

Effect of Fluorination on Dye Aggregation and Photostability
As noted earlier, fluorination of a symmetrical cyanine dye led to decreased dye aggregation and enhanced photostability.12 The effects of fluorination on these properties for the unsymmetrical cyanine dyes are now presented.
Figure 12 illustrates the dependence of the visible absorbance maximum on dye concentration. The slight downward curvature evident in the data for TO, TO-1F and TO-CF3 is characteristic of dye aggregation. In contrast, the absorbance increases linearly for the di- and tetrafluorinated dyes, demonstrating effective suppression of aggregation by fluorination of the benzothiazole side of the dye.
Figure 12.

Plot of maximum UV-vis absorbance versus dye concentration for TO and fluorinated analogues.
The use of unsymmetrical cyanine dyes as fluorescent stains for nucleic acid detection requires that the dyes be photostable.21 To assess the photostability of the new fluorinated TO derivatives, a photobleaching experiment for the dyes was performed as described in the supporting information. The photostability of 5 μM dyes was first tested in buffer solution containing 100 mM sodium chloride. Thiazole orange showed a pronounced drop of its absorbance when it was irradiated with visible light for 10 × 1 min intervals, leading to a 55% total decrease in absorbance (Figure 13A and Table 5). The di- and tetrafluorinated analogues are considerably stabilized against photobleaching, while the trifluoromethyl-substituted dye exhibits minimal stabilization. No photobleaching was observed under the same conditions for any of the dyes in pure methanol solution.
Figure 13.

Absorbance of 5.0 μM dye solutions irradiated with visible light for 1 minute time intervals in either aqueous buffer (A) or 100 μM base pairs CT-DNA (B).
Table 5.
Photobleaching of cyanine dyes in aqueous buffer and in CT-DNA solutions.
a After 10 one-minute irradiation intervals.
The photobleaching experiment was next repeated in the presence of CT-DNA under conditions where the dyes are completely bound. The results are shown in Figure 13B and Table 5 and demonstrate that while all dyes are more photostable when bound to DNA than in solution, TO-p2F and TO-4F are once again more photostable than the other dyes.
DNA Binding
Finally, the effect of fluorination on DNA binding behavior was studied by thermal denaturation experiments. Dyes that bind with higher affinity to double-stranded versus single-stranded DNA induce a stabilization of the former, leading to an elevated melting temperature (Tm). For example, [Poly(dA-dT)2], a double-helical DNA polymer consisting of alternating A-T base pairs dissociates into single strands at Tm = 42.4 °C under the conditions described in the supporting information, but in the presence of TO at a ratio of one TO per 2 DNA base pairs, the melting temperature increases by 22 °C. Repeating the experiment with the fluorinated dyes gives the results shown in Table 6. All five of the TO analogues significantly stabilize the DNA, with the mono- and difluorinated dyes exhibiting similar Tm stabilizations as TO, while slightly lower values are observed for TO-4F and TO-CF3.
Table 6.
DNA duplex stabilization by TO and fluorinated analogues.
| Dye | ΔTm (°C) |
|---|---|
| TO | 22 |
| TO-1F | 22 |
| TO-p2F | 21 |
| TO-4F | 15 |
| TO-CF3 | 14 |
Discussion
The effect of fluorination on the properties of the fluorogenic dye thiazole orange was studied. Replacement of the benzothiazole heterocycle with mono-, para-di-, or tetrafluorobenzothiazole led to progressive reduction in aggregation and improvement in photostability. These results are consistent with what was observed previously for an octafluorinated analogue of the symmetrical cyanine dye S5 (Chart 1). The enhanced photostability of the fluorinated dyes could be due to lower efficiency of singlet oxygen production. In addition, the fluorinated S5 dye exhibited significantly lower reactivity toward singlet oxygen relative to the nonfluorinated dye12 and a similar effect could be operative for the unsymmetrical dyes. Moreover, these substitutions did not substantially impact the DNA-binding ability of the dyes, based on thermal denaturation experiments with [Poly(dA-dT)]2 (Table 6), although the tetrafluorinated dye TO-4F offered slightly less stabilization of the DNA duplex.
In terms of aggregation and photostability, substitution of a trifluoromethyl group on the quinoline heterocycle (TO-CF3) is comparable to monofluorination of the benzothiazole heterocycle (TO-1F), although TO-CF3 offers less stabilization of the [Poly(dA-dT)]2 duplex. Interestingly, TO-CF3 and TO-4F exhibit not only less stabilization of [Poly(dA-dT)]2 but also the lowest fluorescent quantum yields when bound to DNA. One possible explanation for these results is that these two dyes intercalate into the DNA helix in geometries that are less restrictive to twisting in the excited state. For example, insertion of only one of the heterocycles into the helix while leaving the other projecting into one of the grooves could lead to fluorescence enhancements and duplex stabilizations that are weaker than for dyes that intercalate with both heterocycles. A “half-intercalation” binding mode for other unsymmetrical cyanines was also proposed previously by Yarmoluk and coworkers,22 although verification requires high resolution structural studies.
More dramatic differences among the dyes are observed in the optical spectra. Fluorination of the benzothiazole leads to a progressive hypsochromic shift of the absorbance and emission maxima while TO-CF3 exhibits a bathochromic shift. These shifts can be attributed to the effects of fluorine substitution on the frontier molecular orbitals. Inductive effects from fluorine substitution stabilize both the HOMO and LUMO. However, since the HOMO has greater population density on the benzothiazole ring than the LUMO, fluorine substitution on the benzothiazole preferentially stabilizes the HOMO, leading to the observed hypsochromic shift. The opposite is true for trifluoromethyl substitution on the quinoline ring, which preferentially stabilizes the LUMO leading to the observed bathochromic shift.
Clear differences are also observed in the fluorescence quantum yields, where increasing fluorination of the benzothiazole leads to increasing quantum yield in glycerol, whereas TO-CF3 exhibits the lowest quantum yield. This result is again explained by calculations, which indicated that the lowest singlet excited state undergoes an activated crossing from a planar structure to a highly twisted structure. The crossing occurs at a interplanar angle of approximately 60° for TO and the fluorinated analogues. Structures having angles less than 60° should be optically bright, but activated crossing to twisted structures having angles greater than 60° is expected to quench the fluorescence. The activation energy for transition from the planar to twisted conformations was calculated for the substituted dyes. The results indicate that the quantum yields observed in glycerol can be rationalized in terms of the substituent effects on this activation energy. This suggests that twisting through the critical angle of 60° is the dominant quenching mechanism in solution. Regarding the quantum yield in DNA, the observed substituent effects follow a quite different trend, indicating that the DNA environment prevents the dye from adopting a severely twisted conformation, such that the quantum yield is established by some other nonradiative relaxation process. These conclusions are supported by recent ultrafast fluorescence experiments on benzoxazole-based unsymmetrical cyanines reported by Vauthey and coworkers where a rapid (<10 ps) nonradiative relaxation of the dye excited state was observed in water, but that component of the decay disappeared upon intercalation into DNA.23
These results raise an important but subtle point concerning torsionally responsive dyes, including triphenylmethane dyes such as malachite green and the dicyanodihydrofuran dyes developed by Moerner and coworkers20. Our calculations indicate that there is a range of conformations over which these dyes will remain fluorescent, provided twisting through the critical angle is prevented by solvent viscosity or binding in a constraining site, such as between two DNA base pairs.24 Thus, large fluorescence enhancements do not require that the dye be held in a planar conformation, but rather that the dye be rigidified. A striking example of this is provided by the binding of malachite green to an RNA aptamer.25 This binding results in a >1000-fold fluorescence enhancement,26 but a high-resolution NMR structure of the dye-RNA aptamer complex shows that the chromophore is decidedly nonplanar, with a interplanar angle of 57°.27
Implications for Dye Design
Dyes for which the quantum yield is sensitive to the local environment are useful for imaging of biological and other systems. A particularly effective means of achieving this environmental sensitivity is to use dyes that quench due to intramolecular twisting, such as those studied by Willets et al.22 or those described here. The twisting mechanism presented in this work for fluorescence quenching provides a framework in which to consider optimization of dyes for sensing applications. A structural aspect that can be easily altered is the length of the bridge between the heterocycles, however for TO, it is only feasible to increase the length of this bridge. We would expect this to increase the angle at which the barrier occurs since the total twist angle between the heterocycles can be distributed among the various interplanar angles of a longer bridge. Preliminary calculations support this conclusion: replacing the methine bridge of TO with a trimethine bridge results in approximate doubling of the barrier height and the barrier moves to about 80° (data not shown). Thus, dyes with longer bridges are expected to have larger fluorescence quantum yields in fluid solution, so the fluorescence enhancement observed upon binding to DNA or some other molecule of interest will likely be smaller than for the analogous monomethine dye. Ongoing work is directed toward investigating how other substituents on the heterocycles or variations in the heterocycle itself influence the barrier height and position.
Supplementary Material
Full description of dye synthesis and characterization and experimental details for spectroscopy, photobleaching and DNA-binding experiments.
Acknowledgments
Synthesis and spectroscopic experiments were performed by GLS while computational experiments were performed by VE. This work was supported by the National Institutes of Health (1 R33 CA9754101-01 to BAA) and the National Science Foundation (Grant CCF-0330135 to BAA and CHE-0316759 to DY). We also acknowledge the CMU Center for Molecular Analysis, supported by NSF Grant No. DBI-9729351 and the NMR facility, funded in part by NSF Grant No. CHE-9808188.
References
- 1.Mishra A, Behera RK, Behera PK, Mishra BK, Behera GB. Chem Rev. 2000;100:1973–2011. doi: 10.1021/cr990402t. [DOI] [PubMed] [Google Scholar]
- 2.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Bioconjugate Chem. 1993;4:105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- 3.Armitage BA. Top Curr Chem. 2005;253:55–76. [Google Scholar]
- 4.Lee LG, Chen C, Liu LA. Cytometry. 1986;7:508–517. doi: 10.1002/cyto.990070603. [DOI] [PubMed] [Google Scholar]
- 5.Rye HS, Yue S, Wemmer DE, Quesada MA, Haugland RP, Mathies RA, Glazer AN. Nucleic Acids Res. 1992;20:2803–2812. doi: 10.1093/nar/20.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netzel TL, Nafisi K, Zhao M, Lenhard JR, Johnson I. J Phys Chem. 1995;99:17936–17947. [Google Scholar]
- 7.Nygren J, Svanvik N, Kubista M. Biopolymers. 1998;46:39–51. doi: 10.1002/(SICI)1097-0282(199807)46:1<39::AID-BIP4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 8.Carreon JR, Mahon KPJ, Kelley SO. Org Lett. 2004;6:517–519. doi: 10.1021/ol0362818. [DOI] [PubMed] [Google Scholar]
- 9.Babendure J, Liddell PA, Bash R, LoVullo D, Schiefer TK, Williams M, Daniel DC, Thompson M, Taguchi AKW, Lohr D, Woodbury NW. Anal Biochem. 2003;317:1–11. doi: 10.1016/s0003-2697(03)00085-x. [DOI] [PubMed] [Google Scholar]
- 10.Ishiguro T, Saitoh J, Yawata H, Otsuka M, Inoue T, Sugiura Y. Nucleic Acids Res. 1996;24:4992–4997. doi: 10.1093/nar/24.24.4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Svanvik N, Westman G, Wang D, Kubista M. Anal Biochem. 2000;281:26–35. doi: 10.1006/abio.2000.4534. [DOI] [PubMed] [Google Scholar]; b Svanvik N, Nygren J, Westman G, Kubista M. J Am Chem Soc. 2001;123:803–809. doi: 10.1021/ja002294u. [DOI] [PubMed] [Google Scholar]
- 12.Renikuntla BR, Rose HC, Eldo J, Waggoner AS, Armitage BA. Org Lett. 2004;6:909–912. doi: 10.1021/ol036081w. [DOI] [PubMed] [Google Scholar]
- 13.Karunakaran V, Lustres JLP, Zhao L, Ernstring NP, Seitz O. J Am Chem Soc. 2006;128:2954–2962. doi: 10.1021/ja056862n. [DOI] [PubMed] [Google Scholar]
- 14.a Hunt PA, Robb MA. J Am Chem Soc. 2005;127:5720–5726. doi: 10.1021/ja045652s. [DOI] [PubMed] [Google Scholar]; b Sanchez-Galvez A, Hunt P, Robb MA, Olivucci M, Vreven T, Schlegel HB. J Am Chem Soc. 2000;122:2911–2924. [Google Scholar]
- 15.Dewar MJS, Jie C, Yu J. Tetrahedron. 1993;49:5003–5038. [Google Scholar]
- 16.AMPAC, 8, 1992-1994 Semichem, Inc. PO Box 1649 Shawnee, KS 66222.
- 17.Ridley J, Zerner MC. Theor Chim Acta. 1973;32:111–134. [Google Scholar]
- 18.Tomlinson A, Yaron D. J Comp Chem. 2003;24:1782–1788. doi: 10.1002/jcc.10336. [DOI] [PubMed] [Google Scholar]
- 19.Szabo A, Ostlund NS. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. Dover Publications; 1996. [Google Scholar]
- 20.a Willets KA, Callis PR, Moerner WE. J Phys Chem B. 2004;108:10465–10473. [Google Scholar]; b Willets KA, Ostroverkhova O, He M, Twieg RJ, Moerner WE. J Am Chem Soc. 2003;125:1174–1175. doi: 10.1021/ja029100q. [DOI] [PubMed] [Google Scholar]
- 21.Kanony C, Åkerman B, Tuite E. J Am Chem Soc. 2001;123:7985–7995. doi: 10.1021/ja001047n. [DOI] [PubMed] [Google Scholar]
- 22.Yarmoluk SM, Lukashov SS, Ogul'chansky TY, Losytskyy MY, Kornyushyna OS. Biopolymers. 2001;62:219–227. doi: 10.1002/bip.1016. [DOI] [PubMed] [Google Scholar]
- 23.Fürstenburg A, Julliard MD, Deligeorgiev TG, Gadjev NI, Vasilev AA, Vauthey E. J Am Chem Soc. 2006;128:7661–7669. doi: 10.1021/ja0609001. [DOI] [PubMed] [Google Scholar]
- 24.Spielmann HP, Wemmer DE, Jacobsen JP. Biochemistry. 1995;34:8542–8553. doi: 10.1021/bi00027a004. [DOI] [PubMed] [Google Scholar]
- 25.Grate D, Wilson C. Proc Natl Acad Sci USA. 1999;96:6131–6136. doi: 10.1073/pnas.96.11.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babendure JR, Adams SR, Tsien RY. J Am Chem Soc. 2003;125:14716–14717. doi: 10.1021/ja037994o. [DOI] [PubMed] [Google Scholar]
- 27.Flinders J, DeFina SC, Brackett DM, Baugh C, Wilson C, Dieckmann T. ChemBioChem. 2004;5:62–72. doi: 10.1002/cbic.200300701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full description of dye synthesis and characterization and experimental details for spectroscopy, photobleaching and DNA-binding experiments.