

Abstract
While profiling the lipidome of the mouse brain by mass spectrometry, we discovered a novel family of N-acyl phosphatidylserine (N-acyl-PS) molecules. These N-acyl-PS species were enriched by DEAE-cellulose column chromatography, and they were then characterized by accurate mass measurements, tandem mass spectrometry, liquid chromatography/mass spectrometry, and comparison to an authentic standard. Mouse brain N-acyl-PS molecules are heterogeneous and constitute about 0.1 % of the total lipid. In addition to various ester-linked fatty acyl chains on their glycerol backbones, the complexity of the N-acyl-PS series is further increased by the presence of diverse amide-linked N-acyl chains, which include saturated, mono-unsaturated and poly-unsaturated species. N-acyl-PS molecular species were also detected in the lipids of pig brain, mouse RAW264.7 macrophage tumor cells and yeast, but not E. coli. N-acyl-PSs may be biosynthetic precursors of N-acyl serine molecules, such as the recently reported signaling lipid N-arachidonoyl serine from bovine brain. We suggest that a phospholipase D might cleave N-acyl-PS to generate N-acyl serine, in analogy to the biosynthesis of the endocannabinoid N-arachidonoyl ethanolamine (anadamide) from N-arachidonoyl phosphatidylethanolamine.
A comprehensive qualitative and quantitative characterization of lipids in the context of systems biology is crucial to a complete understanding cellular physiology and pathology, and is the rationale behind the Lipid Maps Consortium (www.lipidmaps.org) (1, 2). One of the major aims of the project is to discover novel lipids, with emphasis on those from mouse RAW264.7 macrophage tumor cells. There is ample biochemical and genomic evidence indicating the existence of novel lipids. For instance, radiochemical experiments with high levels of 32Pi indicate the presence of numerous unidentified minor phospholipid species at levels of 0.1 % or less in the total lipids of both prokaryotic and eukaryotic cells (3, 4). In addition, genomic analyses suggest the existence of proteins of unknown function, distantly related in their primary sequences to well-characterized enzymes of lipid metabolism (5). These predicted proteins might be involved in the biosynthesis of some of the minor unknown lipids.
State-of-the-art, high-resolution mass spectrometry (MS) represents a powerful initial approach to the identification and structural characterization of novel lipids. Although MS has long been employed for the characterization of lipid molecules (6, 7), the introduction of electrospray ionization (ESI) (8) and matrix-assisted laser desorption ionization (MALDI) (9) dramatically improved the applicability of MS for lipid analysis. These two soft ionization techniques, together with instrumentation developments, have allowed intact, labile lipid molecules to be analyzed directly by MS with exceedingly high sensitivity and molecular specificity (6, 10, 11), including the detection of trace biosynthetic intermediates (12).
By combining large-scale phospholipid pre-fractionation procedures with high-resolution ESI/MS/MS analysis, we have now discovered a family of novel N-acyl phosphatidylserine (N-acyl-PS) molecules in mouse brain, pig brain, mouse RAW264.7 macrophage tumor cells and yeast. The proposed structures of these N-acyl-PS species were confirmed by comparison with a synthetic standard. In 1970, Nelson provided preliminary evidence for the presence of N-acyl-PS in sheep red blood cells. However, mass spectrometry and NMR were not used to characterize this material, and no follow studies were published. Donahue et al. reported the presence of N-acyl-PS as a major component of Rhodopseudomonas sphaeroides phospholipids in 1982 (13), but Schmid et al. later demonstrated that the proposed R. sphaeroides N-acyl-PS was actually phosphatidyl-Tris, arising by phosphatidyl group transfer to the Tris buffer present in the growth medium (14). To our knowledge, no subsequent reports of N-acyl-PS as a component of biological membranes have appeared. We suggest that N-acyl-PS may function as a precursor of N-acyl L-serine in animal cells, a bioactive signaling lipid recently isolated from bovine brain (15). The biosynthesis, metabolism and function of N-acyl-PS may be analogous to that of N-acyl phosphatidylethanolamine (N-acyl-PE), the precursor of the mammalian endocannabinoid N-arachidonyl ethanolamine (16–18). The unequivocal identification of a family of N-acyl-PS molecular species in brain, macrophages and yeast set the stage for the complete elucidation of its biosynthesis, turnover and function.
Experimental Procedures
Materials
Total porcine brain lipids (Product Number: 131101) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). The N-acyl-PS standard, 1,2-dioleoyl-sn-glycerol-3-phospho-N-nonadecanoyl-L-serine, was synthesized by reacting 1,2-dioleoyl-sn-glycerol-3-phospho-L-serine (Product Number: 840035, Avanti Polar Lipids, Inc.) with the N-hydroxysuccinimide ester of nonadecanoic acid in anhydrous dimethylformamide. The crude product was purified by column chromatography on silica gel. The 1,2-dioleoyl-sn-glycerol-3-phospho-N-nonadecanoyl-L-serine was obtained as a clear oil (1H NMR δ in CDCl3: 0.89 [m, 9H, 3×CH3], 1.25 [m, 70H, 35×CH2], 1.59 [s, br, 6H, beta CH2], 2.16 [s, br, 8H, allylic CH2], 2.29 [m, 6H, alpha CH2], 3.92 [s, br, 2H, sn-3 CH2], 4.00 [s, br, 1H, CH on serine], 4.12 [dd, J = 6.8 and 11.2 Hz, 1H, sn-1 CH2], 4.35 [d, J = 10.6 Hz, 2H, CH2 on serine], 4.64 [s, br, 1H, sn-1 CH2], 5.20 [s, br, 1H, sn-2 CH], 5.34 [m, 4H, alkenyl CH], 7.12 [s, br, 4H, NH4+ counter ion]. P31 NMR δ in CDCl3: 0.11 [s, br]). Chemicals, HPLC-grade solvents, dounce tissue grinders (Potter-Elvehjem style) and Whatman DE-23 DEAE-cellulose anion-exchange resin were purchased from VWR (West Chester, PA). Mouse brains isolated from 18-month E3/3 mice were provided by Dr. Patrick Sullivan (Duke University Medical Center).
Extraction and Anion-exchange Fractionation of Mouse Brain Lipids
Mouse brains were isolated as previously described (19). Half of a frozen mouse brain (~ 0.5 g) was homogenized in 10 mL of ice-cold chloroform/methanol (1:2, v/v) and 2.5 mL of ice-cold phosphate-buffered saline (20) (PBS) using a dounce tissue grinder. The homogenate was then transferred to a glass tube with a Teflon lined cap, vigorously mixed by using a vortex for about 2 minutes, then incubated on ice for 10 minutes. Following centrifugation at 3000 × g at 4 °C for 10 min, the supernatant was transferred to a new glass tube, while the insoluble debris was discarded. The supernatant was then converted to a two-phase Bligh/Dyer system (21) by adding 3.3 mL of chloroform and 3 mL of PBS. After vigorous mixing, the phases were separated by centrifugation at 3000 × g at 4 °C for 10 min, and the lower phase was dried under a stream of nitrogen. The anion-exchange fractionation of mouse brain total lipids was performed as previously described (22, 23). Briefly, the dried mouse brain lipids were re-dissolved in 2 ml of chloroform/methanol/water (2:3:1, v/v/v) and applied to a 1-ml DEAE cellulose column (acetate form) equilibrated with the same solvent mixture (22, 23). Following sample loading, the column was washed with 5-ml of chloroform/methanol/water (2:3:1, v/v/v). The bound lipids were eluted stepwise with 5-ml washes of chloroform/methanol/aqueous ammonium acetate (2:3:1, v/v/v), with increasing ammonium acetate concentrations of 30, 60, 120, 240, and 480 mM as the aqueous component. All fractions were converted to a two-phase Bligh/Dyer system by adding appropriate volumes of chloroform and water. The lower phases were dried under a stream of nitrogen and stored at −20 °C until MS analysis.
High-resolution Electrospray Ionization/Mass Spectrometry
High-resolution ESI mass spectra were acquired on a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (Applied Biosystems, Foster City, CA), equipped with an electrospray source. For ESI/MS analysis, the entire 120 mM fraction from DEAE anion-exchange of mouse brain lipids (prepared as described above) was re-dissolved in 200 μl of chloroform/methanol (2:1, v/v). Typically, 10 μl of this solution was diluted into 200 μl of chloroform/methanol (1:1, v/v) and then infused into the ESI source at 5–10 μl/min. The negative and positive electrospray voltages were set at −4200 V and +5500 V, respectively. Other MS settings were as follows: CUR = 20 psi (pressure), GS1 = 20 psi, DP = −55 V, and FP = −265 V. For MS/MS, collision-induced dissociation was performed with collision energy ranging from 40 V to 70 V (laboratory frame of energy) and with nitrogen as the collision gas. Data acquisition and analysis were performed using the Analyst QS software.
Liquid Chromatography/Mass Spectrometry
LC/MS of lipids was performed using a Shimadzu LC system (comprising a solvent degasser, two LC-10A pumps and a SCL-10A system controller) coupled to a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (as above). LC was operated at a flow rate of 200 μl/min with a linear gradient as follows: 100% of mobile phase A was held isocratically for 2 min and then linearly increased to 100% mobile phase B over 14 min and held at 100% B for 4 min. Mobile phase A consisted of methanol/acetonitrile/aqueous 1 mM ammonium acetate (60/20/20, v/v/v). Mobile phase B consisted of 100% ethanol containing 1 mM ammonium acetate. A Zorbax SB-C8 reversed-phase column (5 μm, 2.1 × 50 mm) was obtained from Agilent (Palo Alto, CA). The post-column splitter diverted ~10% of the LC flow to the ESI source of the mass spectrometer.
Anion-exchange Fractionation of Porcine Brain Total Lipids and Mild Alkaline Hydrolysis of N-acyl-PS
The anion-exchange fractionation of porcine brain total lipids from Avanti Polar Lipids (Part number: 131101) was carried out on an Agilent 1200 HPLC system. The DEAE column (DEAE-5PW, 10 □m, 7.5 mm × 7.5 cm) was from Sigma. The LC flow rate was 2 mL/min. Solvent A consisted of chloroform/methanol/water (2/3/1, v/v/v). Solvent B consisted of chloroform/methanol/480 mM ammonium acetate (2/3/1, v/v/v). Lipid fractionation was carried out by step-elution: after loading 100 mg porcine brain total lipids in 1.5 mL of chloroform/methanol/water (2/3/1, v/v/v) onto the DEAE column, the lipids were eluted with 16-ml steps of chloroform/methanol/aqueous ammonium acetate (2:3:1, v/v), with ammonium acetate concentrations of 0, 30, 60, 120, 240, and 480 mM successively in the aqueous component. Each fraction was then converted to a two-phase Bligh/Dyer system consisting of chloroform/methanol/water (2/2/1.8, v/v/v) by adding appropriate volumes of chloroform and water. The lower phases were dried under a stream of nitrogen. The 120 mM fraction, containing the N-acyl-PS, was subjected to mild alkaline hydrolysis in a 3.8-mL mixture of CHCl3/MeOH/0.38 N NaOH (1:2:0.8, v/v) at room temperature (24 °C) for 1 hour. Following hydrolysis, the reaction mixture was neutralized by adding concentrated HCl (37%) using a glass micropipette, as judged by pH paper. The system was converted to a two-phase Bligh-Dyer mixture consisting of chloroform/methanol/water (2/2/1.8, v/v/v) by adding appropriate volumes of chloroform and water. Following centrifugation at ~500 × g at room temperature for 10 min using a clinical centrifuge, the lower phase was dried under a stream of nitrogen and stored at −20 °C. For LC/MS analysis, the dried hydrolysis products were re-dissolved in 100 μL of MeOH/DMSO (1/1, v/v), of which 10 μL was injected onto a C8 column for LC/MS using the QSTAR XL mass spectrometer as described above.
Analysis of the Level of Brain N-Acyl-PS by Liquid Chromatography Multiple Reaction Monitoring
The level of N-acyl-PS in total porcine brain lipids was estimated by the method of multiple reaction monitoring (MRM) on a 4000 Q-Trap hybrid triple quadrupole linear ion-trap mass spectrometer, equipped with a Turbo V ion source (Applied Biosystems, Foster City, CA). LC-MRM analysis was performed in the negative ion mode with MS settings as follows: CUR = 20 psi (pressure), GS1 = 20 psi, GS2 = 30 psi, IS = −4500 V, TEM = 350 °C, ihe = ON, DP = −70 V, EP = −10 V and CXP = −5 V. The voltage used for collision-induced dissociation was −50 V. To estimate the level of brain N-acyl-PS, a known quantity of synthetic N-acyl-PS standard (Avanti) was added to a defined amount of the total porcine brain lipids dissolved in 1 mL of MeOH/DMSO (1:1, v/v), with the final concentrations of 0.1 ng/μL for the synthetic N-acyl-PS and 0.1 μg/μL for the porcine brain total lipid. Ten μL of the sample solution was then injected onto a C8 column for each LC-MRM analysis, with the LC conditions as described above, but without flow splitting.
Extraction and Anion-exchange Fractionation of Lipids from RAW264.7 Cells
Five 150-mm plates of RAW 264.7 cells were cultured as previously described (24) and were extracted using the method of Bligh and Dyer (21). Each plate of RAW cells at about 90% confluence was washed with 10 ml PBS and then scraped into 5 ml PBS (137 mM NaCl, 0.027 mM KCl, 0.01 mM Na2HPO4, and 0.0018 mM KH2PO4) (20). The cell suspension was centrifuged at 4000 × g to harvest the cells. The cell pellets from 5 plates were combined, re-suspended in 4 ml of PBS, and then transferred in equal volumes (2 mL) to two glass tubes equipped with Teflon lined caps. To each tube, 2.5 ml of chloroform and 5 ml of methanol were added to form a single-phase Bligh-Dyer system. The contents were vigorously mixed on a vortex and then subjected to sonic irradiation in a bath apparatus for 2 minutes. Following incubation at room temperature for 15 min, the single-phase Bligh-Dyer mixtures were centrifuged at ~500 × g for 10 min in a clinical centrifuge to pellet cell debris. Each supernatant was transferred to a fresh tube, followed by addition of 2.5 ml chloroform and 2.5 ml PBS to generate two-phase Bligh-Dyer systems. After mixing, the two tubes were centrifuged as above to resolve the phases. The two lower phases were combined, dried under a stream of nitrogen, and stored at −20 °C. The anion-exchange fractionation of lipids from RAW cells on a DEAE-cellulose column and the negative ion ESI/MS analysis were performed as described above (for the mouse brain lipids).
Identification of N-Acyl-PS in Yeast
Frozen cells of Saccharomyces cerevesiae BY4743 were provided by Dr. Arnold Greenleaf of the Duke University Medical Center. Lipid extraction of the yeast cells was performed using the method of Bligh and Dyer (21). Specifically, about 0.2 g of frozen yeast cell pellet was resuspended in a 3.8-mL mixture of CHCl3/MeOH/PBS (1:2:0.8, v/v) in a glass tube with a Teflon lined cap, was vigorously mixed on a vortex for 2 minutes, and was then subjected to sonic irradiation in a bath apparatus for 10 minutes. Following incubation at room temperature for 15 min, the single-phase Bligh-Dyer mixture was centrifuged at ~500 × g for 10 min in a clinical centrifuge to pellet cell debris. The supernatant was transferred to a fresh tube, followed by the addition of 1 ml chloroform and 1 ml PBS to generate a two-phase Bligh-Dyer system (21). After mixing, the tube was centrifuged as above to resolve the phases. The lower phase was dried under a stream of nitrogen, and stored at −20 °C. For LC-MS (Q-Star XL) and LC-MRM (4000 Q-Trap) analysis, the dried yeast lipid extract was re-dissolved in 100 μL CHCl3. Typically, 10 μL of this solution (in CHCl3) was mixed with 90 μL DMSO/MeOH (1:1, v/v), and 10 μL of the final solution was injected for each analysis, using the LC conditions described above.
Results
Large-scale Pre-fractionation of Anionic Mouse Brain Lipids
Eukaryotic lipids can be separated based on their net negative charge by DEAE-cellulose column chromatography in chloroform/methanol/water (2:3:1, v/v). The column capacity is one to ten mg of lipid per ml of resin, and the process can be scaled from the μg to the g range (22, 23, 25). The ziwtterionic lipids (predominantly phosphatidylethanolamines, phosphatidylcholines, and sphingomyelins) and the uncharged lipids (triacylglcyerols, diacylglycerols, ceramides, dolichols and sterols) emerge in the run-through. These compounds account for about three quarters of the total brain lipids. They can be further fractionated by normal- or reverse-phase chromatography during LC/MS analysis. The anionic lipids (which include the phosphatidylserines, phosphatidylinositols, cardiolipins, sulfatides, phosphatidylglycerols, and phosphatidic acids) bind to the column. These substances are step-eluted with increasing concentrations (30, 60, 120, 240, and 480 mM) of ammonium acetate as the aqueous component of chloroform/methanol/water (2:3:1, v/v). The singly charged phosphtidylinositols, phosphatidyglycerols and phosphatidylserines emerge with 30 to 60 mM ammonium acetate, whereas the more acidic phosphatidic acids, sulfatides and cardiolipins emerge with 60 to 120 mM ammonium acetate. Residual cardiolipins and minor acidic lipids, such as CDP-diacylglycerols and the phosphatidylinositol phosphates, emerge with 240 to 480 mM ammonium acetate. Each fraction contains additional minor lipids, many of which are poorly characterized. Pre-fractionation of biological lipids prior to analysis by ESI/MS or LC/MS reduces signal suppression of the less abundant ions by the major components.
Identification of N-Acyl-PS Molecular Species
Figure 1A shows the negative ion ESI mass spectrum of mouse brain lipids eluting from DEAE cellulose with 120 mM ammonium acetate as the aqueous component of chloroform/methanol/water (2:3:1, v/v). The major peaks were identified by MS/MS as the [M−H]− ions of phosphatidic acids, sulfatides or residual phosphatidyserines, and the [M−H]2− ions of cardiolipins. Several singly charged ions in the range of m/z 1000–1100 could not readily be assigned to known compounds. Among them, the most abundant was seen at m/z 1026.78. It was interpreted as [M−H]−, given the presence of a strong putative [M+H]+ ion at m/z 1028.80 in the positive mode (data not shown). The unknown ion at m/z 1026.78 (Figure 1A) was analyzed by MS/MS (Figure 1B), yielding product ions at m/z 78.96 (PO3−), 96.97 (H2PO4−) and 152.99 (C3H6O5P−) consistent with a glycerophospholipid. The two acyl chains esterified to the glycerol backbone were identified as stearic (C18:0) and oleic (C18:1) acids, as judged by the prominent ions at m/z 283.25 and 281.25, respectively. This assignment is furthermore consistent with the signal at m/z 701.49, which could be a phosphatidic acid anion esterified with C18:0 and C18:1 on its glycerol backbone. The ions at m/z 419.24 and 437.24 would be derived from the phosphatidic acid ion by neutral loss of the C18:1 moiety as a fatty acid (RCO2H) or as a ketene (RCH=CO), respectively. Similarly, the signals at m/z 417.22 and 435.22 would be derived by neutral loss of the C18:0 chain as a fatty acid and as a ketene, respectively.
Figure 1.
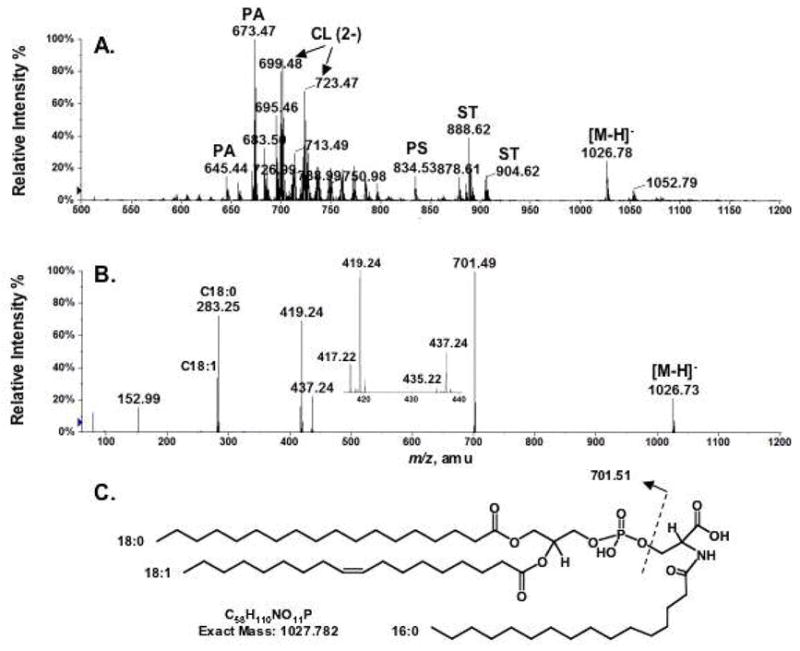
Negative ion ESI/MS analysis and proposed structure of a novel brain lipid with [M−H]− at m/z 1026.78. (A) Negative ion ESI/MS of mouse brain lipids eluting from DEAE cellulose with 120 mM aqueous ammonium acetate. (B) MS/MS analysis of m/z 1026.78. (C) The proposed brain N-acyl-PS structure (1-stearoyl-2-oleoyl-sn-glycerol-3-phospho-N-palmitoyl-serine) for the species with [M−H]− at m/z 1026.78. Abbreviations: PA, phosphatidic acid; CL, cardiolipin; PS, phosphatidylserine; ST, sulfatide.
The positions (sn-1 versus sn-2) of the two fatty acyl chains on the glycerol backbone cannot be determined unequivocally by MS/MS analysis alone. However, Hsu and Turk reported a correlation between the abundances of the fatty acyl-loss product ions and their positions on the glycerol backbone (26, 27). In the MS/MS analysis of phosphatidylserine in the negative mode, they found that the product ions derived from the loss of the fatty acyl substituent at sn-2 position were more abundant than those derived from loss at sn-1 position (26, 27). The expanded mass spectrum (Figure 1B, inset) of the MS/MS of the unknown at m/z 1026.78 shows that the ion at m/z 419.24 is more abundant than the one at m/z 417.22, and that the one at m/z 437.24 is more abundant than the one at m/z 435.22. We conclude that the C18:0 chain is attached predominantly to the sn-1 position and the C18:1 chain to the sn-2 position of the glycerol backbone (26, 27).
The phosphatidic acid anion observed at m/z 701.49 (Figure 1B) was presumably derived during MS/MS from the [M−H]− ion at m/z 1026.73 (Figure 1B) by a neutral loss of 325.24 Da. The odd mass of the neutral loss fragment suggested that it contained an odd-number of nitrogen atoms. The exact mass of the neutral loss, 325.261 Da, was determined by the mass difference between the accurately measured [M−H]− ion mass (1026.773) (Figure 1A) and the theoretical mass (701.512) of the 18:0/18:1 phosphatidic acid anion. The exact mass of the neutral loss suggested an elemental composition of C19H35NO3 (calculated exact mass: 325.262 Da). This elemental composition, combined with the above MS/MS data, suggested N-palmitoyl phosphatidylserine (18:0/18:1) as the structure that gives rise to the signal at m/z 1026.78 (Figures 1A and 1C). N-acyl-PS has a net charge of minus two, consistent with its elution from the DEAE-cellulose column together with cardiolipin.
Comparing Brain N-Acyl-PS with a Synthetic Standard by MS/MS and LC/MS
To confirm the proposed structure, a synthetic N-acyl-PS standard was prepared, as described in the Experimental Procedures. As shown in Figure 2A, the synthetic N-acyl-PS, 1,2-dioleoyl-sn-glycerol-3-phosphoserine-N-nonadecanoate, has the formula of C61H114NO11P and an exact mass of 1067.813 Da. The MS/MS spectrum of the [M−H]− ion of this standard, seen at m/z 1066.81 (Figure 2B), shows a fragmentation pattern that is very similar to that of the novel brain lipid (Figure 1B). The differences in the masses of the product ions are as predicted, based on the differences in the acyl chain compositions of the endogenous and the synthetic compounds.
Figure 2.
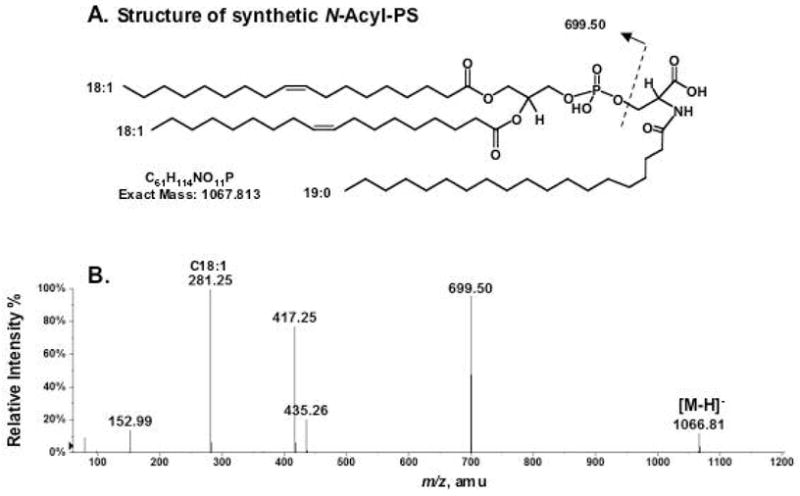
Negative ion ESI/MS analysis of synthetic N-acyl-PS. (A) Structure of the synthetic standard (1,2-dioleoyl-sn-glycerol-3-phospho-N-nonadecanoyl-serine). (B) Negative ion MS/MS analysis of the synthetic N-acyl-PS [M−H]− ion at m/z 1066.81.
MS/MS comparison of the endogenous material with the synthetic N-acyl-PS was also performed in the positive ion mode. The MS/MS analysis of the [M+H]+ ion at m/z 1028.79 yielded prominent product ions (Figure 3A) at m/z 326.26 and at m/z 605.53, consistent with the expected fragmentation scheme for glycerophospholipids. The corresponding product ions for the synthetic standard were observed at m/z 368.33 and 603.55 (Figure 3B), consistent with its structure.
Figure 3.
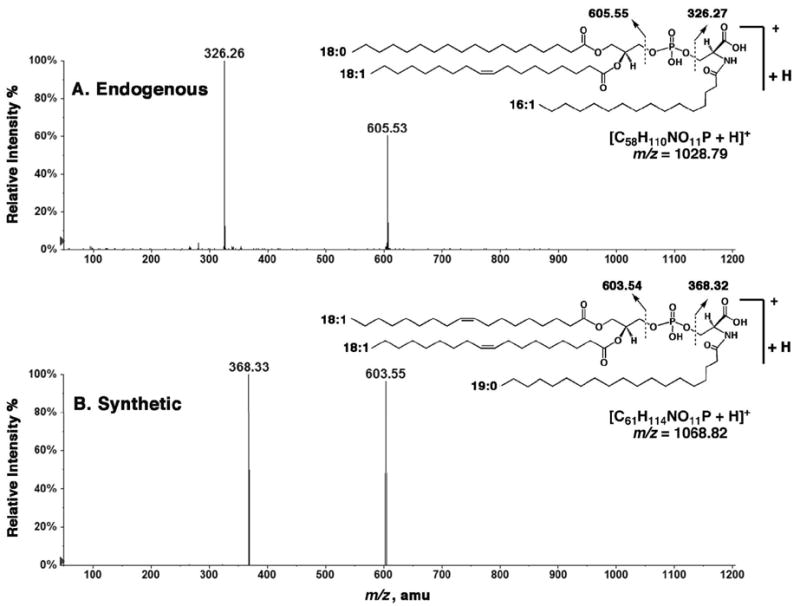
Comparison of the endogenous and synthetic N-acyl-PS molecules by MS/MS of their [M+H]+ ions. (A) MS/MS spectrum of the [M+H]+ ion at m/z 1028.79 (endogenous). (B) MS/MS spectrum of the [M+H]+ ion at m/z 1068.82 (synthetic). The major product ions are indicated on the inset structures.
MS/MS analysis was also performed on the [M+Na]+ ions of both compounds. Figure 4A shows the MS/MS spectrum of the sodium adduct of the endogenous N-acyl-PS [M+Na]+ ion, which is seen at m/z 1050.78 (Figure 4A). In this case the major product ions are observed at m/z 120.98 for [H3PO4+Na]+, at m/z 348.27 for sodiated N-palmitoyl dehydroalanine, at m/z 446.24 for the sodiated N-palmitoyl phosphoserine, at m/z 605.55 for the protonated dehydrated diacylglycerol (C18:0/18:1), at m/z 627.55 for the sodiated dehydrated diacylglycerol (C18:0/18:1), and at m/z 725.50 for the sodiated phosphatidic acid adduct (18:0/18:1). All the expected, corresponding product ions were observed in the MS/MS spectrum of the [M+Na]+ adduct of the synthetic N-acyl-PS standard (Figure 4B).
Figure 4.
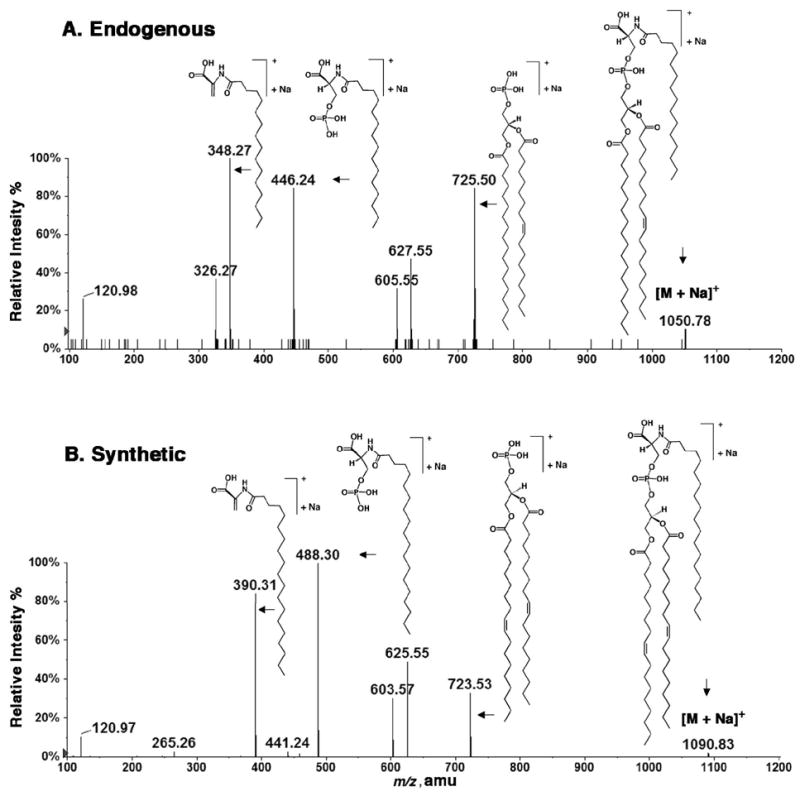
Comparison of the endogenous and synthetic N-acyl-PS molecules by MS/MS of their [M+Na]+ ions. (A) MS/MS spectrum of the [M+Na]+ ion at m/z 1050.78 (endogenous). (B) MS/MS spectrum of the [M+Na]+ ion at m/z 1090.83 (synthetic). The proposed structures of the major product ions are shown.
LC/MS comparison of the endogenous and synthetic N-acyl-PS was performed in the negative ion mode. Figure 5 shows the extracted ion chromatograms (EICs) of the [M−H]− ions derived from the endogenous N-palmitoyl-PS at m/z 1026.8 (Figure 5A) versus those derived from the synthetic standard at m/z 1066.8 (Figure 5B). The retention times of these two species during reverse phase chromatography are consistent with their structural similarity. The synthetic N-acyl-PS, with its slightly longer acyl chains, emerges 1.14 min later than the endogenous N-palmitoyl-PS.
Figure 5.
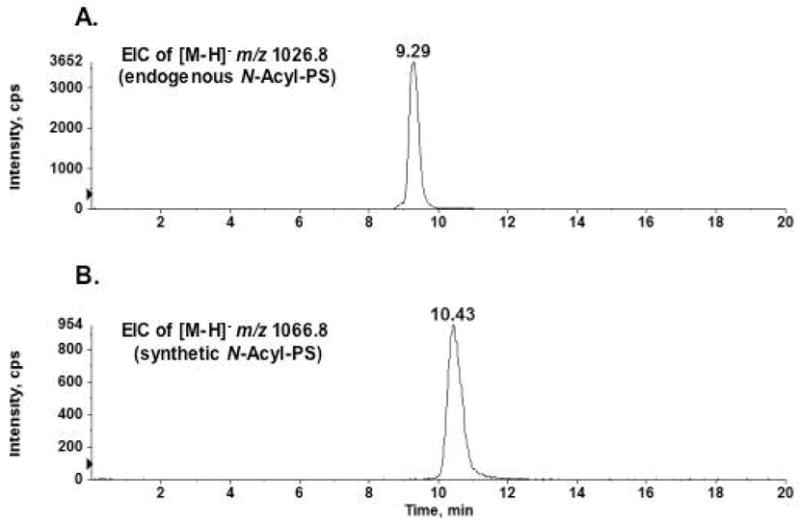
Comparison of endogenous and synthetic N-acyl-PS molecules by LC/MS in the negative ion mode. A) The extracted ion chromatogram (EIC) of the [M−H]− ion at m/z 1026.8 for the endogenous N-acyl-PS. B) The extracted ion chromatogram of the [M−H]− ion at m/z 1066.8 for the synthetic N-acyl-PS.
Presence of Additional N-Acyl-PS Molecular Species Including N-arachidonoyl-PS
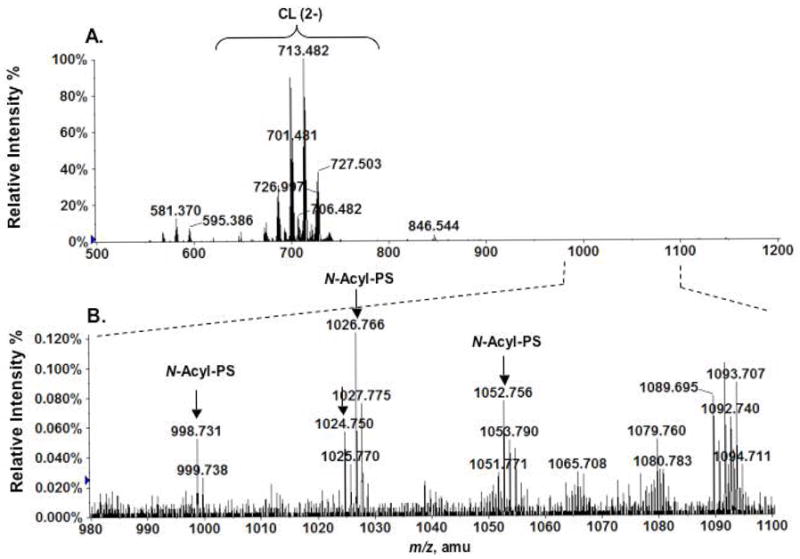
An expansion of the mass spectrum, shown in Figure 1A, in the range of m/z 980–1200 reveals the presence of a series of N-acyl-PS ions in addition to the prominent species at m/z 1026.80 (Figure 6A). When subjected to exact mass measurements and MS/MS analysis, these N-acyl-PS species were found to be complex because of the heterogeneity of their O-and N-linked acyl chains. As illustrated in Figure 1B with the MS/MS of the ion at m/z 1026.80, the various O-linked fatty acyl groups could be assigned based on the masses of the released fatty acid anions. The structures of N-linked acyl chains were deduced from the masses of the neutral losses yielding the phosphatidic acidic anions. The N-acyl-PS molecular species detected in this manner, together with their associated N-acyl chains, are listed in Table 1.
Figure 6.
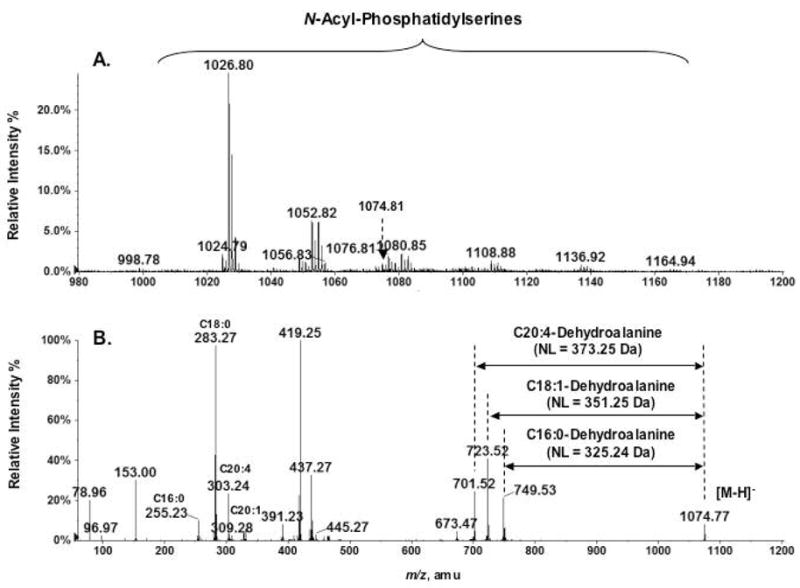
A family of N-acyl-PS molecular species in mouse brain. (A) Expanded negative ion mass spectrum (m/z 980–1200) from Figure 1A, showing a family of putative N-acyl-PS [M−H]− ions. These N-acyl-PS species (see Table 1) were identified by the exact mass measurements and MS/MS analysis. (B) Complex N-acyl chain compositions, as revealed by MS/MS analysis of the peak at m/z 1074.77. The neutral losses in the MS/MS were used to identify the N-acyl chains: 373.25 Da for N-arachidonoyl-PS, 351.25 Da for N-oleoyl-PS, and 325.24 Da for N-palmitoyl-PS.
Table 1. Proposed N-Acyl-PS Molecular Species Detected in Mouse Brain.
The numbers of carbon atoms and double bonds of the N-linked fatty acyl chains were deduced from the neutral losses observed in the negative ion MS/MS spectra of the indicated [M−H]− rounded to nearest 0.1 amu. NL, neutral loss, rounded to nearest integer; only the most prominent NL species are shown for each [M−H]−. We cannot exclude the possibility that the putative odd-chain species actually represent N-acylated phosphatidylthreonines.
| [M−H]− (m/z) | NL | N-acyl chain | [M−H]− (m/z) | NL | N-acyl chain | [M−H]− (m/z) | NL | N-acyl chain |
|---|---|---|---|---|---|---|---|---|
| 998.7 | 297 | C14:0 | 1074.8 | 325 | C16:0 | 1134.8 | 351 | C18:1 |
| 325 | C16:0 | 351 | C18:1 | 379 | C20:1 | |||
| 1012.8 | 311 | C15:0 | 373 | C20:4 | 407 | C22:1 | ||
| 325 | C16:0 | 401 | C22:4 | 435 | C24:1 | |||
| 339 | C17:0 | 1076.8 | 325 | C16:0 | 1136.8 | 325 | C16:0 | |
| 353 | C18:0 | 351 | C18:1 | 351 | C18:1 | |||
| 1024.8 | 323 | C16:1 | 353 | C18:0 | 353 | C18:0 | ||
| 325 | C16:0 | 375 | C20:3 | 379 | C20:1 | |||
| 351 | C18:1 | 1080.8 | 325 | C16:0 | 381 | C20:0 | ||
| 1026.8 | 325 | C16:0 | 351 | C18:1 | 407 | C22:1 | ||
| 1038.8 | 311 | C15:0 | 353 | C18:0 | 435 | C24:1 | ||
| 325 | C16:0 | 379 | C20:1 | 437 | C24:0 | |||
| 337 | C17:1 | 1102.8 | 325 | C16:0 | 1138.8 | 325 | C16:0 | |
| 339 | C17:0 | 351 | C18:1 | 353 | C18:0 | |||
| 351 | C18:1 | 353 | C18:0 | 381 | C20:0 | |||
| 353 | C18:0 | 373 | C20:4 | 409 | C22:0 | |||
| 1040.8 | 311 | C15:0 | 379 | C20:1 | 437 | C24:0 | ||
| 325 | C16:0 | 401 | C22:4 | 465 | C26:0 | |||
| 339 | C17:0 | 1108.8 | 325 | C16:0 | 1164.8 | 379 | C20:1 | |
| 353 | C18:0 | 351 | C18:1 | 381 | C20:0 | |||
| 1048.8 | 325 | C16:0 | 353 | C18:0 | 407 | C22:1 | ||
| 1050.8 | 325 | C16:0 | 379 | C20:1 | 409 | C22:0 | ||
| 351 | C18:1 | 407 | C22:1 | 435 | C24:1 | |||
| 377 | C20:2 | 435 | C24:1 | 437 | C24:0 | |||
| 1052.8 | 325 | C16:0 | 1122.8 | 325 | C16:0 | 463 | C26:1 | |
| 351 | C18:1 | 351 | C18:1 | 465 | C26:0 | |||
| 353 | C18:0 | 353 | C18:0 | 1166.8 | 409 | C22:0 | ||
| 1054.8 | 325 | C16:0 | 421 | C23:1 | 437 | C24:0 | ||
| 353 | C18:0 | 1124.8 | 325 | C16:0 | 465 | C26:0 | ||
| 1072.7 | 325 | C16:0 | 353 | C18:0 | ||||
| 351 | C18:1 | 395 | C21:0 | |||||
| 373 | C20:4 | 423 | C23:0 | |||||
| 399 | C22:5 | 437 | C24:0 |
N-arachidonoyl-PS was detected by MS/MS analysis of the ion at m/z 1074.77 (Figure 6B). The product ions at m/z 255.23, 281.25, 283.27, 303.24, 309.28 are the anions of C16:0, C18:1, C18:0, C20:4 and C20:1 fatty acids respectively, indicating that these are the possible combinations of fatty acyl moieties esterified to the glycerol backbone. The sizes of the neutral loss fragments arising during collisional activation of the isobaric ions at m/z 1074.77 were used to identify the N-acyl chains: 325.24 Da for N-palmitoyl-PS, and 351.25 Da for N-oleoyl-PS, and 373.25 Da for N-arachidonoyl-PS. As discussed below, N-arachidonoyl-PS might be a biosynthetic precursor of the recently discovered signaling lipid N-arachidonoyl serine (15).
Identification and Mild Alkaline Hydrolysis of N-Acyl-PS from Pig Brain
Commercial pig brain lipids (Avanti) were fractionated on a 100 mg scale by chromatography on DEAE-cellulose, as described above for mouse brain lipids. The compounds eluting with 120 mM ammonium acetate as the aqueous component were again enriched in various molecular species of N-acyl-PS (data not shown). To confirm their identity, the partially purified N-acyl-PS was subjected to O-deacylation in chloroform/methanol/0.38 M aqueous NaOH (1:2:0.8, v/v) for 60 minutes at room temperature (Scheme 1), yielding a series of N-acylated glycerophosphoserines (NAGPSs). Figure 7 shows the LC/MS analysis of the O-deacylated pig brain N-acyl-PS. The negative ion mass spectra acquired between 1–1.5 min of reverse phase chromatography (Figure 7A) were consistent with the presence of a series of NAGPS molecules bearing C16:0, C18:1, C20:4, C24:1 or C26:1 fatty acyl chains. The NAGPS species containing arachidonate at m/z 544.263 was a minor, but nevertheless significant, component (Figure 7A, inset). The mass spectra acquired between 2–2.5 min of chromatography (Figure 7B) revealed NAGPSs, containing longer acyl chains (C26:1, C26:0, C28:1 or C30:1). About 30 different N-acyl groups (Table 2) were identified. Representative negative ion MS/MS spectra of NAGPS species, those bearing C16:0, C20:4 or C30:1 chains, are shown in Figure 8. All yielded the identical, expected product ions.
Scheme 1. Mild alkaline hydrolysis of N-acyl-PS.

This N-acyl-PS structure is the most abundant brain N-acyl-PS molecular species.
Figure 7.
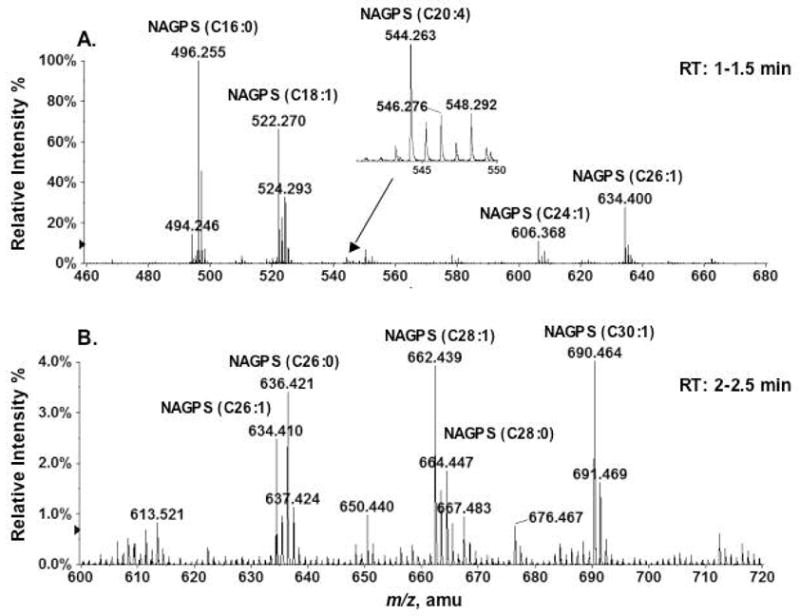
LC/MS detection of N-acyl glycerophosphoserines produced by mild alkaline hydrolysis of porcine brain N-acyl-PS. (A) Mass spectrum (averaged from the spectra acquired during 1–1.5 min) of the NAGPSs with acyl chains of C16:0, C18:1, C24:3 and C26:3. The inset shows the magnified peak near m/z 544 for NAGPS(C20:4), providing additional evidence for the existence of N-arachidonoyl-PS. (B) Mass spectrum (averaged from the spectra acquired during 2–2.5 min) of the NAGPS chromatography with very long acyl chains (C26:3, 26:2, C28:1, and C30:1). The structures of NAGPS(16:0), NAGPS(20:4) and NAGPS(30:1) were confirmed by both MS/MS (Figure 8) and accurate mass measurements (Table 2).
Table 2. Observed and Predicted Exact Masses of the [M−H]− Ions of the mild alkaline hydrolysis products of N-acyl-PS from porcine brain.
The structure of NAGPS(16:0) is shown in Scheme 1. The apparent odd chain species are minor components. We cannot exclude the possibility that they are actually N-acylated phosphatidylthreonines.
| NAGPS | [M−H]− | |
|---|---|---|
| Observed mass | Exact mass | |
| C14:0 | 468.234 | 468.237 |
| C15:0 | 482.250 | 482.252 |
| C16:1 | 494.246 | 494.252 |
| C16:0 | 496.255 | 496.268 |
| C17:1 | 508.263 | 508.268 |
| C17:0 | 510.275 | 510.284 |
| C18:1 | 522.270 | 522.284 |
| C18:0 | 524.293 | 524.299 |
| C20:4 | 544.263 | 544.268 |
| C20:3 | 546.276 | 546.284 |
| C20:2 | 548.292 | 548.299 |
| C20:1 | 550.311 | 550.315 |
| C20:0 | 552.326 | 552.331 |
| C22:6 | 570.283 | 570.284 |
| C22:5 | 572.298 | 572.299 |
| C22:4 | 574.308 | 574.315 |
| C22:3 | 576.321 | 576.331 |
| C22:1 | 578.338 | 578.346 |
| C22:0 | 580.349 | 580.362 |
| C23:1 | 592.361 | 592.362 |
| C24:1 | 606.368 | 606.378 |
| C24:0 | 608.390 | 608.393 |
| C25:1 | 620.398 | 620.393 |
| C25:0 | 622.397 | 622.409 |
| C26:1 | 634.400 | 634.409 |
| C26:0 | 636.421 | 636.425 |
| C27:1 | 648.425 | 648.425 |
| C28:1 | 662.439 | 662.440 |
| C28:0 | 664.447 | 664.456 |
| C30:1 | 690.464 | 690.472 |
Figure 8.
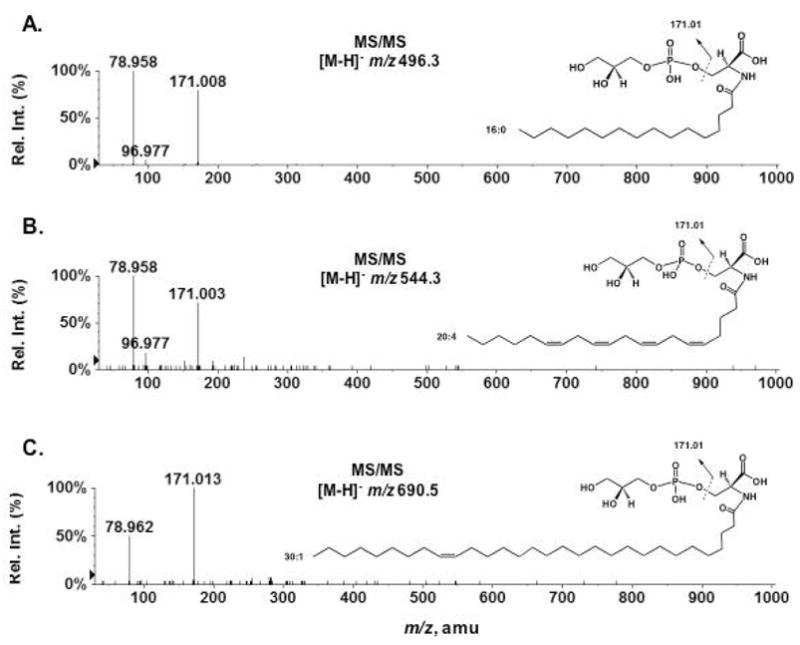
MS/MS analysis of three N-acyl glycerophosphoserines (NAGPSs) produced by mild alkaline hydrolysis of N-acyl-PS. (A) MS/MS of the [M−H]− ion at m/z 496.3 for NAGPS(16:0); (B) MS/MS of the [M−H]− ion at m/z 544.3 for NAGPS(20:4); (C) MS/MS of the [M−H]− ion at m/z 690.5 for NAGPS(30:1). The positions of the double bonds were not determined by MS/MS, but inferred from the literature (6, 10).
Estimation of the Levels of Pig Brain N-Acyl-PS by Liquid Chromatography and Multiple Reaction Monitoring
To estimate the level of the major brain N-acyl-PS, a known quantity of the synthetic N-acyl-PS standard was added to total porcine brain lipids (Avanti), followed by the determination of the ion signal ratio of the endogenous and the synthetic species. This was achieved by LC coupled multiple-reaction-monitoring (MRM) on an ABI 4000 Q-Trap hybrid triple quadrupole linear ion-trap mass spectrometer. Figure 9 shows the LC-MRM chromatograms derived from the most abundant endogenous N-palmitoyl-PS (18:0/18:1) species (the MRM pair of 1026.8/701.6) present in 1 μg of total porcine brain lipid versus 1 ng of the synthetic N-acyl-PS standard (the MRM pair of 1066.8/699.6). Assuming that the signal responses are very similar for these two closely related lipids (Figures 1C and 2A), the peak area ratio of 0.4 indicates that there is about 0.4 ng (or 0.04% by weight) of the most abundant endogenous N-acyl-PS in 1 μg of porcine brain lipids. Since the most abundant N-acyl-PS species accounts for about 40 % of all the N-acyl-PS species, we estimate that the combined N-acyl-PS species make up about 0.1 % of the porcine brain lipid. Although our synthetic standard is not an isotopically labeled analogue, its structure is nevertheless very close to that of the endogenous species (Figs. 1 and 2) and thus should have similar ionization efficiency.
Figure 9.
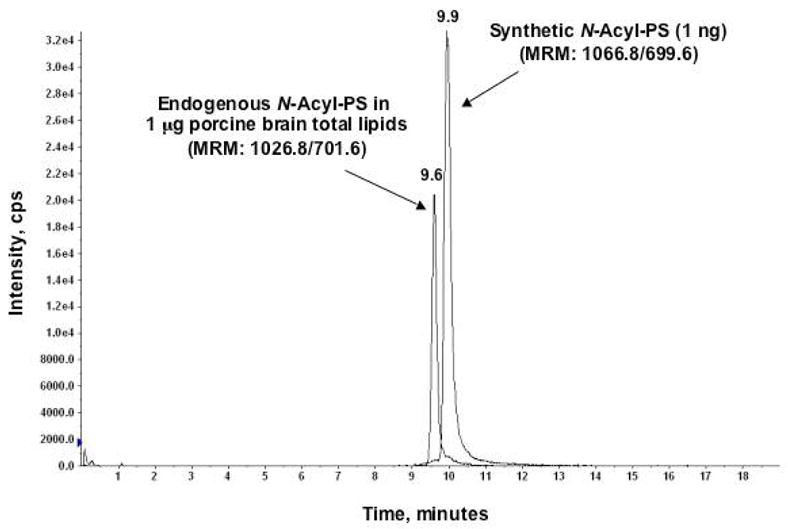
Quantification of the most abundant porcine brain N-acyl-PS species by LC-MRM. The two LC-MRM chromatograms refer to the most abundant endogenous N-acyl-PS species (M.W. 1027.8) present in 1 μg of porcine brain total lipid (MRM pair of 1026.8/701.6) versus 1 ng of added synthetic N-acyl-PS standard (MRM pair of 1066.8/699.6).
Detection of N-Acyl-PS in RAW264.7 and Yeast Cells
By carrying out lipid pre-fractionation in conjunction with high resolution MS analysis, we also detected low levels of N-acyl-PS in the mouse RAW264.7 macrophage cells and yeast. Figure 10A is the negative ion ESI mass spectrum of the 120 mM ammonium acetate fraction of DEAE cellulose fractionated RAW264.7 cell lipids. The signal intensities of the N-acyl-PS ions (Figure 10B) are two orders of magnitude lower than those from brain (compare Figs. 1 and 10). Improved detection of these species was made possible by using LC/MS (data not shown). The major ions arising from the N-acyl-PS molecular species of RAW cells (m/z 998.73, 1024.75, 1026.77, 1052.76 in Figure 10B) are same as those found in mouse brain (Figure 6A). The relatively high levels of N-acyl-PS species in the brain suggest that they may play special roles in the central nervous system.
Figure 10.
Detection of N-acyl-PS molecular species in RAW cells. (A) Negative ion ESI mass spectrum of phospholipids species eluting with 120 mM ammonium acetate from a DEAE cellulose column. (B) Expanded mass spectrum (m/z 980–1100) showing the [M−H]− ions of N-acyl-PS species present in RAW cells.
N-acyl-PS molecular species were also detected in yeast total lipid extracts (data not shown), using LC-MRM on an ABI 4000 Q-Trap hybrid triple quadrupole linear ion-trap mass spectrometer. The major N-acyl-PS species detected are N-palmitoyl-PS (16:0/18:1) (MRM: 998.8 to 673.5), N-myristoyl-PS (16:0/18:1) (MRM: 970.7 to 673.5) and N-stearoyl-PS (16:0/18:1) (MRM: 1026.8 to 673.5). The presence of N-acyl-PS species in yeast should facilitate enzymatic and genetic studies of their biosynthesis.
Discussion
The question of whether or not N-acyl-PS is a naturally occurring phospholipid has remained unanswered for over thirty-five years. Nelson first reported the presence of N-acyl-PS in sheep erythrocytes (28), but his structural analysis was based on infrared spectroscopy, thin-layer chromatography and elemental analysis (28), and it was never followed up with spectroscopic techniques. In 1982, Donohue et al. reported N-acyl-PS as a major phospholipid in Rhodopseudomonas sphaeroides (13). However, this claim was challenged by Schmid et al. who demonstrated conclusively that the material isolated by Donohue et al. was actually phosphatidyl-Tris, unexpectedly generated by microbial incorporation of exogenous Tris buffer into phospholipid (14).
We have now demonstrated the presence of a family of N-acyl-PS molecules in mouse brain, pig brain, RAW macrophage tumor cells and yeast, using high-resolution tandem ESI/MS. Our initial identification of N-acyl-PS was facilitated by chromatographic enrichment of di-anionic phospholipids on a DEAE-cellulose column (Figure 1). The structural elucidation involved accurate mass measurements, tandem MS, and comparison to a synthetic standard. Brain N-acyl-PS consists of a large number of molecular species, given the presence of at least thirty different N-linked acyl chains (Tables 1 and 2). N-palmitoyl-PS is the major component, but there are many additional molecular species, including N-arachidonyl-PS. In aggregate, we estimate that these N-acyl-PS molecules constitute ~ 0.1 % of the total brain lipid or about 0.001% of the total RAW cell lipid, whereas phosphatidylserine typically makes up ~ 7 percent of the RAW phospholipid (4).
The unequivocal identification of a novel collection N-acyl-PS molecular species in mammalian cells and yeast will enable investigations into their biosynthesis, turnover and biological functions. N-acyl-PS is structurally similar to N-acyl phosphatidylethanolamine (N-acyl-PE), which has recently received considerable attention (16–18). First discovered as a novel lipid that accumulates in myocardial infarcts (29), N-acyl-PEs are formed by an enzyme that transfers the sn-1 acyl chain of a glycerophospholipid to the primary amine of PE in a calcium-dependent manner (30–32). The identity of the structural gene(s) encoding the relevant Ca++ dependent transacylase is unknown (16). Importantly, the N-acyl-PEs are biosynthetic precursors of a class of lipophilic signaling molecules, the N-acyl ethanolamines (16, 33), which include the endocannabinoid anandamide (N-arachidonoyl ethanolamine) (34, 35), the anti-inflammatory agent N-palmitoyl ethanolamine (36), the satiety-inducing factor N-oleoyl ethanolamine (37), and the pro-apoptotic lipid N-steroyl ethanolamine (38) (26). Given the N-acyl-PE precedent, we speculate that some of the N-acyl-PS molecular species shown in Table 1 might be hydrolyzed to produce the corresponding N-acyl serines. Recently, the novel signaling lipid, N-arachidonoyl-L-serine was isolated from bovine brain in trace amounts (15). The biological and physiological functions of N-arachidonoyl-L-serine include vasodilation of rat isolated mesenteric arteries and abdominal aorta, and the suppression of LPS-induced TNF-alpha production by RAW macrophage tumor cells (15).
We envisage two possibilities for the formation and metabolism of N-acyl-PS. In the first scenario, N-acyl-PS could arise, as noted above, by a transacylation reaction, similar to the one that generates N-acyl-PE (Scheme 2). The same enzyme might generate N-acyl-PS, but the possibility of a distinct transacylase cannot be excluded. According to Scheme 2, in vitro incubation of 32P-PS with an appropriate phospholipid co-substrate, such as PC (16:0/18:1) should result in the formation of N-palmitoyl-PS, the major N-acyl-PS species seen in brain and RAW cells (Figures 1 and 10). To demonstrate the absolute dependence of N-palmitoyl-PS formation on the presence of the added palmitoyl donor PC (16:0/18:1), the transacylase may have to be partially purified to remove endogenous phospholipids. The turnover of N-acyl-PS by a phosholipase D (Scheme 2) is proposed in analogy to N-acyl-PE turnover (18). This is the most obvious mechanism for the formation of N-acyl serine. However, N-acyl-PS might first be deacylated by a phospholipase A (33, 39) and then converted to N-acyl serine by a phosphodiesterase (not shown).
Scheme 2. Proposed hypothetical pathway for the biosynthesis of N-acyl-PS and its conversion to N-acyl serine.

These reactions are analogous to those involved in the formation of N-acyl-PE and anandamide. PC, phosphatidylcholine; PS, phosphatidylserine; PA, phosphatidic acid. Presumably other glycerophospholipids can substitute as acyl donors. The predominance of the N-palmitoyl moiety in the N-acyl-PS series suggests that the N-acyl chain arises mainly from the sn-1 position of donor glycerophospholipids.
An alternative mechanism for the formation of N-acyl-PS (Scheme 3) involves the formation of N-acyl serine from acyl-coenzyme A and serine, followed by headgroup exchange with phosphatidylcholine (or phosphatidylethanolamine) to generate N-acyl-PS. Exchange reactions of this type are the route by which phosphatidylserine is synthesized in animal cells (40). Two distinct serine exchange enzymes have been identified. One is specific for phosphatidylcholine and the other for phosphatidylethanolamine (not shown) (40). These reactions are also calcium ion dependent. The relevant structural genes have been cloned and the enzymatic activities characterized (41), but N-acyl serine was not tested as a phosphatidyl group acceptor (41). Studies of the biosynthesis and degradation of N-acyl-PS are currently underway in our laboratory to distinguish between these possibilities.
Scheme 3. An alternative hypothetical pathway for the formation of N-acyl-PS.

In animal cells phosphatidylserine is generated by a headgroup exchange reaction involving phosphatidylcholine (or phosphatidylethanolamine) and free serine (40). In principle, N-acyl serine could substitute for serine. PC, phosphatidylcholine.
Acknowledgments
This research was supported by the LIPID MAPS Large Scale Collaborative Grant number GM-069338 from NIH.
We thank Dr. Teresa Garrett, Mr. Ramesh Ghattamaneni and Mr. Reza Kordestani for their expertise and assistance in RAW cell culture and lipid fractionation, and Dr. Robert Murphy for sharing the conditions of liquid chromatography for lipid analysis. This research was supported by the LIPID MAPS Large Scale Collaborative Grant number GM069338 from the NIH.
List of Abbreviations
- ESI/MS
electrospray ionization/mass spectrometry
- LC/MS
liquid chromatography/mass spectrometry
- MRM
multiple reaction monitoring
- MS/MS
tandem mass spectrometry
- N-acyl-PE
N-acyl phosphatidylethanolamine
- N-acyl-PS
N-acyl phosphatidylserine
- NAGPS
N-acyl glycerophosphoserine
- PBS
phosphate-buffered saline
References
- 1.Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Jr, Murphy RC, Raetz CRH, Russell DW, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, VanNieuwenhze MS, White SH, Witztum JL, Dennis EA. A comprehensive classification system for lipids. J Lipid Res. 2005;46:839–862. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Dennis EA, Brown HA, Deems R, Glass CK, Merrill AH, Murphy RC, Raetz CRH, Shaw W, Subramaniam S, Russell DW, van Nieuwenhze M, White SH, Witztum JL, Wooley J. The LIPID MAPS approach to lipidomics. In: Feng L, Prestwich GD, editors. Functional Lipidomics. CRC Press/Taylor and Francis Group; Boca Raton, FL: 2005. pp. 1–15. [Google Scholar]
- 3.Raetz CRH. Molecular genetics of membrane phospholipid synthesis. Annu Rev Genet. 1986;20:253–295. doi: 10.1146/annurev.ge.20.120186.001345. [DOI] [PubMed] [Google Scholar]
- 4.Zoeller RA, Wightman PD, Anderson MS, Raetz CRH. Accumulation of lysophosphatidylinositol in RAW 264.7 macrophage tumor cells stimulated by lipid A precursors. J Biol Chem. 1987;262:17212–17220. [PubMed] [Google Scholar]
- 5.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, Mori H, Perna NT, Plunkett G, 3rd, Rudd KE, Serres MH, Thomas GH, Thomson NR, Wishart D, Wanner BL. Escherichia coli K-12: a cooperatively developed annotation snapshot--2005. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy RC, Fiedler J, Hevko J. Analysis of nonvolatile lipids by mass spectrometry. Chem Rev. 2001;101:479–526. doi: 10.1021/cr9900883. [DOI] [PubMed] [Google Scholar]
- 7.Han X, Yang J, Cheng H, Ye H, Gross RW. Toward fingerprinting cellular lipidomes directly from biological samples by two-dimensional electrospray ionization mass spectrometry. Anal Biochem. 2004;330:317–331. doi: 10.1016/j.ab.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 8.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 9.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 10.Pulfer M, Murphy RC. Electrospray mass spectrometry of phospholipids. Mass Spectrom Rev. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 11.Ivanova PT, Cerda BA, Horn DM, Cohen JS, McLafferty FW, Brown HA. Electrospray ionization mass spectrometry analysis of changes in phospholipids in RBL-2H3 mastocytoma cells during degranulation. Proc Natl Acad Sci U S A. 2001;98:7152–7157. doi: 10.1073/pnas.131195098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan Z, Breazeale SD, Raetz CRH. Extraction and identification by mass spectrometry of undecaprenyl diphosphate-MurNAc-pentapeptide-GlcNAc from Escherichia coli. Anal Biochem. 2005;345:336–339. doi: 10.1016/j.ab.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Donohue TJ, Cain BD, Kaplan S. Purification and characterization of an N-acylphosphatidylserine from Rhodopseudomonas sphaeroides. Biochemistry. 1982;21:2765–2773. doi: 10.1021/bi00540a029. [DOI] [PubMed] [Google Scholar]
- 14.Schmid PC, Kumar VV, Weis BK, Schmid HHO. Phosphatidyl-Tris rather than N-acylphosphatidylserine is synthesized by Rhodopseudomonas sphaeroides grown in Tris-containing media. Biochemistry. 1991;30:1746–1751. doi: 10.1021/bi00221a003. [DOI] [PubMed] [Google Scholar]
- 15.Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Batkai S, Mo FM, Offertaler L, Pacher P, Kunos G, Mechoulam R. N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci U S A. 2006;103:2428–2433. doi: 10.1073/pnas.0510676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 17.Cravatt BF, Lichtman AH. The endogenous cannabinoid system and its role in nociceptive behavior. J Neurobiol. 2004;61:149–160. doi: 10.1002/neu.20080. [DOI] [PubMed] [Google Scholar]
- 18.Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, Sullivan PM. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18:390–398. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Dulbecco R, Vogt M. Plaque formation and isolation of pure lines with poliomyelitis viruses. J Exp Med. 1954;99:167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bligh EG, Dyer JJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 22.Raetz CRH, Kennedy EP. Function of cytidine diphosphate-diglyceride and deoxycytidine diphosphate-diglyceride in the biogenesis of membrane lipids in Escherichia coli. J Biol Chem. 1973;248:1098–1105. [PubMed] [Google Scholar]
- 23.Zhou Z, Lin S, Cotter RJ, Raetz CRH. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy- L-arabinose, phosphoethanolamine and palmitate. J Biol Chem. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 24.Raetz CRH, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Jr, Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, Dennis EA. (Kdo)2-lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Radika K, Raetz CRH. Purification and properties of lipid A disaccharide synthase of Escherichia coli. J Biol Chem. 1988;263:14859–14867. [PubMed] [Google Scholar]
- 26.Hsu FF, Turk J. Charge-remote and charge-driven fragmentation processes in diacyl glycerophosphoethanolamine upon low-energy collisional activation: a mechanistic proposal. J Am Soc Mass Spectrom. 2000;11:892–899. doi: 10.1016/S1044-0305(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 27.Hsu FF, Turk J. Studies on phosphatidylserine by tandem quadrupole and multiple stage quadrupole ion-trap mass spectrometry with electrospray ionization: structural characterization and the fragmentation processes. J Am Soc Mass Spectrom. 2005;16:1510–1522. doi: 10.1016/j.jasms.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 28.Nelson GJ. Studies on the lipids of sheep red blood cells. IV The identification of a new phospholipid N-acyl phosphatidyl serine. Biochem Biophys Res Commun. 1970;38:261–265. doi: 10.1016/0006-291x(70)90706-0. [DOI] [PubMed] [Google Scholar]
- 29.Epps DE, Natarajan V, Schmid PC, Schmid HO. Accumulation of N-acylethanolamine glycerophospholipids in infarcted myocardium. Biochim Biophys Acta. 1980;618:420–430. doi: 10.1016/0005-2760(80)90260-x. [DOI] [PubMed] [Google Scholar]
- 30.Natarajan V, Reddy PV, Schmid PC, Schmid HH. N-Acylation of ethanolamine phospholipids in canine myocardium. Biochim Biophys Acta. 1982;712:342–355. doi: 10.1016/0005-2760(82)90352-6. [DOI] [PubMed] [Google Scholar]
- 31.Reddy PV, Natarajan V, Schmid PC, Schmid HH. N-Acylation of dog heart ethanolamine phospholipids by transacylase activity. Biochim Biophys Acta. 1983;750:472–480. doi: 10.1016/0005-2760(83)90187-x. [DOI] [PubMed] [Google Scholar]
- 32.Natarajan V, Schmid PC, Reddy PV, Zuzarte-Augustin ML, Schmid HHO. Biosynthesis of N-acylethanolamine phospholipids by dog brain preparations. J Neurochem. 1983;41:1303–1312. doi: 10.1111/j.1471-4159.1983.tb00825.x. [DOI] [PubMed] [Google Scholar]
- 33.Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem. 2006;281:26465–26472. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- 34.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 35.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 36.Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9:663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 37.Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–825. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- 38.Maccarrone M, Pauselli R, Di Rienzo M, Finazzi-Agro A. Binding, degradation and apoptotic activity of stearoylethanolamide in rat C6 glioma cells. Biochem J. 2002;366:137–144. doi: 10.1042/BJ20020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N. Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J. 2004;380:749–756. doi: 10.1042/BJ20040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuge O, Nishijima M. Phosphatidylserine synthase I and II of mammalian cells. Biochim Biophys Acta. 1997;1348:151–156. doi: 10.1016/s0005-2760(97)00137-9. [DOI] [PubMed] [Google Scholar]
- 41.Stone SJ, Vance JE. Cloning and expression of murine liver phosphatidylserine synthase (PSS)-2: differential regulation of phospholipid metabolism by PSS1 and PSS2. Biochem J. 1999;342(Pt 1):57–64. [PMC free article] [PubMed] [Google Scholar]