

Abstract

Facile synthesis of C-4 aryl pyrimidinone nucleoside analogues from an easily prepared O4-arylsulfonate derivative of thymidine is reported. Two O4-arylsulfonylthymidine precursors, (4-methylphenyl)sulfonyl and (2,4,6-trimethylphenyl)sulfonyl, were prepared and analyzed for their stabilities. Of the two, the latter possessed suitable stability as well as reactivity for Pd-catalyzed C-C bond forming reactions with a variety of arylboronic acids. These reactions at the C-4 position are non-trivial in comparison with similar reactions at the C-5 position of pyrimidine nucleosides, with hydrolysis of the arylsulfonate precursor being a competing reaction in some cases. There are pronounced solvent influences in these reactions, but successful reactions can be attained by careful control of conditions. Many reactions proceeded efficiently at room temperature and electron-deficient arylboronic acids can also be cross-coupled under suitable conditions. Desilylation of these products was also non-trivial and various conditions were tested. Finally, anti-viral screening was performed with the C-4 aryl pyrimidinone nucleoside analogues, but none possessed any interesting activity. The study represents the first successful synthesis of C-4 aryl pyrimidinone nucleoside analogues by cross coupling of arylboronic acids with an arylsulfonate derived from a pyrimidine nucleoside, as well as antiviral testing of this new class of compounds.
INTRODUCTION
The pharmacological importance and potential pharmaceutical value of nucleoside analogs has fueled the development of novel methods for their synthesis as well as the quest for new compounds.1,2 In this context, metal-catalyzed chemistry offers new directions for nucleoside modification strategies.3
Pd- and Ni-mediated Suzuki-Miyaura reactions of halo or azolyl nucleoside derivatives with arylboronic acids have provided facile access to aryl-linked nucleoside analogues.4 Some of these aryl-nucleoside derivatives have shown anti-HCV as well as cytostatic activity that augments the importance of synthetic access to such compounds.5 In this relation, we have been particularly interested in understanding the basis of the catalysis chemistry as well as reactivity patterns, and have reported C-C bond-forming reactions of 6-bromo and chloropurine nucleosides6 as well as O6-arylsulfonates of 2′-deoxyguanosine.7,8 During the course of our prior work we have demonstrated that, in contrast to simpler halo aromatic systems, C-6 chloropurine nucleosides are highly effective coupling partners in C-C bond forming reactions with arylboronic acids.6 We have also shown that easily synthesized O6-arylsulfonates of 2′-deoxyguanosine are particularly important substrates7,8 since the C-6 chlorination of 2′-deoxyguanosine is not trivial.9 We have additionally demonstrated that 2′-deoxyguanosine O6-arylsulfonates undergo Pd-catalyzed C-C bond forming reactions with arylboronic acids under mild conditions and at room temperature, indicating their excellent reactivity and utility.8
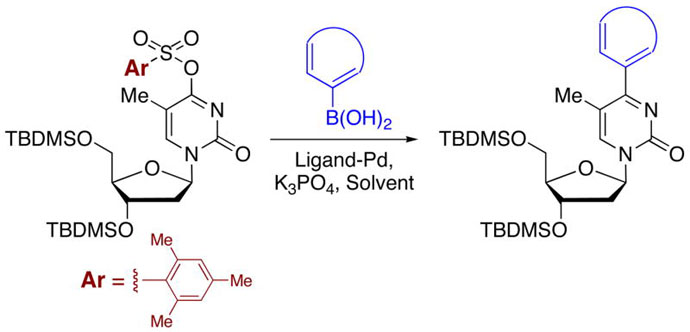
On the basis of these results we were interested in exploring whether O4-arylsulfonate derivatives of pyrimidine nucleosides could be utilized for Pd-catalyzed C-C bond forming reactions with arylboronic acids. Whereas C-5 modifications via such cross coupling are known in the literature,10 to our knowledge, comparable methods for modification at the C-4 are not. Introduction of a leaving group into the C-4 position of a pyrimidine nucleoside places it at the β-position of an α,β-unsaturated system. Although metal-catalyzed C-C bond-forming reactions of enol sulfonates (tosylate and triflate) derived from β-keto amides and lactones with organoboron derivatives are known,11 we have shown that the chemistry of simpler molecules is often not applicable to the more complex nucleosides. Furthermore, while this work was in progress, Negishi and Stille reactions of some uridine O4-(2,4,6-triisopropylphenyl)sulfonate derivatives were reported. That work was specifically prompted by unsuccessful attempts at Suzuki-Miyaura and Heck reactions.12
RESULTS AND DISCUSSION
Our chemistry commenced with the readily available thymidine that was converted to its 3′,5′-bis-O(tert-butyldimethylsilyl) derivative 1 (TBDMSCl, imidazole, DMF, room temperature, 24 h). Conversion of 1 to the O4-arylsulfonates was attempted via two methods. Initially, following a literature procedure 1 was deprotonated with NaH in THF and allowed to react with ArSO2Cl (Ar = 2,4,6-trimethylphenyl and 4-methylphenyl).13 Although products 2 and 3 were available via this route, 2 was contaminated with ~14% of a byproduct that was thought to be the N-sulfonyl derivative but was not rigorously characterized. Also, this procedure was not readily amenable to a larger scale synthesis of 2. An alternative method14 that utilized Et3N, DMAP in CH2Cl2 for sulfonylation with ArSO2Cl was found to be more suitable for the preparation of 2.
Since the O4-(2,4,6-trimethylphenyl)sulfonyl thymidine derivative has been stated as being unstable to storage,13 we wanted to initially ascertain the stabilities of 2 and 3. In solution (CDCl3) both compounds underwent hydrolysis to 1, and 2 was hydrolytically more reactive compared to 3. However, deacidification and drying of the NMR solvent slowed this hydrolysis (~26% at room temperature over 2 weeks), and 2 was stable to storage in the refrigerator (4–5 °C) for 2 months as a solid. Although more stable compared to 2, about 40% hydrolysis of 3 occurred over a 6 month period in the refrigerator (4–5 °C). Overall, 2 was more interesting to us since the relief of steric bulk could be favorable to the oxidative-addition step in the catalysis reactions, and the relative hydrolytic reactivity may be reflective of this. Further, we have had excellent prior results in C-C bond forming reactions of the analogous (2,4,6-trimethyl)sulfonyl derivative of 2′-deoxyguanosine and arylboronic acids.7,8
Initial experiments focused on conditions that we have shown to be successful for Pd-catalyzed C-C bond formation at room temperature.8 Using the catalyst comprised of 10 mol % Pd(OAc)2/20 mol % (2-dicyclohexylphosphino)-1,1′-biphenyl, a reaction of 2 and 2 molar equiv of PhB(OH)2 was conducted at room temperature in the presence of 2 molar equiv of K3PO4in toluene as solvent. This reaction was complete within 15 h and produced a 95% yield of the C-4 aryl product 4a accompanied by a trace amount of 1. When the reaction was conducted for 7 h, the yield of 4a dropped to 84% and the amount of 1 increased. When the ligand was changed to 2-(di-tert-butylphosphino)-1,1′-biphenyl, the reaction yielded only 6% of 4a, 79% of 1 and 15% of unreacted 2 (room temperature, 15 h). Use of Pd(PPh3)4 was also inferior resulting in 33% of 4a, 19% of 1 and 48% of 2 remaining unreacted (NMR analysis of the crude product mixture from a room temperature reaction after 50 h). Although Pd(PPh3)4 has been used to catalyze C-C bond formation of nucleoside derivatives and arylboronic acids,4e,f,5,10b,c the superiority of (2-dicyclohexylphosphino)-1,1′-biphenyl based catalysts for such conversions has been noted by others.15 On the basis of these initial results, the generality of the transformation was assessed, and the results are compiled in Table 1.
Table 1.
Pd-catalyzed C-C bond forming reactions leading to C-4 aryl pyrimidine nucleoside analoguesa
 | |||
|---|---|---|---|
| entry | Ar = | conditions | product: % yield,b (1)c,d |
| 1 |
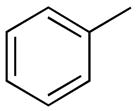
|
PhMe, rt, 15 h | 4a: 95%, (trace) |
| 2 |
|
PhMe, rt, 15 h | 4b: 97%, (trace) |
| 3 |
|
PhMe, rt, 15 h | 4c: 94%, (trace) |
| 4 |
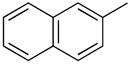
|
PhMe, rt, 15 h | 4d: 90%, (trace) |
| 5 |
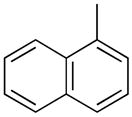
|
PhMe, rt, 15 h | 4e: 91%, (trace) |
| 6 |
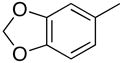
|
PhMe, rt, 15 h | 4f: 84%, (trace) |
| 7 |
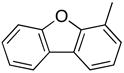
|
PhMe, rt, 15 h | 4g: 49%, (18%) |
| 8 | THP, 50 °C, 15 h | 4g: 47%, (24%) | |
| 9 | THP, 90 °C, 15 h | 4g: 74%, (trace) | |
| 10 |
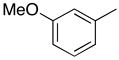
|
PhMe, rt, 15 h | 4h: 39%, (35%) |
| 11 | THP, 50 °C, 15 h | 4h: 54%, (27%) | |
| 12 | THP, 90 °C, 15 h | 4h: 81%, (~20%d) | |
| 13 |
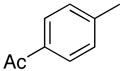
|
PhMe, 90 °C, 18 h | 4i: 59%, (30%) |
| 14 | THP, 90 °C, 15 h | 4i: 77%, (~20%d) | |
| 15 | 1,4-Dioxane, 90 °C, 15 h | 4i: 79%, (~20%d) | |
| 16 |
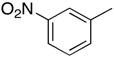
|
PhMe, 90 °C, 15 h | 4j: 0%,d (majord) |
| 17 | THP, 90 °C, 15 h | 4j: 22%,d (~78%d) | |
| 18 | THP, 50 °C, 15 h | 4j: 59%, (20%) | |
| 19 | 1,4-dioxane, 50 °C, 15 h | 4j: 0%,d (majord) | |
Reactions were conducted using 10 mol % Pd(OAc)2, 20 mol % (2-dicyclohexylphosphino)-1,1′-biphenyl, 2 molar equiv K3PO4 and 2 molar equiv ArB(OH)2 (0.14 M nucleoside concentration).
Isolated yields, unless specified otherwise.
The amount of 1 produced was determined either by isolation or from the 1H NMR data of the crude reaction mixture.
On the basis of 1H NMR the amount of 1 produced is an approximate estimation based on integration values relative to 4.
From the data presented in Table 1 it is clear that substrate 2 can be effectively used for C-C bond formation via Pd catalysis. Several reactions proceeded well at room temperature (entries 1–6), which was 24–29 °C during the course of the experimentation. Others (entries 7 and 10) showed progress at room temperature but were not complete. As we have previously reported for reactions of O6-[(2,4,6-trimethylphenyl)sulfonyl]-3′,5′-bis-O-(tert-butyldimethyl-silyl)-2′-deoxyguanosine,8 tetrahydropyran (THP) proved to be an effective solvent for those reactions that were inefficient in PhMe. Reactions of dibenzofuranyl- and 3-methoxyphenylboronic acids proceeded efficiently in THP at 90 °C (entries 9 and 12) but were less efficient at 50 °C (entries 8 and 11). Cross-coupling reactions of electron-deficient arylboronic acids can be generally difficult16 and such boronic acids are also known to undergo competitive protio-deboronation17,18 as well as dimerization.19 We have seen that elevated temperatures and the use of THP as solvent provides reasonable cross-coupling results.8 Consistent with our expectation the reaction of 4-acetylphenylboronic acid with 2 in PhMe at room temperature afforded 16% of 4i, 15% of 1 with 69% of 2 remaining unreacted (by 1H NMR of the crude product mixture). In PhMe at 90 °C, a yield improvement was observed (entry 13), but in THP at 90 °C a 77% yield of 4i was obtained (entry 14). Interestingly, 1,4-dioxane also proved to be a comparably good solvent in this case (entry 15). With 3-nitrophenylboronic acid some other interesting influences were observed. In PhMe at 90 °C, no product formation occurred whereas in THP a small amount of product formation was observed, but conversion to 1 predominated in each case (entries 16 and 17). On the other hand, in THP at 50 °C reasonably good conversion to 4j was observed (entry 18). Replacing THP with 1,4-dioxane abolished the product-forming pathway again resulting in the formation of 1. With some differences, these solvent effects parallel those observed in the C-C bond forming reactions of O6-[(2,4,6-trimethylphenyl)sulfonyl]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine.8 Solvent does seem to play an important, but presently unknown role in these reactions. We feel that coordinating abilities of solvents as well as solvent basicities could have subtle but significant influences in these reactions.
We also briefly tested 3 for its reactivity towards PhB(OH)2. In PhMe at room temperature, after 3 h an 81% yield of 4a (accompanied by ~19% 1) was obtained. However, a comparable reaction after 15 h at room temperature yielded 4a in 95% (and a trace amount of 1). At 50 °C, a 3 h reaction time led to the isolation of 4a in 87% yield with no detectable formation of 1. Thus, 2 also appears to be suitably reactive in these transformations, but a limitation to the use of any specific precursor could be the duration for which the thymidine O4-arylsulfonates can be stored.
Since these novel thymidine derivatives could potentially possess antiviral activity, these compounds were desilylated for testing. The desilylation proved to be non-trivial and reaction of 4a with KF (3–6 molar equiv) in MeOH at 50–80 °C resulted in the formation of some fluorescent byproducts in addition to the desired desilyl compound 5a. Attempt at milder conditions through the use of KF and 18-crown-6 in PhMe resulted in no reaction at room temperature and very slow reaction at elevated temperatures. Therefore, compounds 4a-j were deprotected with KF (3–6 molar equiv) in MeOH at 70–80 °C, and by carefully monitoring the reactions 49–85% yield of the purified products 5a-j were obtained. In contrast to the reasonably successful desilylation of 4d with KF in MeOH (50% yield), use of n-BuN4+F− in THF resulted in the formation of several fluorescent spots. Due to the unanticipated problems encountered in the desilylation of these products, we subsequently briefly investigated conditions for desilylation using 4a as a model substrate. These incuded the use of CsF in CH3CN or CH3CN-H2O,20 buffered n-BuN4+F−,21 as well as tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) in DMF.22,23 From the results shown in Table 2, it appeared that TASF in DMF or CH3CN would be a suitable desilylating agent.
The desilylated compounds were tested for antiviral activity in three types of cell cultures: HEL [Herpes simplex virus-1 (KOS), Herpes simplex virus-2 (G), Vaccinia virus, Vesicular stomatitis virus and Herpes simplex virus-1 (TK− KOS ACVr)], HeLa [Vesicular stomatitis virus, Coxsackie virus B4, Respiratory syncytial virus] and Vero [Parainfluenza-3 virus, Reovirus-1, Sindbis virus, Coxsackie virus B4 and Punta Toro virus]. Activity was also assayed against cytomegalovirus and varicella-zoster in human embryonic lung (HEL) cells. None of the compounds tested possessed a significant level of specific or potent antiviral activity. In all cases the minimal antivirally effective concentration was not more than 5-fold lower than the minimal cytotoxic concentration (most had a minimum cytotoxic concentration of >100 μg/mL). Detailed compilations of the antiviral results are presented in Tables 1–5 of the Supporting Information section.
CONCLUSIONS
In this paper we have demonstrated that an O4-arylsulfonate derivative of thymidine is a suitable substrate for the synthesis of C-4 aryl pyrimidinone nucleoside analogues. To our knowledge, this is first report on successful Suzuki-Miyaura reactions at the C-4 position utilizing an arylsulfonate of a pyrimidine nucleoside. In contrast to facile reactions at the C-5 position, with simpler catalytic systems, reactions at the C-4 are much less trivial. In some cases product formation is accompanied by small amounts of the hydrolysis product and this hydrolysis becomes more significant in cross coupling reactions that are less facile. Nevertheless good to excellent yields were obtained in all reactions studied here. Noteworthy is the fact that, via a Negishi reaction, a 60% yield of compound 4a has been reported in a reaction time of 3 days.12 The slowness of this reaction has been linked to the possibility of steric congestion at the reactive site.12 By contrast, the same compound is attainable within 15 hours at room temperature via the presently described approach, despite the presence of the C-5 methyl group adjacent to the reactive center. This is a representation of the efficiency of the reactions described. In viewing the C-C bond-forming reactions of nucleoside arylsulfonates in general, certain common features emerge. Many reactions can be accomplished at room temperature in toluene. The ligand most generally applicable is 2-(dicyclohexylphosphino)-1,1′-biphenyl usually in combination with Pd(OAc)2. Electron-deficient arylboronic acids, as anticipated, are difficult coupling partners. However, successful reactions in such cases can be attained in THP.
EXPERIMENTAL SECTION
3′,5′-Bis-O-(tert-butyldimethylsilyl)-O4-[(2,4,6-trimethylphenyl)sulfonyl]-thymidine (2)
3′,5′-Bis-O-(tert-butyldimethylsilyl)thymidine 1 (0.942 g, 2.0 mmol) and DMAP (0.024 g, 0.2 mmol) were dissolved in 10 mL of dry, distilled CH2Cl2. The solution was chilled to 0 ºC and Et3N (1.67 mL, 6 molar equiv) was added. After 30 min at this temperature, 2,4,6-trimethylphenylsulfonyl chloride (0.744 g, 3.4 mmol) was added. The reaction mixture was allowed to stir under nitrogen gas at 0 °C for 1hr and then at room temperature for an additional 30 hr. Upon completion, the reaction mixture was poured into a 1:1 mixture of hexanes–EtOAc (40 mL) and filtered through a glass funnel. The filter cake was washed with Et2O and the filtrate was concentrated to yield a brown-yellow oil. Chromatography on silica gel using 2:1 hexanes–Et2O yielded 1.170 g of 2 as a white, foamy solid (89% yield). Rf (silica gel, 2:1 hexanes–Et2O) = 0.33. 1 H NMR (500 MHz, CDCl3): δ 7.95 (s, 1H, H-6), 6.97 (s, 2H, Ar-H), 6.90 (t, 1H, H-1′, J = 6.3), 4.31 (dt, 1H, H-3′, J = 5.9, 3.3), 3.96 (app q, 1H, H-4′, J = 2.9), 3.87 (dd, 1H, H-5′, J = 11.2, 2.4), 3.74 (dd, 1H, H-5′, J = 11.2, 2.4), 2.75 (s, 6H, CH3), 2.46 (ddd, 1H, H-2′, J = 13.7, 5.9, 3.9), 2.29 (s, 3H, CH3), 2.03 (s, 3H, CH3), 1.93 (dt, 1H, H-2′, J = 13.7, 6.5), 0.89, 0.86 (2s, 18H, SiC(CH3)3), 0.09, 0.08, 0.05, 0.04 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 166.0, 153.5, 144.0, 143.0, 140.7, 131.9, 131.7, 103.3, 88.3, 87.2, 71.5, 62.5, 42.3, 25.9, 25.7, 21.1, 18.3, 17.9, 12.3, −4.6, −4.9, −5.4. HRMS calculated for C31H53N2O7SSi2 (M+ + H): 653.3112, found 653.3125.
Typical procedure for the cross coupling of 2 with arylboronic acids (Step 1 under each compound heading)
In an oven-dried, screw-cap vial equipped with a stirring bar were placed Pd(OAc)2 (1.4 mg, 6.1 μmol), 2-(dicyclohexylphosphino)-1,1′-biphenyl (4.3 mg, 12.3 μmol), K3PO4 (26.0 mg, 122.5 μmol), 3′,5′-bis-O-(tert-butyldimethylsilyl)-O4-[(2,4,6-trimethylphenyl)sulfonyl]thymidine 2 (40.0 mg, 61.3 μmol), and the arylboronic acid (2 molar equiv, 122.5 μmol). PhMe or THP or 1,4-dioxane (0.7 mL, see the Results and Discussion section for choice of solvent) was added, the vial was flushed with nitrogen gas, sealed with a Teflon-lined cap, and the mixture was allowed to stir at a suitable temperature (room temperature or 50 ºC or 90 ºC, see the Results and Discussion section). Upon completion, the reaction mixture was filtered through Celite and the residue was washed with CH2Cl2. Evaporation of the filtrate provided the crude product mixture that was purified by column chromatography on silica gel (SiO2, 230–400 mesh) using suitable ratio of hexanes–EtOAc. Details are provided under the individual compound headings. In some cases, the reactions were performed using 100 mg of 2 by appropriate modifications to stoichiometry and concentration.
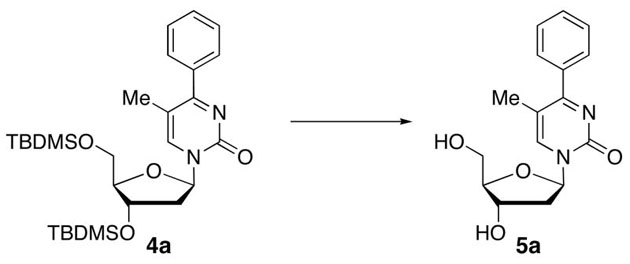
1-(2′-Deoxyribofuranosyl)-5-methyl-4-phenylpyrimidin-2(1H)-one (5a)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-5-methyl-4-phenylpyrimidin-2(1H)-one (4a).
Chromatography with 10:4 hexanes–EtOAc yielded a colorless, oily residue. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.14. 1H NMR (500 MHz, CDCl3): δ 8.08 (s, 1H, H-6), 7.62–7.58 (m, 2H, Ar-H), 7.46–7.42 (m, 3H, Ar-H), 6.33 (t, 1H, H-1′, J = 6.2), 4.40 (dt, 1H, H-3′, J = 5.9, 3.4), 4.04 (app q, 1H, H-4′, J = 2.9), 3.95 (dd, 1H, H-5′, J = 11.5, 2.7), 3.80 (dd, 1H, H-5′, J = 11.5, 2.7), 2.65 (ddd, 1H, H-2′, J = 13.7, 6.4, 3.9), 2.13 (s, 3H, CH3), 2.11 (dt, 1H, H-2′, J = 13.7, 6.7), 0.92, 0.90 (2s, 18H, SiC(CH3)3), 0.12, 0.11, 0.09, 0.08 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 174.6, 155.1, 141.9, 138.1, 129.8, 128.5, 128.1, 110.9, 88.5, 87.4, 71.7, 62.7, 42.6, 25.9, 25.8, 18.4, 18.0, 16.6, −4.5, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4a (18.8 mg, 35.4 μmol) in MeOH (0.3 mL), KF (6.2 mg, 106.2 μmol) was added and the reaction mixture was stirred at 70 °C for 40 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give 9.1 mg (85% yield) of 5a as a colorless, oily residue that upon trituration with Et2O yielded a white, cottony solid. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.12. 1H NMR (500 MHz, CD3OD): δ 8.58 (s, 1H, H-6), 7.64–7.60 (m, 2H, Ar-H), 7.56–7.49 (m, 3H, Ar-H), 6.27 (t, 1H, H-1′, J = 6.0), 4.44 (dt, 1H, H-3′, J = 5.9, 4.4), 4.07 (app q, 1H, H-4′, J = 3.7), 3.92 (dd, 1H, H-5′, J = 12.2, 2.9), 3.81 (dd, 1H, H-5′, J = 12.0, 3.7), 2.60 (ddd, 1H, H-2′, J = 13.7, 6.2, 4.4), 2.28 (dt, 1H, H-2′, J = 13.7, 6.1), 2.17 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 175.2, 155.9, 144.1, 137.8, 130.1, 128.3, 128.3, 112.8, 88.4, 87.8, 70.1, 61.0, 41.5, 15.2. HRMS calculated for C16H19N2O4 (M+ + H): 303.1345, found: 303.1348.
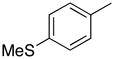
1-(2′-Deoxyribofuranosyl)-5-Methyl-4-[4-(methylsulfanyl)phenyl]pyrimidin-2(1H)-one (5b)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-5-methyl-4-[4-(methylsulfanyl)phenyl]pyrimidin-2(1H)-one (4b).
Chromatography with 10:3 hexanes–EtOAc yielded a pale yellow, oily residue. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.16. 1H NMR (500 MHz, CDCl3): δ 8.05 (s, 1H, H-6), 7.58 (d, 2H, Ar-H, J = 8.2), 7.28 (d, 2H, Ar-H, J = 8.2), 6.32 (t, 1H, H-1′, J = 6.4), 4.40 (dt, 1H, H-3′, J = 6.1, 3.4), 4.03 (app q, 1H, H-4′, J ~ 3.0), 3.95 (dd, 1H, H-5′, J = 11.3, 2.8), 3.80 (dd, 1H, H-5′, J = 11.3, 2.4), 2.64 (ddd, 1H, H-2′, J = 13.5, 6.3, 3.9), 2.52 (s, 3H, SCH3), 2.15 (s, 3H, CH3), 2.10 (dt, 1H, H-2′, J = 13.4, 6.5), 0.92, 0.90 (2s, 18H, SiC(CH3)3), 0.12, 0.11, 0.08, 0.07 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 173.5, 155.1, 142.1, 141.6, 134.2, 129.2, 125.3, 110.9, 88.4, 87.2, 71.6, 63.6, 42.5, 25.9, 25.7, 18.4, 17.9, 16.9, 15.2, −4.6, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4b (80.0 mg, 138.6 μmol) in MeOH (1.0 mL), KF (48.3 mg, 831.9 μmol) was added and the reaction mixture was stirred at 80 °C for 24 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 15:1 CHCl3–MeOH to give a white solid that upon trituration with hexanes–EtOAc yielded 32.8 mg (68% yield) of 5b a pale yellow solid. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.16. 1H NMR (500 MHz, CD3OD): δ 8.55 (s, 1H, H-6), 7.60 (d, 2H, Ar-H, J = 8.5), 7.38 (d, 2H, Ar-H, J = 8.5), 6.26 (t, 1H, H-1′, J = 6.1), 4.43 (dt, 1H, H-3′, J = 6.4, 4.4), 4.06 (app q, 1H, H-4′, J ~ 3.6), 3.92 (dd, 1H, H-5′, J = 12.2, 2.9), 3.81 (dd, 1H, H-5′, J = 12.2, 3.4), 2.59 (ddd, 1H, H-2′, J = 13.7, 6.2, 4.4), 2.56 (s, 3H, SCH3), 2.27 (dt, 1H, H-2′, J = 13.7, 6.1), 2.20 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 174.3, 155.9, 144.1, 142.7, 133.8, 129.1, 125.2, 112.8, 88.4, 87.7, 70.1, 61.1, 41.5, 15.5, 13.8. HRMS calculated for C17H21N2O4S (M+ + H): 349.1222, found: 349.1209.
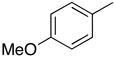
1-(2′-Deoxyribofuranosyl)-4-(4-methoxyphenyl)-5-methylpyrimidin-2(1H)-one (5c)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-4-(4-methoxyphenyl)-5-methylpyrimidin-2(1H)-one (4c).
Chromatography with 10:4 hexanes–EtOAc yielded a pale yellow, oily residue. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.10. 1H NMR (500 MHz, CDCl3): δ 8.03 (s, 1H, H-6), 7.64 (d, 2H, Ar-H, J = 8.8), 6.95 (d, 2H, Ar-H, J = 8.8), 6.33 (t, 1H, H-1′, J = 6.4), 4.40 (dt, 1H, H-3′, J = 5.9, 3.3), 4.02 (app q, 1H, H-4′, J ~ 2.7), 3.95 (dd, 1H, H-5′, J = 11.2, 2.9), 3.86 (s, 3H, OCH3), 3.80 (dd, 1H, H-5′, J = 11.2, 2.4), 2.63 (ddd, 1H, H-2′, J = 13.5, 6.2, 4.0), 2.17 (s, 3H, CH3), 2.09 (dt, 1H, H-2′, J = 13.7, 6.5), 0.92, 0.90 (2s, 18H, SiC(CH3)3), 0.12, 0.11, 0.08, 0.07 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 173.6, 161.2, 155.2, 141.9, 130.6, 130.2, 113.5, 110.9, 88.3, 87.2, 71.6, 62.6, 55.3, 42.5, 25.9, 25.7, 18.4, 17.9, 17.1, −4.6, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4c (83.6 mg, 149.1 μmol) in MeOH (1.0 mL), KF (51.9 mg, 834.3 μmol) was added and the reaction mixture was stirred at 80 °C for 24 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give a white solid that upon trituration with Et2O yielded 32.0 mg (82% yield) of 5c a pale yellow solid. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.15. 1H NMR (500 MHz, CD3OD): δ 8.52 (s, 1H, H-6), 7.66 (d, 2H, Ar-H, J = 8.5), 7.06 (d, 2H, Ar-H, J = 8.5), 6.27 (t, 1H, H-1′, J = 6.0), 4.45–4.42 (m, 1H, H-3′), 4.05 (app q, 1H, H-4′, J =3.6), 3.92 (dd, 1H, H-5′, J = 12.0, 3.2), 3.88 (s, 3H, OCH3), 3.80 (dd, 1H, H-5′, J = 12.2, 3.4), 2.58 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.9), 2.26 (dt, 1H, H-2′, J = 13.7, 6.2), 2.22 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 174.3, 161.9, 155.9, 143.8, 130.5, 129.8, 113.6, 112.8, 88.4, 87.7, 70.1, 61.1, 54.7, 41.5, 15.7. HRMS calculated for C17H21N2O5 (M+ + H): 333.1450, found: 333.1443.
1-(2′-Deoxyribofuranosyl)-5-methyl-4-(2-naphthyl)pyrimidin-2(1H)-one (5d)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-5-methyl-4-(2-naphthyl)pyrimidin-2(1H)-one (4d).
Chromatography with 10:4 hexanes–EtOAc yielded a pale yellow, oily residue. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.20. 1H NMR (500 MHz, CDCl3): δ 8.13 (m, 1H, Ar-H), 8.12 (s, 1H, H-6), 7.91–7.87 (m, 3H, Ar-H), 7.73–7.71 (m, 1H, Ar-H), 7.57–7.51 (m, 2H, Ar-H), 6.36 (t, 1H, H-1′, J = 6.4), 4.42 (dt, 1H, H-3′, J = 6.1, 3.4), 4.05 (app q, 1H, H-4′, J ~ 2.9), 3.97 (dd, 1H, H-5′, J = 11.6, 2.8), 3.82 (dd, 1H, H-5′, J = 11.6, 2.4), 2.67 (ddd, 1H, H-2′, J = 13.4, 6.1, 4.0), 2.20 (s, 3H, CH3), 2.16 (dt, 1H, H-2′, J = 13.4, 6.5), 0.94, 0.90 (2s, 18H, SiC(CH3)3), 0.14, 0.13, 0.09, 0.08 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 174.3, 155.1, 142.1, 135.3, 133.8, 132.7, 128.9, 128.8, 127.8, 127.7, 127.2, 126.5, 125.7, 111.2, 88.4, 87.3, 71.6, 62.6, 42.6, 25.9, 25.8, 18.4, 17.9, 16.9, −4.5, −4.91 −5.4.
Step 2
Desilylation.
To a solution of 4d (30.5 mg, 52.5 μmol) in MeOH (0.5 mL), KF (9.2 mg, 157.5 μmol) was added and the reaction mixture was stirred at 70 °C for 36 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 15:1 CHCl3–MeOH to give a pale yellow residue that upon trituration with hexanes and then Et2O yielded 9.0 mg (49% yield) of 5d a pale yellow powder. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.14. 1H NMR (500 MHz, CD3OD): δ 8.63 (s, 1H, H-6), 8.16 (s, 1H, Ar-H), 8.02–7.98 (m, 2H, Ar-H), 7.97–7.94 (m, 1H, Ar-H), 7.73 (dd, 1H, Ar-H, J = 8.3, 1.5), 7.62–7.57 (m, 2H, Ar-H), 6.30 (t, 1H, H-1′, J = 6.1), 4.46 (dt, 1H, H-3′, J = 6.3, 4.5), 4.08 (app q, 1H, H-4′, J = 3.6), 3.94 (dd, 1H, H-5′, J = 12.2, 2.9), 3.83 (dd, 1H, H-5′, J = 12.2, 3.4), 2.62 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.4), 2.31 (dt, 1H, H-2′, J = 13.7, 6.1), 2.24 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 175.1, 155.9, 144.2, 135.0, 134.2, 133.0, 128.6, 128.5, 128.0, 127.6, 127.4, 126.7, 125.3, 113.1, 88.5, 87.8, 70.1, 61.1, 41.5, 15.4. HRMS calculated for C20H21N2O4 (M+ + H): 353.1501, found: 353.1491.
Desilylation of 4d (39.2 mg, 67.5 mmol) with CsF (51.3 mg, 337.4 mmol) in 10:1 CH3CN–H2O (1.1 mL) at 90 °C over 24 h yielded a 73% yield of 5d after workup and chromatography.
1-(2′-Deoxyribofuranosyl)-5-methyl-4-(1-naphthyl)pyrimidin-2(1H)-one (5e)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-5-methyl-4-(1-naphthyl)pyrimidin-2(1H)-one (4e).
Chromatography with 10:4 hexanes–EtOAc yielded a pale yellow, oily residue. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.16. 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H, H-6), 7.91 (t, 2H, Ar-H, J = 8.9), 7.61 (d, 1H, Ar-H, J = 8.2), 7.54–7.42 (m, 4H, Ar-H), 6.39 (t, 1H, H-1′, J = 6.4), 4.45 (dt, 1H, H-3′, J = 5.8, 3.3), 4.12–4.08 (m, 1H, H-4′), 3.99 (dd, 1H, H-5′, J = 11.6, 2.7), 3.83 (dd, 1H, H-5′, J = 11.6, 2.4), 2.72 (ddd, 1H, H-2′, J = 13.4, 6.1, 3.7), 2.19 (dt, 1H, H-2′, J = 13.4, 6.6), 1.81 (s, 3H, CH3), 0.92, 0.91 (2s, 18H, SiC(CH3)3), 0.123, 0.121, 0.11, 0.10 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 175.9, 155.0, 141.2, 135.7, 133.3, 129.8, 129.3, 128.4, 126.6, 126.1, 125.4, 125.1, 124.9, 112.7, 88.6, 87.6, 71.8, 62.7, 42.6, 25.9, 25.7, 18.3, 17.9, 15.5, −4.6, −4.9, −5.5.
Step 2
Desilylation.
To a solution of 4e (82.0 mg, 141.2 μmol) in MeOH (1.0 mL), KF (49.1 mg, 846.0 μmol) was added and the reaction mixture was stirred at 80 °C for 24 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give a pale yellow residue that upon trituration with Et2O yielded 27.5 mg (55% yield) of 5e a pale yellow powder. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.21. 1H NMR (500 MHz, CD3OD): δ 8.67 (s, 1H, H-6), 8.03–7.98 (m, 2H, Ar-H), 7.63–7.45 (m, 5H, Ar-H), 6.34 (t, 1H, H-1′, J = 6.1), 4.48 (dt, 1H, H-3′, J = 5.9, 4.5), 4.10 (app q, 1H, H-4′, J = 3.6), 3.95 (dd, 1H, H-5′, J = 12.2, 2.9), 3.83 (dd, 1H, H-5′, J = 12.2, 3.9), 2.66 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.9), 2.36 (dt, 1H, H-2′, J = 13.7, 6.1), 1.83 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 176.4, 155.7, 143.7, 135.5, 133.8, 129.9, 129.5, 128.5, 126.9, 126.3, 125.3, 125.1, 124.6, 114.4, 88.5, 88.0, 70.2, 61.1, 41.5, 14.1. HRMS calculated for C20H21N2O4 (M+ + H): 353.1501, found: 353.1516.
1-(2′-Deoxyribofuranosyl)-5-methyl-4-[(3,4-methylenedioxy)phenyl]pyrimidin-2(1H)-one (5f)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-5-methyl-4-[(3,4-methylenedioxy)phenyl]pyrimidin-2(1H)-one (4f).
Chromatography with 10:4 hexanes–EtOAc yielded a light grey solid. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.14. 1H NMR (500 MHz, CDCl3): δ 8.05 (s, 1H, H-6), 7.16 (d, 2H, Ar-H, J = 8.6), 7.15 (s, 1H, Ar-H), 6.86 (d, 2H, Ar-H, J = 8.6), 6.32 (t, 1H, H-1′, J = 6.3), 6.02 (s, 2H, OCH2O), 4.39 (dt, 1H, H-3′, J = 6.1, 3.4), 4.03 (app q, 1H, H-4′, J ~ 2.9), 3.95 (dd, 1H, H-5′, J = 11.5, 2.6), 3.80 (dd, 1H, H-5′, J = 11.5, 2.6), 2.63 (ddd, 1H, H-2′, J = 13.4, 6.2, 3.9), 2.16 (s, 3H, CH3), 2.07 (dt, 1H, H-2′, J = 13.4, 6.3), 0.92, 0.89 (2s, 18H, SiC(CH3)3), 0.12, 0.11, 0.08, 0.07 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 173.5, 155.1, 149.2, 145.6, 142.1, 131.7, 123.5, 110.9, 109.3, 107.9, 101.4, 88.4, 87.3, 71.6, 62.6, 42.5, 25.9, 25.7, 18.4, 18.0, 17.1, −4.6, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4f (76.0 mg, 132.2 μmol) in MeOH (0.8 mL), KF (23.0 mg, 396.6 μmol) was added and the reaction mixture was stirred at 60 °C for 64 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give a pale yellow, oily residue that upon trituration with Et2O yielded 27.4 mg (60% yield) of 5f a pale yellow powder. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.13. 1H NMR (500 MHz, CD3OD): δ 8.53 (s, 1H, H-6), 7.20 (dd, 1H, Ar-H, J = 8.0, 1.5), 7.16 (d, 1H, Ar-H, J = 1.5), 6.95 (d, 1H, Ar-H, J = 8.5), 6.26 (t, 1H, H-1′, J = 6.0), 6.06 (s, 2H, OCH2O), 4.43 (dt, 1H, H-3′, J = 6.4, 4.4), 4.05 (app q, 1H, H-4′, J ~ 3.6), 3.91 (dd, 1H, H-5′, J = 12.2, 2.9), 3.80 (dd, 1H, H-5′, J = 12.0, 3.7), 2.58 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.9), 2.26 (dt, 1H, H-2′, J = 13.7, 6.1), 2.20 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 174.2, 155.9, 149.8, 148.1, 144.0, 131.5, 123.5, 112.7, 108.9, 107.8, 101.9, 88.4, 87.7, 70.1, 61.1, 41.5, 15.6. HRMS calculated for C17H19N2O6 (M+ + H): 347.1243, found: 347.1221.
1-(2′-Deoxyribofuranosyl)-4-(4-dibenzofuranyl)-5-methylpyrimidin-2(1H)-one (5g)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-4-(4-dibenzo-furanyl)-5-methylpyrimidin-2(1H)-one (4g).
Chromatography with 10:4 hexanes–EtOAc yielded an off-white, foamy solid. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.16. 1H NMR (500 MHz, CDCl3): δ 8.18 (s, 1H, H-6), 8.05 (dd, 1H, Ar-H, J = 7.6, 1.2), 7.99 (d, 1H, Ar-H, J = 7.3), 7.6 (dd, 1H, Ar-H, J = 7.6, 1.2), 7.54 (d, 1H, Ar-H, J = 8.3), 7.47-7.45 (m, 2H, Ar-H), 7.37 (t, 1H, Ar-H, J = 7.3), 6.37 (t, 1H, H-1′, J = 6.5), 4.44 (dt, 1H, H-3′, J = 6.4, 3.3), 4.08 (app q, 1H, H-4′, J ~ 2.6), 3.99 (dd, 1H, H-5′, J = 11.7, 2.4), 3.83 (dd, 1H, H-5′, J = 11.7, 2.4), 2.70 (ddd, 1H, H-2′, J = 13.9, 6.3, 3.9), 2.19 (dt, 1H, H-2′, J = 13.7, 6.6), 2.05 (s, 3H, CH3), 0.94, 0.91 (2s, 18H, SiC(CH3)3), 0.14, 0.13, 0.10, 0.096 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 171.9, 156.1, 155.1, 152.6, 141.8, 127.8, 127.5, 124.6, 123.8, 123.1, 123.0, 122.9, 122.2, 120.8, 112.8, 111.7, 88.5, 87.6, 71.7, 62.6, 42.6, 25.9, 25.8, 18.4, 18.0, 15.4, −4.5, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4g (70.0 mg, 112.7 μmol) in MeOH (0.8 mL), KF (19.6 mg, 338.2 μmol) was added and the reaction mixture was stirred at 70 °C for 33 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give a white solid that upon trituration with Et2O yielded 37.1 mg (84% yield) of 5g a white, fluffy solid. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.18. 1H NMR (500 MHz, CD3OD): δ 8.70 (s, 1H, H-6), 8.21 (dd, 1H, Ar-H, J = 7.6, 1.2), 8.11 (d, 1H, Ar-H, J = 6.8), 7.61–7.58 (m, 2H, Ar-H), 7.55–7.51 (m, 2H, Ar-H), 7.45–7.41 (m, 1H, Ar-H), 6.32 (t, 1H, H-1′, J = 6.4), 4.48 (dt, 1H, H-3′, J = 6.4, 4.4), 4.10 (app q, 1H, H-4′, J = 3.6), 3.96 (dd, 1H, H-5′, J = 12.2, 2.9), 3.84 (dd, 1H, H-5′, J = 12.2, 3.4), 2.65 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.9), 2.34 (dt, 1H, H-2′, J = 13.7, 6.1), 2.07 (s, 3H, CH3). 13C NMR (126 MHz, 50 °C, CD3OD): δ 172.4, 156.4, 155.8, 152.6, 143.9, 127.8, 127.1, 125.0, 123.7, 123.4, 123.1, 122.5, 122.4, 120.8, 114.2, 111.4, 88.6, 88.1, 70.3, 61.2, 41.5, 13.9 (since the compound crystallized from MeOH elevated temperature was used for acquiring data). HRMS calculated for C22H21N2O5 (M+ + H): 393.1450, found: 393.1435.
1-(2′-Deoxyribofuranosyl]-4-(3-methoxyphenyl)-5-methylpyrimidin-2(1H)-one (5h)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-4-(3-methoxy-phenyl)-5-methylpyrimidin-2(1H)-one (4h).
Chromatography with 2:1 hexanes–EtOAc yielded a pale yellow, oily product. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.13. 1H NMR (500 MHz, CDCl3): δ 8.08 (s, 1H, H-6), 7.34 (t, 1H, Ar-H, J = 8.1), 7.15–7.14 (m, 2H, Ar-H), 7.00–6.98 (m, 1H, Ar-H), 6.32 (t, 1H, H-1′, J = 6.3), 4.40 (dt, 1H, H-3′, J = 6.4, 3.4), 4.04 (app q, 1H, H-4′, J ~ 2.8), 3.95 (dd, 1H, H-5′, J = 11.5, 2.7), 3.84 (s, 3H, OCH3), 3.80 (dd, 1H, H-5′, J = 11.5, 2.7), 2.65 (ddd, 1H, H-2′, J = 13.7, 6.4, 3.9), 2.12 (s, 3H, CH3), 2.11 (m, 1H, H-2′), 0.917, 0.896 (2s, 18H, SiC(CH3)3), 0.12, 0.11, 0.08, 0.075 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 174.3, 159.4, 154.9, 142.2, 139.0, 129.1, 120.8, 116.1, 113.7, 111.1, 88.5, 87.4, 71.6, 62.6, 55.4, 42.6, 25.9, 25.8, 18.4, 18.0, 16.7, −4.5, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4h (59.0 mg, 105.2 μmol) in MeOH (0.8 mL), KF (18.3 mg, 315.6 μmol) was added and the reaction mixture was stirred at 70 °C for 33 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give a pale yellow, oily residue that upon trituration with Et2O yielded 24.0 mg (69% yield) of 5h an off-white powder. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.12. 1H NMR (500 MHz, CD3OD): δ 8.58 (s, 1H, H-6), 7.42 (t, 1H, Ar-H, J = 7.8), 7.17–7.15 (m, 2H, Ar-H), 7.10–7.08 (m, 1H, Ar-H), 6.27 (t, 1H, H-1′, J = 6.1), 4.44 (dt, 1H, H-3′, J = 5.9, 4.5), 4.07 (app q, 1H, H-4′, J = 3.6), 3.92 (dd, 1H, H-5′, J = 12.2, 2.9), 3.86 (s, 3H, OCH3), 3.81 (dd, 1H, H-5′, J = 12.2, 3.9), 2.60 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.4), 2.27 (dt, 1H, H-2′, J = 13.7, 6.1), 2.16 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 175.1, 159.9, 155.8, 144.1, 139.0, 129.4, 120.5, 115.8, 113.7, 112.8, 88.4, 87.8, 70.1, 61.1, 54.7, 41.5, 15.2. HRMS calculated for C17H21N2O5 (M+ + H): 333.1450, found: 333.1432.
Desilylation of 4h (53.7 mg. 95.7 mmol) through the use of 1.3 M TASF in DMF (0.37 mL, 131.9 mg, 478.7 mmol) in DMF (1 mL) from 0 °C (2 h) to room temperature over 20 h provided a 79% yield of 5h after workup and chromatography.
1-(2′-Deoxyribofuranosyl)-4-(4-acetylphenyl)-5-methylpyrimidin-2(1H)-one (5i)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-4-(4-acetylphenyl)-5-methylpyrimidin-2(1H)-one (4i).
Chromatography with 1:1 hexanes–EtOAc yielded a pale yellow, foamy solid. Rf (silica gel, 1:1 hexanes–EtOAc) = 0.44. 1H NMR (500 MHz, CDCl3): δ 8.13 (s, 1H, H-6), 8.03 (d, 2H, Ar-H, J = 8.3), 7.69 (d, 2H, Ar-H, J = 8.3), 6.32 (t, 1H, H-1′, J = 6.4), 4.40 (dt, 1H, H-3′, J = 6.5, 3.3), 4.05 (app q, 1H, H-4′, J ~ 2.8), 3.96 (dd, 1H, H-5′, J = 11.2, 2.4), 3.80 (dd, 1H, H-5′, J = 11.7, 2.4), 2.66 (ddd, 1H, H-2′, J = 13.2, 6.4, 3.4), 2.65 (s, 3H, COCH3), 2.11 (s, 3H, CH3), 2.11 (dt, 1H, H-2′, J = 13.2, 6.5), 0.92, 0.90 (2s, 18H, SiC(CH3)3), 0.12, 0.11, 0.09, 0.08 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 197.6, 173.4, 154.8, 142.7, 142.1, 137.8, 128.8, 128.1, 110.8, 88.5, 87.5, 71.6, 62.6, 42.6, 29.7, 26.7, 25.9, 25.7, 18.4, 18.0, 16.4, −4.5, −4.9, −5.4.
Step 2
Desilylation.
To a solution of 4i (93.9 mg, 163.9 μmol) in MeOH (0.8 mL), KF (28.6 mg, 491.7 μmol) was added and the reaction mixture was stirred at 60 °C for 30 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give an off-white solid that upon trituration with Et2O yielded 47.3 mg (84% yield) of 5i a yellow solid. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.19. 1H NMR (500 MHz, CD3OD): δ 8.63 (s, 1H, H-6), 8.13 (d, 2H, Ar-H, J = 8.0), 7.74 (d, 2H, Ar-H, J = 7.5), 6.27 (t, 1H, H-1′, J = 6.0), 4.44 (app q, 1H, H-3′, J ~ 5.0), 4.07 (app q, 1H, H-4′, J = 3.6), 3.93 (dd, 1H, H-5′, J = 12.2, 2.9), 3.81 (dd, 1H, H-5′, J = 12.2, 3.4), 2.67 (s, 3H, COCH3), 2.61 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.9), 2.28 (dt, 1H, H-2′, J = 13.7, 6.1), 2.16 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 198.6, 174.2, 155.7, 144.5, 142.1, 138.1, 128.7, 128.3, 112.7, 88.5, 87.9, 70.1, 61.0, 41.5, 25.7, 15.0. HRMS calculated for C18H21N2O5 (M+ + H): 345.1450, found: 345.1448.
1-(2′-Deoxyribofuranosyl)-5-methyl-4-(3-nitrophenyl)pyrimidin-2(1H)-one (5j)
Step 1
Synthesis of 1-[3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyribofuranosyl]-5-methyl-4-(3-nitrophenyl)pyrimidin-2(1H)-one (4j).
Chromatography using sequential elution with 10:1 and then 1:1 hexanes–EtOAc yielded a colorless, oily residue. Rf (silica gel, 10:4 hexanes–EtOAc) = 0.11. 1H NMR (500 MHz, CDCl3): δ 8.47 (br s, 1H, Ar-H), 8.34–8.32 (m, 1H, Ar-H), 8.20 (s, 1H, H-6), 7.99–7.97 (m, 1H, Ar-H), 7.66 (t, 1H, Ar-H, J = 7.8), 6.31 (t, 1H, H-1′, J = 6.0), 4.41 (dt, 1H, H-3′, J = 5.9, 3.4), 4.06 (app q, 1H, H-4′, J = 2.8), 3.97 (dd, 1H, H-5′, J = 11.7, 2.4), 3.81 (dd, 1H, H-5′, J = 11.7, 2.4), 2.67 (ddd, 1H, H-2′, J = 13.7, 6.4, 3.9), 2.16 (s, 3H, CH3), 2.11 (dt, 1H, H-2′, J = 13.7, 6.6), 0.92, 0.90 (2s, 18H, SiC(CH3)3), 0.13, 0.12, 0.09, 0.08 (4s, 12H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 171.8, 154.8, 147.9, 143.3, 139.4, 134.7, 129.5, 124.6, 123.6, 110.5, 88.6, 87.6, 71.6, 62.6, 42.6, 25.9, 25.7, 18.4, 18.0, 16.5, −4.5, −4.9, −5.3.
Step 2
Desilylation.
To a solution of 4j (35.0 mg, 60.8 μmol) in MeOH (0.5 mL), KF (10.6 mg, 182.3 μmol) was added and the reaction mixture was stirred at 70 °C for 30 h. After cooling, silica gel was added to the reaction mixture and the MeOH was evaporated under reduced pressure. The resulting compound-impregnated silica gel was loaded onto a silica gel column and eluted with 10:1 CHCl3–MeOH to give a pale yellow solid that upon trituration with hexanes and then Et2O yielded 9.2 mg (77% yield) of 5j a pale yellow solid. Rf (silica gel, 10:1 CHCl3–MeOH) = 0.15. 1H NMR (500 MHz, CD3OD): δ 8.68 (s, 1H, H-6), 8.52 (br s, 1H, Ar-H), 8.45–8.39 (m, 1H, Ar-H), 8.07–8.04 (m, 1H, Ar-H), 7.79 (t, 1H, Ar-H, J = 7.8), 6.27 (t, 1H, H-1′, J = 6.1), 4.46–4.43 (m, 1H, H-3′), 4.08 (app q, 1H, H-4′, J = 3.4), 3.94 (dd, 1H, H-5′, J = 12.2, 2.9), 3.82 (dd, 1H, H-5′, J = 12.2, 3.9), 2.62 (ddd, 1H, H-2′, J = 13.7, 6.4, 4.9), 2.29 (dt, 1H, H-2′, J = 13.7, 6.1), 2.20 (s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 172.5, 155.7, 148.4, 145.0, 139.3, 134.5, 129.8, 124.6, 123.4, 112.6, 88.5, 88.0, 70.0, 61.0, 41.5, 14.9. HRMS calculated for C16H18N3O6 (M+ + H): 348.1196, found: 348.1198.
Desilylation of 4j (88.8 mg, 154.2 μmol) through the use of 1.3 M TASF in DMF (0.59 mL, 212.5 mg, 771.0 μmol) in DMF (1 mL) from 0 °C (1 h) to room temperature over 24 h provided a 90% yield of 5j after workup and chromatography.
Anti-viral testing
Methodology used for evaluation of the antiviral activity listed in Tables 1–5 of the Supporting Information were performed as per reported procedures.24
SCHEME 1.

Synthesis of two thymidine O4-arylsulfonates
Table 2.
Methods evaluated for the desilylation of 4aa
 | ||
|---|---|---|
| entry | desilylation conditions | Result |
| 1 | KF (3 molar equiv), MeOH, 70°C, 40 h | 85% yieldb |
| 2 | CsF (5 molar equiv), CH3CN, 90 °C, 15 h | Product formation observed, but several byproducts formedc |
| 3 | CsF (5 molar equiv), CH3CN-H2O (10:1), 90 °C, 19 h | Quantitative yielda |
| 4 | n-Bu4N+F− (10 molar equiv)-AcOH (10 molar equiv), THF, rt, 24 h | 77% yieldb |
| 5 | TASF (5 molar equiv), CH3CN, 0 °C to rt, 20 h | 98% yieldb |
| 6 | TASF (5 molar equiv), DMF, 0°C to rt, 17 h | Quantitative yieldb |
See the Experimental Section for additional examples of desilylations involving 4d (CsF), 4h and 4j (both with TASF).
Isolated yield after chromatographic purification.
Product was not isolated.
Acknowledgments
This work was supported by NIH Grant S06 GM008168-24S1 (NIGMS), and in part by NSF Grants CHE-0314326 and CHE-0640417 (M.K.L.). Acquisition of a mass spectrometer was funded by NSF Grant CHE-0520963 (M.K.L.). Infrastructural support via NIH RCMI Grant G12 RR03060 is also gratefully acknowledged. Drs. P. Pradhan and S. Bae are thanked for their assistance.
Footnotes
C-C Bond Formation With A Thymidine Aryl Sulfonate
SUPPORTING INFORMATION PARAGRAPH Results of the anti-viral testing, 1H and 13C NMR spectra of compounds 2, 4a–j and 5a–j. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kisakurek MV, Rosemeyer H, editors. Perspectives in Nucleosides and Nucleic Acid Chemistry. Wiley-VCH; Weinheim, Germany: 2001. [Google Scholar]; (b) Suhadolnik RJ. Nucleosides as Biological Probes. Wiley; New York: 1979. [Google Scholar]
- 2.(a) Ichikawa E, Kato K. Curr Med Chem. 20018:385–423. doi: 10.2174/0929867013373471. [DOI] [PubMed] [Google Scholar]; (b) Zemlicka J. Pharmacol Ther. 200085:251–266. doi: 10.1016/s0163-7258(99)00062-5. [DOI] [PubMed] [Google Scholar]; (c) Ferrero M, Gotor V. Chem Rev. 2000100:4319–4347. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]; (d) Huryn DM, Okabe M. Chem Rev. 199292:1745–1768. [Google Scholar]
- 3.For reviews, please see: Lakshman MK. Curr Org Synth. 20052:83–112.Agrofoglio LA, Gillaizeau I, Saito Y. Chem Rev. 2003103:1875–1916. doi: 10.1021/cr010374q.Hocek M. Eur J Org Chem. 2003:245–254.Lakshman MK. J Organomet Chem. 2002653:234–251.
- 4.For some examples, see: Liu J, Robins MJ. Org Lett. 20057:1149–1151. doi: 10.1021/ol050063s.Western EC, Shaughnessy KH. J Org Chem. 200570:6378–6388. doi: 10.1021/jo050832l.Liu J, Robins MJ. Org Lett. 20046:3421–3423. doi: 10.1021/ol048490d.Western EC, Daft JR, Johnson EM, II, Gannett PM, Shaughnessy KH. J Org Chem. 200368:6767–6774. doi: 10.1021/jo034289p.Hocek M, Holý A, Votruba I, Dvoøáková H. Collect Czech Chem Commun. 200065:1683–1697.Havelková M, Hocek M, Èesnek M, Dvoøák D. Synlett. 1999:1145–1147.
- 5.(a) Ding Y, Girardet J-L, Hong Z, Lai VCH, An H, Koh Y-h, Shaw SZ, Zhong W. Bioorg Med Chem Lett. 200515:709–713. doi: 10.1016/j.bmcl.2004.11.020. [DOI] [PubMed] [Google Scholar]; (b) Hocek M, Holý A, Votruba I, Dvoøáková H. Collect Czech Chem Commun. 200166:483–499. [Google Scholar]
- 6.Lakshman MK, Hilmer JH, Martin JQ, Keeler JC, Dinh YQV, Ngassa FN, Russon LM. J Am Chem Soc. 2001123:7779–7787. doi: 10.1021/ja0107172. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gunda P, Russon LM, Lakshman MK. Angew Chem, Int Ed. 2004;43:6372–6377. doi: 10.1002/anie.200460782. (also see corrigendum Angew Chem., Int Ed. 2005, 44, 1154) [DOI] [PubMed] [Google Scholar]; (b) Lakshman MK, Thomson PF, Nuqui MA, Hilmer JH, Sevova N, Boggess B. Org Lett. 20024:1479–1482. doi: 10.1021/ol025673w. [DOI] [PubMed] [Google Scholar]
- 8.Lakshman MK, Gunda P, Pradhan P. J Org Chem. 200570:10329–10335. doi: 10.1021/jo0513764. [DOI] [PubMed] [Google Scholar]
- 9.Mehta JR, Ludlum DB. Biochim Biophys Acta. 1978521:770–728. doi: 10.1016/0005-2787(78)90316-7. [DOI] [PubMed] [Google Scholar]
- 10.(a) Aucagne V, Berteina-Raboin S, Guenot P, Agrofoglio LA. J Comb, Chem. 20046:717–723. doi: 10.1021/cc049976o. [DOI] [PubMed] [Google Scholar]; (b) Amann N, Pandurski E, Fiebig T, Wagenknecht HA. Chem Eur J. 20028:4877–4883. doi: 10.1002/1521-3765(20021104)8:21<4877::AID-CHEM4877>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (c) Amann N, Wagenknecht HA. Synlett. 2002:687–691. [Google Scholar]
- 11.(a) Yoon-Miller SJP, Opalka SM, Pelkey ET. Tetrahedron Lett. 200748:827–830. [Google Scholar]; (b) Wu J, Zhang L, Luo Y. Tetrahedron Lett. 200647:6747–6750. [Google Scholar]; (c) Wu J, Zhang L, Xia HG. Tetrahedron Lett. 200647:1525–1528. [Google Scholar]; (d) Wu J, Zhu Q, Wang L, Fathi R, Yang Z. J Org Chem. 200368:670–673. doi: 10.1021/jo020640f. [DOI] [PubMed] [Google Scholar]; (e) Yao ML, Deng MZ. J Org Chem. 200065:5034–5036. doi: 10.1021/jo000195t. [DOI] [PubMed] [Google Scholar]
- 12.Hennecke U, Kuch D, Carell T. Synthesis. 2007:929–935. [Google Scholar]
- 13.Bischofberger N. Tetrahedron Lett. 198728:2821–2824. [Google Scholar]
- 14.Allerson CR, Chen SL, Verdine GL. J Am Chem Soc. 1997119:7423–7433. [Google Scholar]
- 15.(a) Meketa ML, Weinreb SM. Org Lett. 20068:1443–1446. doi: 10.1021/ol0602304. [DOI] [PubMed] [Google Scholar]; (b) Èapek P, Pohl R, Hocek M. J Org Chem. 200570:8001–8008. doi: 10.1021/jo051110x. [DOI] [PubMed] [Google Scholar]
- 16.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 17.Kuivila HG, Reuwer JF, Jr, Mangravite JA. J Am Chem Soc. 196486:2666–2670. [Google Scholar]
- 18.Trenkle WC, Barkin JL, Son SU, Sweigart DA. Organometallics. 200625:3548–3551. [Google Scholar]
- 19.(a) Yamamoto Y, Suzuki R, Hattori K, Nishiyama H. Synlett. 2006:1027–1030. [Google Scholar]; (b) Wong MS, Zhang XL. Tetrahedron Lett. 200142:4087–4089. [Google Scholar]
- 20.Cirillo PF, Panek JS. J Org Chem. 199055:6071–6073. [Google Scholar]
- 21.Smith AB, III, Ott GR. J Am Chem Soc. 1996118:13095–13096. [Google Scholar]
- 22.Scheidt KA, Chen H, Follows BC, Chemler SR, Coffey DS, Roush WR. J Org Chem. 199863:6436–6437. [Google Scholar]
- 23.(a) Scheidt KA, Bannister TD, Tasaka A, Wendt MD, Savall BM, Fegley GJ, Roush WR. J Am Chem Soc. 2002124:6981–6990. doi: 10.1021/ja017885e. [DOI] [PubMed] [Google Scholar]; (b) Kurosu M, Marcin LR, Grinsteiner TJ, Kishi Y. J Am Chem Soc. 1998120:6627–6628. [Google Scholar]
- 24.(a) De Clercq E, Holý A, Rosenberg I, Sakuma T, Balzarini J, Maudgal PC. Nature. 1986323:464–467. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. Antimicrob Agents Chemother. 198528:84–89. doi: 10.1128/aac.28.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) De Clercq E, Descamps J, Verhelst G, Walker RT, Jones AS, Torrence PF, Shugar D. J Infect Dis. 1980141:563–574. doi: 10.1093/infdis/141.5.563. [DOI] [PubMed] [Google Scholar]