

Abstract
The remodeling of phosphatidylinositol polyphosphates in cellular membranes by phosphatases and kinases orchestrates the signaling by these lipids in space and time. In order to provide chemical tools to study of the changes in cell physiology mediated by these lipids, three new metabolically-stabilized (ms) analogues of phosphatidylinositol-3-phosphate (PtdIns(3)P were synthesized. We describe herein the total asymmetric synthesis of 3-methylphosphonate, 3-monofluoromethylphosphonate and 3-phosphorothioate analogues of PtdIns(3)P. From differentially protected D-myo-inositol key intermediates, a versatile phosphoramidite reagent was employed in the synthesis of PtdIns(3)P analogues with diacylglyceryl moieties containing dioleoyl, dipalmitoyl and dibutyryl chains. In addition, we introduce a new phosphorlyation reagent, monofluoromethylphosphonyl chloride, which has general applications for the preparation of “pKa-matched” monofluorophosphonates. These ms-PtdIns(3)P analogues exhibited reduced binding activities with 15N-labelled FYVE and PX domains, as significant 1H and 15N chemical shift changes in the FYVE domain were induced by titrating ms-PtdIns(3)Ps into membrane-mimetic dodecylphosphocholine (DPC) micelles. In addition, the PtdIns(3)P analogues with dioleyl and dipalmitoyl chains were substrates for the 5-kinase enzyme PIKfyve; the corresponding phosphorylated ms-PI(3,5)P2 products were detected by radio-TLC analysis.
Introduction
Phosphoinositide (PtdInsPn) signaling networks are dynamically modulated by proteins with lipid recognition motifs as well as kinase, phosphatase, and phospholipase enzymatic activities. Lipid-protein interactions form the cornerstone for many signaling pathways, and the new discipline of functional lipidomics1 is defining many new targets for therapeutic interaction.2 It is now accepted that 3-phosphorylated PtdInsPn lipids -- PtdIns(3)P, PtdIns(3,4)P2, PtdIns(3,5)P2, and PtdIns(3,4,5)P3 -- are intracellular signals in mammalian cells.3 These signaling lipids have been implicated as activators of protein kinase C isoforms, and are putative messengers in cellular signal cascades pertinent to inflammation, cell proliferation, transformation, protein kinesis, and cytoskeletal assembly.4,5
PtdIns(3)P is produced by the action of phosphoinositide-3-kinase (PI 3-K)3,6 on PtdIns, and has cognate binding proteins, kinase and phosphatases that are important in cell physiology. The PI 3-K family of enzymes is an important therapeutic target for prevention of cancer, inflammation, allergy, thrombosis, and autoimmune disorders.5,7–11 PtdIns(3)P is also a substrate for PIKfyve, a 5-kinase required for endomembrane integrity and the formation of multivesicular bodies.12 PtdIns(3)P binds FYVE domains,13,14 and is thus an important player in phagocytosis15 and the biochemistry of vesicular trafficking of proteins.16 PX domains also bind PtdIns(3)P with high affinity, and their spatiotemporal changes mediate important aspects of cell respiration and physiology.17 The 3-phosphatase myotubularin (MTM) and MTM-related (MTMR) proteins18 have been identified as important PtdIns(3)P phosphatases that contribute to lipid remodeling and are commonly mutated in genetic diseases.19 Very recently, PtdIns(3)P phosphatase activity was observed for SapM, a Mycobacterium tuberculosis enzyme required for bacterial viability and inhibition of host cell phagolysosome biogenesis.20 In order to gain deeper insights into these biological pathways, selective reagents that could block binding, inhibit enzyme activity, activate lipid-protein mediate pathways, or act as alternative substrates would be highly desirable. To-date, 3-modified PtdIns(3)P analogues with these properties have not been reported.
In order to create metabolically-stabilized analogues of lysophosphatidic acid (LPA), we prepared two kinds of phosphatase-resistant moieties -- fluoromethylenephosphonates21,22 and phosphorothioates23 -- which were found to be receptor sub-type specific agonists. These two functional groups have second pKa values that are matched to the normal phosphomonoester pKa value of 6.5.24 Phosphonate esters have been used extensively to stabilize naturally-occurring phosphates, replacing the bridging oxygen or one of the phosphate oxygens with a methylene or methyl group. The resulting analogues were less susceptible to either acidic or enzymatic cleavage. Methylphosphonates have been studied as metabolically-stable phosphate mimics, potential enzyme inhibitors, and as probes for the elucidation of biochemical processes,25 but these analogues have significantly higher second pKa values than the native phosphate group. Herein we describe the asymmetric total synthesis of methylphosphonate, fluoromethylphosphonate, and phosphorothioate analogues of PtdIns(3)P phosphatase-resistant analogues of PtdIns(3)P. The utility of these analogues in cellular, molecular, and structural biology will be described in due course.
Results and Discussion
Chemical Synthesis of Phosphatase-Resistant Analogues of Phosphatidylinositol-3-phosphate
Several strategies have been employed for total asymmetric synthesis of phosphoinositides. One approach is the use of enantiopure natural precursors, e.g., D-glucose,26–30 L-chiro-inositol derivatives,31 and quinic acid.32–34 Kinetic resolution or desymmetrization via enantioselective enzymatic acylations and non-enzymatic phosphorylation of protected myo-inositol derivatives is another common strategy.35–38 The separation of diastereomeric derivatives of myo-inositol with chiral auxiliaries is also used in many synthetic routes.39–41 In our early work, we developed the synthesis of enantiomerically-pure PtdInsPn and a variety of derivatives from D-glucose via the Ferrier rearrangement.28,41 In this paper, we have employed the more atom-efficient production of the enantiomeric inositol derivatives via the resolution of myo-inositol by crystallization of diastereomeric D-camphor ketals.40
Each of the phosphorylated phosphatidylinositols synthesized in this work employed the simple and elegant protection scheme developed by Bruzik.40 The inositol 1-position was silylated with the TBDPS group, the phosphomonoester 3-position was protected as a benzoate group, and all the remaining hydroxyl groups were protected as methoxymethyl (MOM)-ethers. In this way, 1-O-(tert-butyldiphenylsilyl)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol was synthesized from myo-inositol in six steps.40
The phosphonochloridates derived from simple phosphonic acid alkyl esters are reliable and readily accessible reagents for the phosphonylation of alcohols to produce the corresponding phosphonates.42,43 (O,O-Dimethyl) methylphosphonyl chloride can be readily prepared from the corresponding methylphosphonate by chlorination with PCl5 at room temperature (rt).44
To prepare methyl fluoromethylphosphonate, we first tried N-F “electrophilic” fluorinating agents to fluorinate dimethyl methylphosphonate 10. The commercially-available fluorinating agents N-fluoro-1,4-diazabicyclooctane bis(tetrafluoroborate) (F-TEDA-BF4) and N-fluorobenzenesulfonimide (NFSI) were chosen for investigation.45 A number of procedures for deprotonation of methylphosphonate, and its subsequent monofluorination, were evaluated. The only positive result was obtained using F-TEDA-BF4 in anhydrous THF solvent to fluorinate the sodium enolate, but the product yields were too low (5%) to be practical. Thus, as illustrated in Scheme 3, we developed a new method to prepare methyl fluoromethylphosphonic chloride 9.
Scheme 3.

Synthesis of (fluoro)methylphosphonate chloride. Reagents and conditions: (a) PCl5, benzene; (b) (CH2O)n, K2CO3, CH3OH; (c) Tf2O, 2,6-lutidine, CH2Cl2; (d) TBAF, THF; (e) t-BuNH2; Dowex resin; then (ClCO)2, DMF, CH2Cl2; (f) n-BuLi, THF; then Selectfluor; (g) DAST, CH2Cl2.
Dimethyl hydroxymethylphosphonate 6 was prepared in quantitative yield by the reaction of dimethyl phosphite with paraformaldehdye and anhydrous potassium carbonate in methanol.46 Direct fluorination of 6 with DAST at −78°C gave a low yield (5%). In contrast, activation of the alcohol 6 as triflate 7 (with 2,6-lutidine as base), followed by nucleophilic displacement with tetra-butylammonium fluoride (TBAF) in THF proved to be a safe and simple route to the protected monofluoromethylphosphonate 8.47 Direct chlorination of dialkyl phosphonate with oxalyl chloride48 was unsuccessful. However, a two-step protocol was successful. Thus, aminolysis of 8 with tert-butyl amine afforded the desired phosphonic acid monomethyl ester tert-butylamine salt in quantitative yield.43 The tert-butylamine salt was then converted to the free monomethyl phosphonic acid by passage through a Dowex® acidic resin. The monomethyl phosphonate was then treated with oxalyl chloride and catalytic DMF in CH2Cl2 initially at 0°C, followed by stirring for 2 h at rt.49 Analysis of a reaction aliquot by 31P NMR demonstrated complete conversion. Concentration in vacuo eliminated the volatile reagents and left the desired phosphonochloridate 9 as an oily yellow residue. The product was then used without further purification, as attempts at vacuum distillation resulted in decomposition.
Returning to the inositol ring intermediates, the secondary alcohol 10 was phosphorylated with methyl (fluoromethyl)phosphonyl chloride in the presence of t-BuOK and gave the protected 3-(fluoromethyl)phosphonate 11a in good yield (46–48%). Using amine bases commonly employed for this type of reaction, e.g., triethylamine and N-methylimidazole, resulted in poorer yields. The silyl group in the 1-position was removed with the neutral reagent TBAF-HOAc, and the resulting alcohols 11 were treated with one of the three diacylglyceryl phosphoramidite reagents 19a–19c in the presence of tetrazole followed by mild oxidation with n-Bu4NIO4 to give the fully protected PtdIns(3)P derivatives 14. Using the mild oxidation reagent n-Bu4NIO4 avoided the undesired epoxidation of the oleic acyl double bond.
To preparate the PtdIns(3)P diesters, different phosphoramidites with preattached long, medium, and short diacyl chains were prepared (Scheme 5). For evaluation in different biological and biophysical systems, we prepared the dioleoyl, dipalmitoyl, and dibutyryl reagents. Following our previously reported procedures, the 3-O-PMB-sn-(2R)-glycerol was esterified with various fatty acids using DCC/DMAP to give compounds in high yields.26 Oxidative removal of the PMB protective group with DDQ gave the corresponding 1,2-diacyl-sn-(2S)-glycerols in good yields without significant acyl migration. Although the 1,2-diacyl-sn-(2S)-glycerol was reasonably stable, slow purification on silica gel facilitated acyl chain migration. Therefore, rapid flash chromatography was essential to obtain homogeneous 1,2-diacyl-sn-(2S)-glycerols without acyl migration. Finally, coupling these alcohols with methyl N,N-diisopropylchlorophosphoramidite and cyanoethyl N,N-diisopropylchlorophosphoramidite gave phosphorylation reagents 19 and 20 in high yields.
Scheme 5.
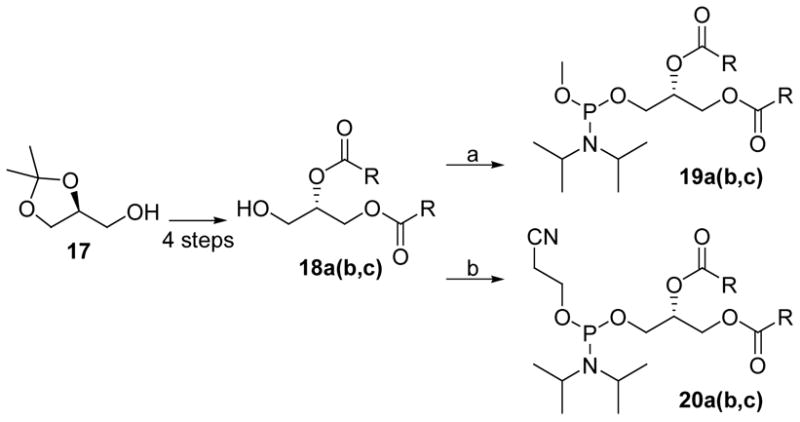
Synthesis of glyceryl phosphoramidites. Reagents and conditions: (a) methyl N,N-diisopropylchlorophosphoramidite, DIPEA, CH2Cl2; (b) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, DIPEA, CH2Cl2.
The removal of protective groups from the phosphate and hydroxyl of the fully-protected inositol lipid intermediates was performed as follows. First, TMSBr was used to deprotect the phosphate methyl esters.40 The fully protected derivatives were dissolved in TMSBr and CH2Cl2 (1:1, v:v) under strictly anhydrous conditions, and stirred at rt for 1 h. After concentration in vacuo, the residue was dissolved in a methanol-water mixture (95:5, v:v), and then stirred for an additional 30 min to hydrolyze the silyl phosphate esters. After completely drying the reaction mixture in vacuo, ethanethiol was added to remove all MOM groups and provide the final products in >98% purity.40
The reported methods of preparation of phosphorothioates vary considerably. Some methods that are suitable for specific compounds utilize conditions that would preclude application to the synthesis of derivatives bearing more labile functional groups. For example, methyl and ethyl esters have often been used as protecting groups; deprotection is then accomplished with TMSI or TMSBr. However, this approach fails to give practical yields with phosphonothioate or phosphorothioate derivatives.50 Deprotection of the dibenzyl esters of phosphonothioic acids has been achieved with sodium in liquid ammonia51 but we sought milder reaction conditions. We turned to the cyanoethyl ester (CE), which is widely used for the synthesis of phosphonothioate or phosphorothioate acids. The CE group can be deprotected under mild basic conditions.23,52 Primary, secondary, and tertiary amine, e.g., triethylamine and t-butylamine, will readily remove the CE protection from phosphates and phosphorothioates at rt.
Alcohol 10 was then phosphorylated employing phosphoramidite methodology. The resulting phosphoramidite triester was oxidized with elemental sulfur to yield the corresponding phosphorothioate triester. TBAF was frequently used to deprotect the TBDPS protective group. However, we found TBAF not only removed the TBDPS ether, but simultaneously removed the CE group, despite the fact that the TBAF reagent had been neutralized with 1 equiv of acetic acid. Apparently the basicity of TBAF was sufficient to cleave the CE phosphate linkage. Thus, to selectively deprotect the TBDPS group, we selected the HF- pyridine complex, which in Py-THF solution selectively removed the TBDPS ether. This deprotection reaction was very slow, and required three weeks to reach completion. Importantly, the reaction was conducted in a Teflon container. The reaction did not occur in common glassware, which was damaged in the process. The reaction rate could not be increased without also accelerating side reactions. Finally, TASF (tris(dimethylamino)sulfonium difluorotrimethylsilicate) was also explored for this deprotection50,53, but TASF was unable to cleave the secondary alcohol silyl ether 21.
Three phosphoramidite reagents 20a–20c were prepared by the reaction of 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite with diacylglycerol preattached with different acyl chains using N,N-diisopropylethylamine (DIPEA) as the base.54 The air-sensitive phosphoramidites were purified by rapid flash chromatography using a basic solvent system. The phosphoramidites were then condensed with secondary alcohol 22 in the presence of 1H-tetrazole to yield the phosphoramidite intermediates.
Oxidation of phosphoramidite triester proved to be problematic. The commonly used oxidation reagents, including MCPBA, n-Bu4NIO4 and H2O2, oxidized both the phosphoramidite triester and the 3-position phosphorothioate, even at low temperatures (−78°C to −20°C). Indeed, the phosphorothioate is readily oxidized to the corresponding phosphate when exposed to standard oxidation reagents. Therefore, a mild oxidation reagent was required to selectively oxidize the phosphoramidite triester without overoxidation of the phosphorothioate. Recently, t-BuOOSi(CH3)3 and t-BuOOH have been utilized to selectively oxidize phosphoramidite triesters.55,56 Using t-BuOOH gave complete oxidation of the phosphoramidite triester to phosphate without overoxidation of the phosphorothioate. The CE groups on the phosphorothioate were removed by using triethylamine (TEA) plus bistrifluoromethylsilylacetamide (BTFSA) in anhydrous acetonitrile.52 BTFSA was added to prevent the phosphorothioate anion from undergoing re-alkylation. The cleavage of the O-silyl derivatives was achieved by aqueous hydrolysis at neutral pH to give the MOM ether-protected intermediate. The MOM groups were removed using ethanethiol-BF3 at rt to give the final products.57
Consistent with the analytical TLC behavior of these molecules, each of the final amphiphilic ms-PtdIns(3)P derivatives could be purified by conventional chromatography using a CHCl3-MeOH-NH4OH solvent system on silica gel.58 The 1H NMR of the PtdIns(3)P analogues illustrated the curious solubility of these compounds and the intrinsic difficulty in obtaining high-quality NMR spectroscopic data.27 Only broad, poorly resolved 1H NMR resonances could be detected in the single-solvent systems CDCl3 or CD3OD. In contrast, in (CDCl3-CD3OD, 3:2, v:v), the 1H NMR resonances for the head group and acyl chains were well resolved.
Binding to FYVE and PX Domains
In cellular membranes, PtdIns(3)P is specifically recognized by a number of protein binding partners including FYVE and PX domains.14,59 To test whether the PtdIns(3)P analogues are able to bind the physiological targets, we investigated interactions of human EEA1 FYVE and yeast Vam7 PX domains by NMR spectroscopy. Significant 1H and 15N chemical shift changes in the FYVE domain were induced by titrating C16-PtdIns(3)P-CH2F 16b embedded in membrane-mimetic dodecylphosphocholine (DPC) micelles (Fig. 1A). These perturbations paralleled chemical shift changes seen in the FYVE domain as C4-PtdIns(3)P-enriched DPC micelles were titrated in (Fig. 1B). Thus the PtdIns(3)P analog and unmodified lipid are accommodated by the same binding pocket consisting of four Arg and two His residues of the FYVE domain.59 However, the resonance perturbations observed during PtdIns(3)P-CH2F titration were smaller in magnitude indicating weaker binding. These results reveal the importance of two negatively charged oxygen atoms of the 3-phosphate group of PtdIns(3)P and their hydrogen bonding potential for the strong anchoring of the inositol ring by the basic residues of the FYVE domain. Similarly, when the PX domain was treated with PtdIns(3)P-CH2F 16b(c) which was pre-bound to the mixed diheptanoyl phosphatidylcholine (DHPC)/CHAPS micelles, the observed chemical shift changes mirrored those seen upon binding of unmodified PtdIns(3)P. Addition of PtdIns(3)P(S) 24b(c) and PtdIns(3)P-CH3 15b(c) lipids or titrating the soluble C4- forms of PtdIns(3)P analogs resulted in considerably smaller chemical shift changes in 1H-15N HSQC spectra of the proteins, although the pattern of resonance perturbations remained essentially unchanged (Fig. 1C,D and data not shown).
Figure 1.

PtdIns(3)P analogs are recognized by the FYVE and PX domains. Superimposed 1H-15N HSQC NMR spectra of (A, B, upper left and right) 0.2 mM EEA1 FYVE domain and (C,D, lower left and right) 0.2 mM Vam7 PX domain collected before and after addition of (A) 4 mM C16-PtdIns(3)P-CH2F 16b and 250 mM d38-DPC; (B) 1 mM C4-PtdIns(3)P and 250 mM d38-DPC; (C) 2 mM C4-PtdIns(3)P-CH2F 16c in 100 mM DHPC and 33 mM CHAPS; (D) 2 mM C4-PtdIns(3)P in 100 mM DHPC and 33 mM CHAPS. Directions of the chemical shift changes are indicated by arrows.
PIKfyve uses ms-PtdIns(3)P analogs as substrates
PtdIns(3,5)-P2 and PtdIns(5)-P could be produced in vitro by two mammalian enzymes: type I PIP kinase60 and PIKfyve.61 Genetic and biochemical evidence has recently accumulated to implicate PIKfyve as the principal enzyme responsible for their biosynthesis in the cellular context.62 We now provide evidence for specific and high affinity interactions between the PIKfyve and metabolically-stabilized analogues of PtdIns(3)P in vitro. PIKfyve activity was assayed as described previously by the γ-32P-ATP-dependent phosphorylation of PtdIns. It was found that each of the three PtdIns(3)P analogues having dioleyl and dipalmitoyl chains were substrates for the kinase PIKfyve, as phosphorylated products were detected by radio-TLC analysis (Figure 2). Since (monofluoro)methyl phosphonate and phosphorothioate are less polar than the phosphate group, the resulting 5-32P-ms-PtdIn(3,5)P2 analogues migrated faster than the unmodified 5-32P-PtdIns(3,5)P2 (Figure 2). Thus, the stabilized phosphorothioate and phosphonate groups at the 3-position of these PtdIns(3)P analogues are recognized by the non-classical FYVE motif within the PIKfyve catalytic domain.63
Figure 2.
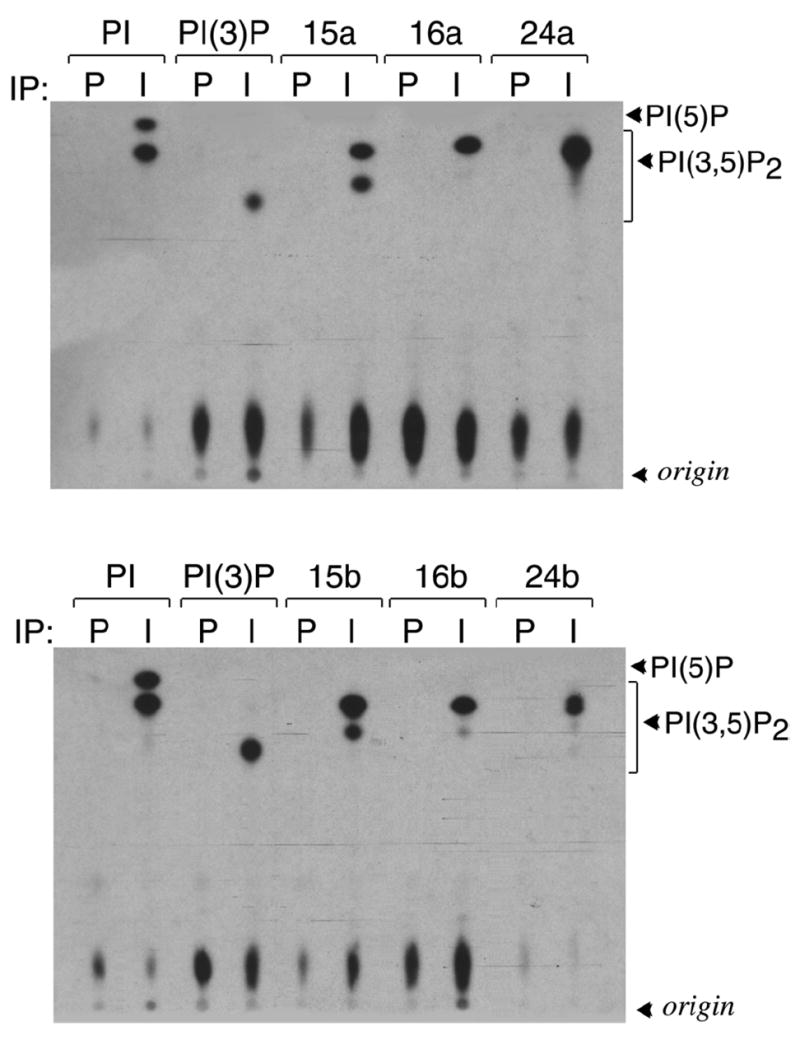
Radio-TLC shows that PtdIns(3)P analogs are recognized by and phosphorylated by PIKfyve. Refer to experimental section for details.
Conclusions
In summary, we have developed a general approach to the synthesis of methylphosphonate, monofluoromethylphosphonate and phosphorothioate analogues of PtdIns(3)P. This strategy also is applicable to the synthesis of analogues of all other PtdInsPn and InsPn. In addition, our method enables synthesis of both saturated and unsaturated acyl analogues of PtdInsPn. These PI(3)P analogues exhibited reduced binding activities with 15N-labeled FYVE and PX domains as significant 1H and 15N chemical shift changes in the FYVE domain were induced by titrating ms-PtdIns(3)P in membrane-mimetic dodecylphosphocholine (DPC) micelles. In addition, these PtdIns(3)P analogues with dioleyl and dipalmitoyl chains were recognized by PIKfyve, as phosphorylated ms-PtdIns(3,5)P2 products were detected by radio-TLC analysis. The metabolically-stabilized analogues of PtdIns(3)P reported herein provide new tools for the elucidation of the roles of these phosphoinositide monophosphates in cell signaling. Specific applications of these analogues in cell and molecular biology will be presented in due course.
Experimental Section
General
Chemicals were purchased from Aldrich and Acros Chemical Corporation and used without prior purification. Solvents were reagent-grade and distilled before use: CH2Cl2 was distilled from CaH2 and THF was distilled from sodium wire. TLC used precoated silica gel aluminum sheets (EM SCIENCE silica gel 60F254). Flash chromatography (FC) employed Whatman 230~400 mesh ASTM silica gel. NMR spectra were recorded on a Varian INOVA 400 at 400 MHz (1H), 101 MHz (13C), 162 MHz (31P) and 376 MHz (19F) at 25 °C. Chemical shifts are reported in ppm with TMS as internal standard (δ = 0.00); 31P, 85% H3PO4 (δ=0.00); 19F, CFCl3 (δ=0.00). Low- and high-resolution mass spectra were obtained on HP5971A MSD and Finnigan MAT95 double focusing mass spectrometer (MS) instruments, respectively. Dibutyryl and dipalmitoyl PtdIns(3)P were obtained from Echelon Biosciences (Salt Lake City, UT).
Methyl methylphosphonyl chloride (4)
To 12.87 g (0.104 mol) of dimethyl methylphosphonate in 30 mL of anhydrous benzene was added 21.6 g (0.104 mol) of PCl5 at 0 °C. The solution was stirred vigorously for 1 h at 0 °C. The solvent was removed under vacuum. Vacuum distillation (80 °C/22 mmHg) gave 12.1 g (0.094 mol, 94%) of homogeneous product. 1H NMR (CDCl3): δ 3.72 (d, J = 6.0 Hz, 2H), 3.57 (d, J = 10.5 Hz, 6H). 13C NMR (CDCl3): δ 56.40 (s), 54.78 (s), 53.14 (s), 53.07 (s). 31P NMR (CDCl3): δ 28.18 (s). MS (CI) m/z 129.0 (M++1, 8.32). HRMS (CI), M++1, Found: 128.9872; calcd for C2H6ClO2P, 128.9871.
Dimethoxyphosphinylmethyl triflates (7)
To a stirred solution of dimethoxy (hydroxymethyl)phosphonate (30.6 mmol) and 2,6-lutidine (37.6 mmol) in anhydrous CH2Cl2 (50 mL) at −50 °C under N2 was added trifluoromethanesulfonic anhydride (35.5 mmol) dropwise. The resulting mixture was allowed to warm to 0 °C over a period of 1.5 h, whereupon the dark brown solution was diluted with ether (300 mL). The precipitates formed were removed by filtration. The ethereal solution was successively washed with water, 1 N HCl, and brine and then dried over Na2SO4. After concentration, a yellow oil was obtained, which was used in the next step without further purification.
Dimethyl fluoromethylphosphonate (8)
A solution of the triflate 7 (5.08 g, 0.015 mol) in THF (22 mL) was cooled to 0 °C before 20 mL (0.02 mol) of a 1 M solution of tetrabutylammonium fluoride in THF was added dropwise. The solution was stirred at 0 °C for 1 h. Solvents were then removed and CH2Cl2 (35 mL) was added. The organic layer was washed (H2O, 3 × 8 mL), dried (MgSO4) and evaporated to a crude oil. This was purified by FC, using EtOAc–hexane (1:1) as eluent, to give 8 as a pale yellow oil (1.67 g, 67%). 1H NMR (CDCl3): δ 4.68 (dd, J = 46.8, 4.8 Hz, 2H), 3.81 (d, J = 10.8 Hz, 6 H). 31P NMR (CDCl3): δ 19.83 (d, J = 63.0 Hz). 19F NMR (CDCl3): δ −250.74 (dt, J = 61.7, 47.0 Hz). MS (CI) m/z 143.0 (M++1, 100.00). HRMS (CI), M++1, Found: 143.0262; calcd for C3H9FO3P, 143.0267.
Methyl fluoromethylphosphonate chloride (9)
Dimethyl fluoromethylphosphonate 8 (0.221 g, 0.592 mmol) was dissolved in t-butylamine (8 mL, 76 mmol) and heated at reflux overnight. The reaction was concentrated and gave 0.256 g of product as a white salt (quantitative yield). Methyl fluoromethylphosphonate t-butylamine salt (0.290 g, 0.670 mmol) was dissolved in CHC13 and treated with cation exchange resin (Dowex 50W-X8 (H+ form) 200–400 mesh). The Dowex® resin was removed by filtration and the filtrate was concentrated in vacuo to give 0.241 g (quantitative yield) of the oily phosphonate. Next, oxalyl chloride (0.132 g, 1.04 mmol) was added dropwise to a solution of the methyl fluoromethylphosphonic acid (0.240 g, 0.668 mmol) and DMF (2.5 μL, 0.033 mmol) dissolved in CH2Cl2 at 0 °C. The solution was stirred at 0 °C for 20 min and then warmed to rt and stirred for 1.5 h. The reaction was concentrated, dissolved in toluene (2 mL), and then re-concentrated in vacuo to remove the volatile reagents. This gave phosphonochloridate 9 as a yellow oil, which was then used immediately for reaction with the 3-hydroxy-containing protected inositol derivatives. 1H NMR (CDCl3): δ 4.62 (dd, J = 46.8, 4.4 Hz, 2H), 3.74 (d, J = 11.2 Hz, 3 H). 13C NMR (CDCl3): δ 75.72 (dd, J = 180.2, 168.7 Hz), 53.14 (d, J = 6.1 Hz). 31P NMR (CDCl3): δ 19.80 (d, J = 63.0 Hz). 19F NMR(CDCl3): δ −250.98 (dt, J = 62.8, 47.0 Hz). MS (CI) m/z 147.0 (M++1, 100.00). HRMS (CI), M++1, Found: 146.9759 Calcd for C2H6ClFO2P, 146.9778.
1D-1-O-(tert-Butyldiphenylsilyl)-3-(methyl methylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (11a)
t-BuOK (18 mg, 0.163 mmol, 1.4 equiv) was added to a stirred solution of (69 mg, 0.116 mmol) 10 and methyl methylphosphonate chloride (18 mg, 0.139 mmol, 1.2 eq.) in CH2Cl2 (1 mL) at 0°C, then stirred 2 h at rt and the reaction was complete. A saturated aq solution of NH4Cl (1 mL) was added, stirred 10 min, the aqueous phase was extracted with CH2Cl2 (3× 5 mL), the organic solution was dried with anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude product was purified by chromatography (n-hexane-acetone, 2:1, v:v) to afford a colorless liquid (37 mg, 0.054 mmol, 46%). 1H NMR (CDCl3): δ 7.72-7.67 (m, 4H), 7.41-7.36 (m, 6H), 4.98 (d, J = 4.4 Hz, 1H), 4.91 (d, J = 4.4 Hz, 1H), 4.79 (d, J = 4.4 Hz, 1H), 4.75 (m, 3H), 4.57 (d, J = 4.8 Hz, 1H), 4.26 (d, J = 4.8 Hz, 1H), 3.92-3.85 (m, 4H), 3.86 (d, J = 6.0 Hz, 3H), 3.42-3.32 (m, 12H), 3.20 (d, J = 23.0 Hz, 2H), 1.34 (d, J = 17.6 Hz, 3H), 1.06 (s, 9H). 13C NMR (CDCl3): δ 135.93 (s), 135.81 (s), 133.80 (s), 133.77 (s), 132.51 (s), 132.48 (s), 129.95 (s), 129.86 (s), 129.80 (s), 128.01 (s), 127.89 (s), 127.71 (s), 127.69 (s), 98.93 (s), 98.35 (s), 98.30 (s), 98.22 (s), 97.46 (d, J = 14.58 Hz), 82.04 (d, J = 8.45 Hz), 81.28 (d, J = 5.43 Hz), 78.88 (d, J = 34.49 Hz), 74.84 (s), 70.49 (s), 56.05 (dd, J = 62.05, 24.54 Hz), 27.07 (s). 31P NMR (CDCl3): δ 33.89 (s), 33.04 (s). MS (CI) m/z 687.1 (M++1, 79.67), 655.1 (M+-OCH3, 100.00). HRMS (CI), M++1, Found: 687.2962; calcd for C32H52O12PSi, 687.2966.
1D-3-(Methyl methylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (12a)
A solution of (24 mg, 0.035 mmol) in THF (1 mL) was treated with tetrabutylammoniumfluoride trihydrate (16 mg, 0.049 mmol) at rt. After stirring for 18 h, the reaction was complete (monitored by TLC). The solvent was evaporated under reduced pressure and the crude product was purified by passage through a short silica gel column (n-hexane-acetone, 3:1, v:v) to afford 12 mg of a colorless liquid (0.027 mmol, 77%). 1H NMR (CDCl3): δ 4.79-4.64 (m, 8H), 4.17 (t, J = 10.4 Hz, 1H), 4.10 (d, J = 20.4 Hz, 1H), 3.98 (dd, J = 11.6, 4.4 Hz, 1H), 3.89 (dd, J = 21.2, 9.6 Hz, 1H), 3.69 (dd, J = 20.0, 9.2 Hz, 1H), 3.58 (td, J = 9.2, 1.2 Hz, 1H), 3.42 (m, 1H), 3.41-3.34 (m, 15H), 1.46 (dd, J = 18.0, 3.6 Hz, 3H). 13C NMR (CDCl3): δ 98.52 (s), 98.35 (s), 98.31 (s), 98.26 (s), 98.17 (s), 98.11 (s), 97.97 (s), 82.93 (s), 79.11 (d, J = 16.09 Hz), 78.41 (d, J = 6.94 Hz), 76.50 (d, J = 6.13 Hz), 74.82 (dd, J = 29.06, 6.84 Hz), 70.40 (d, J = 5.33 Hz), 52.03 (dd, J = 40.63, 6.13 Hz), 10.97 (dd, J = 146.52, 23.73 Hz). 31P NMR (CDCl3): δ 34.60 (s), 33.36 (s). MS (CI) m/z 449.1 (M++1, 100.00). HRMS (CI), M++1, Found: 449.1779; calcd for C16H34O12P, 449.1788.
1D-O-(1,2-Di-O-oleoyl-sn-(2S)-glycerol-3-O-methylphospho)-3-(methyl methylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (13a)
N,N-Diisopropyl-O-methyl-O-(di-(2S)-oleoyl-sn-glycerol)phosphonamidite (0.187 g, 0.135 mmol) was added under an argon atmosphere to a solution of 12a (110 mg, 0.089 mmol) and 1H-tetrazole (26 mg, 0.374 mmol) in 4 mL dry CH2Cl 2/THF (1/1, v/v). After stirring for 20 h at rt, oxidation was performed with (n-C4H9)4NIO4 (78 mg, 0.180 mmol) at −20°C for 1 h. The reaction mixture was warmed to rt for an additional 30 min, and after aqueous work-up, the crude product was purified by FC on silica gel (n-hexane/acetone, 3/1, v/v) to give 77 mg of a homogeneous colorless oil (0.068 mmol, 76%). 1H NMR (CDCl3): δ 5.32 (m, 2H), 5.21 (m, 1H), 4.84-4.71 (m, 8H), 4.32-4.09 (m, 8H), 3.96-3.89 (m, 2H), 3.77-3.72 (m, 6H), 3.41-3.33 (m, 12H), 2.29 (m, 4H), 1.96 (m, 8H), 1.55 (m, 4H), 1.52 (dd, J = 17.6, 6.8 Hz, 3H), 1.21 (m, 42H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.15 (s), 172.73 (s), 129.91 (s), 129.66 (s), 98.91 (s), 98.54 (s), 98.45 (s), 98.37 (s), 98.03 (s), 97.85 (s), 79.13 (d, J = 13.78 Hz), 74.24 (dd, J = 31.48, 6.13 Hz), 69.37 (s), 65.80 (s), 65.52 (s), 61.61 (s), 56.78 (s), 56.65 (s), 56.56 (s), 56.51 (s), 55.76 (s), 54.81 (s), 53.75 (s), 52.18 (dd, J = 35.30, 6.13 Hz), 34.08 (s), 33.96 (s), 31.88 (s), 29.65 (s), 29.63 (s), 29.61 (s), 29.45 (s), 29.31 (s), 29.25 (s), 29.22 (s), 29.09 (s), 29.06 (s), 24.78 (s), 22.64 (s), 14.07 (s), 11.03 (dd, J = 148.83, 12.27 Hz). 31P NMR (CDCl3): δ 34.60 (s), 33.49 (s), 0.74 (s), 0.52 (d, J = 7.61 Hz). MS (CI) m/z 1145.7 (M++1, 100.00). HRMS (CI), M++1, Found: 1145.6897; calcd for C56H107O19P2, 1145.6882.
1D-O-(1,2-di-O-oleoyl-sn-(2S)-glycerol-3-phospho)-3-(methylphosphonate)-myo-inositol (15a)
The phosphate 13a (22 mg, 0.019 mmol) in 5 mL flask was dried in vacuo, and then anhydrous TMSBr (0.2 mL) and CH2Cl2 (0.2 mL) were added into the flask. The solution was stirred at rt for 30 min. TMSBr and volatile products were evaporated under high vacuum during 6 h. The residue was dissolved in MeOH-H2O (95:5, 1.0 mL) and stirred for 30 min at rt. The solution was thoroughly concentrated for an additional 3 h under high vacuum. Ethanethiol (1 mL) was added, the solution was kept at rt for 1 h, and then concentrated to yield the crude product. Chromatography on silica gel (CHCl3-CH3OH-NH4OH (2.0 M), 65:25:3, v:v:v) provided 16 mg of pure product 15a (0.017 mmol, 89%). 1H NMR (CDCl3/CD3OD, 3/2, v/v): δ 5.32 (m, 2H), 5.21 (m, 1H), 3.93 (dd, J = 12.0, 3.6 Hz, 1H), 3.82 (m, 2H), 3.73-3.60 (m, 5H), 3.30 (m, 2H), 2.33 (m, 4H), 1.96 (m, 8H), 1.55 (m, 4H), 1.52 (dd, J = 17.6, 6.8 Hz, 3H), 1.21 (m, 42H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3/CD3OD, 3/2, v/v): δ 173.28 (s), 172.89 (s), 129.25 (s), 128.98 (s), 97.13 (s), 73.70 (s), 70.87 (s), 70.46 (s), 69.98 (s), 69.33 (s), 64.39 (s), 61.60 (s), 54.88 (s), 42.53 (s), 42.46 (s), 33.30 (s), 31.24 (s), 29.26 (s), 29.06 (s), 28.83 (s), 28.64 (s), 28.60 (s), 28.56 (s), 28.53 (s), 28.45 (s), 28.43 (s), 28.39 (s), 26.46 (s), 24.17 (s), 21.96 (s), 13.08 (s), 10.62 (d, J = 140.80 Hz). 31P NMR (CDCl3/CD3OD, 3/2, v/v): δ 32.56 (s), 32.40 (s), −0.51 (s). MS (ESI) m/z 963.53 (M++Na, C46H86NaO15P2, 100.0). HRMS(MALDI), M++Na, Found: 963.5312; calcd for C46H86NaO15P2, 963.5340.
1D-O-(1,2-Di-O-palmitoyl-sn-(2S)-glycerol-3-O-methylphospho)-3-(methyl methylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (13b)
N,N-diisopropyl-O-methyl-O-(di-(2S)-palmitoyl-sn-glycerol)phosphonamidite (91 mg, 0.125 mmol) was added under an argon atmosphere to a solution of 12a (37 mg, 0.083 mmol) and 1H-tetrazole (25 mg, 0.349 mmol) in 4 mL of dry CH2Cl 2-THF (1:1, v:v). After stirring for 20 h at rt, oxidation was performed with (n-C4H9)4NIO4 (69 mg, 0.160 mmol) at −20 °C for 1 h. The reaction mixture was warmed to rt for an additional 30 min, and after aqueous work-up, the crude product was chromatographed on silica gel (n-hexane-acetone, 3:1, v:v) to give 67 mg pure 13b as a colorless oil (0.061 mmol, 73%). 1H NMR (CDCl3): δ 5.21 (m, 1H), 4.84-4.71 (m, 8H), 4.34-4.07 (m, 8H), 3.96-3.89 (m, 2H), 3.77-3.72 (m, 6H), 3.41-3.33 (m, 12H), 2.29 (m, 4H), 1.55 (m, 4H), 1.52 (dd, J = 17.6, 6.8 Hz, 3H), 1.21 (m, 48H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.19 (s), 172.80 (s), 98.90 (s), 98.53 (s), 98.43 (s), 98.35 (s), 98.03 (s), 97.83 (s), 79.13 (d, J = 13.78 Hz), 77.21 (s), 76.90 (s), 76.35 (s), 74.24 (dd, J = 31.48, 6.13 Hz), 69.44 (s), 69.35 (s), 65.75 (s), 65.57 (s), 61.60 (s), 56.77 (s), 56.64 (s), 56.55 (s), 56.50 (s), 55.75 (s), 53.75 (s), 52.18 (dd, J = 35.30, 6.13 Hz), 34.08 (s), 33.96 (s), 31.88 (s), 29.65 (s), 29.63 (s), 29.61 (s), 29.45 (s), 29.31 (s), 29.25 (s), 29.22 (s), 29.09 (s), 29.06 (s), 24.78 (s), 22.64 (s), 14.07 (s), 11.03 (dd, J = 148.83, 12.27 Hz). 31P NMR (CDCl3): δ 34.60 (d, J = 8.74 Hz), 33.51 (d, J = 6.64 Hz), 0.74 (s), 0.53 (d, J = 8.74 Hz). MS (CI) m/z 1093.7 (M++1, 4.58). HRMS (CI), M++1, Found: 1093.6583; calcd for C52H103O19P2, 1093.6569.
1D-O-(1,2-Di-O-palmitoyl-sn-(2S)-glycerol-3-phospho)-3-(methylphosphonate)-myo-inositol (15b)
The phosphate 13b (29 mg, 0.027 mmol) in 5 mL flask was thoroughly dried in vacuo, and then anhydrous TMSBr (0.2 mL) and CH2Cl2 (0.2 mL) were added. The solution was stirred at rt for 30 min. TMSBr and volatile products were evaporated under high vacuum during 6 h. The residue was dissolved in MeOH-H2O (95:5, 1.0 mL) and stirred for 30 min at rt. The solution was thoroughly concentrated for an additional 3 h under high vacuum. Ethanethiol (1 mL) was added, the solution was kept at rt for 1 h, and then concentrated to yield the crude product. Chromatography on silica gel (CHCl3-CH3OH-NH4OH (2.0 M), 65:25:3, v:v:v) provided 22 mg of pure product 15b (0.025 mmol, 92%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.27 (m, 1H), 3.93 (dd, J = 12.0, 3.6 Hz, 1H), 3.82 (m, 2H), 3.73-3.60 (m, 6H), 3.30 (m, 2H), 2.33 (m, 4H), 1.62 (m, 4H), 1.56 (dd, J = 17.6, 6.8 Hz, 3H), 1.27 (m, 48H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3/CD3OD, 3/2, v/v): δ 173.17 (s), 172.75 (s), 98.20 (s), 73.80 (s), 70.97 (s), 70.55 (s), 70.02 (s), 69.74 (s), 64.52 (s), 61.81 (s), 54.97 (s), 33.45 (s), 33.42 (s), 31.36 (s), 29.32 (s), 29.12 (s), 28.85 (s), 28.64 (s), 28.70 (s), 14.01 (s), 10.58 (d, J = 138.20 Hz). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 32.45 (s), 32.30 (s), 0.74 (s), −0.55 (s). MS (ESI) m/z 911.50 (M++Na, C42H82NaO15P2, 100.00). HRMS (MALDI), M++Na, Found: 911.5045; calcd for C42H82NaO15P2, 911.5027.
1D-O-(1,2-di-O-butanoyl-sn-(2S)-glycerol-3-O-methylphospho)-3-(methyl methylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (13c)
To a solution of alcohol 12a (17 mg, 0.038 mmol) in dry THF (0.5 mL), was added N,N-diisopropyl-O-(methyl)-O-di-O-butanoyl-sn-(2S)-glycerol)phosphonamidite (25 mg, 24 μL, 0.125 mmol) and 1H-tetrazole (26 mg, 0.374 mmol). The mixture was stirred at rt for 16 hour. Then oxidation was performed with (n-C4H9)4NIO4 (76 mg, 0.174 mmol) at −20 °C for 1 h. The reaction mixture was warmed-up to rt for additional 30 min. The solution was diluted with methylene chloride (20 mL) and washed with 10% sodium bisulfite. The organic layer was concentrated and the residue was chromatographed on silica gel (n-hexane-acetone, 1:1, v:v) to give 25 mg of pure 13c as colorless oil (0.033 mmol, 87%). 1H NMR (CDCl3): δ 5.23 (m, 1H), 4.84-4.74 (m, 8H), 4.34-4.07 (m, 8H), 3.96-3.89 (m, 2H), 3.77-3.72 (m, 6H), 3.41-3.33 (m, 12H), 2.31 (m, 4H), 1.62 (m, 4H), 1.52 (dd, J = 17.6, 6.8 Hz, 3H), 0.93 (t, J = 7.2 Hz, 3H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 173.03 (s), 172.90 (s), 98.90 (s), 98.45 (s), 97.98 (s), 97.85 (s), 98.03 (s), 79.20 (m), 74.28 (d, J = 31.48 Hz), 69.36 (s), 65.65 (d, J = 23.03 Hz), 61.57 (s), 56.79 (s), 56.57 (s), 55.78 (s), 54.59 (s), 52.20 (dd, J = 35.30, 6.13 Hz), 35.93 (s), 35.83 (s), 18.26 (s), 13.57 (s), 11.03 (dd, J = 148.83, 12.27 Hz). 31P NMR (CDCl3): δ 34.65 (s), 33.54 (s), 0.75 (s), 0.52 (d, J = 7.61 Hz). MS (ESI) m/z 757.41 (M++1, 100.00). HRMS (MALDI), M++Na, Found: 779.2627; calcd for C28H54NaO19P2, 779.2632.
1D-O-(1,2-Di-O-butanoyl-sn-(2S)-glycerol-3-phospho)-3-(methylphosphonate)-myo-inositol (15c)
The phosphate 13c (12 mg, 0.016 mmol) was dried and reacted with TMSBr and then hydrolyzed in aqueous MeOH as described above for 15a. Ethanethiol (1 mL) was added, the solution was kept at rt for 1 h, and then concentrated to yield the crude product. Chromatography on silica gel (CHCl3-CH3OH-NH4OH (2.0 M), 65:25:3, v:v:v) provided 8 mg of pure product 15c (0.016 mmol, 94%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.27 (m, 1H), 4.74 (s, 1H), 4.36 (dd, J = 12.0, 3.2 Hz, 1H), 4.28 (m, 1H), 4.18-4.06 (m, 5H), 3.78 (m, 2H), 3.23 (t, J = 9.2 Hz, 1H), 2.28 (m, 4H), 1.58 (m, 7H), 0.99 (m, 6H). 13C NMR (CDCl3-CD3OD, 3:2, v:v): δ 174.32 (s), 173.94 (s), 98.25 (s), 78.74 (m), 78.36 (m), 76.19 (m), 74.75 (s), 71.99 (s), 71.57 (s), 71.08 (s), 70.32 (s), 65.56 (s), 62.60 (s), 56.07 (s), 36.42 (s), 36.29 (s), 18.72 (s), 13.73 (d, J = 3.84 Hz). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 36.57 (s), 36.42 (s), 3.47 (s), 3.31 (s). MS (ESI) m/z 575.13 (M++Na, C18H34NaO15P2). HRMS (MALDI), M++Na, Found: 575.1296; calcd for C18H34NaO15P2, 575.1271.
1D-1-O-(tert-Butyldiphenylsilyl)-3-(methyl fluoromethylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (11b)
t-BuOK (230 mg, 2.31 mmol, 1.4 eq.) was added to a stirred solution of 980 mg (1.65 mmol) of 10 and methyl fluoromethylphosphonate chloride (1.99 mmol, 1.2 eq.) in CH2Cl2 (10 mL) at 0°C; stirring was continued for 2 h at rt to complete the reaction. A saturated aqueous solution of NH4Cl (1 mL) was added, stirred 10 min, and the aqueous phase was extracted with CH2Cl2 (3× 5 mL). The combined organic phases were dried with anhydrous Na2SO4 and the solvent was removed in vacuo. The crude product was purified by chromatography (n-hexane-acetone, 3:1, v:v) to afford 552 mg of 11b as a colorless liquid (0.784 mmol, 48%). 1H NMR (CDCl3): δ 7.70-7.65 (m, 4H), 7.42-7.34 (m, 6H), 4.97 (d, J = 2.0 Hz, 1H), 4.96 (d, J = 2.4 Hz, 1H), 4.90 (d, J = 6.0 Hz, 1H), 4.76 (m, 3H), 4.62 (m, 1H), 4.50 (m, 2H), 4.46 (m, 1H), 3.98-3.73 (m, 5H), 3.52 (m, 2H), 3.43-3.23 (m, 14H), 1.05 (s, 9H). 13C NMR (CDCl3): δ 135.99 (s), 135.85 (s), 133.79 (s), 133.76 (s), 132.48 (s), 130.08 (s), 129.97 (s), 129.91 (s), 129.89 (s), 128.10 (s), 127.99 (s), 127.81 (s), 127.77 (s), 99.14 (s), 99.09 (s), 98.86 (s), 98.67 (s), 98.38 (s), 97.78 (s), 97.55 (s), 78.85 (m), 78.04 (s), 77.76 (s), 76.42 (m), 75.38 (s), 75.31 (s), 75.23 (s), 73.54 (d, J = 7.68 Hz), 56.68 (d, J = 3.84 Hz), 56.63 (s), 56.43 (s), 53.10 (d, J = 6.16 Hz), 52.46 (d, J = 6.97 Hz), 27.13 (s), 27.10 (s), 19.11 (s), 19.08 (s). 31P NMR (CDCl3): δ 19.57 (d, J = 61.88 Hz), 18.91 (d, J = 64.15 Hz). 19F NMR (CDCl3): δ −249.64 (m). MS (ESI) m/z 705.3 (M++1, 100.00). HRMS (MALDI), M++Na, Found: 727.2685; calcd for C32H50FNaO12PSi, 727.2691.
1D-3-(Methyl fluoromethylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (12b)
A solution of 11b (40 mg, 0.057 mmol) in THF (1 mL) was treated with tetrabutylammoniumfluoride trihydrate (36 mg, 0.114 mmol) and acetic acid (7 μL, 0.114 mmol) at rt. After stirring for 12 h, TLC indicated that the reaction was complete; solvent was evaporated in vacuo and the crude product was purified by passage through a short silica column (n-hexane-acetone, 1:1, v:v) to afford 15 mg of 12b as a colorless liquid (0.032 mmol, 56%). 1H NMR (CDCl3): δ 4.86-4.68 (m, 10H), 4.32 (m, 1H), 4.17 (m, 1H), 3.98 (m, 1H), 3.87 (dd, J = 10.8, 3.2 Hz, 3H), 3.65 (t, J = 9.6 Hz, 1H), 3.41-3.34 (m, 12H), 3.20 (m, 2 H). 13C NMR (CDCl3): δ 98.73 (s), 98.66 (s), 98.48 (s), 98.38 (s), 98.18 (s), 83.40 (s), 83.00 (s), 78.72 (d, J = 90.09 Hz), 78.33 (d, J = 90.09 Hz), 76.10 (d, J = 10.00 Hz), 70.43 (d, J = 6.16 Hz), 58.68 (s), 56.65 (s), 56.52 (s), 56.37 (s), 56.12 (s), 55.91 (s), 55.85 (s), 53.52 (s), 53.11 (s). 31P NMR (CDCl3): δ 34.60 (d, J = 62.05 Hz), 33.36 (d, J = 65.12 Hz). 19F NMR (CDCl3): δ −249.36 (dt, J = 60.19, 45.88 Hz), −250.08 (dt, J = 62.78, 45.88 Hz). MS (CI) m/z 467.2 (M++1, 100.00). HRMS (MALDI), M++Na, Found: 489.1540; calcd for C16H32FNaO12P, 489.1508.
1D-O-(1,2-Di-O-oleoyl-sn-(2S)-glycerol-3-O-methylphospho)-3-(methyl fluoromethylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (14a)
N,N-diisopropyl-O-methyl-O-(di-(2S)-oleoyl-sn-glycerol)phosphoramidite (77 mg, 0.093 mmol) was added under an argon atmosphere to a solution of 12b (29 mg, 0.062 mmol) and 1H-tetrazole (0.73 mL, 3% wt, 0.249 mmol) in 4 mL dry CH3CN-THF (1:1, v:v). After stirring for 20 h at rt, oxidation was performed with (n-C4H9)4NIO4 (40 mg, 0.093 mmol) at −20°C for 1 h. The reaction mixture was warmed-up to rt for additional 30 min, and after aqueous work-up the crude product was purified by FC (n-hexane-acetone, 2:1, v:v) to give 60 mg of pure product 14a as colorless oil (0.052 mmol, 83%). 1H NMR (CDCl3): δ 5.30 (m, 4H), 5.21 (m, 1H), 4.84-4.67 (m, 10H), 4.32-4.09 (m, 8H), 3.93 (m, 2H), 3.86 (d, J = 11.2 Hz, 3H), 3.78 (dd, J = 10.8, 3.2 Hz, 3H), 3.41 (m, 12H), 2.29 (m, 4H), 1.96 (m, 8H), 1.57 (m, 4H), 1.26 (m, 42H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.16 (s), 172.78 (s), 129.96 (s), 129.64 (s), 98.86 (s), 98.70 (s), 98.42 (s), 98.18 (s), 97.93 (s), 78.94 (d, J = 6.16 Hz), 76.39 (m), 75.40 (d, J = 22.32 Hz), 75.30 (d, J = 22.42 Hz), 69.34 (m), 65.82 (s), 65.51 (s), 61.56 (s), 56.73 (s), 56.56 (s), 55.83 (s), 55.56 (s), 54.80 (d, J = 6.16 Hz), 54.60 (d, J = 8.08 Hz), 53.60 (d, J = 6.06 Hz), 53.25 (d, J = 6.16 Hz), 34.04 (s), 33.92 (s), 31.85 (s), 29.70 (s), 29.67 (s), 29.47 (s), 29.26 (s), 29.17 (s), 29.15 (s), 29.08 (s), 29.05 (s), 29.02 (s), 27.16 (s), 27.12 (s), 24.75 (s), 22.63 (s), 14.06 (s). 31P NMR (CDCl3): δ 19.53 (d, J = 63.02 Hz), 19.13 (d, J = 67.39 Hz), 0.57 (s). 19F NMR (CDCl3): δ −249.23 (m), −250.08 (m). MS (ESI) m/z 1163.7 (M++1, 100.00). HRMS (MALDI), M++Na, Found: 1185.6608; calcd for C56H105FNaO19P2, 1185.6607.
1D-O-(1,2-Di-O-oleoyl-sn-(2S)-glycerol-3-phospho)-3-(fluoromethylphosphonate)-myo-inositol, (16a)
The phosphate 14a (32 mg, 0.028 mmol) was dried and reacted with TMSBr and then hydrolyzed in aqueous MeOH and dried as described above for 15a. Ethanethiol (1 mL) was added, the solution was kept at rt for 1 h, and concentrated to yield the crude product. Chromatography on silica gel (CHCl3-CH3OH-NH4OH (2.0 M), 65:25:3, v:v:v) provided 20 mg of pure product 16a (0.021 mmol, 75%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.29 (m, 4H), 5.21 (m, 1H), 4.78 (m, 1H), 4.73 (m, 1H), 4.66 (m, 1H), 4.14 (m, 4H), 3.77 (m, 2H), 3.38 (m, 2H), 2.28 (m, 4H), 1.96 (m, 8H), 1.56 (m, 4H), 1.24 (m, 42H), 0.83 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3-CD3OD, 3:2, v:v): δ 174.26 (s), 173.87 (s), 130.27 (s), 129.97 (s), 98.10 (s), 71.70 (m), 71.24 (m), 70.12 (s), 70.04 (s), 65.37 (m), 62.43 (m), 56.01 (s), 34.36 (s), 30.03 (s), 29.80 (s), 29.60 (s), 29.58 (s), 29.42 (m), 27.47 (s), 27.44 (s), 25.12 (s), 22.9 (s), 14.22 (s). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 21.25 (d, J = 63.02 Hz), 21.10 (d, J = 67.39 Hz), 3.61 (s), 3.36 (s). 19F NMR (CDCl3-CD3OD, 3:2, v:v): δ −245.96 (m). MS (ESI) m/z 1015.6 (M++Na, C46H91FN2NaO15P2). HRMS (MALDI), M++Na, Found: 1015.5803; calcd for C46H91FN2NaO15P2, 1015.5776.
1D-O-(1,2-Di-O-palmitoyl-sn-(2S)-glycerol-3-O-methylphospho)-3-(methyl fluoromethylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (14b)
N,N-diisopropyl-O-methyl-O-(di-(2S)-palmitoyl-sn-glycerol)phosphoramidite (90 mg, 0.123 mmol) was added under an argon atmosphere to a solution of 12b (38 mg, 0.082 mmol) and 1H-tetrazole (1.0 mL, 3% wt, 0.320 mmol) in 4 mL dry CH3CN-THF (1:1, v:v). After stirring for 20 h at rt, oxidation was performed with (n-C4H9)4NIO4 (60 mg, 0.123 mmol) at −20°C for 1 h. The reaction mixture was warmed to rt for an additional 30 min, and after aqueous work-up the crude product was purified by FC (n-hexane-acetone, 2:1, v:v) to give 49 mg of pure 14b as colorless oil (49 mg, 0.044 mmol, 54%). 1H NMR (CDCl3): δ 5.22 (m, 1H), 4.85-4.69 (m, 10H), 4.34-4.10 (m, 8H), 3.95 (t, J = 9.6 Hz, 2H), 3.87 (d, J = 10.8 Hz, 1H), 3.79 (d, J = 11.2 Hz, 1H), 3.43-3.39 (m, 12H), 2.29 (m, 4H), 1.58 (m, 8H), 1.23 (m, 48H), 0.86 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.19 (s), 172.80 (s), 98.90 (s), 98.53 (s), 98.43 (s), 98.35 (s), 98.03 (s), 97.83 (s), 79.13 (d, J = 13.78 Hz), 77.21 (s), 76.90 (s), 76.35 (s), 74.24 (dd, J = 31.48, 6.13 Hz), 69.44 (s), 69.35 (s), 65.75 (s), 65.57 (s), 61.60 (s), 56.77 (s), 56.64 (s), 56.55 (s), 56.50 (s), 55.75 (s), 53.75 (s), 52.18 (dd, J = 35.30, 6.13 Hz), 34.08 (s), 33.96 (s), 31.88 (s), 29.65 (s), 29.63 (s), 29.61 (s), 29.45 (s), 29.31 (s), 29.25 (s), 29.22 (s), 29.09 (s), 29.06 (s), 24.78 (s), 22.64 (s), 14.07 (s), 11.03 (dd, J = 148.83, 12.27 Hz). 31P NMR (CDCl3): δ 19.53 (d, J = 61.88 Hz), 0.74 (s). 19F NMR (CDCl3): δ −249.09 (m). MS (ESI) m/z 1133.6 (M++1, 100.00). HRMS (MALDI), M++Na, Found: 1133.6325; calcd for C52H101FNaO19P2, 1133.6294.
1D-O-(1,2-Di-O-palmitoyl-sn-(2S)-glycerol-3-phospho)-3-(fluoromethylphosphonate)-myo-inositol (16b)
The phosphate 14b (35 mg, 0.032 mmol) was dried, treated with TMSBr, the resulting ethers hydrolyzed in aqueous MeOH, and the crude product dried as described above for 15a. Ethanethiol (1 mL) was added, the solution was kept at rt for 1 h, and concentrated to yield the crude product. FC on silica gel gel (CHCl3-CH3OH-NH4OH (2.0 M), 65:25:3, v:v:v) provided 21 mg of 16b (0.022 mmol, 67%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.21 (m, 1H), 4.78 (m, 1H), 4.73 (m, 1H), 4.66 (m, 1H), 4.14 (m, 4H), 3.77 (m, 2H), 3.38 (m, 2H), 2.28 (m, 4H), 1.56 (m, 4H), 1.24 (m, 48H), 0.83 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3-CD3OD, 3:2, v:v): δ 174.26 (s), 173.87 (s), 98.10 (s), 71.70 (m), 71.24 (m), 70.12 (s), 70.04 (s), 65.37 (m), 62.43 (m), 56.01 (s), 34.36 (s), 30.03 (s), 29.80 (s), 29.60 (s), 29.58 (s), 29.42 (m), 27.47 (s), 27.44 (s), 25.12 (s), 22.9 (s), 14.22 (s). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 21.25 (d, J = 63.02 Hz), 21.10 (d, J = 67.39 Hz), 3.61 (s), 3.36 (s). 19F NMR (CDCl3-CD3OD, 3:2, v:v): δ −245.96 (m). MS (ESI) m/z 959.63 (M++NH4, C42H91FN3O15P2, 100.00). HRMS (MALDI), M++Na, Found: 958.5884; calcd for C42H91FN3O15P2, 958.5909.
1D-O-(1,2-Di-O-butanoyl-sn-(2S)-glycerol-3-O-methylphospho)-3-(methyl fluoromethylphosphonate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (14c)
To a solution of alcohol 12b (12 mg, 0.026 mmol) in dry THF (0.5 mL) was added N,N-diisopropyl-O-(methyl-O-di-butanoyl-sn-(2S)-glycerol)phosphoramidite (145 mg, 0.039 mmol) and 1H-tetrazole (0.31 mL, 3% wt, 0.104 mmol). The mixture was stirred at rt for 16 h. Then oxidation was performed with (n-C4H9)4NIO4 (17 mg, 0.039 mmol) at −20°C for 1 h. The reaction mixture was warmed to rt for an additional 30 min. The solution was diluted with methylene chloride (20 mL) and washed with 10% sodium bisulfite. The organic layer was concentrated and the residue was purified by FC (n-hexane-acetone, 2:1, v:v) to give 14 mg of pure 14c as colorless oil (0.018 mmol, 70%). 1H NMR (CDCl3): δ 5.23 (m, 1H), 4.85-4.69 (m, 10H), 4.34-4.07 (m, 8H), 3.94 (t, J = 10.0 Hz, 2H), 3.87 (t, J = 9.6 Hz, 3H), 3.97 (dd, J = 5.6, 4.4 Hz, 3H), 3.42-3.37 (m, 12H), 2.29 (m, 4H), 1.62 (m, 4H), 0.93 (t, J = 7.2 Hz, 3H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 173.06 (s), 172.90 (s), 98.89 (s), 98.73 (s), 98.46 (s), 97.96 (s), 79.01 (s), 76.44 (s), 75.54 (s), 69.34 (s), 65.85 (s), 61.56 (s), 56.81 (s), 56.59 (s), 55.87 (s), 35.94 (s), 35.83 (s), 18.28 (s), 13.60 (s), 13.54(s). 31P NMR (CDCl3): δ 19.55 (dd, J = 60.75, 3.24 Hz), 19.11 (dd, J = 63.99, 5.34 Hz), 0.74 (s), 0.68 (s), 0.59 (s), 0.56 (s). 19F NMR (CDCl3): δ −249.26 (m), −250.01 (m). MS (CI) m/z 775.46 (M++1, 100.00). HRMS (CI), M++1, Found: 775.2751; calcd for C28H54FO19P2, 775.2719.
1D-O-(1,2-Di-O-butanoyl-sn-(2S)-glycerol-3-phospho)-3-(fluoromethylphosphonate)-myo-inositol (16c)
The phosphate 14c (17 mg, 0.022 mmol) was dried, treated with TMSBr, the silyl ethers hydrolyzed in aqueous MeOH, and the crude product dried as described above for 16a. Ethanethiol (1 mL) was added, the solution was kept at rt for 1 h, and concentrated and the crude product was chromatographed on silica gel gel (CHCl3-CH3OH-NH4OH (2.0 M), 65:25:3, v:v:v) to give pure product as a white powder (9 mg, 0.015 mmol, 68%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.18 (m, 1H), 4.77 (m, 1H), 4.66 (dd, J = 12.0, 5.2 Hz, 1H), 4.43-4.15 (m, 2H), 4.12-4.07 (m, 4H), 3.95 (m, 1H), 3.74 (m, 1H), 3.15 (m, 2H), 2.23 (m, 4H), 1.56 (m, 4H), 0.86 (m, 6H). 13C NMR (CDCl3): δ 174.46 (s), 174.07 (s), 130.44 (s), 130.16 (s), 74.70 (d, J = 9.16 Hz), 71.51 (s), 71.15 (s), 70.36 (d, J = 7.64 Hz), 65.61 (s), 62.77 (s), 34.61 (s), 34.49 (s), 32.40 (s), 30.23 (s), 30.00 (s), 29.81 (s), 29.77 (s), 29.72 (s), 29.70 (s), 29.61 (s), 29.55 (s), 27.65 (s), 25.34 (s), 23.13 (s), 14.32 (s). 31P NMR (CDCl3/CD3OD, 3/2, v/v): δ 21.49 (s), 21.09 (s), 3.35 (s), 3.32 (s). 19F NMR (CDCl3-CD3OD, 3:2, v:v): δ −246.28 (m). MS (ESI) m/z 571.26 (M++1, 18.00). HRMS(MALDI), M++Na, Found: 593.1199 Calcd for C18H33FNaO15P2, 593.1176.
N,N-Diisopropyl-O-cyanoethyl-O-(di-oleoyl-sn-(2S)-glycerol)phosphonamidite (20a)
A solution of 53 mg of 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.226 mmol) and 35 mg (47 μL, 0.271 mmol) of DIPEA in 3 mL anhydrous CH2Cl2 was cooled to 0°C under nitrogen. To this solution, 140 mg (0.226 mmol) 1,2-di-oleoyl-sn-(2S)-glycerol was added with vigorous stirring. After stirring at 0°C for 1 h and stirring at rt for 3 h under nitrogen, the solution was separated from the white precipitate. FC on silica gel (hexane-EtOAc-TEA, 100:10:1, v:v:v) gave 96 mg of 20a (0.118 mmol, 52%) as colorless oil. 1H NMR (CDCl3): δ 5.32 (m, 4H), 5.17 (m, 1H), 4.32 (m, 1H), 4.13 (m, 1H), 3.85-3.52 (m, 6H), 2.61 (t, J = 6.8 Hz, 2H), 2.28 (m, 4H), 1.98 (m, 8H), 1.58 (m, 4H), 1.26 (m, 42H), 1.14 (m, 12H), 0.86 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.35 (s), 172.96 (s), 129.99 (s), 129.69 (s), 62.36 (s), 61.77 (s), 61.62 (s), 58.57 (s), 58.48 (s), 58.30 (s), 43.20 (s), 43.08 (s), 34.28 (s), 34.08 (s), 33.36 (s), 31.88 (s), 29.75 (s), 29.70 (s), 29.51 (s), 29.31 (s), 29.19 (s), 29.12 (s), 28.96 (s), 27.20 (s), 27.16 (s), 24.89 (s), 24.86 (s), 24.60 (s), 24.53 (s), 22.66 (s), 20.38 (s), 20.31 (s), 14.10 (s). 31P NMR (CDCl3): δ 150.61 (s), 150.46 (s). MS (CI) m/z 821.6 (M++1, C48H90N2O6P, 41.22). HRMS(CI), M++1, Found: 821.6562 Calcd for C48H90N2O6P, 821.6537.
N,N-Diisopropyl-O-cyanoethyl-O-(di-palmitoyl-sn-(2S)-glycerol)phosphonamidite (20b)
A solution of 38 mg (0.162 mmol) of 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite and 25 mg (34 μL, 0.194 mmol) of DIPEA in 3 mL anhydrous CH2Cl2 was cooled to 0°C under nitrogen. To this solution, (92 mg, 0.162 mmol) 1,2-di-palmitoyl-sn-(2S)-glycerol was added with vigorous stirring. After stirring at 0°C for 1 h and stirring at rt for 3 h under nitrogen, the solution was separated from the white precipitate. FC on silica gel (hexane-EtOAc-TEA, 100:10:1, v:v:v) gave 67 mg of 20b (0.087 mmol, 54%) as a white solid. 1H NMR (CDCl3): δ 5.16 (m, 1H), 4.31 (m, 1H), 4.15 (m, 1H), 3.85-3.54 (m, 6H), 2.60 (t, J = 6.8 Hz, 2H), 2.29 (m, 4H), 1.59 (m, 4H), 1.26 (m, 48H), 1.26 (m, 42H), 1.14 (m, 12H), 0.85 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.36 (s), 172.97 (s), 117.50 (s), 70.56 (d, J = 3.84 Hz) 62.34 (s), 61.76 (s), 61.61 (s), 61.44 (s), 58.56 (s), 58.48 (s), 58.38 (s), 58.29 (s), 43.19 (s), 43.06 (s), 34.29 (s), 34.10 (s), 33.35 (s), 31.90 (s), 29.67 (s), 29.63 (s), 29.61 (s), 29.47 (s), 29.34 (s), 29.28 (s), 29.12 (s), 29.09 (s), 24.90 (s), 24.87 (s), 24.59 (s), 24.52 (s), 22.66 (s), 20.37 (s), 20.29 (s), 14.09 (s). 31P NMR (CDCl3): δ 150.60 (s), 150.45 (s). MS (CI) m/z 769.6 (M++1, 22.44). HRMS (CI), M+, Found: 768.6141; calcd for C44H85N2O6P, 768.6145.
N,N-Diisopropyl-O-cyanoethyl-O-(di-butanoyl-sn-(2S)-glycerol)phosphonamidite (20c)
A solution of (220 mg, 0.931 mmol) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite and (144 mg, 200 μL, 1.117 mmol) DIPEA in 5 mL anhydrous CH2Cl2 was cooled to 0°C under nitrogen. To this solution 1,2-di-butanoyl-sn-(2S)-glycerol (216 mg, 0.931 mmol) was added with vigorous stirring. After stirring at 0°C for 1 h and stirring at rt for 3 h under nitrogen, the solution was separated from the white precipitate. FC on silica gel (hexane:EtOAc:TEA, 100:10:1, v:v:v) gave 285 mg of pure 20c (0.661 mmol, 71%) as a colorless oil. 1H NMR (CDCl3): δ 5.07 (m, 1H), 4.22 (m, 1H), 4.05 (m, 1H), 3.76-3.47 (m, 6H), 2.54 (t, J = 6.4 Hz, 2H), 2.19 (m, 4H), 1.53 (m, 4H), 1.06 (m, 12H), 0.84 (t, J = 7.2 Hz, 3H), 0.81 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 172.78 (s), 172.43 (s), 117.32 (s), 70.34 (dd, J = 7.68, 3.84 Hz), 62.01 (d, J = 1.52 Hz), 61.51 (s), 61.35 (s), 61.21 (s), 58.28 (d, J = 7.68 Hz), 58.09 (d, J = 8.48 Hz), 42.90 (s), 42.77 (s), 35.82 (s), 35.62 (s), 24.28 (s), 24.22 (s), 20.08 (s), 20.02 (s), 18.07 (s), 18.04 (s), 13.31 (s), 13.27 (s). 31P NMR (CDCl3): δ 150.38 (s), 150.27 (s). MS (CI) m/z 433.3 (M++1, 100.00). HRMS (CI), M+, Found: 432.2408; calcd for C20H37N2O6P, 432.2389.
1D-1-O-(tert-Butyldiphenylsilyl)-3-(dicyanoethyl phosphothionate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (21)
Di(2-cyanoethyl) diisopropylphosphorodiamidite (28 mg, 0.104 mmol) was added under an argon atmosphere to a solution of 10 (56 mg, 0.094 mmol) and 1H-tetrazole (0.48 mL, 3% wt in CH3CN, 0.207 mmol) in 1 mL dry CH3CN. After stirring at rt for 2 h, sulfur (100 mg) and CS2/pyridine (1.0 mL, 1/1, v/v) were added. After stirring at rt for 2 h, the reaction mixture was filtered and the filtrate was washed with brine, dried over Na2SO4, and concentrated. FC (EtOAc-hexane, 1:3, v:v) gave 68 mg of 21 as a colorless oil (0.085 mmol, 91%). 1 H NMR (CDCl3): δ 7.71-7.65 (m, 4H), 7.44-7.34 (m, 6H), 4.97 (d, J = 6.4 Hz, 1H), 4.92 (d, J = 6.0 Hz, 1H), 4.85 (d, J = 6.4 Hz, 1H), 4.78 (d, J = 7.2 Hz, 1H), 4.73 (d, J = 6.4 Hz, 1H), 4.53 (d, J = 6.8 Hz, 1H), 4.43 (d, J = 6.8 Hz, 1H), 4.22-3.86 (m, 8H), 3.44-3.34 (m, 12H), 3.23 (s, 3H), 2.67-2.58 (m, 4H), 1.06 (s, 9H). 13C NMR (CDCl3): δ 136.00 (s), 135.89 (s),133.88 (s), 132.60 (s), 129.89 (s), 128.16 (s), 127.82 (s), 116.39 (s), 99.19 (s), 98.86 (s), 98.45 (s), 97.71 (s), 78.91 (s), 78.76 (s), 78.16 (d, J = 5.45 Hz), 77.88 (s), 77.24 (d, J = 6.16 Hz), 73.52 (s), 62.37 (d, J = 3.84 Hz), 61.98 (d, J = 3.13 Hz), 56.74 (s), 56.72 (s), 55.59 (s), 27.15 (s), 19.22 (s), 19.17 (s), 19.14 (s), 19.07 (s), 18.98 (s). 31P NMR (CDCl3): δ 68.69 (s). MS (CI) m/z 797.3 (M++1, 43.15). HRMS (CI), M+, Found: 796.2845; calcd for C36H53N2O12PSSi, 796.2826.
1D-3-(Dicyanoethyl phosphothionate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (22)
A solution of 35 mg (0.035 mmol) of 21 in THF (1 mL) was added along with anhydrous pyridine (0.4 mL) and hydrogen fluoride pyridine complex (70%, 0.2 mL) at rt into a Teflon container. After stirring for 3 weeks, the reaction was complete as determined by TLC; the reaction was then diluted with ethyl acetate (30 mL), and washed with 10% sodium bicarbonate (8 mL × 2). The organic layer was washed with brine, dried over Na2SO4, and concentrated. FC (acetone-hexane, 1:3, v:v) gave 21 mg of 22 as a colorless liquid (0.038 mmol, 86%). 1H NMR (CDCl3): δ 4.85-4.71 (m, 8H), 4.34 (m, 5H), 4.20 (s, 1H), 4.10 (s, 1H), 4.01 (t, J = 10.0 Hz, 1H), 3.64 (t, J = 10.0 Hz, 1H), 3.44 (m, 14H), 2.75 (m, 4H). 13C NMR (CDCl3): δ 115.48 (s), 108.75 (s), 97.73 (s), 97.50 (s), 97.14 (s), 82.50 (s), 78.21 (d, J = 1.62 Hz), 77.34 (d, J = 6.87 Hz), 75.57 (d, J = 6.97 Hz), 69.43 (s), 61.72 (d, J = 3.84 Hz), 61.47 (d, J = 3.94 Hz), 55.76 (s), 55.39 (s), 55.12 (s), 54.99 (s), 18.48 (s), 18.45 (s), 18.39 (s), 18.37 (s). 31P NMR (CDCl3): δ 68.12 (s). MS (CI) m/z 559.2 (M++1, 17.87). HRMS (CI), M+, Found: 558.1686; calcd for C20H35N2O12PS, 558.1648.
1D-O-(1,2-Di-O-oleoyl-sn-(2S)-glycerol-3-O-cyanoethylphospho)-3-(dicyanoethyl phosphothionate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (23a)
To a solution of 12 mg of alcohol 22 (0.022 mmol) in dry THF (0.5 mL) was added N,N-diisopropyl-O-cyanoethyl-O-(di-oleoyl-sn-(2S)-glycerol)phosphonamidite (25 mg, 0.030 mmol) and 1H-tetrazole (6 mg, 0.26 mL, 0.088 mmol). The mixture was stirred at rt for 16 h. Oxidation was then performed with t-BuOOH (9.9 mg, 11 μL, 0.110 mmol) at rt for 1 h. The solution was diluted with CH2Cl2 (20 mL) and washed with 10% sodium bisulfite. The organic layer was concentrated and the residue purified by FC (acetone-hexane, 1:3, v:v) to give 25 mg of 23a as a colorless oil (0.019 mmol, 88%).1H NMR (CDCl3): δ 5.35 (m, 4H), 5.22 (m, 1H), 4.83-4.74 (m, 8H), 4.42-4.19 (m, 12H), 4.10 (m, 2H), 3.42 (m, 12H), 2.78 (m, 6H), 2.29 (m, 4H), 1.99 (m, 8H), 1.57 (m, 4H), 1.28 (m, 42H), 0.85 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.23 (s), 172.84 (s), 172.81 (s), 130.00 (s), 129.67 (s), 116.63 (s), 116.49 (s), 98.91 (s), 98.93 (s), 98.55 (s), 98.49 (s), 98.01 (s), 97.96 (s), 79.17 (s), 76.40 (m), 75.70 (d, J = 19.29 Hz), 69.28 (d, J = 5.35 Hz), 66.09 (d, J = 5.35 Hz), 65.98 (d, J = 5.45 Hz), 62.89 (d, J = 3.84 Hz), 62.70 (m), 62.30 (d, J = 5.35 Hz), 62.24 (d, J = 4.55 Hz), 61.60 (s), 56.64 (s), 56.62 (s), 56.59 (s), 56.03 (s), 34.10 (s), 33.94 (s), 31.87 (s), 29.73 (s), 29.70 (s), 29.50 (s), 29.29 (s), 29.21 (s), 29.19 (s), 29.12 (s), 29.08 (s), 29.06 (s), 27.19 (s), 27.16 (s), 24.79 (s), 22.65 (s), 19.69 (s), 19.62 (s), 19.44 (s), 19.36 (s), 14.09 (s). 31P NMR (CDCl3): δ 67.86 (s), 67.70 (s), −0.84 (s), −1.25 (s). MS (CI) m/z 1294.3 (M+, 40.74), 1262.2 (M+-OCH4, 100.00). HRMS (CI), M+, Found: 1293.6830 Calcd for C62H109N3O19P2S, 1293.6851.
1D-O-(1,2-Di-O-oleoyl-sn-(2S)-glycerol-3- phospho)-3-(phosphothionate)-myo-inositol, (24a)
To a solution of 21 mg of compound 23a (0.016 mmol) in CH3CN (1.0 mL) under N2 was added triethylamine (0.5 mL) followed by the addition of bistrifluoromethylsilylacetamide (0.50 mL). After 24 h, the reaction mixture was concentrated and the residue was dissolved in 30 mL of 8 mM ammonium acetate (pH 7.1). The water phase was lyophilized and white powder was obtained. The anhydrous white powder was dissolved in ethanethiol (1 mL) and treated with boron trifluoride diethyl etherate (13 μL). After 3 h, the reaction was stopped by adding dry triethylamine (20 μL). The thiol was removed by evaporation and the semi-solid residue was dissolved in ammonium acetate buffer. FC (CHCl3-CH3OH-NH4OH (2.0 M): 9:7:2, v:v:v) provided 15 mg of pure 24a (0.015 mmol, 93%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.32 (m, 4H), 5.24 (m, 1H), 4.20 (m, 3H), 4.04-3.95 (m, 5H), 3.77 (m, 2H), 3.20 (m, 2H), 2.29 (m, 4H), 2.00 (m, 8H), 1.57 (m, 4H), 1.27 (m, 42H), 0.86 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3-CD3OD, 3:2, v:v): δ 175.15 (s), 174.88 (s), 130.71 (s), 130.43 (s), 76.82 (s), 75.73 (s), 75.13 (s), 72.63 (s), 72.27 (s), 71.61 (s), 71.42 (s), 64.53 (s), 63.86 (s), 34.95 (s), 34.83 (s), 32.60 (s), 30.50 (s), 30.47 (s), 30.44 (s), 30.19 (s), 30.06 (s), 30.00 (s), 29.97 (s), 29.90 (s), 29.87 (s), 29.82 (s), 27.91 (s), 27.88 (s), 25.67 (s), 25.57 (s), 23.35 (s), 14.58 (s). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 48.96 (s), 0.74 (s). MS (ESI) m/z 981.64 (M+-NH4+, 100.00). HRMS (MALDI), M++2NH4++H+, Found: 1046.6530; Calcd for C45H102NO15P2S, 1046.6568.
1D-O-(1,2-Di-O-pamitoyl-sn-(2S)-glycerol-3-O-cyanoethylphospho)-3-(dicyanoethyl phosphothionate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (23b)
To a solution of alcohol 22 (53 mg, 0.095 mmol) in dry THF (0.5 mL), was added N,N-diisopropyl-O-cyanoethyl-O-(di-palmitoyl-sn-(2S)-glycerol)phosphonamidite (102 mg, 0.133 mmol) and 1H-tetrazole (1.12 mL, 3% wt, 0.380 mmol). The mixture was stirred at rt for 16 hour. Then oxidation was performed with t-BuOOH (60 μL, 0.380 mmol) at rt for 1 h. The solution was diluted with methylene chloride (20 mL) and washed with 10% sodium bisulfite. The organic layer was concentrated and the residue was purified by FC (acetone-hexane, 1:3, v:v) to give 95 mg of pure product (0.077 mmol, 81%) as colorless oil. 1 H NMR (CDCl3): δ 5.24 (m, 1H), 4.83-4.72 (m, 7H), 4.39 (m, 1H), 4.31-4.13 (m, 12H), 3.93 (m, 2H), 3.44 (m, 2H), 3.40 (m, 12H), 2.77 (m, 6H), 2.28 (m, 4H), 1.56 (m, 4H), 1.21 (m, 48H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3): δ 173.23 (s), 172.84 (s), 116.61 (s), 116.48 (s), 98.87 (s), 98.51 (s), 98.45 (s), 97.97 (s), 97.92 (s), 79.14 (s), 76.34 (m), 75.77 (s), 75.56 (s), 69.29 (s), 66.11 (m), 62.71 (d, J = 3.84 Hz), 62.20 (d, J = 12.32 Hz,), 61.60 (s), 58.31 (s), 56.80 (s), 56.55 (s), 55.99 (s), 34.09 (s), 33.94 (s), 31.86 (s), 29.64 (s), 29.60 (s), 29.45 (s), 29.30 (s), 29.26 (s), 29.08 (s), 29.06 (s), 24.78 (s), 22.63 (s), 19.66 (s), 19.58 (s), 19.40 (s), 19.32 (s), 14.06 (s). 31P NMR (CDCl3): δ 67.84 (s), 67.68 (s), −0.88 (s), −1.28 (s). MS (ESI) m/z 1242.8 (M++1, 18.00). HRMS (MALDI), M++Na, Found: 1264.6463 Calcd for C58H105N3NaO19P2S, 1264.6436.
1D-O-(1,2-Di-O-palmitoyl-sn-(2S)-glycerol-3-phospho)-3-(phosphothionate)-myo-inositol (24b)
To a solution of 35 mg of compound 23b (0.028 mmol) in CH3CN (1.0 mL) under N2 was added triethylamine (0.5 mL) followed by the addition of bistrifluoromethylsilylacetamide (0.50 mL). After 24 h, the reaction mixture was concentrated and the residue was dissolved in 30 mL of 8mM ammonium acetate (pH 7.1). The water phase was lyophilized and white powder was obtained. The anhydrous white powder was dissolved in ethanethiol (2 mL) and treated with boron trifluoride diethyl etherate (14 μL). After 3 h, the reaction was stopped by adding dry triethylamine (30 μL). The thiol was removed by evaporation and the semi-solid residue was dissolved in ammonium acetate buffer. FC (CHCl3-CH3OH-NH4OH (2.0M): 9:7:2, v:v:v) provided 22 mg of pure 24b (0.022 mmol, 79%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.27 (m, 1H), 3.93 (dd, J = 12.0, 3.6 Hz, 1H), 3.82 (m, 2H), 3.73-3.60 (m, 5H), 3.30 (m, 2H), 2.33 (m, 4H), 1.62 (m, 4H), 1.56 (dd, J = 17.6, 6.8 Hz, 3H), 1.27 (m, 48H), 0.84 (t, J = 7.2 Hz, 6H). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 48.96 (s), 0.74 (s). MS (ESI) m/z 981.64 (M++ Na, 17.00). HRMS (MALDI), M++Na+, Found: 980.5410; Calcd for C41H89N3NaO15P2S, 980.5387.
1D-O-(1,2-Di-O-butanoyl-sn-(2S)-glycerol-3-O-cyanoethylphospho)-3-(dicyanoethyl phosphothionate)-2,4,5,6-O-tetrakis(methoxymethylene)-myo-inositol (23c)
To a solution of alcohol 22 (35 mg, 0.063 mmol) in dry THF (0.5 mL) was added N,N-diisopropyl-O-cyanoethyl-O-(di-butanoyl-sn-(2S)-glycerol)phosphonamidite (38 mg, 0.088 mmol) and 1H-tetrazole (0.74 mL, 3% wt, 0.252 mmol). The mixture was stirred at rt for 16 h. Then oxidation was performed with t-BuOOH (33 μL, 0.300 mmol) at rt for 1 h. The solution was diluted with CH2Cl2 (20 mL) and washed with 10% sodium bisulfite. The organic layer was concentrated and the residue was chromatographed (acetone-hexane, 1:3, v:v) on silica gel to give pure product as a colorless oil (49 mg, 0.054 mmol, 86%). 1H NMR (CDCl3): δ 5.26 (m, 1H), 4.83-4.74 (m, 9H), 4.40 (d, J = 2.4 Hz, 1H), 4.35-4.12 (m, 12H), 3.79 (m, 2H), 3.45-3.38 (m, 12H), 2.76 (m, 6H), 2.31 (m, 4H), 1.67 (m, 4H), 0.95 (t, J = 7.2 Hz, 3H), 0.91 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 173.07 (s), 172.68 (s), 116.63 (s), 116.50 (s), 98.91 (s), 98.89 (s), 98.48 (s), 98.00 (s), 97.94 (s), 79.16 (m), 76.41 (m), 75.69 (d, J = 19.91 Hz), 69.27 (d, J = 6.13 Hz), 66.14 (dd, J = 12.97, 7.64 Hz), 62.90 (d, J = 3.82 Hz), 62.70 (s), 62.38 (d, J = 4.63 Hz), 62.22 (d, J = 4.20 Hz), 61.53 (s), 56.84 (s), 56.61 (s), 56.02 (s), 35.94 (s), 35.80 (s), 19.68 9s), 19.61 (s), 19.43 (s), 19.35 (s), 18.27 (s), 13.58 (s), 13.54 (s). 31P NMR (CDCl3): δ 67.83 (s), 67.70 (s), −0.90 (s), −1.30 (s). MS (ESI) m/z 906.41 (M++1, 30.00). HRMS (MALDI), M++Na+NH4+, Found: 946.2996; calcd for C34H61N4NaO19P2S, 946.3024.
1D-O-(1,2-Di-O-butanoyl-sn-(2S)-glycerol-3-phospho)-3-(phosphothionate)-myo-inositol (24c)
To a solution of 20 mg of compound 23a (0.022 mmol) in CH3CN (1.0 mL) under N2 was added triethylamine (0.5 mL) followed by the addition of bistrifluoromethylsilylacetamide (0.50 mL). After 24 h, the reaction mixture was concentrated and the residue was dissolved in 30 mL of 8mM ammonium acetate (pH 7.1). The water phase was lyophilized and white powder was obtained. The anhydrous white powder was dissolved in ethanethiol (1 mL) and treated with boron trifluoride diethyl etherate (11 μL). After 3 h, the reaction was stopped by adding dry triethylamine (20 μL). The thiol was removed by evaporation and the semi-solid residue was dissolved in ammonium acetate buffer. FC (CHCl3-CH3OH-NH4OH (2.0M): 9:7:2, v:v:v) provided 11 mg of pure 24c (0.018 mmol, 80%). 1H NMR (CDCl3-CD3OD, 3:2, v:v): δ 5.26 (m, 1H), 3.93 (m, 2H), 3.82 (m, 2H), 3.73-3.60 (m, 5H), 3.30 (m, 2H), 2.31 (m, 4H), 1.67 (m, 4H), 0.95 (t, J = 7.2 Hz, 3H), 0.91 (t, J = 7.2 Hz, 3H). 31P NMR (CDCl3-CD3OD, 3:2, v:v): δ 48.96 (s), 0.74 (s). MS (ESI) m/z 624.27 (M+ + 3, 21.00), 667.28 (M+ + 2 + 2Na+, 100.00). HRMS (MALDI), M++Na+, Found: 644.1652; Calcd for C17H41N3O15P2S, 644.1631.
Protein Expression and Purification
DNA fragments encoding residues 1325–1410 of human EEA1 FYVE and residues 2–122 of yeast Vam7 PX were cloned in pGEX-KG and pGEX-2T vectors (Amersham). The 15N-labeled proteins were expressed in E. coli BL21 (DE3) pLysS and BL21 Codon Plus RP strains in minimal media supplemented with 15NH4Cl (Cambridge Isotope). Bacteria were harvested by centrifugation after induction with IPTG (0.5 mM) and lysed by French press. The glutathione S-transferase (GST)-fusion FYVE and PX were purified on a glutathione sepharose 4B column (Amersham). The GST tag was cleaved with thrombin (Sigma). The proteins were further purified by FPLC and concentrated in Millipore concentrators (Millipore). The buffers were exchanged into 20 mM d11-Tris (FYVE) or 50 mM potassium phosphate (PX), pH 6.8, 100–200 mM KCl, 1–20 mM perdeuterated dithiothreitol, 50 μM 4-amidinophenylmethane sulfonyl fluoride, 1 mM NaN3, and 7% 2H2O.
NMR spectroscopy and titration of PtdIns(3)P analogs
NMR spectra were recorded at 25°C on Varian INOVA 500 MHz spectrometer. The 1H-15N heteronuclear single quantum coherence (HSQC) spectra of 0.2–0.3 mM uniformly 15N-labeled FYVE and PX domains were collected while dibutanoyl (C4)- or dipalmitoyl (C16)-PtdIns(3)P analogs (Echelon Biosciences) (up to 4 mM) embedded in micelles consisting of d38-DPC (250 mM) (Cambridge Isotopes) or DHPC/CHAPS (100 mM/17 mM) (Avanti/Anatrace) were added stepwise.
Phosphorylation of PtdIns(3)P analogues by PIKfyve
Phosphatase-resistant dipalmitoyl and dioleoyl analogues of PtdIns(3)P and di-C16 PtdIns 3-P (Echelon Biosciences, Inc.) were separately prepared as 0.5 mM aqueous stocks and stored at −80 °C. Just before use as substrates in the phosphorylation reaction with PIKfyve immunoprecipitates, a 60 μl aliquot of the PtdIns(3)P, the stabilized analogues, or soybean PtdIns (Avanti Polar Lipids, Inc.) were evaporated down to dryness with absolute ethanol (2 × 200 μl) under a stream of dry N2. Lipids were then reconstituted in an equal volume of lipid buffer (20 mM pH,7.5, HEPES, 1 mM EDTA) by sonication (2 × 30 s) in a bath sonicator at room temperature. A 10 μl aliquot of each 0.5 mM phosphoinositide reconstituted in lipid buffer was then phosphorylated with immunoprecipitates of PIKfyve immune (R7069) or preimmune sera derived from PC-12 cell lysates prepared in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% w/v Nonidet P-40, 0.5% w/v sodium deoxycholate) plus protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5μg/ml aprotinin, 1μg/ml pepstatin and 1 mM benzamidine) and immunoadsorbed onto 10 μl of packed protein A-sepharose beads, as previously described.61 The reaction in 50 μl of assay buffer (50 mM, pH 7.5, Tris-HCl/2.5 mM MnCl2/2.5 mM MgCl2) containing 10 μCi of [γ-32P]ATP (50 μM) was incubated for 15 min at 37°C before stopping with 200 μl of 1 N HCl and extracting with 160 μl of 1:1 (v/v) CHCl3-CH3OH. The lower chloroform layer containing the 32P-labeled lipid product was collected by centrifuging for 30 sec at 5000 rpm in a microfuge and quickly rinsed twice with 100 μl of 1:1 (v/v) 1N HCl- CH3OH before spotting 50 μl onto a silica-gel glass TLC plate (20 × 20-cm × 0.25 mm layer thickness, Merck) and developing up to the top (4–5 hrs) at room temperature with 65:35 (v/v) n-propanol-2 M acetic acid. Following exposure with X-omat autoradiography film for appropriate length of time the radiolabeled spots were scraped into glass scintillation vials and deacylated by methylaminolysis at 54 °C for 50 min by standard protocol.61 Recovery of the aqueously soluble deacylation products were then analyzed by HPLC on a Whatman 5-μm Partisphere SAX (H2PO4−) column as formerly described61 and the 32P detected with a Radiomatic 525TR online flow scintillation analyzer and FLO-ONE radiochromatography software (Packard Instrument Co., Downers Grove, IL) by Cerenkov emission in the low-energy tritium channel.
Supplementary Material
Experimental procedures and characterization for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Scheme 1.

Metabolic interconversions of PtdIns(3)P.
Scheme 2.

Synthesis of enantiomerically-pure D-myo-inositol intermediate.
Scheme 4.
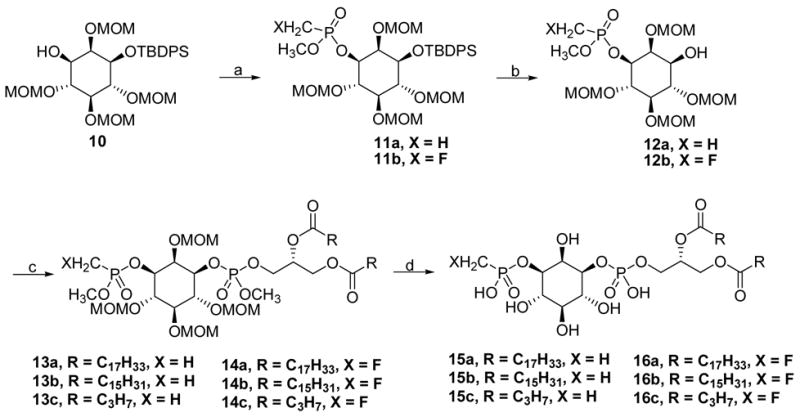
Synthesis of 3-(fluoro)methylphosphonate analogues of PtdIns(3)P. Reagents and conditions: (a) 4 or 9, t-BuOK, CH2Cl2; (b) TBAF-3H2O, HOAc, THF; (c) 19a–19c, 1H-tetrazole, THF/CH3CN; then n-Bu4NIO4, CH3CN; (d) TMSBr, CH2Cl2; then CH3OH/H2O; then EtSH.
Scheme 6.
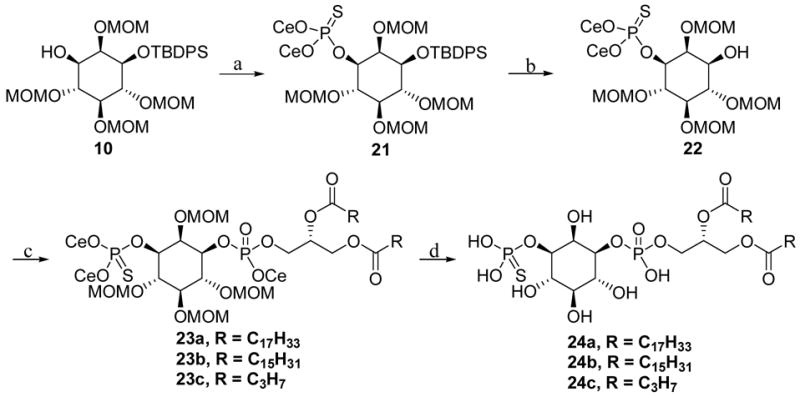
Synthesis of 3-phosphorothioate analogues of PtdIns(3)P. Reagents and conditions: (a) di(2-cyanoethyl) diisopropylphosphorodiamidite, 1H-tetrazole, CH3CN; then S8, CS2/Py;.(b) HF.Py-Py, THF; (c) 20a–20c, 1H-tetrazole, THF/CH3CN; t-BuOOH, CH3CN; (d) TEA, TFBSA, CH3CN; then NH4OAc/H2O; then EtSH, BF3.Et2O.
Scheme 7.

Phosphorylation of PtdIns(3)P analogues by PIKfyve.
Acknowledgments
We thank the NIH (NS29632), the American Cancer Society RSG0513601 (TGK), and the American Diabetes Association and NIH (DK58058) (AS) for financial support of this work.
References
- 1.Feng L, Ferguson CG, Neilsen PO, Chakravarty L, Rzepecki P, Prestwich GD. In: Functional Lipidomics. Feng L, Prestwich GD, editors. Dekker-CRC Press; New York: 2005. pp. 189–210. [Google Scholar]
- 2.Prestwich GD. Chem Biol. 2004;11:619–637. doi: 10.1016/j.chembiol.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 4.Yin HL, Janmey PA. Annual Review of Physiology. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 5.Wymann MP, Bjorklof K, Calvez R, Finan P, Thomas M, Trifilieff A, Barbier M, Altruda F, Hirsch E, Laffargue M. Biochem Soc Trans. 2003;31:275–280. doi: 10.1042/bst0310275. [DOI] [PubMed] [Google Scholar]
- 6.Anderson KE, Jackson SP. International Journal of Biochemistry & Cell Biology. 2003;35:1028–1033. doi: 10.1016/s1357-2725(02)00270-4. [DOI] [PubMed] [Google Scholar]
- 7.Drees BE, Mills GB, Rommel C, Prestwich GD. Expert Opin Therapeut Pat. 2004;14:703–732. [Google Scholar]
- 8.Wetzker R, Rommel C. Curr Pharmaceut Design. 2004;10:1915–1922. doi: 10.2174/1381612043384402. [DOI] [PubMed] [Google Scholar]
- 9.Wymann MP, Zvelebil M, Laffargue M. Trends in Pharmacological Sciences. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- 10.Jackson SP, Yap C, Anderson K. Biochem Soc Trans. 2004;32:387–392. doi: 10.1042/bst0320387. [DOI] [PubMed] [Google Scholar]
- 11.Workman P. Biochem Soc Trans. 2004;32:393–396. doi: 10.1042/bst0320393. [DOI] [PubMed] [Google Scholar]
- 12.Ikonomov OC, Sbrissa D, Fligger J, Foti M, Carpentier JL, Shisheva A. Mol Biol Cell. 14;4581–4591:2003. doi: 10.1091/mbc.E03-04-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kutateladze T, Ogburn K, Watson W, de Beer T, Emr S, Burd C, Overduin M. Molec Cell. 1999;3:805–811. doi: 10.1016/s1097-2765(01)80013-7. [DOI] [PubMed] [Google Scholar]
- 14.Gaullier JM, Simonsen A, D’Arrigo A, Bremnes B, Stenmark H, Aasland R. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- 15.Gillooly DJ, Simonsen A, Stenmark H. J Cell Biol. 2001;155:15–17. doi: 10.1083/jcb.200109001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petiot A, Faure J, Stenmark H, Gruenberg J. J Cell Biol. 2003;162:971–979. doi: 10.1083/jcb.200303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato T, Overduin M, Emr SD. Science. 2001;294:1881–1885. doi: 10.1126/science.1065763. [DOI] [PubMed] [Google Scholar]
- 18.Wishart MJ, Dixon JE. Trends Cell Biol. 2002;12:579–585. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- 19.Laporte J, Bedez F, Bolino A, Mandel JL. Human Molecular Genetics. 2003;12:R285–R292. doi: 10.1093/hmg/ddg273. [DOI] [PubMed] [Google Scholar]
- 20.Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Proc Natl Acad Sci USA. 2005;202:4033–4038. doi: 10.1073/pnas.0409716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y, Qian L, Prestwich GD. J Org Chem. 2003;68:5320–5330. doi: 10.1021/jo020729l. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Qian L, Prestwich GD. Organic Lett. 2003;5:2267–2270. doi: 10.1021/ol034597+. [DOI] [PubMed] [Google Scholar]
- 23.Qian L, Xu Y, Hasegawa Y, Aoki J, Mills GB, Prestwich GD. J Med Chem. 2003;46:5575–5578. doi: 10.1021/jm034207p. [DOI] [PubMed] [Google Scholar]
- 24.Prestwich GD, Xu Y, Qian L, Gajewiak J, Jiang G. Biochem Soc Trans. 2005;33:1357–1361. doi: 10.1042/BST0331357. [DOI] [PubMed] [Google Scholar]
- 25.Schmitt L, Spiess B, Schlewer G. Tetrahedron Lett. 1993;34:7059–7060. [Google Scholar]
- 26.Chen J, Profit AA, Prestwich GD. J Org Chem. 1996;61:6305–6312. doi: 10.1021/jo960895r. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Feng L, Prestwich GD. J Org Chem. 1998;63:6511–6522. doi: 10.1021/jo972046p. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Prestwich GD. J Org Chem. 1998;63:430–431. doi: 10.1021/jo972046p. [DOI] [PubMed] [Google Scholar]
- 29.Peng J, Prestwich GD. Tetrahedron Lett. 1998;39:3965–3968. [Google Scholar]
- 30.Thum O, Chen J, Prestwich GD. Tetrahedron Lett. 1996;37:9017–9020. [Google Scholar]
- 31.Qiao L, Hu Y, Nan F, Powis G, Kozikowski AP. Org Lett. 2000;2:115–117. doi: 10.1021/ol991188y. [DOI] [PubMed] [Google Scholar]
- 32.Reddy KK, Saady M, Falck JR. J Org Chem. 1995;60:3385–3390. [Google Scholar]
- 33.Reddy KK, Ye J, Falck JR, Capdevila JH. Bioorg Med Chem Lett. 1997;7:2115–2116. [Google Scholar]
- 34.Reddy KK, Rizo J, Falck JR. Tetrahedron Lett. 1997;38:4729–4730. [Google Scholar]
- 35.Sculimbrene BR, Xu Y, Miller SJ. J Am Chem Soc. 2004;126:13182–13183. doi: 10.1021/ja0466098. [DOI] [PubMed] [Google Scholar]
- 36.Sculimbrene BR, Miller SJ. J Am Chem Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- 37.Gou DM, Chen CS. J Chem Soc, Chem Commun. 1994;18:2125–2126. [Google Scholar]
- 38.Wang DS, Chen CS. J Org Chem. 1996;61:5905–5910. [Google Scholar]
- 39.Bruzik KS, Tsai MD. J Am Chem Soc. 1992;114:6361–6374. [Google Scholar]
- 40.Kubiak RJ, Bruzik KS. J Org Chem. 2002:960–968. doi: 10.1021/jo0206418. [DOI] [PubMed] [Google Scholar]
- 41.Prestwich GD. Acc Chem Res. 1996;29:503–513. [Google Scholar]
- 42.Denmark SE, Chen CT. J Org Chem. 1994;59:2922–2924. [Google Scholar]
- 43.Malachowski WP, Coward JK. J Org Chem. 1994;59:7625–7634. [Google Scholar]
- 44.Esalthazor M, Flores RA. J Org Chem. 1980;45:529–531. [Google Scholar]
- 45.Lal GS, Pez GP, Syvret RG. Chem Rev. 1996;96:1737–1755. doi: 10.1021/cr941145p. [DOI] [PubMed] [Google Scholar]
- 46.Jeanmarie T, Hervaud Y, Boutevin B. Phosphorous, Sulfur and Silicon. 2002;177:1137–1145. [Google Scholar]
- 47.Hamilton CJ, Roberts SM. J Chem Soc, Perkin Trans. 1;1999:1051–1056. [Google Scholar]
- 48.Deussen HJ, Danielsen S, Breinholt J, Borchert TV. Bioorg Med Chem. 2000;8:507–513. doi: 10.1016/s0968-0896(00)00002-x. [DOI] [PubMed] [Google Scholar]
- 49.Hirschmann R, Yager KM, Taylor CM, Witherington J, Sprengeler PA, Phillips BW, Moore W, Smith ABI. J Am Chem Soc. 1997;119:8177–8190. [Google Scholar]
- 50.Borecka B, Chojnowski J, Cypryk M, Michalski J, Zielinska J. J Organomet Chem. 1979;171:17–34. [Google Scholar]
- 51.Piettre SR, Raboisson P. Tetrahedron Lett. 1996;37:2229–2232. [Google Scholar]
- 52.Heeb NV, Nambiar KP. Tetrahedron Lett. 1993;39:6193–6196. [Google Scholar]
- 53.Kurosu M, Marcin LR, Grinsteiner TJ, Kishi Y. J Am Chem Soc. 1998;120:6627–6628. [Google Scholar]
- 54.Uhlman E, Engels J. Tetrahedron Lett. 1986;27:1023–1026. [Google Scholar]
- 55.Salamonczyk GM. Tetrahedron Lett. 2003;44:7449–7453. [Google Scholar]
- 56.Sekine M, Tsuruoka H, Iimura S, Kusuoku H, Wada T. J Org Chem. 1996;61:4087–4100. doi: 10.1021/jo952073k. [DOI] [PubMed] [Google Scholar]
- 57.Kubiak RJ, Bruzik KS. Bioorg & Med Chem Lett. 1997;7:1231–1234. [Google Scholar]
- 58.Auger KR, Carpenterg CL, Cantley LC, Varticovski L. J Biol Chem. 1989;264:20181–20184. [PubMed] [Google Scholar]
- 59.Kutateladze T, Overduin M. Science. 2001;291:1793–1796. doi: 10.1126/science.291.5509.1793. [DOI] [PubMed] [Google Scholar]
- 60.Tollas KF, Rameh LE, Ishihara H, Shibashi Y, Chen J, Prestwich GD, Cantley LC, Carpenter CL. J Biol Chem. 1998;273:18040–18046. doi: 10.1074/jbc.273.29.18040. [DOI] [PubMed] [Google Scholar]
- 61.Sbrissa D, Ikonomov OC, Shisheva A. J Biol Chem. 1999;274:21589–21597. doi: 10.1074/jbc.274.31.21589. [DOI] [PubMed] [Google Scholar]
- 62.Shisheva A. Cell Biology International. 2001;25:1201–1206. doi: 10.1006/cbir.2001.0803. [DOI] [PubMed] [Google Scholar]
- 63.Sbrissa D, Ikonomov OC, Shisheva A. J Biol Chem. 2002;277:6073–6079. doi: 10.1074/jbc.M110194200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org